

Abstract
Introduction
Extracellular vesicles (EVs) from human Alzheimer's disease (AD) biospecimens contain amyloid beta (Aβ) peptide and tau. While AD EVs are known to affect brain disease pathobiology, their biochemical and molecular characterizations remain ill defined.
Methods
EVs were isolated from the cortical gray matter of 20 AD and 18 control brains. Tau and Aβ levels were measured by immunoassay. Differentially expressed EV proteins were assessed by quantitative proteomics and machine learning.
Results
Levels of pS396 tau and Aβ1–42 were significantly elevated in AD EVs. High levels of neuron‐ and glia‐specific factors are detected in control and AD EVs, respectively. Machine learning identified ANXA5, VGF, GPM6A, and ACTZ in AD EV compared to controls. They distinguished AD EVs from controls in the test sets with 88% accuracy.
Discussion
In addition to Aβ and tau, ANXA5, VGF, GPM6A, and ACTZ are new signature proteins in AD EVs.
Keywords: Alzheimer's disease, amyloid beta peptide, cortical gray matter, extracellular vesicles, machine learning, microtubule‐associated protein tau, proteome
1. BACKGROUND
Alzheimer's disease (AD) is the most common form of dementia affecting nearly 50 million people worldwide. Neuropathologically, disease is characterized by amyloid plaques formed by extracellular aggregation of amyloid beta (Aβ) peptide and intracellular accumulation of neurofibrillary tangles (NFTs). These are formed in brain tissue by the hyperphosphorylated and misfolded microtuble‐associated protein tau. 1 , 2 As AD progresses, Aβ and tau aggregates spread throughout the brain in a spatiotemporal manner. 3 , 4 Aβ deposition is most prominent in the frontal, anterior/posterior cingulate, lateral parietal, and lateral temporal regions. Tau pathology, as classified by Braak and Braak, occurs in six histopathological stages. These correspond to tauopathy stages of AD. In stages I and II, NFTs appear in the entorhinal cortex and hippocampus, while in stage III and IV, higher densities extend beyond the entorhinal cortex and hippocampus to the neocortex. In the final V‐VI stages, pathological tau deposits are present throughout the hippocampus. 3
Extracellular vesicles (EVs), including exosomes (50 to 150 nm), ectosomes/microvesicles (150 to 1000 nm), and apoptotic bodies (1000 to 5000 nm) are released from neurons, glia, and various other neural cells into the extracellular space. 5 , 6 , 7 These vesicles contain multiple forms of nucleic acids (microRNA, ncRNA, mRNA, DNA, among others), lipids and proteins that are transferred from cell to cell, and are found in blood, urine, and cerebrosprinal fluid (CSF). 8 , 9 In the central nervous system (CNS), it has been reported that AD‐associated pathogenic proteins in brain EVs including tau and Aβ oligomers play important roles in AD pathogenesis. 9 , 10 , 11 , 12 , 13 Moreover, it has been reported that inhibition of EV synthesis reduced amyloid plaque deposition in the mouse model of AD, and stimulation of EV secretion increased intracellular transfer of prion protein in vitro. 10 , 11 EVs are involved in the extracellular enzymatic degradation of Aβ and promote both Aβ aggregation and clearance by microglia. 12 , 13 Moreover, models of neuron‐to‐neuron transfer of tau seeds by EVs were reported. 14 , 15 , 16 , 17 Our own prior work demonstrated that microglia spread tau by EV secretion and that reducing EV synthesis significantly reduces tau propagation. 18 One mechanism centers around bridging integrator 1 (BIN1), which is associated with the progression of tau pathology and observed by its abilities to alter tau clearance and by promoting the release of tau‐enriched microglial EVs. 19
While EVs recovered from human and mouse brain tissues were examined by morphology, proteomics, and RNA analyses, 20 , 21 , 22 , 23 no comprehensive and quantitative proteomics databases have yet been acquired for AD human brain tissues. Herein, we provide the first proteomic profiling of EVs isolated from post mortem AD and cognitively impaired control brain tissues. The analyses were combined with machine learning and quantitation of Aβ and tau species by epitope‐specific enzyme‐linked immunosorbent assay (ELISA). Machine learning identified and distinguished protein signatures of AD brain‐derived EVs from controls with high degrees of accuracy.
