

Abstract
Polyelectrolyte complex (PEC) nanoparticles assembled from plasmid DNA (pDNA) and polycations such as linear polyethyleneimine (lPEI) represent a major non-viral delivery vehicle for gene therapy tested thus far. Efforts to control the size, shape and surface properties of pDNA/polycation nanoparticles have been primarily focused on fine-tuning the molecular structures of the polycationic carriers and on assembly conditions such as medium polarity, pH, and temperature. However, reproducible production of these nanoparticles hinges on the ability to control the assembly kinetics, given the non-equilibrium nature of the assembly process and nanoparticle composition. Here we adopt a kinetically controlled mixing process, termed flash nanocomplexation (FNC), that accelerates the mixing of pDNA solution with polycation lPEI solution to match the PEC assembly kinetics through turbulent mixing in a microchamber. This achieves explicit control of the kinetic conditions for pDNA/lPEI nanoparticle assembly as demonstrated by the tunability of nanoparticle size, composition, and pDNA payload. Using a combined experimental and simulation approach, we prepared pDNA/lPEI nanoparticles having an average of 1.3 to 21.8 copies of pDNA per nanoparticle and average size of 35 to 130 nm in a more uniform and scalable manner than bulk mixing methods. Using these nanoparticles with well-defined compositions and sizes, we showed the correlation of pDNA payload and nanoparticle formulation composition with the transfection efficiencies in vitro and in vivo. These nanoparticles exhibited long-term stability at −20°C for at least 9 months in a lyophilized formulation, validating scalable manufacture of an off-the-shelf nanoparticle product with well-defined characteristics as a gene medicine.
Keywords: gene delivery, DNA/polycation nanoparticle, polyelectrolyte complex, kinetic control, turbulent mixing, linear polyethyleneimine, transfection
Polyelectrolyte complex (PEC) formation has been widely used to assemble particulate vehicles for delivery of a wide range of macromolecular therapeutics including plasmid DNA (pDNA), messenger RNA (mRNA), small interfering RNA (siRNA), proteins, and peptides. As a promising non-viral gene delivery approach, pDNA molecules are condensed and packaged into PEC nanoparticles using a polycationic carrier in an aqueous solution. The assembled pDNA/polycation nanoparticles facilitate transport and access to target cells and cellular compartments, and protect pDNA from enzymatic degradation.1 The in vivo fate and delivery efficiency of the nanoparticles, as revealed by many recent works, are dependent on nanoparticle characteristics such as size range and distribution,2 morphology,3 surface properties, composition, and structure.4 However, it has been challenging to determine a detailed understanding of the relationship between the characteristics of nanoparticles and their interactions with biological systems. This is largely due to the lack of sufficient control over the PEC assembly kinetics that dictates the characteristics of the assembled nanoparticles.
The polymeric nature of the assembly components, for example, pDNA as the polyanion and lPEI as the polycation, indicates slower diffusion rates of these chains comparing with their electrostatic complexation rate. As a result, the PEC assembly often yields non-equilibrium, kinetically arrested complex structures. During the assembly process, the transient and local concentration profiles of different components determine how each PEC assembly initiates, propagates, and terminates to form a distinct nanoparticle. Control over these kinetic conditions is only possible when mixing is faster than the assembly process to allow distribution of the assembly components in a homogenous manner before each nanoparticle starts to assemble. Homogeneous mixing not only ensures that PEC nanoparticles are produced with uniform characteristics, but also provides the opportunity to control the size, surface properties, and composition of the nanoparticles through manipulation of the parameters of the inputs that go into the assembly system. Such an assembly condition implies that the characteristic mixing time (τM), within which different assembly components are mixed homogenously, must be reduced to less than the characteristic assembly time (τA), over which the PEC nanoparticle assembly occurs. The conventional mixing methods, such as pipetting and vortexing, cannot fulfill this requirement.
Mixing occurs through diffusion of assembly components across the interfaces of different flows. The most common approach to achieve small τM is by shortening the diffusion path. This can be achieved by both laminar flow and turbulent flow set-ups. In a laminar flow set-up, mixing is achieved as different flow paths are introduced within a small compartment. Due to manufacturing difficulties, engineering approaches such as hydrodynamic focusing5–6 and droplet confinement7 have been developed to further increase surface-to-volume ratio. In a turbulent flow set-up, turbulent eddies enable rapid flow break-down to tiny dimensions to facilitate diffusion. Flow turbulence can be delivered by T connectors,8 Tesla mixers and herring-bone mixers,9 coaxial jet mixers,10–11 confined impinging jets (CIJ),12–13 and multi-inlet vortex mixers (MIVM).14–16 Various degrees of success have been achieved using these devices to prepare drug-loaded nanoparticles with more uniform characteristics compared to those produced with conventional methods, owing to a higher degree of control of mixing kinetics of the assembly components. We recently reported the production of pDNA/lPEI nanoparticles by turbulent mixing in a CIJ mixer to demonstrate the scalability of the method and feasibility of controlling the size,17 but the kinetics of mixing and nanoparticle assembly during this process has not been fully analyzed.
Kinetically restricted assembly has been well illustrated for self-assembly of amphiphilic polymeric micelles where nanoparticle formation can be tuned by changing solvent mixing rate vs. polymer aggregation and drug partition rates in mixed solvents in a process called flash nanoprecipitation (FNP).18 FNP uses turbulent mixing in an MIVM or CIJ mixer to mix two opposing jets carrying miscible solvents in a time shorter than the characteristic time for hydrophobic chain aggregation. Uniform nanoparticles can be produced as a result of homogenous super-saturation conditions.19–20 By varying kinetic conditions under such mixing status, a diffusion-limited and fusion-dominated aggregation mechanism for nanoparticle formation21 and a quantitative model for predicting nanoparticle size have been proposed.22 Here we adopt a similar analysis to uncover the kinetic control aspects of pDNA/polycation PEC nanoparticle assembly.
In this work, we demonstrate the kinetic control of PEC assembly and nanoparticle formation using a turbulent mixing approach in a CIJ mixer, which is termed flash nanocomplexation (FNC). The diffusion kinetics of polyelectrolytes pDNA and linear polyethyleneimine (lPEI) in FNC is significantly different from that of the solvent and polymer in FNP, as the complexation kinetics mediated by polyelectrolyte charge neutralization is faster than hydrophobic aggregation of the polymer chain segments in FNP, and the PEC occurs in aqueous medium absent of organic solvent mixing that occurs in FNP. These factors contribute to the unique process and additional challenges for kinetic control of PEC assembly into nanoparticles in FNC. For this study, we selected in vivo-jetPEI® as the testing carrier due to its high transfection efficiency in vivo as the benchmark for non-viral carriers, its availability in GMP quality, and its molecular simplicity as a polycation with uniform charge density. Using PEC nanoparticles of in vivo-jetPEI® and plasmids of typical sizes of 4 to 7 kb as a model system, we examined the mixing flow regimen in a CIJ mixer using fluid dynamic simulations and analyzed the requirements of achieving kinetic control over the PEC assembly process. We demonstrated exquisite control over the pDNA/in vivo-jetPEI® nanoparticle composition through manipulation of kinetic conditions, and characterized the effect of nanoparticle size and composition on their transfection efficiency in vitro and in vivo. We also analyzed the advantages of pDNA/in vivo-jetPEI® nanoparticles assembled under kinetically controlled conditions in terms of their transfection efficiency and translational potentials for non-viral gene therapy.
RESULTS AND DISCUSSIONS
Control of τM by turbulent mixing in CIJ and mixing theory of pDNA/lPEI system.
In the FNC setup used for this study and our previous paper,17 two opposing flows with the same flow rate carrying pDNA and lPEI solutions, respectively, impinge in a CIJ microchamber with a volume of 27 μL that breaks down the two flows into turbulent structures at an appropriate flow rate (Q). These turbulent structures can be described using an average dimensional scale denoted as the Kolmogorov length scale η,23 that is determined by the volume-averaged turbulent energy dissipation rate :
| (1) |
where is spatial coordinates within the microchamber, and υ is the average kinematic viscosity of solution mixture in the efflux. Since it is difficult to determine experimentally, we adopted a simulation approach to obtain the distribution of throughout the microchamber with regard to each flow rate input, and then calculate the . Instead of approximating ε through dividing energy input by the mass over which the energy is dissipated as in a previous work on the CIJ device,12 we generated a numeric solution of ε using a finite-difference method (see Experimental Section),24–25 based on the mass and momentum conservation functions of viscous fluids, the Navier-Stokes equations, and the actual CIJ device dimensions.
The simulation results are demonstrated by the instantaneous velocity isosurface and Q-criterion vortex isosurface distributions sampled at a representative time point t = 110 ms upon impinging, and the time-averaged turbulent kinetic energy (TKE) distribution profiles for Q = 1, 10 and 20 mL/min as shown in Fig. 1A. It shows a clear transition from a laminar mixing pattern to a turbulent mixing pattern that occurs around a flow rate of 10 mL/min. With a flow rate of 20 mL/min representing a condition for turbulent mixing, the two jet flows changed their direction upon impinging by 90° to move radially in parallel that formed a disk-like structure of the velocity isosurface (Fig. 1A–2 and Video S1). During this parallel movement, oscillations occurred due to intrinsic flow instability and caused twisting and deformation of the flows. On the periphery of the disk-like structure, the velocity isosurface broke apart and vorticial structures were shed that broke the flows into the turbulent mixing flow structures (Fig. 1A–3 and Video S2). The turbulent flow structures are illustrated in a simplified model shown in Fig. 1B–1, in which the pDNA and lPEI solution flows are separated on a scale of the Kolmogorov length η (Fig. 1B–2).
Figure 1. Turbulent mixing of pDNA and lPEI solutions in a confined impinging jet (CIJ) microchamber.
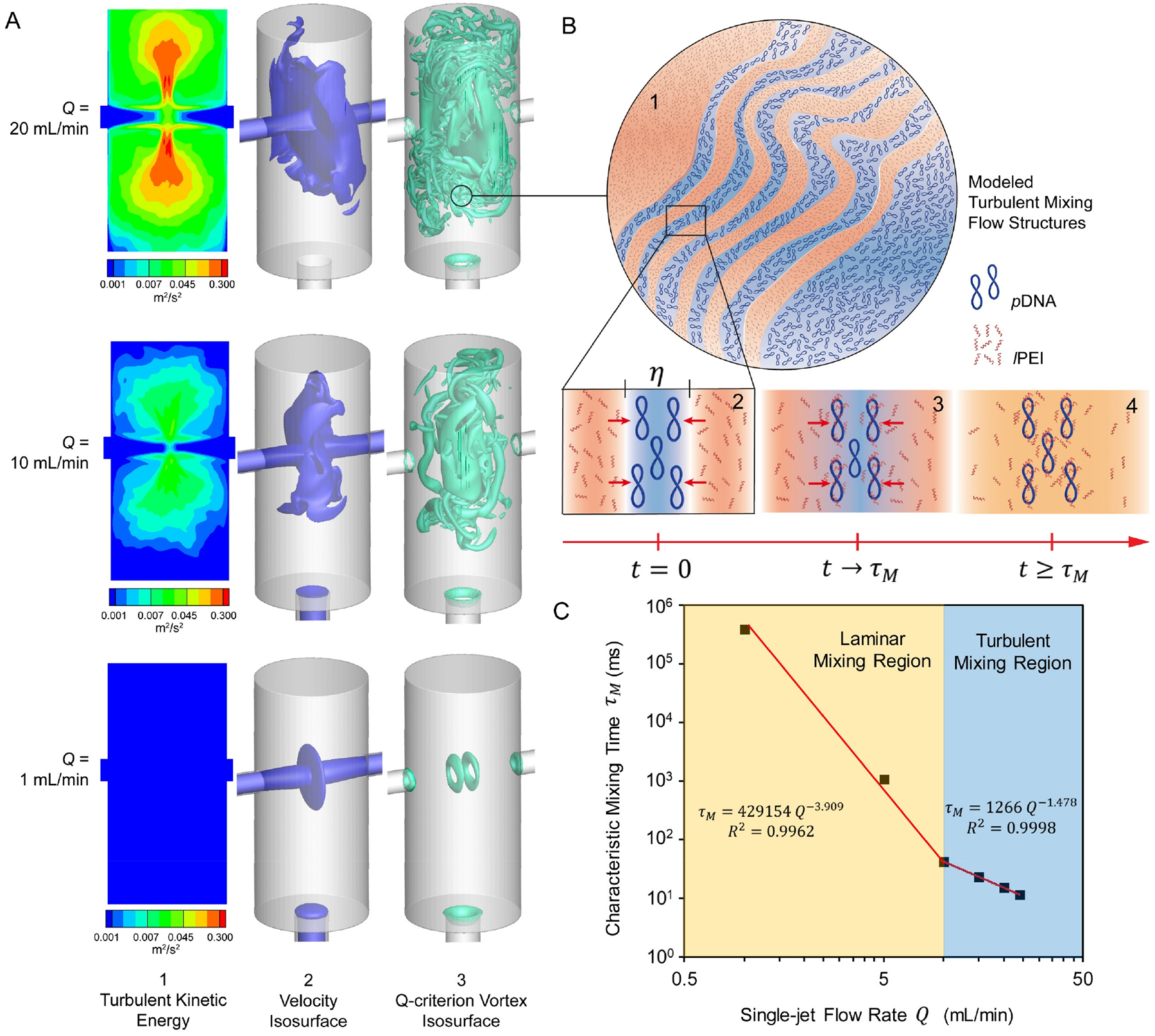
(A) Simulated flow fields based on the actual CIJ mixer dimensions: (1) Time-averaged turbulent kinetic energy (TKE) distribution, (2) Instantaneous velocity isosurface sampled at t = 110 ms, and (3) Instantaneous Q-criterion vortex isosurface sampled at t = 110 ms; (B) Illustrations of (1) the turbulent mixing flow structures generated at a flow rate of 20 mL/min, (2) separation of the pDNA and lPEI solution flows at the Kolmogorov length scale η, (3) diffusion of lPEI molecules into the pDNA flow regions, and (4) uniform mixing as defined by the input concentration profile of pDNA and lPEI across a time scale of τM; (C) Correlation between the characteristic mixing time τM and flow rate Q, as fitted from simulation results of 6 different flow rates.
