

Abstract
Glutathione (GSH)/GSH reductase (GSR) and thioredoxin/thioredoxin reductase (TXNRD) are two major compensating thiol-dependent antioxidant pathways that maintain protein dithiol/disulfide balance. We hypothesized that functional deficiency in one of these systems would render cells dependent on compensation by the other system for survival, providing a mechanism-based synthetic lethality approach for treatment of cancers. The human GSR gene is located on chromosome 8p12, a region frequently lost in human cancers. GSR deletion was detected in about 6% of lung adenocarcinomas in The Cancer Genome Atlas database. To test whether loss of GSR sensitizes cancer cells to TXNRD inhibition, we knocked out or knocked down the GSR gene in human lung cancer cells and evaluated their response to the TXNRD inhibitor auranofin. GSR deficiency sensitized lung cancer cells to this agent. Analysis of a panel of 129 NSCLC cell lines revealed that auranofin sensitivity correlated with the expression levels of the GSR, glutamate-cysteine ligase catalytic subunit (GCLC), and NAD(P)H quinone dehydrogenase 1 (NQO1) genes. In NSCLC patient-derived xenografts with reduced expression of GSR and/or GCLC, growth was significantly suppressed by treatment with auranofin. Together these results provide a proof of concept that cancers with compromised expression of enzymes required for GSH homeostasis or with chromosome 8p deletions that include the GSR gene may be targeted by a synthetic lethality strategy with inhibitors of TXNRD.
Keywords: Chromosome 8p deletions, glutathione reductase, thioredoxin reductase, synthetic lethality, lung cancer, gold compounds
Introduction
Glutathione (GSH)/GSH reductase (GSR) and thioredoxin (TXN)/thioredoxin reductase (TXNRD) are two parallel, compensating thiol-dependent antioxidant pathways that regulate and maintain cellular thiol redox homeostasis and protein dithiol/disulfide balance (1,2). Both GSH and TXN catalyze the reduction of disulfide bonds in proteins to dithiols, which is critical for maintaining protein functions (Fig. 1A). Oxidized GSH (GSSG) and TXN generated during this catalytic process are converted to their reduced states by GSR and TXNRD, respectively, both using NADPH generated in the pentose phosphate cycle as the hydrogen donor. Functional deficiency in one of these systems will render cells dependent on compensation by the other system for survival. In GSR-deficient yeasts (3) and Drosophila (4), the TXN/TXNRD pathway is required for the reduction of GSSG to GSH. Similarly, GSH acts as a backup for human TXNRD1 to reduce TXN, thereby preventing cell death induced by gold compound–mediated TXNRD1 inhibition (5). Combined inhibition of the GSH and TXN pathways has led to synergistic cancer cell death (6,7). Because synthetic lethality interactions occur significantly more frequently among genes involved in similar biological processes, such as those in parallel or compensating pathways (8), we hypothesized that loss and/or reduced expression of enzymes required for GSH homeostasis, including GSR and glutamate-cysteine ligase (the first rate-limiting enzyme of GSH synthesis), in a subgroup of cancers predisposes them to vulnerability to treatment with TXNRD inhibitors, providing a mechanism-based synthetic lethality therapy for these cancers.
Figure 1.

GSR gene deletions in human cancers from TCGA database. (A) GSH/GSR and TXN/TXNRD pathways in regulating protein dithiol/disulfide balance. Both GSH and TXN catalyze the reduction of disulfide bonds in proteins to dithiols, which is critical for maintaining protein functions. (B) Alterations in GSR and other selected genes in lung adenocarcinomas. The mutation frequencies are shown on the left side of the graph. Deletions of the GRS gene overlap mostly with the deletion of TUSC3, a gene located in chromosome 8p22, indicating co-occurrence of GSR deletion and chromosome 8p loss. (C) Frequencies of GSR gene deletions in different types of cancers. The data were obtained by using the cBioPortal for Cancer Genomics.
The human GSR gene is located on chromosome 8p12, a region frequently lost in lung cancers (9,10) and other types of cancers, including breast (11), head and neck (12), prostate (13), bladder (14), and gastric/colorectal (10) cancers and lymphoma (15). Accumulating evidence has shown that loss of chromosome 8p increases tumor invasiveness and metastasis potential (16–18) and confers resistance to chemotherapy and poor survival in cancer patients (13,18). Targeted deletion of chromosome 8p yielded altered lipid metabolism and increased autophagy, which trigger invasiveness and confer tumor growth under stress conditions (18). Unfortunately, therapeutic strategies for cancers with chromosome 8p loss have yet to be developed. Loss of the GSR gene may offer a unique opportunity for treatment of cancer in a subgroup of patients whose cancer has chromosome 8p aberrations.
To explore the concept of targeting cancers in which expression of enzymes required for GSH homeostasis is reduced or lost, we searched The Cancer Genome Atlas (TCGA) database for genetic alterations and found that deep GSR deletion was detected in 6% of lung adenocarcinomas, 3.5% of squamous cell lung cancers, and 4-9% of prostate, breast, liver, ovarian, and colon cancers, suggesting that loss of the GSR gene occurs in a subset of human cancers. We then tested whether loss of GSR in cancer cells will result in their vulnerability to auranofin, a TXNRD inhibitor (19). Our results show that reduced expression of enzymes required for GSH homeostasis sensitized cancer cells to TXNRD inhibitor auranofin,
Materials and Methods
Cell lines and cell culture
Human non–small cell lung cancer (NSCLC) cell lines, including A549, H2023, H1993 and DFC1024, were obtained from Dr. John Minna’s laboratory (University of Texas Southwestern Medical Center) and propagated in monolayer culture in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. Cell lines were tested for mycoplasma with LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich, St. Louis, MO) and authenticated per routine at our institution’s Characterized Cell Line Core using a short tandem repeat DNA fingerprint assay with a PowerPlex 16 HS kit (Promega, Madison, WI).