2. METHODS
2.1. Brain sample acquisitions
Two cohorts of brains were used in this study. The first cohort was obtained from the University of Nebraska Medical School (11 AD and 9 control) and the Greater Los Angeles Veteran's Affairs Hospital (9 AD and 9 control) as part of NIH NeuroBioBank, which were matched for age and sex. The second cohort was obtained from the NIH NeuroBioBank (22 AD and 18 control). All the AD and control cases were neuropathologically diagnosed and matched for age and sex. The Institutional Review Board at the University of Nebraska Medical School, Greater Los Angeles Veteran's Affairs Hospital and the National Institutes of Health (NIH) NeuroBioBank approved the brain acquisitions provided by informed consent.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (PubMed) sources, meeting abstracts, and presentations. The quantitative proteomics and machine learning of extracellular vesicles (EVs) isolated from Alzheimer's disease (AD) brain tissue has never been published.
Interpretation: This is the first AD brain‐derived EV quantitative proteomics study. The AD brain‐derived EVs were enriched with pathogenic proteins, such as amyloid beta (Aβ), tau, apolipoprotein E (APOE),and α‐synuclein. ANXA5, VGF, GPM6A, and ACTZ were selected by machine learning quantitative proteomics datasets, which can distinguish AD and control brain‐derived EVs with 88% accuracy. Moreover, a validation cohort study shows that the expression level ANXA5 was significantly elevated in AD brain‐derived EVs.
Future directions: Validation of these proteins in EVs isolated from plasma and cerebrospinal fluids to monitor the progression of AD. Evaluation of these molecules for their roles in the spread and propagation of tau as therapeutic targets.
2.2. Purification of EVs from human brain samples
0.5 g of largely gray matter tissue from the frontal cortex of deceased AD or control cases were processed for EV extraction based on reported method with some modifications (see supporting information for detailed methods). 21
2.3. Protein concentrations
The bicinchoninic acid (BCA) assay was used to determine protein concentration for each sample using Pierce BCA protein assay kit (# 23225 Pierce) (see supporting information for detailed methods).
2.4. Enzyme‐linked immunosorbent assay
ELISAs were performed to assess levels of t‐tau, p‐tau, Aβ1‐40 and Aβ1‐42, and ANXA5 (see supporting information for detailed methods).
2.5. Nanoparticle tracking analysis
All samples were diluted in dfPBS at least 1:1000 or more to get particles within the target reading range for the Nanosight 300 machine (Malvern Panalytical Inc) (see supporting information for detailed methods).
2.6. Transmission electron microscopy
The EV isolated from AD or control brain tissue were analyzed by transmission electron microscopy (TEM; see supporting information for detailed methods).
2.7. Mass spectrometry
The EV samples were subjected to chemical treatment, tryptic digestion, and liquid chromatography (LC)‐ electrospray ionization (ESI) tandem mass‐spectroscopy (MS/MS) analysis (see supporting information for detailed methods).
2.8. Sequence database
The raw LC‐MS/MS data were converted into mZML format using ProteoWizard msConvert and analyzed using using PeaksDB and PeaksPTM using Peaks Studio version 8.0 (Bioinformatics Solutions, Inc., Waterloo, ON, Canada) against the Uniprot/Swissprot database for Homo sapiens with a 0.1% false discovery rate (FDR) and at least two unique peptides (see supporting information for detailed methods). 24
2.9. Statistical analyses
Statistical analysis was conducted using IBM SPSS software version 25 and GraphPad Prism6. Between‐group comparisons were analyzed by Student's t‐test, nonparametric Mann‐Whitney U, or one‐way analysis of variance followed by Bonferroni correction for multiple comparisons. The Gene Ontology of identified proteins were elucidated by DAVID Bioinformatics Resources 6.8. (https://david.ncifcrf.gov). The Venn diagram was generated using Venny_2.1 (http://bioinfogp.cnb.csic.es/tools/venny/).