Figure 2. Effect of characteristic mixing time τM on pDNA/lPEI nanoparticle assembly.
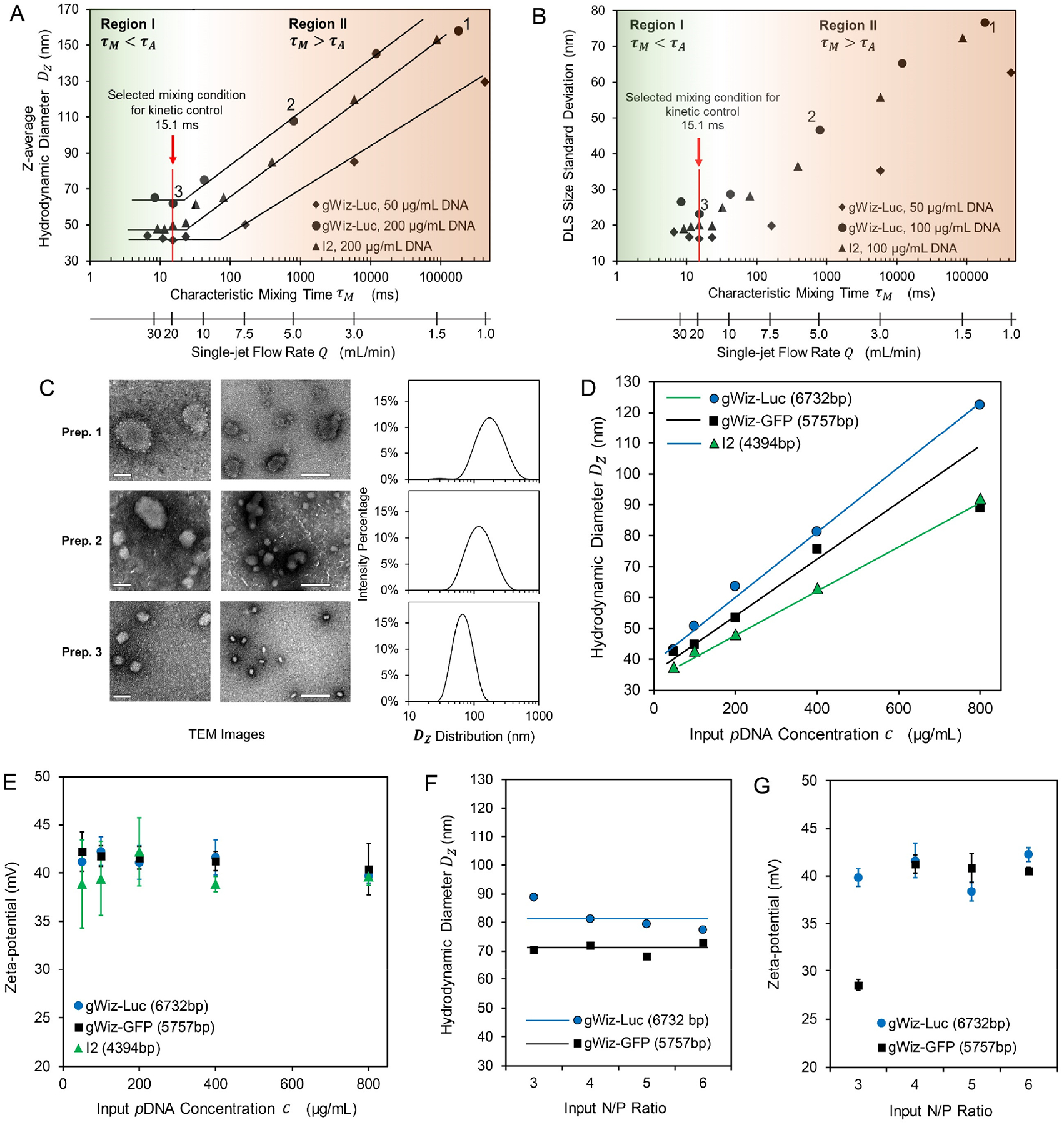
(A, B) Effect of mixing kinetics profile (τM and flow rate Q) on the average nanoparticle size Dz (A) and uniformity shown as the DLS size standard deviation (B). The mixing kinetics scale is divided into two regions: Region I (τM < τA) and Region II (τM > τA). Labels 1, 2 and 3 denote three representative preparations generated from three different mixing conditions; (C) Transmission electron microscopy (TEM) images and DLS profiles of the three sets of nanoparticles prepared at Q = 1.25 mL/min, τM = 1.8 × 105 ms (Prep. 1), Q = 5 mL/min, τM = 790 ms (Prep. 2), and Q = 20 mL/min, τM = 15 ms (Prep. 3). Scale bar = 50 nm (for left panel) and 200 nm (for right panel); (D, E) Effect of input pDNA concentration and plasmid size on the average nanoparticle size Dz (D) and zeta potential (E) prepared by τM = 15 ms; (F, G) Effect of N/P ratio on the average nanoparticle size Dz (F) and zeta potential (G) prepared by τM = 15 ms. For the conditions tested, the size profile and zeta potential of pDNA/lPEI nanoparticles did not vary with the N/P ratio from 4 to 6.
Figure 3. Compositions of the FNC-assembled pDNA/lPEI nanoparticles.
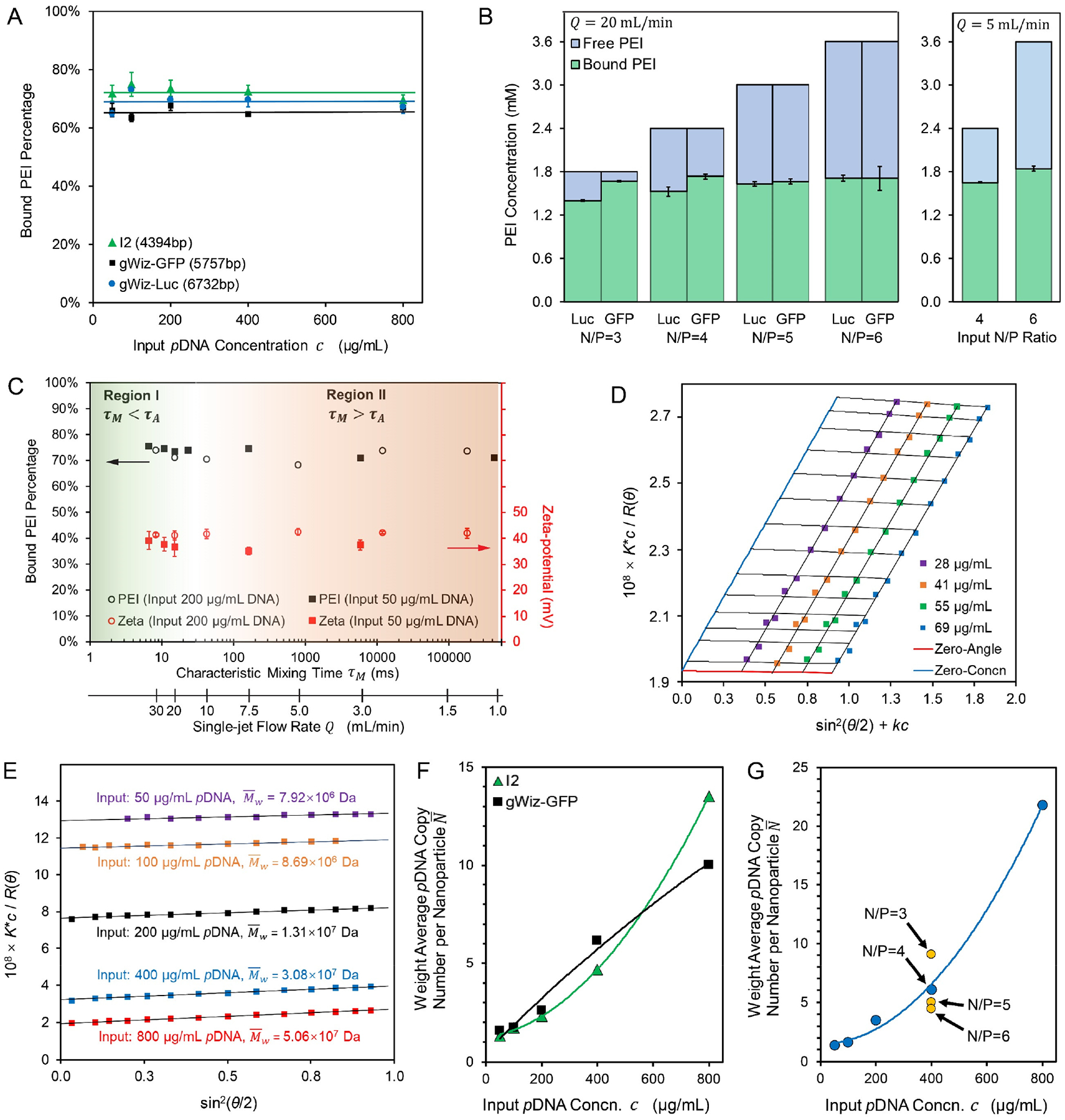
(A) The fraction of bound lPEI and the composition of the assembled nanoparticles remained similar when nanoparticles were prepared at different input pDNA concentrations or with different plasmids; (B) Bound vs. free lPEI amount and proportions with an input N/P ratio from 3 to 6 for gWiz-Luc and gWiz-GFP nanoparticle formulations assembled under a turbulent mixing condition (Q = 20 mL/min, τM = 15 ms < τA) and a laminar mixing condition (Q = 5 mL/min, τM= 790 ms > τA). Labels: Luc and GFP for gWiz-Luc and gWiz-GFP plasmid nanoparticles, respectively; (C) Bound lPEI fraction and zeta-potential of nanoparticles prepared with 50 or 200 μg/mL of gWiz-Luc pDNA under different flow rates, suggesting that all gWiz-Luc/lPEI nanoparticles share the same average composition; (D) A representative Zimm plot for I2/lPEI nanoparticles with a molar mass of 5.32 × 107 Da, also showing the second viral coefficient A approaching zero; (E) Representative Debye plots for gWiz-GFP/lPEI nanoparticles prepared by varying input pDNA concentration for I2 plasmid; (F, G) Calculated weight average pDNA copy numbers per nanoparticle (N) for preparations from lPEI and I2 or gWiz-GFP plasmids at N/P = 4 (F) or gWiz-Luc plasmid at different concentrations and N/P ratios (G).
Mixing occurs as molecular diffusion across the interfaces between the pDNA and lPEI flows in the turbulent structures. The diffusion is asymmetric for the two species. The dimensions of a pDNA are much larger with a molecular weight on an order of millions of Dalton. Due to steric restrictions imposed by the double-helix structure, they assume a rod shape in aqueous solutions with a low ionic strength, decreasing their diffusivity to a magnitude of 10−8 cm2/s in the solution.26 The charged lPEI molecules in an aqueous solution at pH 3.5 (kept consistent in this study) with a molecular weight of 22 kDa have a diffusivity estimated to be about 10−5 cm2/s,26 which is 3 orders of magnitude higher than that of pDNAs. Therefore, the diffusion distance for a pDNA is approximately times shorter than that of a lPEI molecule within the same period of time. Therefore, we approximated the time scale for the mixing process of this pDNA/PEI system, i.e. the characteristic mixing time τM, as the diffusion time for an lPEI molecule to cross half of the Kolmogorov length scale η (Fig. 1B–2,3,4):
| (2) |
where DlPEI is the diffusion coefficient of lPEI molecules in the solution mixture.
Figure 4. Assembly of pDNA/lPEI PEC nanoparticles.