Gene knockout and knockdown in lung cancer cells
A549–GSR-knockout cells were generated by using the CRISPR/Cas9 genome editing system. Briefly, GSR-specific guide RNA (gRNA) expression vectors were generated as described (20). LentiCas9-Blast (plasmid 52962) and gRNA cloning vector lentiGuide-Puro (plasmid 52963) were obtained from Addgene (Cambridge, MA). The CRISPR guide sequences targeting GSR were GATCATGCATGAATTCAGAG (gRNA1) and GTGAGGGTAAATTCAATTGG (gRNA2). A549 cells were co-transfected with lentiCas9-Blast and gRNA expression vectors. The transfected cells were selected using puromycin (2 μg/mL) for 48 h. Isolated single colonies were analyzed by Western blot. The colonies without GSR expression were subjected to detection of genomic mutations by DNA sequencing.
For small-interfering RNA (siRNA)-mediated gene knockdown, scramble control siRNA or siRNA targeting the GSR or the glutamate-cysteine ligase catalytic subunit (GCLC) gene (all, Sigma-Aldrich, St Louis, MO) was transfected into cells with oligofectamine (Sigma-Aldrich) according to the manufacturer’s instructions. Plasmids for lentiviral vectors expressing cDNA or short-hairpin RNA (shRNA) used in this study were obtained from Open Biosystems through our institution’s shRNA and ORFeome Core facility. Lentiviral vectors were packaged in 293 cells after co-transfection with lentiviral packaging plasmids by using Fugene 6 as directed by the manufacturer (Promega). The medium from the transfected 293 cells was filtered through a sterile 0.22-μm filter and used to infect target cells in the presence of 8 μg/mL polybrene. After selection with puromycin (1-2 μg/mL) or blasticidin (2-10 μg/mL), the cells were pooled together for subsequent studies. The proportion of stably transfected cells (expressing GFP or RFP in the lentiviral backbone) was usually >85% after selection.
Cell viability assay
The cell viability was determined using the sulforhodamine B assay as previously described (21) or using a CellTiter-Glo assay kit (Promega) according to the manufacturer’s instructions. Cell viability following siRNA transfection was assayed as follows: cells were seeded in 96-well plates at 2000 cells/per well for 24 h. siRNA transfection was performed with 50 μL/well siRNA transfection mix in OptiMEM containing 0.2 μL of oligofectamine and 200 nM of siRNA; after 4 h incubation at 37°C, another 50 μL of regular culture medium containing 20% FBS was added to each well. Auranofin was added 12 h after siRNA transfection, and cell viability was assayed 72 h later. The percentage of viable cells was determined relative to the viability of the controls, which was set at 100%. Each experiment was performed in triplicate and repeated three times. Number of apoptotic cells (sub-G1) were measured using fluorescence-activated cell sorting after annexin V/propidium iodide staining.
Western blot analysis
Antibodies for detecting human GSR, GCLC, glutamate-cysteine ligase modifier subunit (GCLM), TNXRD1, NAD(P)H quinone dehydrogenase 1 (NQO1), and beta-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) or Abcam (Cambridge, MA). Cells were harvested after stable selection or at 48 h after siRNA transfection. In brief, cells were washed in phosphate-buffered saline solution (PBS), collected, and then subjected to lysis in RIPA buffer containing proteinase inhibitor cocktail and phosphatase inhibitor cocktail (Roche, Indianapolis, IN). The lysates were subjected to centrifugation at 10,000g at 4°C for 10 min and the supernatants removed. The total protein (30 μg) was fractioned by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and electrophoretically transferred to Immobilon-FL PVDF membranes. The membrane was blocked with blocking buffer (Li-Cor, Lincoln, NE) at room temperature for 1 h, then incubated with goat anti-human P53 IgG (1:1000) for 2 h or with the primary antibody at 4°C overnight. After washing with PBS containing 0.05% Tween 2 (PBST), the membrane was incubated with IRDye infrared secondary antibody (Li-Cor) for 1 h at room temperature. The membrane was washed with PBST again, and protein expression was detected with the Odyssey Infrared Imaging System (Lincoln, NE).
Animal experiments
Animals were maintained and experiments carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health Publication 85-23) and the institutional guidelines of MD Anderson Cancer Center. Patient-derived xenografts (PDXs) were established from clinical surgically resected specimens or from pleural fluid as we previously reported (22), except that non-obese diabetic/severe combined immunodeficiency (NOD-SCID) mice with null mutations of the gene encoding the interleukin-2 receptor γ (NSG) obtained from Jackson Laboratory (Bar Harbor, Maine) were used instead of regular NOD-SCID for generation and passaging of PDXs. This study is approved by Institutional Animal Care and Use Committee and Institutional Review Board at The University of Texas MD Anderson Cancer Center. All clinical samples were collected with informed consent from the patients. Each PDX was subcutaneously inoculated into the dorsal flank of nude mice. When the tumors had grown to 3-5 mm in diameter, the mice were assigned randomly into two groups (n=5/group) and treated with intraperitoneal administration of auranofin (10 mg/kg) in a solvent comprising 2% dimethylsulfoxide (DMSO), 10% ethanol, and 5% polyethylene glycol 400 or the solvent without auranofin as controls. Treatment was administered daily for the first week, then twice a week for the duration of the experiment. Tumor growth was monitored every 2 to 3 days and was normalized to tumor volume at the beginning of treatment, which was set to 1. Subcutaneous tumors were measured with calipers, and tumor volume was calculated according to the formula V = ab2/2, where a is the largest diameter and b is the smallest. The experiment was ended and the mice euthanized when the largest tumors reached 15 mm in diameter. This in vivo experiment was performed three times.