2.10. Machine learning
The protein biomarkers to distinguish patients with AD from controls were selected using least absolute shrinkage and selection operator (LASSO) on the proteomics data from the training set (n = 21), in which each patient's true state is labeled. An ensemble machine learning classifier to evaluate the performance of the selected proteins was developed as described (see supporting information for detail methods). 25 , 26 The machine learning generated model's performance was evaluated on a separate, user‐blinded test set (n = 17).
3. RESULTS
A total of 38 patient samples, composed of 20 AD (15 M, 5 F, mean age: 75.0) and 18 control (14 M, 4 F, mean age: 75.7) cases were used in study for EV biological and proteomics analyses. A total of 78 patient samples, composed of 42 AD (25 M, 17 F, mean age: 81.0) and 36 control (22 M, 14 F, mean age: 79.0) cases, were included for validation analysis (Table 1). There were no statistical differences in the demographics between AD and controls with the exception of post mortem intervals (PMI) of the validation set, which will be discussed in the validation study (see Figure 4E).
TABLE 1.
Patient information
| Discoverer set | |||
|---|---|---|---|
| For proteomics | AD (n = 20) | Control (n = 18) | P a |
| Age, mean | 75.0 ± 9.12 | 75.7 ± 8.88 | .997 |
| Sex (male, female) | 15 M, 5F | 14 M, 4F | |
| PMI, mean | 12.5 ± 8.68 | 17.8 ± 7.57 | .0675 |
| Validation set | |||
|---|---|---|---|
| For ELISA | AD (n = 42) | Control (n = 36) | P |
| Age, mean | 81.0 ± 11.64 | 79.0 ± 11.59 | .1533 |
| Sex (male, female) | 25 M, 17F | 22 M, 14F | |
| PMI, mean | 9.0 ± 7.25 | 17.3 ± 8.28 | .0069 |
The statistical significance of the differences were calculated using Mann‐Whitney test.
FIGURE 4.
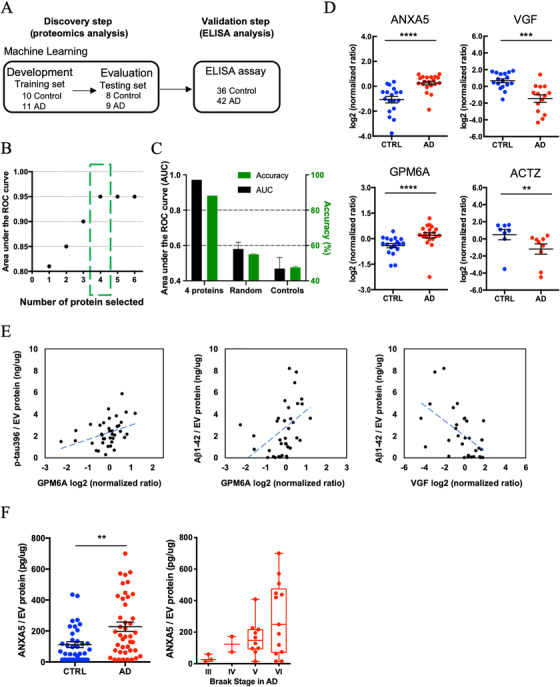
Machine learning model to identify Alzheimer's disease (AD) brain‐specific extracellular vesicles (EV) biomarker molecules: A, Workflow for machine learning approach. The training set is fed into linear discriminant analysis (LDA) to generate LDA vectors, which are applied to the blinded test set for classification. The predicted molecules are validated from the validation cohort by a commercial enzyme‐linked immunosorbent assay (ELISA). B, The best performing panel based on the area under the reciever operating characteristic (ROC) curve using the least absolute shrinkage and selection operator (LASSO)algorithm was selected in the training set. Y‐axis; the area under the ROC curve, x‐axis; proteins selected by the LASSO algorithm. The green box shows the selected protein with high area under the curve (AUC) for the blinded test set. C, The accuracy for four selected using the test set. Randomly selected control: accuracy = 55%, AUC = 0.58. Shuffling control: accuracy = 47.6%, AUC = 0.47. D, A scatter plot of log2 (normalized ratio) as measured by proteomics per selected candidate protein in machine learning. (ANXA5: P = 4.58E‐06, log2 fold change = 1.1, VGF: P = 5.30E‐04, log2 fold change = −1.6, GPM6A: P = 5.37E‐04, log2 fold change = 0.6, ACTZ: P = 4.44E‐03, log2 fold change = −1.5.) The t test was caluculated by Mann‐Whitney test. E, Scattered plot of candidate molecules and AD pathogenic molecule in brain‐derived EVs. Left: GPM6A and pS396 tau (r = 0.380, P = .019 using two‐tailed t‐test). Center: GPM6A and Aβ1‐42 (r = 0.387, P = .016). Right: VGF and Aβ1‐42 (r = −0.538, P = .002). F, Left: Significant difference in total ANXA5 to total EV protein between AD and CTRL group by ELISA (P = .0042 by Mann‐Whitney test) (AD = 42, CTRL = 36). Right: Braak stage‐dependent increase in the ANXA5 expression level to total EV protein in AD‐dependent manner