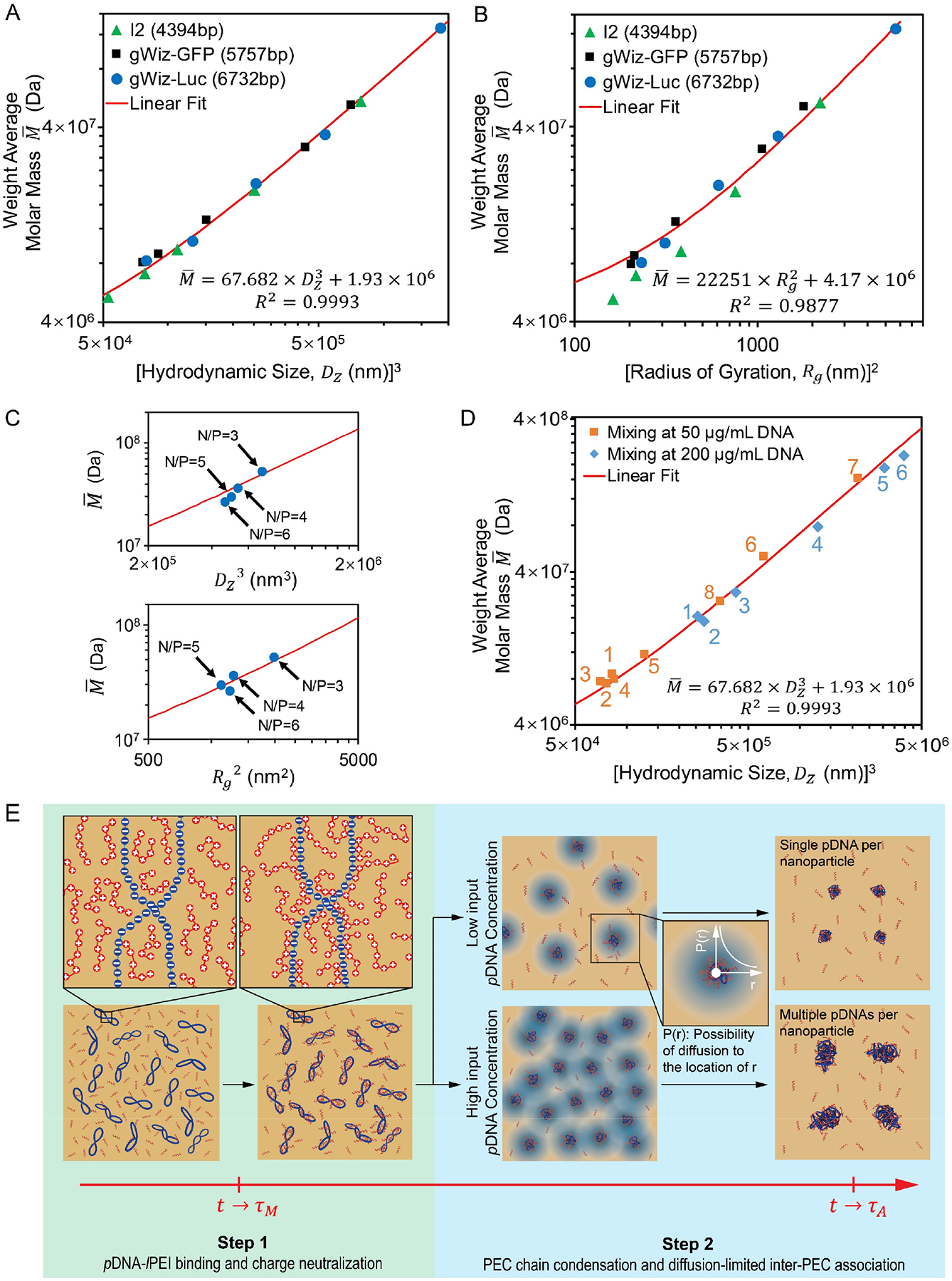
(A, B) Correlation of nanoparticle average molar mass and size (A) and radius of gyration (B) for nanoparticles assembled under turbulent mixing condition (Q = 20 mL/min, τM = 15 ms). Each data point in (A) and (B) represents an independent formulation batch; (C) Application of the linear fits from Eq. 5 (Upper panel) and Eq. 6 (Bottom panel) to nanoparticles formulated with different N/P ratios at Q = 20 mL/min; (D) Correlation of nanoparticle average molar mass and size for nanoparticles produced by different mixing conditions, i.e. with different τM. For input pDNA concentration of 25 μg/mL (orange), label 1 to 8 represent τM of 7, 11, 15, 23, 163, 5855, 4 × 105 ms, and pipetting respectively; for 100 μg/mL (blue), label 1 to 6 represent τM of 8, 15, 42, 795, 104 and 2 × 105 ms, respectively; (E) The proposed two-step pDNA/lPEI PEC nanoparticle assembly model under turbulent mixing condition (τM < τA).
These analyses showed that a higher mixing efficiency can be achieved by a smaller η as a result of greater turbulence generated by higher flow rate Q. Using this simulation framework, we quantitatively analyzed the relationship between τM and Q, and identified a powered τM ~ Q relationship (Fig. 1C) as shown in Eqs. 3 and 4, which was used to calculate all corresponding τM values for Q inputs for the CIJ device used in this study.
| (3) |
| (4) |
Effect of characteristic mixing time τM on the outcomes for pDNA/lPEI nanoparticle assembly.
Next, we experimentally examined the effect of flow rate Q on FNC assembly of pDNA/lPEI nanoparticles when impinging gWiz-Luc pDNA (6.7 kb) solution and in vivo-jetPEI® solution. The following parameters were maintained constant for this study: the pH of the lPEI solution was kept consistent at 3.5 to maintain the same protonation degree of lPEI at around 75%, and thus the same charge density for lPEI,27 and the concentrations of the input pDNA and in vivo-jetPEI® solutions were maintained at 200 μg/mL and 106 μg/mL, respectively. When increasing Q, i.e. decreasing τM, the size (z-average hydrodynamic diameter, Dz) given by dynamic light scattering (DLS) measurement of the nanoparticles decreased until it reached a plateau of 65 nm at Q~15 mL/min (Fig. 2A).
When the concentrations of pDNA and lPEI were both proportionally reduced by 4-fold, or the pDNA size reduced from 6.7 to 4.4 kb (I2 plasmid, see Table S1), the measured DLS nanoparticle sizes followed the same trend. The critical Q above which consistent DLS size for the nanoparticles was obtained was lowered from ~ 15 to ~ 8.5 mL/min (critical τM,0 increased from ~ 20 to ~ 85 ms) by the lowered input pDNA concentration. The DLS size standard deviation (a reflection of peak width, as defined in Experimental Section) of the nanoparticles showed the same dependency on Q (Fig. 2B), indicating increased uniformity of the nanoparticles as τM decreases. The flow rate-dependent average size and uniformity were confirmed by transmission electron microscopy (TEM) observations (Fig. 2C). Based on these findings, we could divide the field of characteristic mixing time τM or flow rate Q into two regions:
Region I corresponds to the kinetic condition where the average DLS size and uniformity remained constant independent of Q or τM. This indicates that the mixing conditions within the microchamber have reached the maximum degree of homogeneity to allow the assembly to occur uniformly, so that all nanoparticles have a similar assembly path. This assembly process has a time scale defined as the characteristic assembly time (τA), and with τM < τA, almost all pDNA/lPEI nanoparticles are assembled under the same defined conditions (concentrations of pDNA and lPEI, temperature, medium pH, ionic strength, etc.). In other words, the assembly components pDNA and lPEI can be mixed at a rate that is faster than nanoparticle formation to initiate nanoparticle assembly nearly “simultaneously” and in nearly the same microenvironment. As discussed above, DlPEI ≫ DDNA, and it is lPEI molecules that primarily diffuse into pDNA flow regions, resulting in homogeneous distribution of lPEI molecules to the vicinity of pDNA molecules. This establishes uniform initial kinetic conditions defined by the input concentration profiles of pDNA and lPEI (Fig. 1B–4).
Region II corresponds to the kinetic condition where τM > τA such that the molecular mixing process occurs on a time scale that is greater than the nanoparticle assembly process. Under this condition, nanoparticle assembly happens in a heterogeneous manner as mixing progresses, and partially formed nanoparticles can further associate with late-arrival molecules in a poorly defined manner. This leads to less uniform and larger nanoparticle sizes, or even aggregates; and compositions of the nanoparticles are dependent on the flow rate and mixing condition. As the flow rate increases and approaches the critical condition allowing τM = τA, the assembly mixture is closer to turbulent mixing structure (Fig. 1A,B), and is becoming more homogenous. Therefore, the mixture composition becomes closer to the input concentration profiles of pDNA and lPEI. For example, when comparing Preparation 1 (Q = 1.25 mL/min and τM = 1.8 × 105 ms, based on Fig. 1C and Eq. 4), Preparation 2 (Q = 5 mL/min and τM = 7.9 × 102 ms, Eq. 4), and Preparation 3 (Q = 20 mL/min and τM = 15 ms, Eq. 3), the nanoparticle ensembles showed the characteristics consistent with this analysis (Fig. 2A,C).
As the flow rate increases, i.e. as τM decreases, the flow mixing profile undergoes a transition from the laminar to turbulent mixing, which coincides with the nanoparticle assembly transition from Region II (τM > τA) to Region I (τM < τA). This shows the capability of turbulent mixing in a CIJ device to match the solution mixing time scale with the pDNA/lPEI nanoparticle assembly time scale by varying the flow rates from 1 to 50 mL/min.
Effect of pDNA concentration and N/P ratio on pDNA/lPEI nanoparticle assembly.
When assembly occurs under the kinetic conditions defined in Region I, the assembly concentration profiles of pDNA and lPEI are defined by the input concentrations (i.e. pDNA concentrations and N/P ratio in the impinging solutions). This provides an opportunity to examine the effect of assembly concentration profiles on nanoparticle characteristics. We selected a flow rate of 20 mL/min (τM = 15 ms, as labeled in Fig. 2A,B) for this comparison. As shown in Fig. 2D, an increase in pDNA concentration resulted in increase of nanoparticle size, relatively narrow size distribution and low PDI (0.12 – 0.16) of these formulations (Fig. S1A,B). TEM observations (Fig. S2A) confirmed the kinetically controlled mixing and uniform assembly of the pDNA/lPEI nanoparticles. On the other hand, when the input pDNA concentration was fixed at 400 μg/mL, nanoparticles assembled at different ratios of nitrogen (in lPEI) to phosphorous (in pDNA) (N/P) gave similar size (Fig. 2F and Fig. S2B), indicating that the plasmid was compacted most effectively under this mixing condition independent of the initial lPEI concentration, and to a maximum compaction degree even when the N/P ratio was reduced to 3. This observation further confirms the effectiveness of the turbulent mixing with τM < τA in maximizing the access of lPEI molecules to complex with pDNA during the assembly process. In addition, zeta-potential assessments revealed a similar surface charge around +40 mV for all formulations regardless of the plasmid used, or input pDNA concentration (Fig. 2E) or N/P ratio (Fig. 2G). This suggests that the nanoparticle surfaces are similar and consist of excess amount of lPEI molecules.
Conventional mixing methods for preparation of pDNA/polycation nanoparticles, such as pipetting, provide a mixing time on an order of seconds, therefore falling into Region II on this kinetics scale. The method of pipetting followed by vortexing (Table S2) using small volumes of solutions generated nanoparticles with average sizes and uniformity that are similar to those of FNC preparations with a flow rate Q < 1.5 mL/min. There was no clear dependence of the nanoparticle size on input pDNA concentration (Fig. S3). Moreover, there was a higher degree of variability in size (Fig. S3) as a result of different pipetting procedures (Table S2).
Average nanoparticle composition and free lPEI measurement.
Full complexation of pDNA by lPEI is achieved with an N/P ratio greater than 3; therefore, assembly with an N/P ≥ 3 would result in an excess of unbound or free lPEI in the nanoparticle suspension.28 To assess the actual composition of the assembled nanoparticles, we first characterized the amount of free lPEI according to a published protocol.29 When the pDNA/lPEI nanoparticles were assembled under the turbulent mixing condition (Q = 20 mL/min and τM = 15 ms) as defined in Fig. 2, all nanoparticle formulations with different pDNA concentration inputs had the same bound vs. free lPEI compositions, as long as the input N/P ratio was fixed at 4 (Fig. 3A). The amount of bound lPEI was in the region of 70%, which corresponded to an N/P ratio of 2.7 in the nanoparticles. When adjusting the input N/P from 3 to 6 with a consistent pDNA concentration (Fig. 3B left panel), the amount of lPEI bound to nanoparticles was consistent regardless of the input N/P ratio. This indicates that the amount of lPEI bound to pDNA was the same among the nanoparticles prepared under different preparation conditions; and this average composition corresponds to an N/P ratio of 2.74 ± 0.14 (average ± standard deviation in n = 28 individual preparations with different input pDNA concentrations and/or N/P ratios) in the nanoparticles. These resultant “overcharging” nanoparticles are consistent with the fact that not all charged groups are accessible to participate in charge neutralization in the process of PEC formation.30
There was a minor difference between the two plasmids tested in that gWiz-Luc seemed to result in slightly lower amount of bound lPEI as N/P ratio decreased. Nonetheless, the overall conclusion is clear that the binding of lPEI to pDNA to form PEC is not affected by the concentrations of either pDNA or lPEI, nor by the input N/P ratio.
Charge neutralization is not a rate-limiting step for PEC nanoparticle assembly.