Statistical analysis
The half-maximal effective dose (IC50) values were calculated with the best-fit dose-response model selected by the residual standard error using the drexplorer package (23). The mRNA levels determined by microarray assay were available for 76 of 129 NSCLC cell lines tested for auranofin sensitivity. Association between gene expression and IC50 value was assessed using the Spearman rank correlation. All statistical analyses were performed using the R software. P values were obtained using computed t-statistics. The significance of the in vivo animal study data was determined by using the Mann-Whitney U test for two treatment groups (two tailed). A P <0.05 was considered statistically significant.
Results
Deep deletion of GSH homeostasis genes in human cancers
To test the concept of targeting cancers with compromised GSH homeostasis, we searched the TCGA database for alterations of the GSR, GCLC, and GCLM genes using the cBioPortal website (http://cBioPortal.org). We found that deep GSR deletion was detected in 6% of lung adenocarcinomas, 3.5% of squamous cell lung cancers, and 4-9% of prostate, breast, liver, ovarian, and colon cancers (Fig. 1B, C). In both lung adenocarcinoma and squamous cell cancer, most deletions of GSR are concurrent with deletions of TUSC3, a gene located in chromosome 8p22, indicating coexistence of GSR deletion and chromosome 8p loss. Deletions of the GCLC and GCLM genes, which encode the two protein subunits of glutamate-cysteine ligase, were detected occasionally in lung adenocarcinoma or squamous cell cancer (about 1%). Deletion of GCLM was relatively more frequent (about 2-3%) in prostate and pancreatic cancers. This result indicates that there is a subset of human cancers that are likely compromised in GSH homeostasis. Deletion of the GSR gene in a lung adenocarcinoma did not overlap significantly with epidermal growth factor receptor (EGFR) mutation or amplification but did overlap TP53 and KRAS gene mutations, suggesting that available targeted agents for cancer are not effective in cancers with GSR, GCLC, or GCLM mutation.
Loss of GSR sensitizes cancer cells to TXNRD inhibitor auranofin
To test whether loss of enzymes required for GSH homeostasis sensitizes cancer cells to TXNRD inhibition, we knocked out the GSR gene in auranofin-resistant A549 lung cancer cells using CRISPR/Cas9 technology with two GSR-specific gRNAs (Fig. 2A). We identified five A549 clones with loss of GSR expression by Western blot analysis and verified genomic deletions in both alleles of the GSR gene using PCR-mediated genomic sequencing of the target sequence in one of the clones (Fig. 2B). A dose-dependent cell viability assay on two of the five clones demonstrated that knockout of the GSR gene led to a dramatic increase in the sensitivity of A549 cells to auranofin (Fig. 2C). According to annexin V/propidium iodide staining, the rate of auranofin-induced apoptosis was markedly higher in A549/GSR−/− cells (16.4%) than in A549 cells wild-type for GSR (3.8%) (Fig. 2D–F). A similar dose-dependent cytotoxic effect was observed for another TXNRD inhibitor aurothioglucose (5) in A549/ GSR−/− cells but not in the parental A549 cells (Supplement Figure 1).
Figure 2.
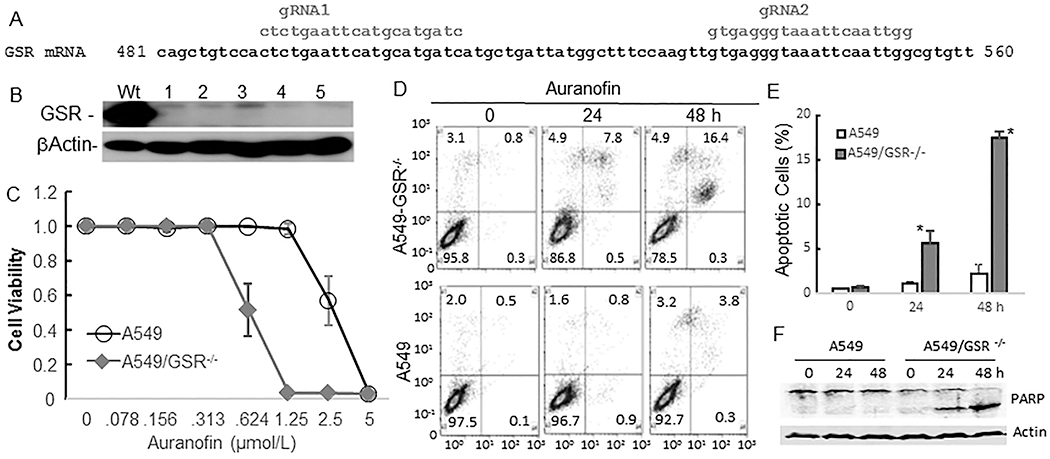
Effect of GSR knockout on auranofin activity in lung cancer cells. (A) Schema of CRISPR/Cas9-mediated knockout of the GSR gene. (B) Western blot verification of GSR-knockout clones 1-5. Wt, controls. (C) Dose responses of A549 and A549/GSR−/− cells to auranofin treatment. (D-E) Apoptosis in response to treatment with auranofin in A549 and A549/GSR−/− cells. * indicates P< 0.05 between two cell lines. (F) PARP cleavage detected by Western blot analysis. β-actin is used as loading control.