3.1. Biochemical characterization of brain‐derived EVs
The experimental workflow is summarized in Figure 1A. The EV samples were isolated from 20 AD and 18 sex‐ and age‐matched cognitively unimpaired controls using the discontinuous sucrose gradient ultracentrifugation as previously described with modifications (see Materials and Methods). This technique of EV isolation has been successfully used to isolate EVs from frozen mouse brain tissues. 21 , 27 In addition, we performed the quantitative proteomics analysis of human brain tissue homogenates and purified EV samples. The tetraspanins and ESCRT proteins were enriched in the EV samples, and contamination of non‐EV molecules such as nucleus, mitochondria, ER, and Golgi‐related proteins as indicated in MISEV2018 guidelines 5 were diminished in the EV samples (Table S1 in supporting information). To determine the purity of the EV preparations, EV samples were analyzed for their size and number by nanoparticle tracking analysis (NTA). The concentration of EVs derived from AD and control brains were not significantly different (P = .6075). The mode size distribution for EVs was significantly different and peaked at 122 nm for AD and 131 nm for controls (P = .0095) (Figure 1B). The EVs isolated from frozen brain tissue showed cap‐shaped morphology by transmission electron microscopy (TEM; Figure 1C). We next measured the concentration of total tau (t‐tau) and p‐tau at threonine 181 (pT181 tau), serine 199 (pS199 tau), and serine 396 (pS396 tau) in lysed EVs by ELISA. The levels of t‐tau, pT181 tau, and pS199 tau showed no significant differences between AD and controls (t‐tau: P = .398, pT181 tau: P = .7235, and pS199 tau: P = .4384; Figure 1D and Table S1). Conversely, pS396 tau was significantly increased in AD‐brain derived EVs over controls (pS396 tau: P = .0375; Figure 1D and S1). Moreover, we observed a significant increase in Aβ1‐42 in AD‐derived EVs over controls (P < .0001), but not in Aβ1‐40 (P = .119; Figure 1E and Table S1). The amount of pS396 and Aβ1‐42 in the brain homogenate tissue were quantified by ELISA, and we performed the bivariate correlation analysis between the homogenate samples and EV samples purified from the same AD brain samples. There is a significant difference in pS396 tau and Aβ1‐42 levels in the brain tissue homogenate between AD and controls (pS396 tau; P < .0001, Aβ1‐42; P = .0001). In addition, there is a significant positive correlation between the brain tissue homogenate and the EVs. The results suggest increased pS396 and Aβ1‐42 level in the brain tissue might be related with elevation of pS396 and Aβ1‐42 in the EVs (pS396; r = 0.4987, P = .0050, Aβ1‐42; r = 0.5632, P = .0005; Figure 1F).
FIGURE 1.