We found that this consistent minimal bound N/P ratio for pDNA neutralization was also true for nanoparticles prepared under non-turbulent mixing conditions (Fig. 3B right panel and Fig. 3C). The surface charges (i.e. zeta-potentials) measured for nanoparticles prepared under different mixing conditions also remained the same (Fig. 3C). Since lPEI content in the nanoparticles and zeta-potential are directly related to the charge neutralization and complex formation processes, our findings, highlighted in Fig. 3A–C, suggest that the charge neutralization and pDNA-lPEI binding are not rate limiting steps for nanoparticle assembly. In other words, charge neutralization occurs at a rate much faster than condensation and chain folding of the pDNA/lPEI PECs into nanoparticles, i.e. it occurs on a time scale that is much shorter than the total characteristic assembly time τA.
We then could model the pDNA/lPEI PEC nanoparticle assembly process achieved under kinetically controlled mixing conditions into two distinct steps, which is also in agreement with several literature reports:31–32 Step 1: Charge neutralization step, in which lPEI molecules bind to the pDNA as soon as they diffuse into the vicinity of pDNA molecules forming pDNA/lPEI PECs, is not rate-limiting. In this study, regardless of input pDNA concentrations or N/P ratios, lPEI complexed with pDNA consistently at an N/P ratio of ~ 2.7. Step 2: PEC chain assembly, where the neutralized pDNA/lPEI complexes undergo condensation or compaction through folding33–34 that significantly reduces the complex volume, occurs. This is the rate limiting step, such that the time scale for Step 2 is much larger than that of Step 1. Therefore, the characteristic assembly time τA is primarily determined by the completion time of Step 2. When the neighboring pDNAs or PECs are close enough to diffuse into each other during the assembly process before the structure is stabilized by repulsion from net positively surface charges, compaction and assembly involving multiple PECs could occur, resulting in multiple pDNAs being packaged into a single distinct nanoparticle.
Characterization of the average pDNA copy number in each pDNA/lPEI nanoparticle.
Given that we have prepared nanoparticles with narrow size distribution and consistent composition, we aimed at characterizing the molar mass of the nanoparticles using the static light scattering (SLS) technique. With a fixed in-nanoparticle pDNA/lPEI mass ratio (Fig. 3A), we assumed that the refractive increment (dn/dc) value of the nanoparticles is constant and follows the additive rule.35 By measuring the intensity of scattered light to obtain Rayleigh scattering ratio with regard to each scattering angle and each nanoparticle mass concentration, and extrapolating concentration and angle-dependence curves to zero concentration and zero angle on a Zimm plot, we can characterize the weight average molar mass of nanoparticles , from which the weight average copy number of pDNA per nanoparticle () can be calculated (see Experimental Section).36–37 A representative Zimm plot for pDNA/lPEI nanoparticles with an is shown in Fig. 3D. For all nanoparticles measured by this method (Fig. 3D and Fig. S5A,B,C), the Zimm plot analyses indicate that the second virial coefficient (A2) of these nanoparticles approaches zero. This finding implies that the solvent (water) and temperature (25°C) conditions used for SLS measurement satisfies the θ condition, i.e. the PEC-solvent interaction cancels out the Vander Waals interaction and volume expansion of the PEC chains such that the PEC chain compaction occurs in a random packing manner. This θ condition significantly simplifies the measurement of average molar mass since we could ignore the concentration dependence of the light scattering behavior of these nanoparticles and measure the Rayleigh ratios at a fixed concentration, and apply calculations using the Debye plots (Fig. 3E and Fig. S5D,E,F). We found that for pDNA/lPEI nanoparticles prepared under a turbulent mixing condition (Q = 20 mL/min) varied significantly depending on the input pDNA concentrations: 1.3 to 13.5 for I2 pDNA (Fig. 3F); 1.6 to 10.0 for gWiz-GFP pDNA (Fig. 3F); and 1.4 to 21.8 for gWiz-Luc pDNA (Fig. 3G). On the other hand, varying input N/P ratio showed minor changes in the average number of plasmids in each nanoparticle. When input N/P ratio changed from 3 to 6 for input of 400 μg/mL pDNA, decreased from 9.2 for N/P = 3 to 6.1 for N/P = 4, 5.0 for N/P=5, and 4.4 for N/P=6 (Fig. 3G and Fig. S5F).
Correlation of DLS size and molar mass of pDNA/lPEI nanoparticles.
When the measured weight average molar masses of nanoparticles prepared under different conditions for all three plasmids were plotted against their hydrodynamic volume dimensions (i.e. Dz3) (Fig. 4A), a common linear correlation emerged:
| (5) |
where Dz is the z-average size as measured by DLS of the nanoparticle suspension, and is the weight average molar mass of the nanoparticles given by SLS. Such a “universal” fit for various nanoparticles independent of the plasmid and conditions used for nanoparticle assembly suggest that the PEC assembly units and compaction degree of these nanoparticles are similar. More specifically, Eq. 5 suggests that these nanoparticles have the same apparent hydrodynamic density of 67.7 Da/nm3, i.e. pDNAs are condensed to the same degree no matter how many of them are packed into a single nanoparticle.
Similarly, we also identified another composition-size correlation that the weight average molar mass of the nanoparticles is linearly proportional to the second power of radius of gyration (i.e. Rg2) (Fig. 4B):
| (6) |
This correlation fits well for nanoparticles with Dz between 50 and 130 nm, and the deviation from experimental data points increases as the size goes smaller than 50 nm. This linear relationship between and Rg2 further confirms that these pDNA/lPEI nanoparticles are assembled and characterized under the θ condition; and the PEC units assume random packing behavior under the solvent and temperature conditions tested.37 If we consider each PEC chain formed in Step 1 (a pDNA with all its bound lPEI) as a packing unit (i.e. a PEC unit) for nanoparticle assembly in Step 2, a pDNA/PEI nanoparticle can be modeled as an entity consisted of either one or multiple PEC units. The nanoparticle assembly follows a quantized combination pattern.
Nanoparticles generated by N/Ps varying from 4 to 6 have a similar molar mass, while those generated by N/P = 3 are heavier in molar mass but still fall into the two linear fits of Eq. 5 and Eq. 6 (Fig. 4C). It is presumed that with input N/P larger than 2.7, where lPEI is in excess to the amount required to sufficiently compact pDNAs, quantized combination stays valid.
This model is further supported by the fact that nanoparticles prepared under laminar mixing condition (τM > τA) also follow the same correlation of Eq. 5 (Fig. 4D). It is remarkable that nanoparticles with lower uniformity (i.e. broader distribution) appeared to have the same apparent hydrodynamic density as that of more uniform nanoparticles with fewer copies of pDNA per nanoparticle. This analysis is consistent with the hypothesis that PEC units formed in Step 1 are the building blocks for nanoparticle assembly, and they are compacted and associated in a similar manner as random folding of PEC unit chains in the solution under the θ condition.
Modeling pDNA/lPEI nanoparticle assembly kinetics in FNC under turbulent mixing condition.
Based on these findings and the nanoparticle assembly model mentioned above, we could analyze the assembly kinetics under τM < τA condition to understand the concentration-dependent mechanism for determining pDNA copy number per nanoparticle (Fig. 4E). The rate of pDNA-lPEI binding (i.e. PEC unit formation) in Step 1 is much faster than the rate of PEC compaction and association in Step 2 (as a conclusion from Fig. 3A,B,C), such that τStep 1≪ τStep 2, and τA ≅ τStep 2. The characteristic assembly time τA is presumably influenced by intrinsic properties of the polyelectrolytes involved in nanoparticle assembly, such as plasmid length, lPEI structure and molecular weight, stoichiometric and steric nature of pDNA-lPEI binding, etc. Upon generation of the turbulent flow structures (defined as t = 0), mixing occurs primarily by lPEI molecules diffusing into the pDNA solution regions, and lPEI diffusion proceeds on a time course of τM. The fast lPEI binding onto pDNAs happens with an N/P ratio of ~ 2.7 as lPEIs diffuse. When t ≥ τM, mixing completes and results in almost all pDNAs bound with the same amount of lPEIs, forming uniform PEC units that are about to proceed to Step 2 as the building blocks for nanoparticle assembly. The assembly occurs under the θ condition (Fig. 3D, Fig. 4B, and Eq. 6), where PEC chain-chain interaction cancels the PEC-solvent interaction. There is no additional barrier for multi-PEC chain folding and association as opposed to single PEC chain folding. As a result, the compaction of PEC units ends with the same condensation degree regardless of the number of pDNA involved in the assembly of a single nanoparticle (i.e. regardless of the final ). An is possible when multiple PEC units are brought into contact by diffusion for multi-PEC chains to be compacted into a single nanoparticle. Therefore, the number of PEC units that is involved in the assembly of a single nanoparticle is dictated primarily by PEC diffusion within the time course of τA. A higher input pDNA concentration results in a higher pDNA concentration in the pDNA flow regions in the turbulent flow structures, thus a lower average distance between pDNA molecules in the solution, such that more PEC units could be associated within a time scale similar to τA as a result of a shorter diffusion distance between them. Therefore, it is possible to explicitly control the number of pDNA to be packaged into a single nanoparticle under the kinetically controlled mixing conditions defined in the FNC process (i.e., when τM < τA).
Based on the analysis above, if the input pDNA concentration is sufficiently low, such that the average distance between any two pDNA molecules is too large for them to diffuse into each other over the time scale of τA, single plasmid-containing nanoparticles can be produced. With the correlation of weight average molar mass and nanoparticle size (Fig. 4A), the extrapolated size limit when c → 0 falls between 30 and 40 nm for the plasmids (4.4 kb to 6.7 kb) tested in this study (Fig. 2D), represents a typical size for pDNA/lPEI nanoparticles containing only one pDNA per nanoparticle. This small size and single-pDNA payload were never achieved by pipetting, for which mixing kinetics were poorly controlled, as shown in Fig. S3 with the lowest input pDNA concentration of 25 μg/mL.
Transfection efficiency of pDNA/lPEI nanoparticles with different copy numbers of pDNA per nanoparticle.
Using gWiz-Luc plasmid at different concentrations, we generated nanoparticles in our FNC device at a flow rate of 20 mL/min with (input c = 100 μg/mL), 3.5 (input c = 200 μg/mL), 6.1 (input c = 400 μg/mL) and 21.8 (input c = 800 μg/mL). With this series of nanoparticles, we examined the effect of on the in vitro and in vivo transfection efficiency of these nanoparticles. It is important to note that the sizes of these nanoparticles are also different, even though surface charges (zeta potentials) and compositions (bound and free lPEI fractions) are the same (Table S4).
Previous reports demonstrate dependence of cellular uptake on nanoparticle size due to differences in surface contact, avidity and trafficking kinetics.2 We used 3H-labelled pDNA to assemble nanoparticles (see Experimental Section) with different to assess their cellular uptake over a 4-h incubation period with PC3 prostate cancer cells. Our data showed no significant difference among these nanoparticle groups (Fig. 5A). As we fixed the total pDNA dose (0.6 μg per 1 × 104 cells with 5 × 104 per well in a 24-well plate) for the transfection test, the measured total fraction of nanoparticle uptake (out of the total dosed nanoparticles) is a function of the total number of nanoparticles available per cell and uptake rate. Presumably, a formulation with higher has fewer nanoparticles in number and thus higher cellular uptake rate. In vitro transfection efficiency experiments in the PC3 cancer cell line showed that and 21.8 had similar transfection efficiency levels and that these were much higher than those of either or 3.5 (Fig. 5B). With the similar uptake pDNA amount for each time point considered, and 21.8 may have higher efficiencies in intracellular delivery process, such as endosomal escape, pDNA dissociation, and nuclear transport. In previous literature reports, pDNA/lPEI nanoparticles typically give transfection and transgene activity in the lung following intravenous (i.v.) injection.38 Consistent with the in vitro findings, showed an appreciably lower transfection efficiency than other formulations, and there was a perceived trend for nanoparticles with , 6.1, and 21.8 that a higher gave better transfection efficiency (Fig. 5C and Fig. S7) in vivo in lungs of healthy BALB/c mice.
Figure 5. Transfection process and efficiency of pDNA/lPEI nanoparticles with different numbers of pDNA per particle.