We observed similar results when we knocked down the GSR and/or GCLC gene in H2023 and DFC1024 NSCLC cells via transfection of either siRNA or lentivirus-mediated shRNA. Knockdown of either GSR or GCLC sensitized these cells to auranofin (Fig. 3A–H). In contrast, enforced expression of the GSR gene in H1993 cells increased viable cells after treatment with auranofin at the doses of 0.16 – 0.62 μmol/L (Supplement Figure 2). These results provided proof-of concept evidence that cancer cells with loss of or reduced expression of enzymes required for GSH homeostasis can be selectively eliminated by treatment with a TXNRD antagonist.
Figure 3.
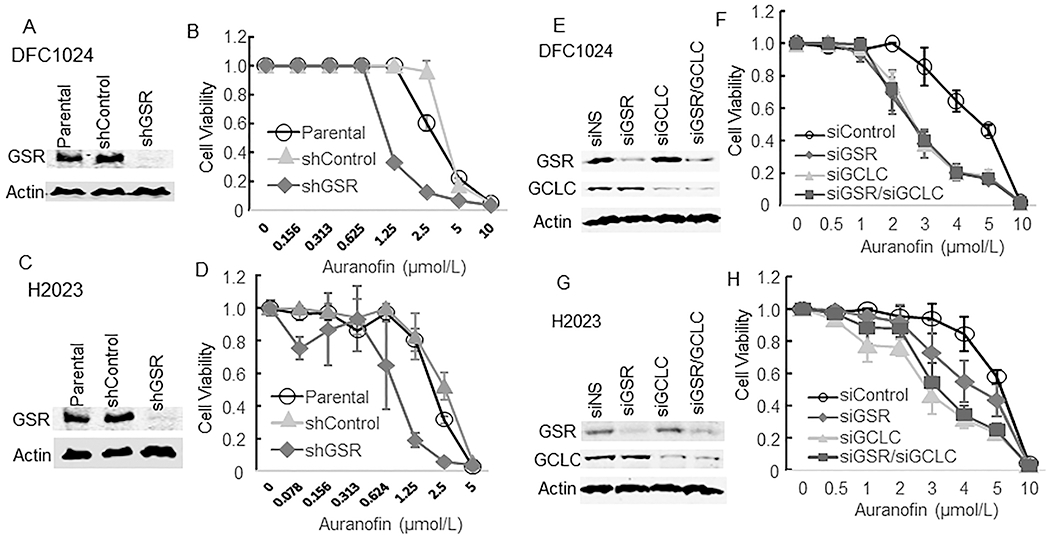
Auranofin sensitivity in lung cancer cells after knockdown of GSR and/or GCLC genes with shRNA (A-D) or siRNA (E-H). Western blot (A, C, E, G) analysis confirmed knockdown of GSR and/or GCLC in DFC1024 and H2023 lung cancer cell lines. (B, D, F, H) Auranofin sensitivity of the knockdown cells and of the parental and control cells (shControl and siControl) was determined by a quadruplicate assay. The values represent mean ± SD.
Levels of enzymes required for GSH homeostasis are associated with sensitivity to auranofin in NSCLC cell lines
Our previous study showed that auranofin’s anticancer activity was inversely correlated with TXNRD1 expression and enzymatic activity in 10 NSCLC cell lines (24). To further investigate potential predictive biomarkers of NSCLC sensitivity to auranofin, dose responses to auranofin were examined in 129 NSCLC cell lines. The cells were treated with different concentrations of auranofin ranging from 78 nM to 5 μM. Dose-dependent cell viability was determined using a CellTiter-Glo assay kit. These cells had varying sensitivity to auranofin. Twenty-eight of the 129 cell lines had an IC50 less than 1 μM (Fig. 4A) (Supplemental Table 1). Illumina mRNA expression profiling data on 11,544 unique genes available for 76 of these 129 NSCLC cell lines were analyzed by the Spearman rank correlation test to determine the correlation between auranofin response and mRNA levels in these cell lines. The result showed that the levels of GSR (r = 0.43, P = 7×10−5), GCLC (r =0.44, P = 3.7×10−5), TXNRD1 (r = 0.44, P = 4×10−5), NQO1 (r = 0.52, P = 8.5×10−7), and glucose-6-phosphate dehydrogenase (G6PD; r = 0.44, P = 4.1×10−5) gene expression were all significantly correlated with auranofin activity in these cell lines (Supplemental Table 2). In addition, Western blot analysis of the expression of GSR, GCLC, GCLM, NQO1, and TXNRD1 in 22 of these NSCLC cell lines (12 auranofin-sensitive and 10 auranofin-resistant) demonstrated dramatic differences in the expression of GSR, GCLC, and NQO1 proteins in these 22 cell lines (Fig. 4B–C). The auranofin-sensitive cancer cell lines expressed low levels of these three genes, whereas the auranofin-resistant cell lines expressed high levels of these genes, suggesting that the levels of these three genes can be used as a signature to distinguish auranofin-sensitive and -resistant NSCLCs.