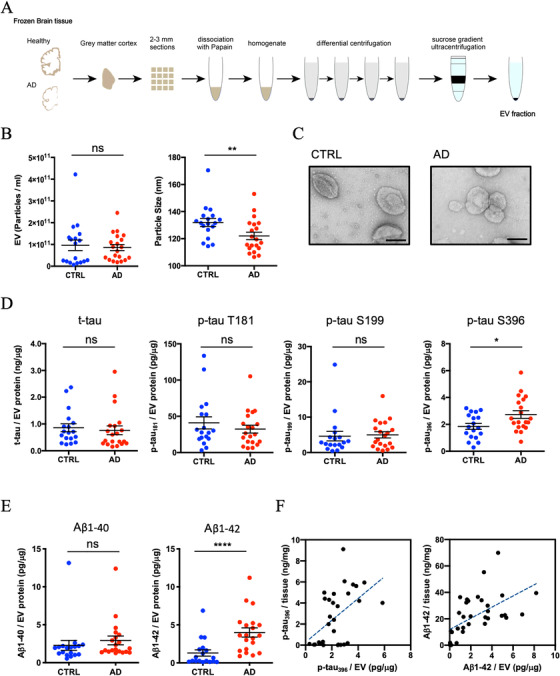
Biophysical and biochemical characterization of extracellular vesicles (EVs) isolated from Alzheimer's disease (AD) and control (CTRL)brain tissues: A, Schematic of extracellular vesicle isolation protocol from human frozen brain tissue (see supporting information for detailed methods). B, Left: particle numbers of brain‐derived EV fraction from control (CTRL) or AD by nanoparticle tracking analysis. P = .6075 by Mann‐Whitney test. Right: Particle size of brain‐derived EV fraction. P = .0095 by Mann‐Whitney test. C, Transmission electron microscopy image of frozen human brain‐derived EVs. Scale bar = 100 nm. Left: CTRL, Right: AD. D, Comparison of total tau and tau phosphorylated at threonine 181, serine 199, and serine 396 in EVs. pS 396 tau; P = .0375 by Mann‐Whitney test. E, Comparison of amyloid beta 1‐40 or 1‐42 in EVs. Aβ1‐42; P < .0001 by Mann‐Whitney test. F, Scattered plot of brain tissue homogenates and brain‐derived EVs. Left: pS396 tau (r = 0.4897, P = .005 using two‐tailed t‐test), Right: Aβ1‐42 (r = 0.5632, P = .0005 using two‐tailed t‐test)
3.2. Proteomic profiling of brain‐derived EVs
We performed a label‐free Nano‐LC‐MS/MS analysis of 38 EV samples for proteomic profiling. Across both cohorts, a total of 1088 proteins were identified with at least two unique peptides (Figure 2A and Tables S3 and S4 in supporting information). There were 940 proteins identified in control EVs and 1000 proteins identified in AD EVs. Among them, 852 proteins were detected in both groups, with 88 proteins unique to the controls and 148 proteins unique to the AD group (Figure 2A). The common, AD‐unique, and control‐unique proteins were tested for properties pertaining to the “cellular component” and “pathway” ontologies by Gene Ontology analysis in the Database for Annotation, Visualization and Integrated Discovery (DAVID). Among the 852 shared proteins, 60.9% were found to be included in the extracellular exosome ontology (Figure 2B). The 148 proteins unique to the AD group were linked to mitochondria metabolism known to be dysfunctional in AD brain 28 (Figure 2B). Interestingly, in pathway analysis by DAVID, neurodegenerative disorders, including AD and Parkinson's and Huntington's diseases were enriched in common and unique proteins (Figure 2C). Figure 2D shows the peptides identified in AD group (not all AD patients) by Nano‐LC‐MS/MS, which covered 55.1% of tau (1‐441), 9.1% of amyloid‐beta precursor protein (APP 1‐770), 74.3% of alpha‐synuclein (SNCA 1‐140) and 6.3% of apolipoprotein E (APOE 1‐317). Post‐translational modification (PTM) analysis detected six phosphorylation sites (pT181, pS198, pS199, pS202, pT231, and pS404) on tau (Figure 2D, red bold letters). Notably, Aβ sequence was identified in APP fragments. Figure 2E shows the AD pathway from KEGG pathway analysis based on 68 proteins identified in the AD group, which are designated with red stars. The list of AD pathway‐related proteins is provided in Table S5 in supporting information. Proteins known to play an important role in AD pathogenesis, such as APP, APOE, tau, and NADH‐ubiquinone oxidoreductase (Cx I‐V), were all identified in the AD group, although they were not unique to this group.