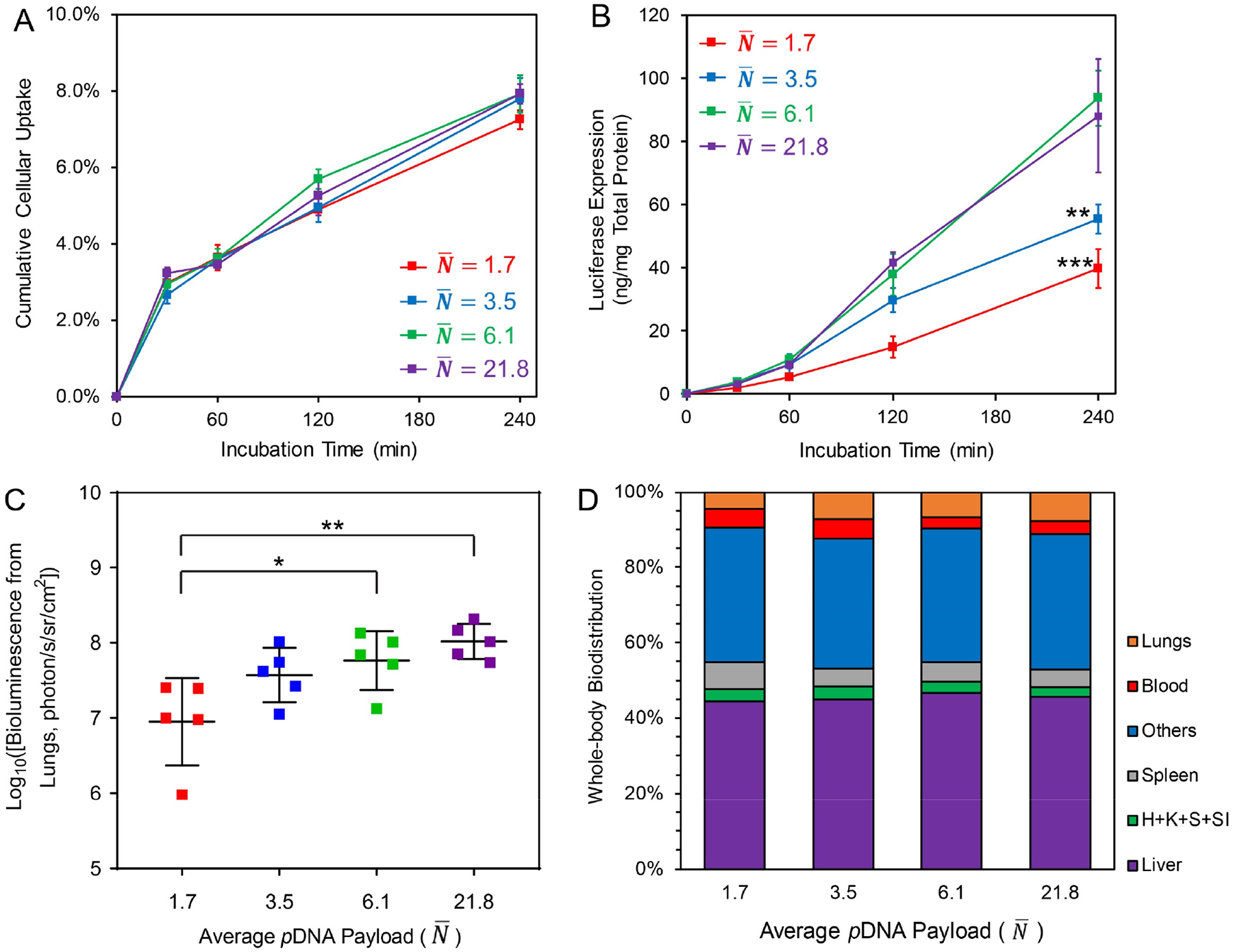
(A) Cellular uptake quantitative assay of nanoparticles prepared with 3H-labeled pDNA in PC3 prostate cell line following a 4-h incubation period (pDNA dosage = 0.6 μg/104 cells); (B) The in vitro transfection efficiency of nanoparticles with different in PC3 cells with a 4-h incubation (pDNA dosage = 0.6 μg/104 cells). The asterisks denote significance level when comparing with nanoparticle group; (C) The in vivo transfection efficiency (bioluminescence radiance) in the lung of BALB/c mice at 12 h post i.v. injection of nanoparticles (dose = 30 μg pDNA per mouse); (D) Whole-body biodistribution of nanoparticles at 1-h post i.v. injection of 3H-labeled nanoparticles containing 30 μg pDNA per mouse. Labels: H: heart, K: kidneys, S: stomach, SI: small intestine. For statistical analysis, *p < 0.05, **p < 0.01, and ***p < 0.001 from one-way ANOVA and multiple comparisons (GraphPad Prism 8).
A biodistribution study was conducted in BALB/c mice by i.v. injection of 3H-labeled nanoparticles at a dose of 30 μg pDNA/mouse. Mice were sacrificed at 1 h following dosing of the nanoparticles, and major organs and blood samples were collected and weighted. The biological samples were solubilized, and the solutions were subjected to liquid scintillation assessment to quantify 3H-labeled pDNA in the samples. The results revealed a rapid distribution (> 95%) of the nanoparticles into organs and tissues within 1 h for all formulations. The distribution patterns of these nanoparticles were similar, except that those with an of 1.7 resulted in fewer nanoparticles deposited in the lung (Fig. 5D and Fig. S8A) and the clearance via the spleen was more significant (Fig. S8C). For all groups, even though 42–45% of the total dose ended up in the liver, compared with 5–8% of the dose to the lung (Fig. S8B), there was no detectable level of transgene expression in the liver. This was probably due to the rapid clearance and degradation of nanoparticles by the Kupffer cells in the liver.39
The smaller difference in in vivo delivery efficiency among these nanoparticles was likely due to the fact that pDNA/lPEI nanoparticles interact strongly with serum components and aggregate rapidly following i.v. injection, leading to entrapment in the lung microvasculature and substantial uptake by endocytic cells in the liver and lung,40 which would mask the differences of the payload capacity as a result of controlling . Identifying nanoparticles (e.g. PEGylated nanoparticles) with a lower tendency for opsonization and aggregation, and understanding the mechanism of serum coating will help to better reveal the detailed effects of nanoparticles with defined composition and size on transfection efficiency in vivo, as a future direction.
Effect of pDNA payload and the ratio of bound vs. free PEI content on the in vitro and in vivo transfection efficiency of pDNA/lPEI nanoparticles prepared under kinetically controlled conditions.
The pilot study described above revealed that nanoparticles with of 6 or higher showed better transfection efficiency in vitro and in vivo than nanoparticles with lower plasmid payload. We next examined nanoparticles prepared under defined turbulent mixing conditions (Q = 20 mL/min, τM = 15 ms < τA) at different N/P ratios and compared them with those prepared by poorly mixed condition (Q = 5 mL/min, τM = 790 ms ≫ τA). Two series of nanoparticles were prepared with gWiz-Luc plasmid, with characteristics shown in Table 1.
Table 1.
pDNA/lPEI nanoparticles prepared with different mixing conditions and N/P ratios
| Nanoparticle Code | Plasmid | Q (mL/min) | Input N/P | Z-average Diameter* Dz (nm) | Weight Average |
|---|---|---|---|---|---|
| W1 | gWiz-Luc | 20 | 3 | 88.2 ± 30.0 | 9.1 |
| W2 | 4 | 81.2 ± 25.5 | 6.1 | ||
| W3 | 5 | 79.3 ± 40.4 | 5.0 | ||
| W4 | 6 | 77.5 ± 30.1 | 4.4 | ||
| W5 | 5 | 3 | 153.0 ± 61.9 | 40.7** | |
| W6 | 4 | 158.9 ± 64.5 | 45.6** | ||
| W7 | 5 | 148.1 ± 62.8 | 37.0** | ||
| W8 | 6 | 155.5 ± 72.0 | 42.8** | ||
| W9 | Pipetting | 6 | 171.2 ± 86.3 | 57.0** | |
| P1 | PEG-fLuc | 20 | 3 | 81.4 ± 21.2 | 8.1** |
| P2 | 4 | 86.0 ± 25.1 | 9.5** | ||
| P3 | 5 | 82.7 ± 28.4 | 8.5** | ||
| P4 | 6 | 81.0 ± 26.6 | 8.0** | ||
| P5 | 5 | 3 | 146.9 ± 57.8 | 45.7** | |
| P6 | 4 | 151.1 ± 60.2 | 49.7** | ||
| P7 | 5 | 145.6 ± 54.7 | 44.5** | ||
| P8 | 6 | 134.1 ± 46.6 | 34.9** |
We first tested all nanoparticles prepared with gWiz-Luc plasmid in the PC3 cancer cell line (Fig. 6A). For the two sets of FNC nanoparticles (W1–W4 and W5–W8), a higher N/P ratio (i.e. a higher free PEI fraction as a result) yielded a higher transfection efficiency, which was in agreement with previous literature reports.38, 41 Nanoparticles carrying lower payload showed a better performance than those nanoparticles with a higher payload; and the differences were statistically significant at N/P = 4 and 6. We then administered the same sets of nanoparticles to BALB/c mice and monitored their transfection efficiencies in the lung at 12, 24 and 48 h post-injection time points. The results (Fig. 6B and Fig. S10) showed a similar pattern to the in vitro experiments. Transgene expression activity was low for nanoparticles prepared at an N/P ratio of 3 for both low-payload and high-payload nanoparticles. With an N/P = 4, nanoparticles with a low payload () were more effective than nanoparticles with a higher payload (). The transgene activities of the nanoparticles prepared at N/P = 5 and 6, regardless of the payload level, , showed similar luciferase expression levels to the N/P = 4, formulation.
Figure 6. Transgene expression of pDNA/lPEI nanoparticles produced under kinetically controlled conditions with different N/P ratios and payload levels ().
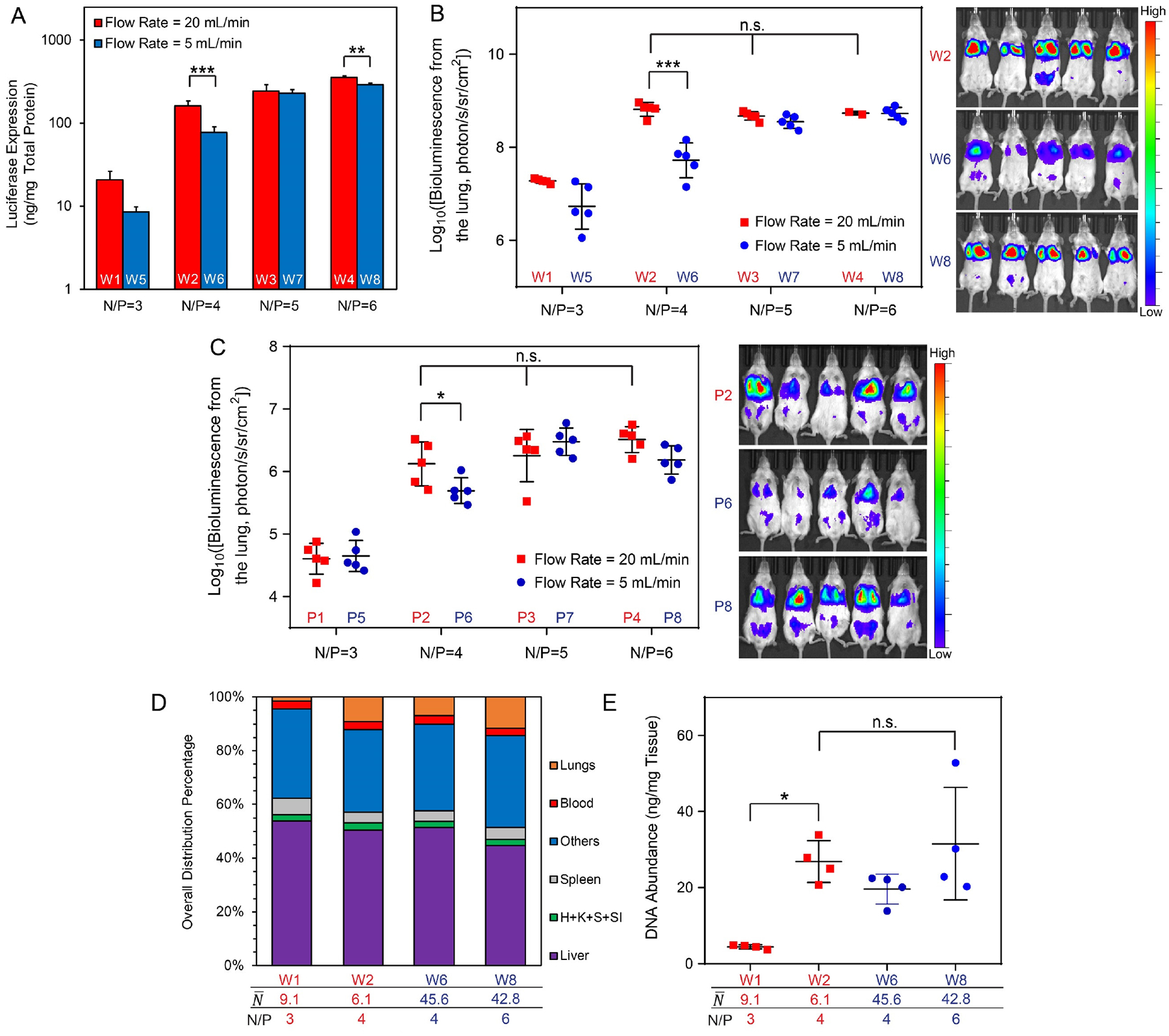
(A) In vitro transfection efficiencies of nanoparticles (W1–W8, see Table 1) in PC3 cancer cell line (dose = 0.6 μg gWiz-Luc plasmid/104 cells); (B) In vivo transfection efficiency in the lung in healthy BALB/c mice at 12 h post i.v. injection of nanoparticles (W1–W8, see Table 1) containing 40 μg gWiz-Luc plasmid per mouse (left) and representative IVIS images of groups with significant differences in transgene expression (right); (C) In vivo transfection efficiency in the lung of an LL/2 metastasis model in the NSG mice at 48 h post injection of nanoparticles (P1-P8, see Table 1) containing 40 μg PEG-Luc plasmid per mouse (left) and representative IVIS images of groups with significant differences in transgene expression (right); (D) Whole-body biodistributions in BALB/c mice at 1 h post injection of nanoparticles (W1, W2, W6, W8) containing 40 μg 3H-labeled gWiz-Luc plasmid per mouse. Labels: H: heart, K: kidneys, S: stomach, SI: small intestine; (E) Biodistributions to the lung of mice shown in (D); For statistical analysis, n.s. denotes no statistical significance with p > 0.05, *p < 0.05, **p < 0.01, and ***p < 0.001 from one-way or two-way ANOVA and multiple comparisons.