Figure 4.
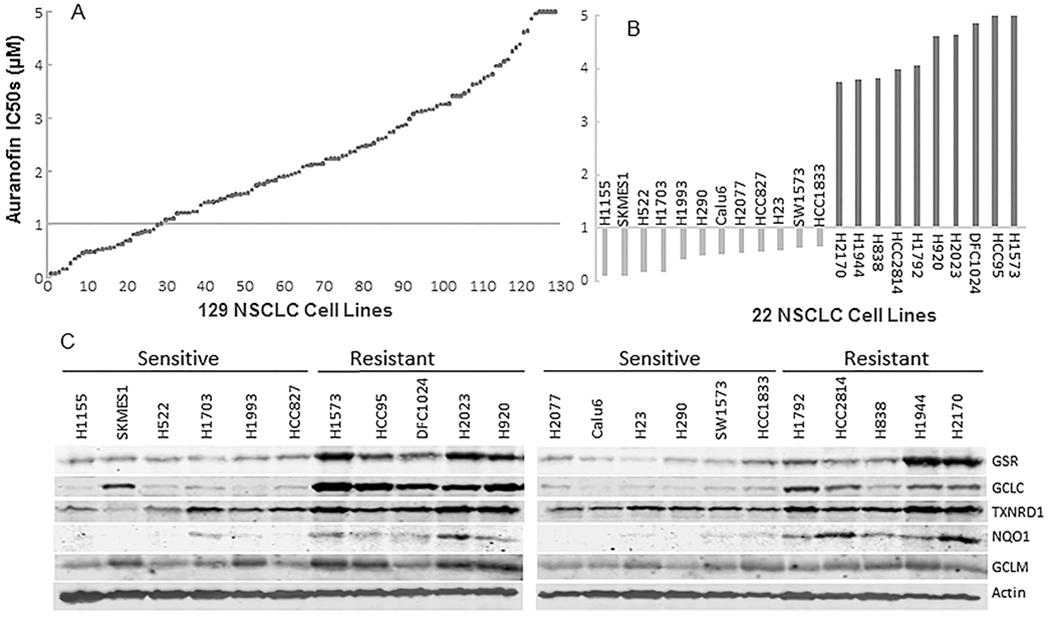
Auranofin sensitivity and GSR and GCLC gene expression in NSCLC cell lines. (A) The IC50 of auranofin in 129 NSCLC cell lines. (B) The IC50 of auranofin in 12 auranofin-sensitive NSCLC cell lines and 10 auranofin-resistant NSCLC cell lines. (C) Western blot analysis of GSR, GCLC, GCLM, NQO1, and TXNRD1 expression in auranofin-sensitive and -resistant NSCLC cells. Note: the expression of GSR, GCLC, and NQO1 was highly associated with auranofin sensitivity.
Notably, expression of the GSR and GCLC genes are concurrently low in the auranofin-sensitive NSCLC cell lines. We searched the TCGA database to see if the expression of GSR and GCLC genes is correlated in human primary lung cancers. We found that the GSR gene was co-expressed with the SRXN1, TXNRD1, GCLM, G6PD, NQO1, and GCLC genes in both lung adenocarcinomas and squamous cell lung cancers, with Pearson r values ranging from 0.76 to 0.50 (Supplemental Table 3), indicating that expression of these genes occurs concurrently in primary lung cancers.
Auranofin suppresses growth of lung cancer PDX models with reduced expression of GSR and/or GCLC in vivo
Some recent studies demonstrated that PDXs have response rates similar to those of primary tumors for several anticancer drugs (25,26). We established a panel of PDXs from surgical or pleural fluid NSCLC specimens. Our previous study, which carried out targeted sequencing of 200 cancer-related genes in 23 NSCLC PDXs, revealed that 93% of the mutations detected in primary tumors also occurred in their corresponding PDXs, indicating that PDXs can recapitulate the mutations in primary tumors (22).
We examined GSR and GCLC expression in 19 NSCLC PDXs (F1-F3 generations) using Western blot analysis. The results demonstrated that six of the PDXs had low GSR and GCLC expression (Fig. 5A). In two of these PDXs, Western blot analysis revealed that the levels of GSR and GCLC expression were consistent in the F1 and F3 generations. We then performed an experiment to determine the in vivo activity of auranofin in two PDXs (PDX-2 and PDX-12) with relative low levels of GSR and GCLC expression, and in one PDX model with relatively high GSR and GCLC expression (PDX-1). The results demonstrated that treatment with auranofin resulted in significant suppression of the growth of both GSR/GCLC low PDX models in vivo (P<0.05 at 2 weeks after the treatment began), but not in the GSR/GCLC high model (Fig. 5B–D). Treatment with auranofin led to 65% and 68% inhibition of PDX-2 and PDX-12 tumor growth, respectively, compared with that in control mice. Weight loss was not detectable in PDX-12 and PDX-1 mice and not significant in PDX-2 mice. Together, these findings indicate that treatment of NSCLC PDX-bearing mice with auranofin was effective in GSR/GCLC low tumors and well tolerated.
Figure 5.