FIGURE 2.
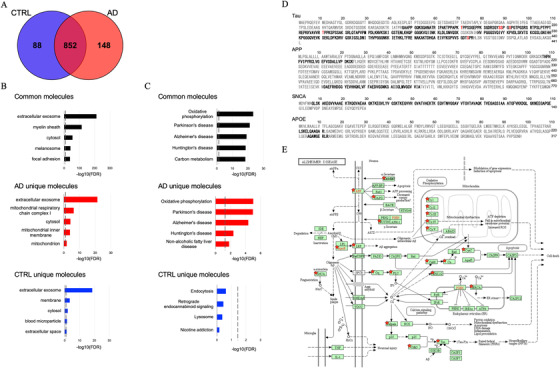
Proteomics profiling of extracellular vesicles (EVs) isolated from Alzheimer's disease (AD) and control (CTRL) brains: A, Venn diagram representing the number of EV proteins differentially identified in CTRL and AD. B, Gene ontology (GO) analysis using DAVID Bioinformatics Resources 6.8. The GO term of Top 5 Cellular Component with ‐log10 (FDR P‐value). C, The GO term of Top 5 Pathway Ontology with ‐log10 (FDR ‐value). D, Sequence coverage of identified tryptic fragment peptide from AD‐related protein (tau, APP, SNCA, APOE) in AD group by LC‐MS/MS analysis. Identified peptides and phosphorylation sites are shown in black and red bold, respectively. E, KEGG pathway of AD. The 68 proteins identified in the AD group are highlighted by red stars
3.3. Analysis of label‐free quantitative proteomics comparison of AD and control brain‐derived EVs
Label‐free quantitative proteomics analysis was performed using PEAKS studio software. A total of 949 proteins were quantified (Figure 3A and Table S6 in supporting information). The 934 quantified proteins were common between AD and control groups. Between these groups, three proteins were uniquely expressed in the controls, while 12 proteins were uniquely expressed in the AD group. The principal component analysis (PCA) showed a marginal separation of the two groups (Figure 3B). Figure 3C shows the volcano plot of the common 289 proteins that were detected in > 50% of the group (AD: n > 10 and controls: n > 9). Among these proteins, 15 proteins were significantly upregulated and three proteins were significantly downregulated in AD compared to the control group (as determined by P < 0.05 and log2 fold change threshold of > 1 or < −1; Figure 3C and Table 2). The expression levels of 18 molecules identified in the AD group relative to the control group are displayed in a heatmap (Figure 3D). We next searched for brain cell‐type–specific molecules within the EV proteomics dataset using the mouse proteomics dataset as a reference. 29 The top 100 ranked cell type–specific molecules, which have at least two‐fold change in concentration in the cell type of interest over the other cell types, were tested with our EV proteomics dataset (Figure 3E). The distribution of these markers indicates that in the human brain, 49% of the identified molecules were related to neuronal origin, whereas the other 50% of EV proteins are related to glial origin, including microglia, astrocytes, and oligodendrocytes. Moreover, using label‐free quantitative value, differences in the expression of cell type–specific marker molecules between AD and controls were seen (Figure 3F). Interestingly, neuron‐specific molecules were enriched in the control group (Figure 3F, blue), while glia‐specific marker molecules were enriched in the AD group (Figure 3F, red). These results suggest that glia may proliferate upon inflammatory response or accelerate their turnover, leading to enhanced generation of EVs in AD brains, which may be attributed to enhanced gliosis often seen in AD brains.
FIGURE 3.