We then sought to confirm these findings in an LL/2 lung metastasis model in NSG mice. Tumor-specific transfection and expression were investigated through preparations of pDNA/lPEI nanoparticles using a custom-built plasmid construct encoding luciferase gene driven by a tumor-specific PEG-3 promoter as we have reported previously.42–43 We prepared the same set of nanoparticles (P1–P8) as described above under different assembly conditions using the FNC setup (Table 1). Tumor inoculation was conducted by injection of 200 μL PBS containing 5 × 105 LL/2 tumor cells into immunodeficient NSG mice 7 days prior to nanoparticle dosage. In this mouse model, tumor cells primarily engraft in the lung.44 Mice with successful tumor engraftment in the lung were selected and randomly grouped for this experiment. Mice were imaged at 48 h following nanoparticle injection by IVIS imaging. The tumor-specific gene expression in the lungs of the engrafted NSG mice showed a similar transgene expression pattern as that in healthy BALB/c mice transfected by gWiz-Luc plasmid (Fig. 6C and Fig. S11). Nanoparticles prepared with higher N/P ratios (4 to 6) showed higher transfection efficiencies than those prepared at N/P = 3, though the lower payload nanoparticles () performed better than higher payload nanoparticles () at N/P = 4. Transgene expression levels in the lung was comparable among nanoparticles with N/P = 5 or 6. There was a perceived trend (however p > 0.05) that for lower payload nanoparticles generated by a flow rate of 20 mL/min, higher N/P ratio resulted in greater average transgene expression level in the lung.
We also characterized the biodistribution of four selected nanoparticle formulations from this series with the biggest differences in their transfection efficiencies: nanoparticles with lower payload and lower N/P ratios (Table 1, W1: N/P = 3 and , W2: N/P = 4 and ), and nanoparticles with higher payload and higher N/P ratios (Table 1, W6: N/P = 4 and , W8: N/P = 6 and ). The 3H-labeled nanoparticles were injected intravenously at the same dose as used for Fig. 6B in BALB/c mice. More than 95% of the injected nanoparticle dose was distributed to tissues and organs within 1 h (Fig. 6D). Compared with the other nanoparticle formulations, W1 nanoparticles had the lowest fraction of nanoparticles distributed into the lung (1.4%) (Fig. 6D,E) with the highest levels of distribution to the liver (54%, though with no statistical significance) and spleen (6%) (Fig. S12), correlating with the lowest transfection efficiency in the lung. W2 and W8 showed similar levels of distribution to the lung (Fig. 6D,E), which correlated with similar transgene expression levels in these two formulations (Fig. 6C). However, although W6 had a similar biodistribution profile to W2, it gave a significantly lower transfection efficiency. This may be attributed to less efficient intracellular delivery efficiency as shown in the in vitro study (Fig. 6A), plus delivery of 7.5-fold fewer nanoparticles to the lung. For lower payload nanoparticle formulations, the increased number of nanoparticles may facilitate more frequent transfection events in a higher number of cells. On the other hand, the smaller size of these nanoparticles may positively influence nanoparticle trafficking in the tissue and the access to tumor tissues. Comparing W6 and W8 nanoparticles, even though their payloads ( and 42.8) and sizes (Dz = 158.9 and 155.5 nm) were similar, a higher level of free lPEI (Fig. 3B, 1.75 mM vs. 0.74 mM) led to a lower level of clearance by the liver (Fig. 6D and Fig. S12A). Together with a higher intracellular delivery efficiency (Fig. 6A), this factor may be responsible for a relatively higher transgene expression activity in the lung.
Scale-up production of an off-the-shelf, lyophilized pDNA/lPEI nanoparticles.
Successful clinical translation of non-viral DNA delivery gene therapy depends on a high delivery and transfection efficiency, good biocompatibility, scalable production processes, prolonged storage stability, and good performance consistency (i.e. low batch-to-batch variability). Current methods for systemic delivery of pDNA through PEC nanoparticles rely on on-site mixing of the therapeutic pDNA and in vivo-jetPEI® solutions in the clinic immediately before administration (i.e. a process that is essentially similar to manual pipetting, as W9 in Table 1). As could be expected, defining the reproducibility of nanoparticle formation and performance consistency of such a process would be difficult (Fig. S2). The preparation of nanoparticles by the FNC process that we have reported here offers a continuous, and therefore highly scalable and reproducible method.15–17 With a single bench-model device, 0.5 grams of pDNA, which is equivalent to 12,500 doses at 40 μg pDNA/mouse, could be packaged into pDNA/lPEI nanoparticles within one hour. The resulting nanoparticle suspension can be subjected to an optimized lyophilization protocol for storage in a powder form (Fig. 7A) that includes 9.5% w/w trehalose as a cryoprotectant agent. Lyophilized pDNA/lPEI nanoparticles were stable for at least 9 months when stored at −20°C. Upon reconstitution, the size, PDI, zeta-potential, PEI recovery, and DNA recovery at each time point were consistent with the freshly prepared sample (Fig. 7B). Reconstitution requires only the addition of water, free of aggressive processing such as sonication or vortexing. A clear suspension free of aggregation is generated after less than 1 minute at room temperature (Fig. 7A and Video S3). No vortexing is required. The reconstituted pDNA/lPEI nanoparticles are stable at 4°C for at least 4 days.
Figure 7. Scale-up production of off-the-shelf pDNA/lPEI nanoparticles and the long-term storage stability.
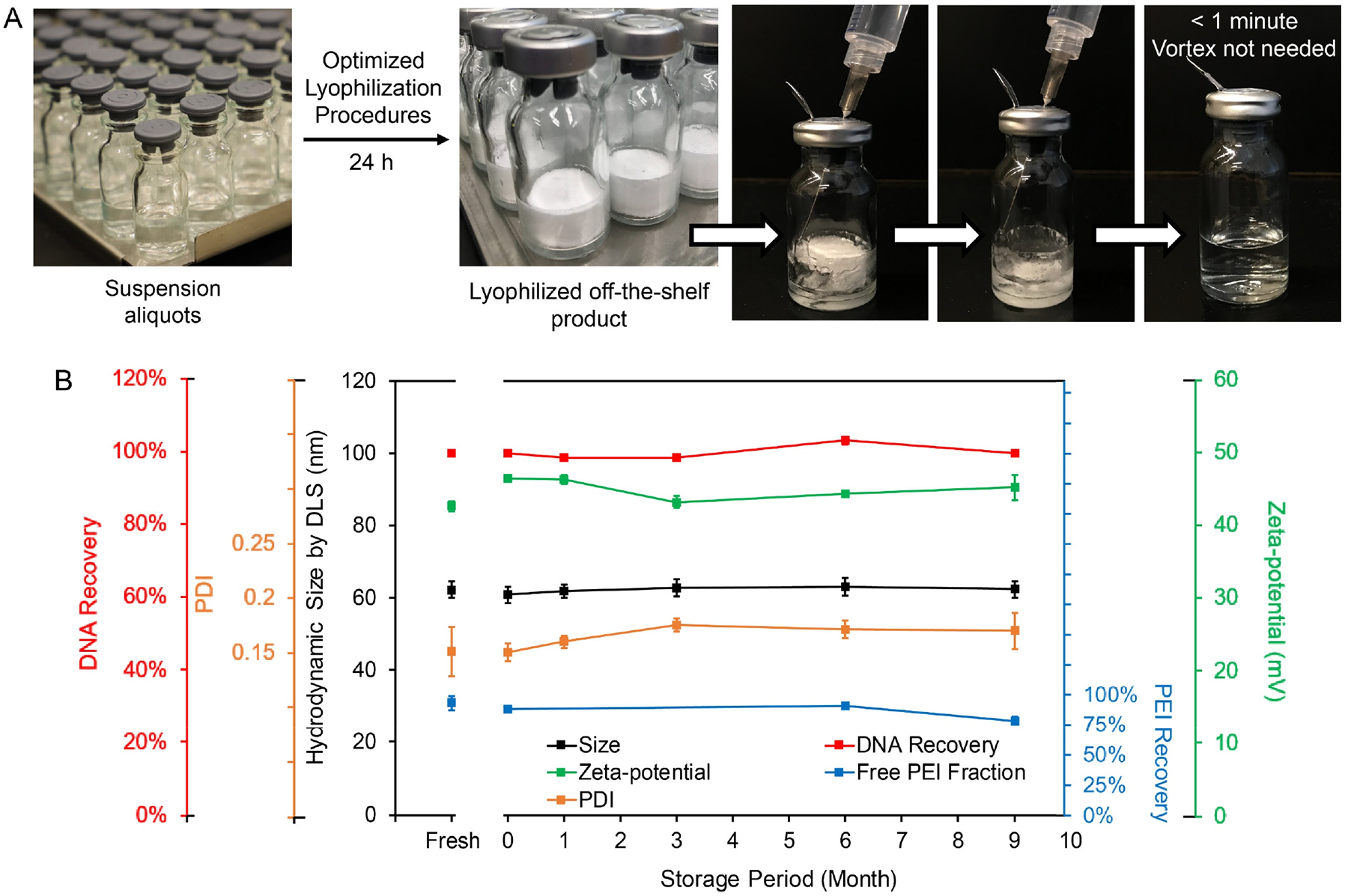
(A) Lyophilization and reconstitution of nanoparticles prepared using FNC setup; (B) Nanoparticle characteristics upon reconstitution of lyophilized nanoparticles stored at −20°C at Months 0, 1, 3, 6 and 9. Month 0 represents a reconstituted sample right after completion of lyophilization.
CONCLUSIONS
In this study, we combined a simulation method and an experimental approach to achieve a detailed understanding of the kinetics of mixing in the CIJ microchamber used for FNC assembly of pDNA/lPEI nanoparticles. The CIJ microchamber allows us to correlate the flow rates of the input pDNA and lPEI solutions with the characteristic mixing time of the mixing process. By controlling the mixing kinetics that enables the turbulent mixing of the solutions in the microchamber, we have demonstrated an explicit control over the nanoparticle size, surface charge and composition with high uniformity and scalability. We have used the static light scattering measurements and composition analysis of the FNC-assembled pDNA/lPEI nanoparticles, to reveal a “universal” correlation between the nanoparticle hydrodynamic size and pDNA payload per nanoparticle. This suggests that pDNA neutralization and compaction were achieved to the same degree for nanoparticles assembled with different pDNA payloads under different conditions. These findings not only allowed for finer composition control of pDNA/lPEI nanoparticles beyond control over nanoparticle size, but also provided experimental evidence to probe the kinetic process of pDNA/lPEI nanoparticle assembly. We confirmed that the charge neutralization between pDNA and lPEI molecules in forming PEC units is not a rate-limiting step, and the characteristic assembly time is primarily determined by chain folding and compaction of the PEC units. The number of pDNA packaged into each nanoparticle is primarily determined by the average diffusion distance of PEC units (i.e. the local pDNA concentration). By controlling the input pDNA concentration from 50 to 800 μg/mL under kinetically controlled conditions by FNC, an average of 1.3 to 21.8 pDNA can be assembled in to each nanoparticle, which correlates with the average hydrodynamic size of 35 to 130 nm.
The pDNA/lPEI nanoparticles with an N/P ratio of 3 to 6 can be produced with excellent shelf stability. These well-defined nanoparticles enabled the investigation of the effect of pDNA payload and formulation composition on their transfection efficiency. In both healthy and tumor bearing mouse models tested in this study, a payload of 6–10 plasmids per particle was found to be optimal for transgene expression in the lung following i.v. administration, which correlated well with their in vitro transfection activities.
These nanoparticles can be produced in a scalable manner as an off-the-shelf, lyophilized formulation that maintains its stability for at least 9 months when stored at −20 °C. Moreover, this formulation is easy to reconstitute and administer. Since this method is not specifically dependent on the carrier structure or on plasmid length and type, it should be generally applicable to many other potential polycation carriers. Thus, this FNC production process offers distinct technical advantages towards the clinical translation of non-viral nanoparticle vehicles for gene delivery.
EXPERIMENTAL SECTION
Simulation of turbulent mixing in the confined impinging jet (CIJ) micromixer.