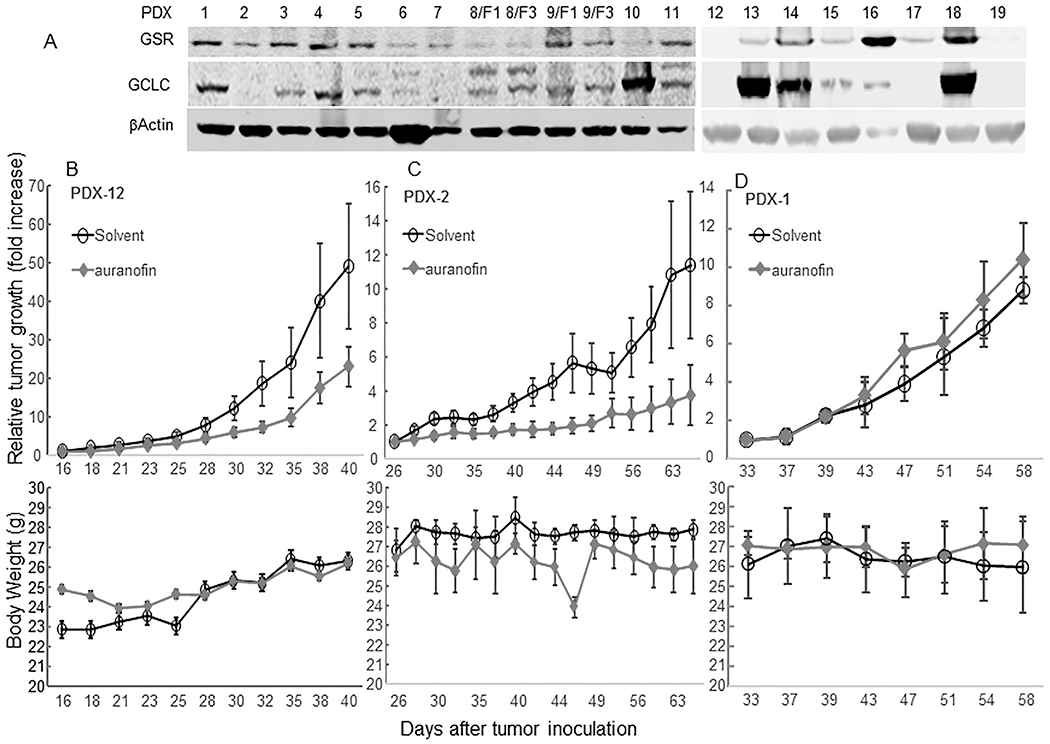
In vivo activity of auranofin in NSCLC PDX models. (A) Western blot analysis of GSR and GCLC expression in 19 NSCLC PDX specimens. Mice bearing a PDX-12 (B), PDX-2 (C), and PDX-1 (D) were given auranofin (10 mg/kg) or a solvent (control) for the time periods shown. Upper panels show tumor growth after treatment started. The values are presented as the means ± SD (n = 3/group for PDX-1, and n=5/group for PDX-2 and -12). The mean tumor volume in the auranofin group was not significantly different from that in solvent group in PDX-1 model, but was significantly lower than that in the solvent group in both PDX-2 and -12 models (P <0.01) according to analysis of variance with a repeated measurement module. Lower panels show that auranofin caused no significant weight loss in the mice.
Discussion
Our results support the hypothesis that cancers with loss and/or reduced expression of enzymes required for GSH homeostasis, including GSR and GCLC, can be targeted using a synthetic lethality strategy with TXNRD inhibitors. These results were in consistence with reports by others that combined inhibition of GSH and TXN antioxidant pathways leads to a synergistic cancer cell death in vitro and in vivo (6,27). Synthetic lethality therapy has been exploited for treatment of cancers with mutations in the KRAS (28,29), BRCA (30,31), and TP53 (32) genes. The success of using poly(ADP-ribose) polymerase (PARP) inhibitors in treatment of BRCA-mutant cancers (33,34) has demonstrated the clinical feasibility of targeting cancers via synthetic lethality. Because the GSR gene is located in chromosome 8p, a region that is frequently deleted in several types of human cancers, this strategy may applied to cancers whose chromosome 8p deletion involves the GSR gene.
Chromosome 8p deletions detected by analysis for loss of heterozygosity (LOH) have been frequently reported in cancers of lung, colon, breast, prostate, or liver. Chromosome fragile sites located at 8p22 (FRA8B) and at 8p12 (within the NRG1 gene) have been proposed to account for the frequent 8p aberrations observed in cancers (35). The GSR gene is located between these sites, suggesting that its loss might be common in cancers with chromosome 8p deletions. Efforts have been made to identify possible tumor suppressor genes within the 8p region. A study that used shRNA-mediated knockdown of the mouse orthologs of the 21 genes frequently deleted on human chromosome 8p as detected in human cancers found that multiple genes on chromosome 8p can cooperatively inhibit tumorigenesis in a murine hepatocellular carcinoma model, and that their co-suppression can synergistically promote tumor growth (36). A recent study of 8p LOH established in human mammary epithelial cell line MCF10A by using TALEN-based genomic engineering revealed that 8p LOH triggers invasiveness and resistance to chemotherapeutic drugs and confers tumor growth under stress conditions due to increased autophagy (18), providing molecular insights into the poorer clinical outcomes of cancer patients with a chromosome 8p deletion.