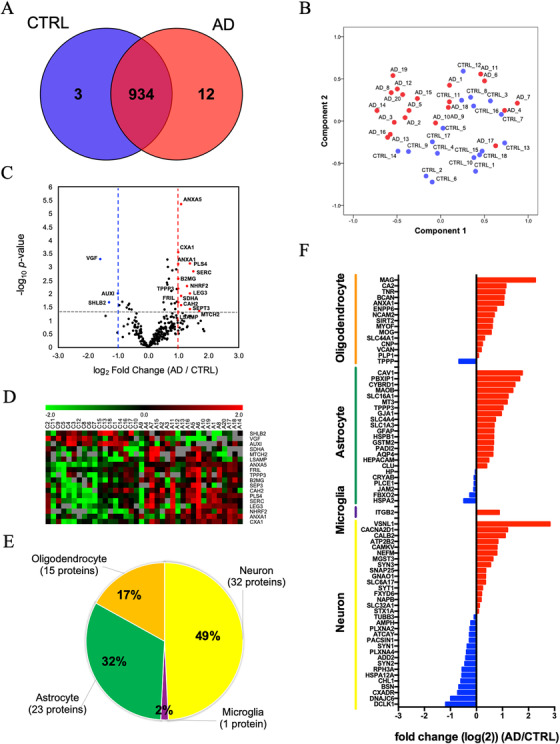
Label‐free quantitative proteomics comparison of Alzheimer's disease (AD) brain‐derived extracellular vesicles (EVs) and control (CTRL) brain‐derived EVs: A, Venn diagram representing the number of EV proteins differentially expressed in CTRL and AD. B, A principal component analysis (PCA). CTRL; blue symbols, AD; red symbols. C, Volcano plot showing a degree of differential expression of EV proteins in AD compared with CTRL. X‐axis; log transformed fold change in expression, y‐axis; log‐transformed P‐values. Gray dot lines: 0.1 P‐value and 1‐ or ‐1‐fold change cutoff. D, Heat map representation of the up‐ and downregulated proteins in AD. Red shows upregulated proteins, and green shows downregulated proteins. E, Enrichment of brain cell type–specific markers in brain‐derived EV proteins. Yellow: Neuron, purple: microglia, green: astrocytes, orange: oligodendrocytes. The parentheses show the number and percentage of identified cell type‐specific protein. F, Comparison of the cell type–specific protein in AD brain‐derived EV and CTRL EV. The red bar shows higher expression in AD. Blue bar indicates higher expression in CTRL
TABLE 2.
The list of up‐ and downregulated protein in expression between Alzheimer's disease and control
| UniProt ID | Protein name | log2(Fold change) (AD/CTRL) a | ‐log10(P‐value) b |
|---|---|---|---|
| P08758 | ANXA5 | 1.1 | 5.34 |
| P17302 | CXA1 | 1.0 | 3.54 |
| O15240 | VGF | −1.6 | 3.28 |
| Q9NRQ2 | PLS4 | 1.4 | 3.11 |
| P04083 | ANXA1 | 1.0 | 3.10 |
| Q9Y617 | SERC | 1.5 | 2.83 |
| P61769 | B2MG | 1.0 | 2.55 |
| Q15599 | NHRF2 | 1.3 | 2.27 |
| Q9BW30 | TPPP3 | 1.0 | 2.18 |
| P17931 | LEG3 | 1.4 | 2.00 |
| O75061 | AUXI | −1.0 | 2.00 |
| P31040 | SDHA | 1.1 | 1.90 |
| P02792 | FRIL | 1.0 | 1.67 |
| Q9NR46 | SHLB2 | −1.3 | 1.67 |
| P00918 | CAH2 | 1.1 | 1.55 |
| Q9UH03 | SEPT3 | 1.4 | 1.42 |
| Q9Y6C9 | MTCH2 | 1.7 | 1.33 |
| Q13449 | LSAMP | 1.0 | 1.30 |
The log2 fold change were calculated by the protein intensity which was analyzed by PEAKS software.
The statistical significance of the differences were calculated using student's t test.