Basic assumptions for simulation include: (1) Due to the low volume fraction of pDNA and PEI molecules in the flow, their presences do not affect the flow field; (2) Upon PEC formation, the kinematic viscosity of the efflux is close to water as the major viscosity contributor pDNA is already turned into a condensed form. Simulations are performed by solving the incompressible Navier-Stokes equations:
| (7) |
| (8) |
where is the fluid velocity vector, p is the pressure, p and p are the density and kinematic viscosity of water, respectively. They are solved by a fractional step method:
| (9) |
| (10) |
where and . A second order Crank-Nicolson method is used for the time discretization of convection and diffusion terms, and a second order central finite difference method is used for spatial discretization. A sharp-interface immersed boundary method is used to resolve the geometry on the non-body conformal Cartesian grid.24–25
Preparations of pDNA/in vivo-jetPEI® polyelectrolyte complex (PEC) nanoparticles.
All CIJ devices were manufactured by Johns Hopkins University Whiting School of Engineering machine shop based on a CIJ design reported previously.12 In vivo-jetPEI® (purchased from Polyplus-transfection, France) was used as received and diluted by ultrapure water to desired concentrations corresponding to different input N/P ratios from 3 to 6, when investigating the effect of input N/P ratio on nanoparticles with input pDNA concentration of 400 μg/mL. The pH of the solutions (regardless of the concentration) was adjusted to 3.5 by 1 M NaOH or 1 M HCl solutions before use. The plasmids (Table S1, purchased from Aldevron, US or constructed by lab of Dr. Pomper) were diluted by ultrapure water to a concentration range from 50 to 800 μg/mL when investigating the effect of input pDNA concentration on nanoparticle assembly. The pDNA/lPEI nanoparticles were formulated by injecting the two working solutions into the CIJ chamber at the preset flow rates using a high-pressure syringe pump (New Era Pump Systems, US). The pDNA/lPEI nanoparticles were subjected to downstream characterizations and applications directly. When an isotonic condition is required, the pDNA and lPEI working solutions were prepared in 9.5% (w/w) trehalose instead of water. The nanoparticle formulations prepared by the FNC process were stable for at least one month in room temperature. For preparations of pDNA/lPEI nanoparticles (W9, Table 1) through pipetting, the procedure B3 was used as shown in Table S2.
Characterizations of the pDNA/lPEI nanoparticle preparations.
The dynamic light scattering (DLS) measurements were conducted using a Malvern ZEN3690 Zetasizer at 25°C to assess the z-average hydrodynamic diameter (Dz). We followed the definitions in DLS ISO 13321 to use the DLS size standard deviation to evaluate the uniformity of the nanoparticles with different Dz in Fig. 2A. Whenever there is a single peak for the nanoparticles measured, the DLS size standard deviation is directly related to the polydispersity index (PDI) and Dz that:
| (11) |
The zeta-potential measurements were conducted on the same Zetasizer by phase analysis light scattering (PALS) in a low-salt buffer (5 mM NaCl with a conductivity of 0.6 mS/cm) for all nanoparticle suspensions to improve the reliability of the measurements.
The static light scattering (SLS) measurements were conducted on a Wyatt DAWN HELEOS 18-angle laser light scattering photometer, equipped with a laser source with the wavelength of 658 nm and a fused silica flow cell as the optical compartment. The machine was calibrated with all the laser detectors normalized against an isotropic scatter (3 nm dextran, MW 9000–11000, Sigma, US). The pDNA/lPEI nanoparticle suspensions were diluted to appropriate concentrations and introduced into the flow cell through a filter with size cut-off of either 450 nm or 1000 nm. Each sample was run at a flow rate of 200 μL/min for 5 min to establish stable signals from the detectors. Data collection was followed for 5 min to give time-averaged intensities of each detector. The pDNA/lPEI nanoparticle suspensions exiting the flow cell were collected and subjected to DLS and DNA recovery assessments (Fig. S4) to confirm no loss of nanoparticles in the flow cells. The Zimm or Debye plots of the nanoparticles were generated using the Wyatt ASTRA 6.1 software. Three independent runs were conducted for each sample.
In order to calculate the weight average molar mass of the nanoparticles from SLS measurements, the pDNA/lPEI nanoparticles were modeled as copolymers consisting of two components,36 as directed by light scattering theories.37 The radius of gyration assessment was calculated regardless of possible variations of the nanoparticle morphology if form factors of the experiments largely fall below 1 (as examined in Table S3). To determine the refractive index increment (dn/dc) of the pDNA/lPEI nanoparticles, we followed the additive rule described previously:35
| (12) |
where wpDNA and wPEI are the weight fraction of pDNA and lPEI complexed in nanoparticles, respectively. The dn/dc values are available by plugging in input pDNA concentrations and bound lPEI fraction from results of free lPEI measurements. Based on the proposed models for nanoparticle assembly, for each one of pDNA molecule, all the associated bound lPEI have a molar mass of:
| (13) |
where γ is bound lPEI fraction given by the free lPEI assessment; cm(lPEI) and cm(pDNA) are input mass concentrations of lPEI and pDNA for each formulation, respectively; and MpDNA is the molecular weight of the pDNA used. The weight average pDNA copy number per nanoparticle can be calculated by:
| (14) |
where is the weight average molar mass of the nanoparticles given by SLS.
Transmission electron microscopy (TEM).
Carbon-coated copper grids (Electron Microscopy Services, US) were subjected to plasma treatment (N2 glow discharge) for 30 sec to render the surface hydrophilic. The pDNA/lPEI nanoparticle suspensions were then loaded on the grid and incubated for 20 min. Excess suspension was removed, and an aliquot of 10 μL of 2% (w/v) uranyl acetate solution was added and incubated for 1 min. After removing the solution, the grids were allowed to be dried under hood for 24 h before imaging on a transmission electron microscope (Tecnai 12 Twin, Field Electron and Ion Company) operated at 100 kV. All images were taken by a Megaview III wide-angle camera (EMSIS GmbH, Germany).
Measurement of free lPEI in nanoparticle suspension.
The method was adapted from a previous report.29 A 500-μL aliquot of nanoparticle suspension was added into a Vivaspin 500 centrifugal concentrator (PES, MWCO of 100,000, Sartorius, Germany), and then centrifuged for 1 min at 7200 rpm. The filtrate was analyzed using the Protein Red Advanced Protein Assay (PRAPA, purchased from Cytoskeleton, US) accordingly to the manufacturer’s protocol. Each assay was conducted in quadruplicate. The lPEI reacts with the assay solution to generate products that have absorbance at a wavelength of 590 nm, through which the lPEI concentrations can be calculated against a linear standard curve.
In vitro transfection activity.
PC3 cancer cells were seeded in 24-well plates with a density of 5 × 104 cells/well. After 24-h culture, the medium in each well was aspirated. An aliquot of 50-μL pDNA/lPEI nanoparticle suspension containing 3 μg pDNA was added into 500 μL fresh medium, vortexed for 20 sec to mix, and the whole mixture was added into each well. The cells were incubated with the pDNA/lPEI nanoparticles for 1 to 4 h. The cells were then washed by PBS twice and incubated in 0.5 mL of fresh medium/well for 24 h. Before analysis, medium was removed and 100 μL of reporter lysis buffer (Promega, US) was added to each well. The cells were then subjected to two freezing-thawing cycles, and the lysate was analyzed using a luciferase assay kit (Promega, US). Transgene expression level was normalized against the total protein amount obtained by a protein assay kit (Pierce BCA reagents, Thermo Scientific, US). Pure luciferase was used as a standard. The transfection efficiency was plotted in terms of the amount of luciferase (ng) expressed per mg of total protein in the lysate. Transfection and analysis were conducted in quadruplicate for each group tested.
In vivo transfection efficiency.
The in vivo experimental procedures were approved by the Johns Hopkins Institutional Animal Care and Use Committee. The pDNA/lPEI nanoparticles were injected intravenously (i.v.) via mouse lateral tail vein at a dose of 30 or 40 μg pDNA per mouse at a concentration of 200 μg pDNA/mL. For groups with a lower input pDNA concentration in preparation, the nanoparticle suspensions were concentrated to 200 μg pDNA/mL by Amicon Ultra-2 centrifugal filter unit with a MWCO of 3,000 to concentrate both the PECs and the free PEI molecules with the same ratio. In vivo bioluminescence imaging was performed using the IVIS® Spectrum (PerkinElmer, US) and the images were processed with Living Image Software (PerkinElmer, US). The region of interest (ROI) quantitative analysis results have good correlations with luciferase protein abundance found in lungs that presents a linear relationship (Fig. S9), so tissue homogenization was not widely adopted for monitoring the kinetics of the transgene activities. Preliminary tests revealed that the transgene expression level (luciferase concentration in healthy lung tissues or tumor cells in the lung) peaks at around 12 and 48–72 h post injection for healthy BALB/c mouse model (Jackson Laboratory, US) and LL/2 metastasis in NSG mouse model (Johns Hopkins University Animal Core), respectively. IVIS assessment time points were set accordingly, with the mice anesthetized by isoflurane and imaged by IVIS system upon i.p. injection of 100 μL of 30 mg/mL D-luciferin (Gold Biotechnology, US) solution and 5-min diffusion period. For LL/2 tumor model, inoculation was done through i.v. injection of 200 μL PBS solution containing 5 × 105 cancer cells, 7 days prior to pDNA/lPEI nanoparticle dosing.
Cellular uptake and biodistribution studies.
The 3H-labeled PEC nanoparticles were prepared according to a previous report.45 The pDNA was labeled by methylation using S-adenosyl-L[methyl-3H]-methionine (3H-SAM, purchased from PerkinElmer, US. Application of radioactive materials in this study was approved and monitored by the Johns Hopkins University Radiation Safety Office) as the source prior to nanoparticle preparations. This labeling technique does not affect the nanoparticle assembly process. The advantage of this labeling technique lies with the capability of assessing absolute quantity of labeled pDNAs in biological samples through counting disintegration events per minute (DPM), thus ideal for cellular uptake and biodistribution analyses of these nanoparticles. Within a working range, DPM is linearly proportional to labeled pDNA quantity in the assay (Fig. S6). To label pDNAs, water, 10× NEB buffer (New England Biolabs, US), 3H-SAM (PerkinElmer, US), pDNA (1 mg/mL) and M. Sssl enzyme (New England Biolabs, US) were added at a ratio of 12:2:2:1:1 (v/v) into a 50-mL tube. The solution was mixed well, incubated for reaction at 37 °C for 2 h and quenched by heating to 65 °C for > 30 min. The reaction mixture was diluted by EB buffer, with labeled pDNA purified using a QIAprep Spin Miniprep kit (Qiagen, US), and finally mixed with nonlabelled pDNA to give the working solution for pDNA/lPEI nanoparticle assembly. For cellular uptake experiments, the same dose of pDNA/lPEI nanoparticles were used as that in the in vitro transfection experiment described above. At each time point, cells were washed twice with PBS and harvested. For the biodistribution study, the same dosage and formulation concentration were applied using the same protocol as described above for in vivo transfection experiments. At 1-h post injection, the animals were sacrificed with tissues harvested and weighed. Sufficient SOLVABLE solubilization fluid (PerkinElmer, US) was added to tissues and incubate at 70 °C for 48 h. The tissue lysate was mixed well, and 100 μL of each sample (n = 3 independent measurements) was added into 4 mL of Ultima Gold scintillation cocktail fluid (PerkinElmer, US) in 7-mL scintillation vials. DPM was assessed by a Tri-Carb 2200CA liquid scintillation analyzer (Packard Instrument Company, US) in a measurement time course of 5 min.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Cancer Targeting Systems Inc., the National Institutes of Health (R01 EB018358, P41 EB024495 and P50 CA058236), and Maryland Advanced Research Computing Center. The authors thank Dr. Will West from Cancer Targeting Systems, Ms. Beihang Yu from University of California Santa Barbara (on SLS analysis), Dr. Michael Bevan and Ms. Elena Alexandra Garcia from Johns Hopkins University (on SLS instrumentation), and Dr. Michael McCaffery from Johns Hopkins Integrated Imaging Center (on TEM analysis) for their helpful discussions and technical assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
List of plasmid DNAs used in this study; Procedures to prepare PEC nanoparticles by bulk-mixing (pipetting); Examinations of form factors for static light scattering (SLS) experiments; Summary of the characteristics of the nanoparticles with different weight average pDNA copy number per nanoparticle () tested in vitro and in vivo; The size distributions of PEC nanoparticles formulated with different input pDNA concentrations and input N/P ratios with τM < τA; Supplementary TEM images for nanoparticles prepared by different input pDNA concentrations and N/P ratios; Nonuniform PEC nanoparticles produced by pipetting method without tunability of size by input pDNA concentrations; Determination of pDNA concentration in PEC nanoparticle suspensions; Supplementary SLS data for nanoparticles prepared by a flow rate of 20 mL/min with different input pDNA concentrations and N/P ratios; Standard curve for quantitative assessment of absolute amount of 3H-labeled pDNA in biological samples; In vivo transfection efficiency in lungs upon dosing of PEC nanoparticles with different weight average copy numbers of pDNA per nanoparticle (); Biodistribution of dosed PEC nanoparticles with different weight average copy numbers of pDNA per nanoparticle (); Correlation between IVIS region of interest (ROI) quantitative results and luciferase abundance in tissue; In vivo transfection efficiencies of PEC nanoparticles with different pDNA payloads and PEI compositions prepared by kinetically controlled conditions in healthy BALB/c mice; In vivo transfection efficiencies of PEC nanoparticles with different pDNA payload and PEI compositions prepared by kinetically controlled conditions in a LL/2 lung metastasis model on NSG mice; Tumor-specific transfection and expression efficiencies of PEC nanoparticles with different pDNA payloads and PEI compositions prepared by kinetically controlled conditions in a LL/2 lung metastasis model on NSG mice; Supplementary biodistribution data of PEC nanoparticle formulations with significant findings in transfection and transgene activities; Video of development of velocity isosurface in the CIJ chamber under a flow rate of 20 mL/min; Video of development of vorticial structures (Q-criterion vortex isosurface) in the CIJ chamber under a flow rate of 20 mL/min; Video of demonstration of the reconstitution process of lyophilized pDNA/in vivo-jetPEI® nanoparticles.