The genetic interaction between GSH/GSR and TXN/TXNRD pathways may provide a therapeutic opportunity for cancers with compromised GSH homeostasis or with a chromosome 8p deletion that includes the GSR gene. Intriguingly, gene expression data from our lung cancer cell lines and from TCGA database indicate that the expression of the GSR, GCLC and TXNRD1 genes is highly correlated in both lung adenocarcinomas and squamous cell lung cancers. The underlying mechanism is not yet clear. Nevertheless, the TCGA data showed that overexpression of the GSR, GCLC and TXNRD1 were highly enriched in KEAP1 mutant lung adenocarcinoma (P =1.2 x10−8, 1.0 x 10−6 and 1.4 x 10−9, respectively) and in NFE2L2 mutant lung squamous cell cancer (P =1.5 x 10−3, 4.7 x 10−4 and 6.1 x10−9, respectively), supporting the roles of the KEAP1/NRF2 pathway in regulation of the expression of these oxidative stress-associated genes (37). It is possible that, unlike mutations in KEAP1/NRF2 pathways, low expression of GSR alone is not sufficient to trigger TXNRD1 overexpression.
In conclusion, we report that cancer cells with reduced or lost expression of the GSR gene have increased sensitivity to the TXNRD1 inhibitor auranofin, a gold complex that has been used to treat rheumatoid arthritis since the 1980s (38). The mechanism of auranofin-mediated immunomodulation in patients with rheumatoid arthritis is not entirely clear, although the primary molecular target of auranofin and other gold-containing drugs is documented to be TXNRD (19), the only enzyme that catalyzes the reduction of TXN with electrons from NADPH (39). The median inhibitory concentration (IC50) of auranofin for inhibiting TXNRD is about 9 nM, but it is 15 μM for the closely related enzyme GSR (40). Long-term use of auranofin is well tolerated in both juvenile and elderly patients (41,42). The discovery of auranofin’s activity against Entamoeba histolytica infection also led to quick regulatory approval of auranofin in the U.S. for the treatment of amebiasis (43). Our previous study demonstrated that auranofin had single-agent activity in vitro and in vivo in a subset of lung cancer cells via inhibition of multiple key nodes in the PI3K/AKT/mTOR axis (24). It is not yet clear whether the similar mechanism occurs in vivo. Studies by others have shown that auranofin effectively induces apoptosis in chemoresistant cancer cells (44,45). Auranofin and sirolimus combination therapy is currently under a phase II trial for treatment of ovarian cancer (trial registration NCT03456700). The correlations between auranofin sensitivity and expression levels of the GSR, GCLC, and NQO1 genes suggest that these three genes might be used as a predictive signature for identifying the patient subgroups whose tumors may respond to the treatment with a TXNRD inhibitor.
Supplementary Material
Significance.
Findings demonstrate that lung cancers with compromised expression of enzymes required for glutathione homeostasis, including reduced GSR gene expression, may be targeted by thioredoxin/thioredoxin reductase (TXNRD) inhibitors.
Acknowledgment
We would like to thank Uma Giri for technical assistance Kathryn Hale and the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editorial review of the manuscript.
Funding
This work was supported in part by the National Institutes of Health through grants R01CA190628, Specialized Program of Research Excellence (SPORE) grant CA070907, The University of Texas PDX Development and Trial Center grant U54CA224065, and The University of Texas MD Anderson Cancer Center support grant P30CA016672 (used the Flow Cytometry and Cellular Imaging Facility, Gene Editing/Cellular Model Core Facility, shRNA and ORFeome facility, and Research Animal Support Facility); and by endowed funds to The University of Texas MD Anderson Cancer Center, including the Lung Cancer Moon Shot Program and the Sister Institution Network Fund.
Footnotes
Conflicts of Interest
The authors declare that they have no competing interests.
Ethics Approval and Consent to Participate
Not applicable
References
- 1.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med 2014;66:75–87 [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Hyde AS, Simpson MA, Barycki JJ. Emerging regulatory paradigms in glutathione metabolism. Adv Cancer Res 2014;122:69–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan SX, Greetham D, Raeth S, Grant CM, Dawes IW, Perrone GG. The thioredoxin-thioredoxin reductase system can function in vivo as an alternative system to reduce oxidized glutathione in Saccharomyces cerevisiae. J Biol Chem 2010;285:6118–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, et al. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 2001;291:643–6 [DOI] [PubMed] [Google Scholar]
- 5.Du Y, Zhang H, Lu J, Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem 2012;287:38210–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression.[Erratum appears in Cancer Cell. 2015 Feb 9.27(2):314] Cancer Cell 2015;27:211–22 [DOI] [PubMed] [Google Scholar]
- 7.Mandal PK, Schneider M, Kolle P, Kuhlencordt P, Forster H, Beck H, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res 2010;70:9505–14 [DOI] [PubMed] [Google Scholar]
- 8.Fang B Development of synthetic lethality anticancer therapeutics. J Med Chem 2014;57:7859–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wistuba II, Behrens C, Virmani AK, Milchgrub S, Syed S, Lam S, et al. Allelic losses at chromosome 8p21-23 are early and frequent events in the pathogenesis of lung cancer. Cancer Res 1999;59:1973–9 [PubMed] [Google Scholar]