H.-Q.M., Y.H., H.-W.L., I.M., M.G.P., C.G.U., and C.A.C. are co-inventors of U.S. Patent Applications covering the method and plasmid DNA/polycation nanoparticles as described in this paper. M.P. is a co-founder of Cancer Targeting Systems (CTS), Inc. that has licensed the technology described here. Other authors declare no competing financial interest.
REFERENCES
- (1).Shi B; Zheng M; Tao W; Chung R; Jin D; Ghaffari D; Farokhzad OC Challenges in DNA Delivery and Recent Advances in Multifunctional Polymeric DNA Delivery Systems. Biomacromolecules 2017, 18, 2231–2246. [DOI] [PubMed] [Google Scholar]
- (2).Hickey JW; Santos JL; Williford J-M; Mao H-Q Control of Polymeric Nanoparticle Size to Improve Therapeutic Delivery. J. Controlled Release 2015, 219, 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Williford J-M; Santos JL; Shyam R; Mao H-Q Shape Control in Engineering of Polymeric Nanoparticles for Therapeutic Delivery. Biomater. Sci 2015, 3, 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Blanco E; Shen H; Ferrari M Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lu M; Ho Y-P; Grigsby CL; Nawaz AA; Leong KW; Huang TJ Three-Dimensional Hydrodynamic Focusing Method for Polyplex Synthesis. ACS Nano 2014, 8, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu M; Ozcelik A; Grigsby CL; Zhao Y; Guo F; Leong KW; Huang TJ Microfluidic Hydrodynamic Focusing for Synthesis of Nanomaterials. Nano Today 2016, 11, 778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Juul S; Nielsen CJF; Labouriau R; Roy A; Tesauro C; Jensen PW; Harmsen C; Kristoffersen EL; Chiu Y-L; Frøhlich R; Fiorani P; Cox-Singh J; Tordrup D; Koch J; Bienvenu A-L; Desideri A; Picot S; Petersen E; Leong KW; Ho Y-P; Stougaard M; Knudsen BR Droplet Microfluidics Platform for Highly Sensitive and Quantitative Detection of Malaria-Causing Plasmodium Parasites Based on Enzyme Activity Measurement. ACS Nano 2012, 6, 10676–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kasper JC; Schaffert D; Ogris M; Wagner E; Friess W The Establishment of An Up-scaled Micro-mixer Method Allows the Standardized and Reproducible Preparation of Well-defined Plasmid/LPEI Polyplexes. Eur. J. Pharm. Biopharm 2011, 77, 182–185. [DOI] [PubMed] [Google Scholar]
- (9).Feng Q; Sun J; Jiang X Microfluidics-mediated Assembly of Functional Nanoparticles for Cancer-related Pharmaceutical Applications. Nanoscale 2016, 8, 12430–12443. [DOI] [PubMed] [Google Scholar]
- (10).Liu D; Cito S; Zhang Y; Wang C-F; Sikanen TM; Santos HA A Versatile and Robust Microfluidic Platform Toward High Throughput Synthesis of Homogeneous Nanoparticles with Tunable Properties. Adv. Mater 2015, 27, 2298–2304. [DOI] [PubMed] [Google Scholar]
- (11).Liu D; Zhang H; Cito S; Fan J; Mäkilä E; Salonen J; Hirvonen J; Sikanen TM; Weitz DA; Santos HA Core/Shell Nanocomposites Produced by Superfast Sequential Microfluidic Nanoprecipitation. Nano Lett. 2017, 17, 606–614. [DOI] [PubMed] [Google Scholar]
- (12).Johnson BK; Prud’homme RK Chemical Processing and Micromixing in Confined Impinging Jets. AIChE J. 2003, 49, 2264–2282. [Google Scholar]
- (13).Liu Y; Fox RO CFD Predictions for Chemical Processing in A Confined Impinging-jets Reactor. AIChE J. 2006, 52, 731–744. [Google Scholar]
- (14).Liu Y; Cheng C; Liu Y; Prud’homme RK; Fox RO Mixing in A Multi-inlet Vortex Mixer (MIVM) for Flash Nano-precipitation. Chem. Eng. Sci 2008, 63, 2829–2842. [Google Scholar]
- (15).He Z; Santos JL; Tian H; Huang H; Hu Y; Liu L; Leong KW; Chen Y; Mao H-Q Scalable Fabrication of Size-controlled Chitosan Nanoparticles for Oral Delivery of Insulin. Biomaterials 2017, 130, 28–41. [DOI] [PubMed] [Google Scholar]
- (16).He Z; Hu Y; Nie T; Tang H; Zhu J; Chen K; Liu L; Leong KW; Chen Y; Mao H-Q Size-controlled Lipid Nanoparticle Production Using Turbulent Mixing to Enhance Oral DNA Delivery. Acta Biomater. 2018, 81, 195–207. [DOI] [PubMed] [Google Scholar]
- (17).Santos JL; Ren Y; Vandermark J; Archang MM; Williford J-M; Liu H-W; Lee J; Wang T-H; Mao H-Q Continuous Production of Discrete Plasmid DNA-Polycation Nanoparticles Using Flash Nanocomplexation. Small 2016, 12, 6214–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Saad WS; Prud’homme RK Principles of Nanoparticle Formation by Flash Nanoprecipitation. Nano Today 2016, 11, 212–227. [Google Scholar]
- (19).Nikoubashman A; Lee VE; Sosa C; Prud’homme RK; Priestley RD; Panagiotopoulos AZ Directed Assembly of Soft Colloids through Rapid Solvent Exchange. ACS Nano 2016, 10, 1425–1433. [DOI] [PubMed] [Google Scholar]
- (20).Zhang C; Pansare VJ; Prud’homme RK; Priestley RD Flash Nanoprecipitation of Polystyrene Nanoparticles. Soft Matter 2012, 8, 86–93. [Google Scholar]
- (21).Johnson BK; Prud’homme RK Mechanism for Rapid Self-Assembly of Block Copolymer Nanoparticles. Phys. Rev. Lett 2003, 91, 118302. [DOI] [PubMed] [Google Scholar]
- (22).Pagels RF; Edelstein J; Tang C; Prud’homme RK Controlling and Predicting Nanoparticle Formation by Block Copolymer Directed Rapid Precipitations. Nano Lett. 2018, 18, 1139–1144. [DOI] [PubMed] [Google Scholar]
- (23).A First Course in Turbulence; Tennekes H; Lumley JL; Lumley J; MIT Press: Massachusetts, 1972. [Google Scholar]
- (24).Mittal R; Dong H; Bozkurttas M; Najjar FM; Vargas A; von Loebbecke A A Versatile Sharp Interface Immersed Boundary Method for Incompressible Flows with Complex Boundaries. J. Comput. Phys 2008, 227, 4825–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Seo JH; Mittal R A Sharp-interface Immersed Boundary Method with Improved Mass Conservation and Reduced Spurious Pressure Oscillations. J. Comput. Phys 2011, 230, 7347–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Clamme JP; Azoulay J; Mely Y Monitoring of the Formation and Dissociation of Polyethylenimine/DNA Complexes by Two Photon Fluorescence Correction Spectroscopy. Biophys. J 2003, 84, 1960–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Curtis KA; Miller D; Millard P; Basu S; Horkay F; Chandran PL Unusual Salt and pH Induced Changes in Polyethylenimine Solutions. PLoS One 2016, 11, e0158147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yue Y; Jin F; Deng R; Cai J; Chen Y; Lin MCM; Kung H-F; Wu C Revisit Complexation between DNA and Polyethylenimine — Effect of Uncomplexed Chains Free in the Solution Mixture on Gene Transfection. J. Controlled Release 2011, 155, 67–76. [DOI] [PubMed] [Google Scholar]
- (29).Bertschinger M; Chaboche S; Jordan M; Wurm FM A Spectrophotometric Assay for the Quantification of Polyethylenimine in DNA Nanoparticles. Anal. Biochem 2004, 334, 196–198. [DOI] [PubMed] [Google Scholar]
- (30).Berret J-F Evidence of Overcharging in the Complexation between Oppositely Charged Polymers and Surfactants. J. Chem. Phys 2005, 123, 164703. [DOI] [PubMed] [Google Scholar]
- (31).Barreleiro PCA; Lindman B The Kinetics of DNA−Cationic Vesicle Complex Formation. J. Phys. Chem. B 2003, 107, 6208–6213. [Google Scholar]
- (32).Santhiya D; Dias RS; Dutta S; Das PK; Miguel MG; Lindman B; Maiti S Kinetic Studies of Amino Acid-Based Surfactant Binding to DNA. J. Phys. Chem. B 2012, 116, 5831–5837. [DOI] [PubMed] [Google Scholar]
- (33).Osada K; Shiotani T; Tockary TA; Kobayashi D; Oshima H; Ikeda S; Christie RJ; Itaka K; Kataoka K Enhanced Gene Expression Promoted by the Quantized Folding of pDNA within Polyplex Micelles. Biomaterials 2012, 33, 325–332. [DOI] [PubMed] [Google Scholar]
- (34).Takeda KM; Osada K; Tockary TA; Dirisala A; Chen Q; Kataoka K Poly(ethylene glycol) Crowding as Critical Factor To Determine pDNA Packaging Scheme into Polyplex Micelles for Enhanced Gene Expression. Biomacromolecules 2017, 18, 36–43. [DOI] [PubMed] [Google Scholar]
- (35).Dai Z; Wu C How Does DNA Complex with Polyethylenimine with Different Chain Lengths and Topologies in Their Aqueous Solution Mixtures? Macromolecules 2012, 45, 4346–4353. [Google Scholar]
- (36).Dautzenberg H; Koetz J; Linow KJ; Philipp B; Rother G Static Light Scattering of Polyelectrolyte Complex Solutions In Macromolecular complexes in chemistry and biology; Dubin P, Bock J, Davis R, Schulz DN, Thies C, Eds.; Springer Science & Business Media: Berlin, 2012; pp 119–133. [Google Scholar]
- (37).Light Scattering by Polymer Solutions In Polymer chemistry; Hiemenz PC; Lodge TP; CRC Press Taylor and Francis: New York, 2007; pp 289–325. [Google Scholar]
- (38).Boeckle S; von Gersdorff K; van der Piepen S; Culmsee C; Wagner E; Ogris M Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J. Gene Med 2004, 6, 1102–1111. [DOI] [PubMed] [Google Scholar]
- (39).Tsoi KM; MacParland SA; Ma X-Z; Spetzler VN; Echeverri J; Ouyang B; Fadel SM; Sykes EA; Goldaracena N; Kaths JM; Conneely JB; Alman BA; Selzner M; Ostrowski MA; Adeyi OA; Zilman A; McGilvray ID; Chan WCW Mechanism of Hard-nanomaterial Clearance by the Liver. Nat. Mater 2016, 15, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ogris M; Brunner S; Schüller S; Kircheis R; Wagner E PEGylated DNA/transferrin-PEI Complexes: Reduced Interaction with Blood Components, Extended Circulation in Blood and Potential for Systemic Gene Delivery. Gene Ther. 1999, 6, 595. [DOI] [PubMed] [Google Scholar]
- (41).Klauber TCB; Søndergaard RV; Sawant RR; Torchilin VP; Andresen TL Elucidating the Role of Free Polycations in Gene Knockdown by siRNA Polyplexes. Acta Biomater. 2016, 35, 248–259. [DOI] [PubMed] [Google Scholar]
- (42).Bhang H-EC; Gabrielson KL; Laterra J; Fisher PB; Pomper MG Tumor-specific Imaging through Progression Elevated Gene-3 Promoter-driven Gene Expression. Nat. Med 2010, 17, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Minn I; Bar-Shir A; Yarlagadda K; Bulte JWM; Fisher PB; Wang H; Gilad AA; Pomper MG Tumor-specific Expression and Detection of A CEST Reporter Gene. Magn. Reson. Med 2015, 74, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bertram JS; Janik P Establishment of A Cloned Line of Lewis Lung Carcinoma Cells Adapted to Cell Culture. Cancer Lett. 1980, 11, 63–73. [DOI] [PubMed] [Google Scholar]
- (45).Williford J-M; Archang MM; Minn I; Ren Y; Wo M; Vandermark J; Fisher PB; Pomper MG Critical Length of PEG Grafts on lPEI/DNA Nanoparticles for Efficient in Vivo Delivery. ACS Biomater. Sci. Eng 2016, 2, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.