- 10.Emi M, Fujiwara Y, Nakajima T, Tsuchiya E, Tsuda H, Hirohashi S, et al. Frequent loss of heterozygosity for loci on chromosome 8p in hepatocellular carcinoma, colorectal cancer, and lung cancer. Cancer Res 1992;52:5368–72 [PubMed] [Google Scholar]
- 11.Armes JE, Hammet F, de SM, Ciciulla J, Ramus SJ, Soo WK, et al. Candidate tumor-suppressor genes on chromosome arm 8p in early-onset and high-grade breast cancers. Oncogene 2004;23:5697–702 [DOI] [PubMed] [Google Scholar]
- 12.Li X, Lee NK, Ye YW, Waber PG, Schweitzer C, Cheng QC, et al. Allelic loss at chromosomes 3p, 8p, 13q, and 17p associated with poor prognosis in head and neck cancer. J Natl Cancer Inst 1994;86:1524–9 [DOI] [PubMed] [Google Scholar]
- 13.El Gammal AT, Bruchmann M, Zustin J, Isbarn H, Hellwinkel OJ, Kollermann J, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res 2010;16:56–64 [DOI] [PubMed] [Google Scholar]
- 14.Adams J, Williams SV, Aveyard JS, Knowles MA. Loss of heterozygosity analysis and DNA copy number measurement on 8p in bladder cancer reveals two mechanisms of allelic loss. Cancer Res 2005;65:66–75 [PubMed] [Google Scholar]
- 15.Martinez-Climent JA, Vizcarra E, Sanchez D, Blesa D, Marugan I, Benet I, et al. Loss of a novel tumor suppressor gene locus at chromosome 8p is associated with leukemic mantle cell lymphoma. Blood 2001;98:3479–82 [DOI] [PubMed] [Google Scholar]
- 16.Diep CB, Kleivi K, Ribeiro FR, Teixeira MR, Lindgjaerde OC, Lothe RA. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosomes Cancer 2006;45:31–41 [DOI] [PubMed] [Google Scholar]
- 17.Macartney-Coxson DP, Hood KA, Shi HJ, Ward T, Wiles A, O’Connor R, et al. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer 2008;8:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Y, Crowther J, Pastor T, Abbasi AL, Baietti MF, De TM, et al. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell 2016;29:751–66 [DOI] [PubMed] [Google Scholar]
- 19.Schuh E, Pfluger C, Citta A, Folda A, Rigobello MP, Bindoli A, et al. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. Journal of Medicinal Chemistry 2012;55:5518–28 [DOI] [PubMed] [Google Scholar]
- 20.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu H, Wang L, Gao W, Meng J, Dai B, Wu S, et al. IGFBP2/FAK pathway is causally associated with dasatinib resistance in non-small cell lung cancer cells. Mol Cancer Ther 2013;12:2864–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao C, Wang L, Peng S, Cao M, Li H, Hu J, et al. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett 2015;357:179–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong P, Coombes KR, Johnson FM, Byers LA, Diao L, Liu DD, et al. drexplorer: A tool to explore dose-response relationships and drug-drug interactions. Bioinformatics 2015;31:1692–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Hu J, Wu S, Wang L, Cao X, Zhang X, et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016;7:3548–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318–25 [DOI] [PubMed] [Google Scholar]
- 26.Owonikoko TK, Zhang G, Kim HS, Stinson RM, Bechara R, Zhang C, et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med 2016;14:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz DR, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE 2012;7:e48175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res 2008;68:7403–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Scie USA 2011;108:8773–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21 [DOI] [PubMed] [Google Scholar]
- 31.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase.[Erratum appears in Nature. 2007 May 17;447(7142):346] Nature 2005;434:913–7 [DOI] [PubMed] [Google Scholar]
- 32.Baldwin A, Grueneberg DA, Hellner K, Sawyer J, Grace M, Li W, et al. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc Natl Acad Sci USA 2010;107:12463–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 2014;15:852–61 [DOI] [PubMed] [Google Scholar]
- 34.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res 2015;21:4257–61 [DOI] [PubMed] [Google Scholar]
- 35.Birnbaum D, Adelaide J, Popovici C, Charafe-Jauffret E, Mozziconacci MJ, Chaffanet M. Chromosome arm 8p and cancer: a fragile hypothesis. Lancet Oncol 2003;4:639–42 [DOI] [PubMed] [Google Scholar]
- 36.Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci USA 2012;109:8212–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Namani A, Matiur Rahaman M, Chen M, Tang X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018;18:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernhard GC. Auranofin therapy in rheumatoid arthritis. J Lab Clin Med 1982;100:167–77 [PubMed] [Google Scholar]
- 39.Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta 2009;1790:495–526 [DOI] [PubMed] [Google Scholar]
- 40.Rubbiani R, Kitanovic I, Alborzinia H, Can S, Kitanovic A, Onambele LA, et al. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J Med Chem 2010;53:8608–18 [DOI] [PubMed] [Google Scholar]
- 41.Marcolongo R, Mathieu A, Pala R, Giordano N, Fioravanti A, Panzarasa R. The efficacy and safety of auranofin in the treatment of juvenile rheumatoid arthritis. A long-term open study. Arthritis Rheum 1988;31:979–83 [DOI] [PubMed] [Google Scholar]
- 42.Glennas A, Kvien TK, Andrup O, Clarke-Jenssen O, Karstensen B, Brodin U. Auranofin is safe and superior to placebo in elderly-onset rheumatoid arthritis. Br J Rheumatol 1997;36:870–7 [DOI] [PubMed] [Google Scholar]
- 43.Debnath A, Parsonage D, Andrade RM, He C, Cobo ER, Hirata K, et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat Med 2012;18:956–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Medic 2007;42:872–81 [DOI] [PubMed] [Google Scholar]
- 45.Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res 2014;74:2520–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.