3.4. Machine learning to identify distinctive AD brain‐derived EV proteins
To discover a combination of protein molecules that can accurately distinguish AD EVs from controls, the label‐free quantitative proteomics dataset was analyzed using a machine learning method (Figure 4A). For this purpose, we split the proteomics dataset into an unblinded training subset (AD: n = 11; control: n = 10) and a blinded testing subset (AD: n = 9; control: n = 8). The ensemble machine learning model was built using only the data from the training subset, and then the accuracy of the diagnosis was determined using only the blinded testing set. We found that a panel that included annexin‐A5 (ANXA5), Neurosecretory protein VGF (VGF), neuronal membrane glycoprotein M6‐a (GPM6A), and alpha‐centractin (ACTZ), selected by the LASSO algorithm, resulted in an area under the ROC curve (AUC) of 0.95 within the training set (Figure 4B and Table S7 in supporting information). We then examined the accuracy in the independent blinded test set using the four proteins in the dotted green box (Figure 4B). Using this model, we achieved an 88% accuracy (AUC = 0.97) in identifying AD patients using the panel that consisted of the four proteins (Figure 4C). Further, we ran two control experiments that randomly selected four proteins from a total possible 949 proteins to form the diagnosis panel (repeated 20 times, AUC = 0.58, accuracy = 55%) and shuffling the true labels of the subjects within the training set (AUC = 0.47, accuracy = 48%). The control's AUC was significantly worse than using the four‐protein panel's AUC (P < .001) (Figure 4C). Figure 4D shows the scatter plot of four selected proteins, which were significantly differentially expressed between AD and control groups (Table S8 in supporting information). Next, we assessed the correlation of expression of these candidate molecules to the levels of p‐tau and Aβ1–42 in EVs by Pearson's correlation analysis. There was a significantly positive correlation between GPM6A and pS396 tau, and between GPM6A and Aβ1‐42 levels (pS396 tau: r = 0.380, P = .019; Aβ1‐42: r = 0.387, P = .016), and a significantly negative correlation between VGF and Aβ1‐42 levels (r = −0.538, P = .002) (Figure 4E and Table S9 in supporting information). ANXA5 expression level was significantly increased in AD brain‐derived EVs compared to the control group in the validation cohort as determined by ELISA (P = .0042, 42 AD and 36 control cases, Figure 4F). Although there was statistical difference in PMI between AD and control cases in this cohort (Table 1), Pearson's correlation analysis of PMI and ANXA5 levels showed no significant correlation (r = −0.165, P = .149). Thus, this is not due to the difference in PMI between the two groups. Interestingly, ANXA5 expression level shows a tendency to increase along with Braak stages in an AD‐dependent manner (Figure 4F). Therefore, ANXA5 is a potential EV molecule for both distinguishing AD and control EVs and as a surrogate marker for Braak stage.
4. DISCUSSION
The biophysical properties of EVs isolated from unfixed post mortem human brain tissues, quantitative analysis of tau and Aβ species, and label‐free quantitative proteomic profiling by Nano‐LC‐MS/MS analyses were performed. pS396 tau and Aβ1‐42 levels were significantly increased in AD brain‐derived EVs compared to controls. A total of 1088 unique proteins from brain‐derived EVs were found to be enriched as extracellular exosomes molecules. We also quantified 949 proteins by label‐free quantitative proteomic analysis, which were enriched in neuron‐specific molecules in controls and glial cell type‐specific molecules in the AD group. We used the feature selection algorithm LASSO to select a panel of protein biomarkers that could accurately diagnose AD, including ANXA5, VGF, GPM6A, and ACTZ. It is important to note that the feature selection algorithm we used identifies the best panel of biomarkers for diagnosis, but is not necessarily a list of the most informative individual biomarkers. LASSO has the property that if there were, for example, two excellent biomarkers for AD, but which correlated highly to one another, only one of these biomarkers would be included in the panel because including both would bring only redundant information. 30 Using the validation cohort with the larger sample size, the increased protein level of ANXA5 in the AD group, compared to controls, was confirmed by ELISA.
Previous reports indicate pT181 tau to be an early PTM associated with AD, pS199 tau modification is thought to promote tau accumulation, and pS396 tau modification is associated with tau seeding activity and aggregation. 31 , 32 The PTM of tau at either pT181, pS199, or/and pS396 may facilitate the recruitment of tau in EVs, but neither pT181 nor pS199 tau is enriched in AD brain‐derived EVs. This was unexpected because both pT181 and pS396 tau are elevated in neuron‐derived exosomes in plasma samples obtained from AD and prodromal AD cases. 33 One potential explanation is that phosphorylation of specific sites on tau may be more preferentially incorporated into EV presumably via their ubiquitination, which is necessary for their multivesicular body (MVB) incorporation. In general, protein phosphorylation induces ubiquitination of lysine residues proximal to the phosphorylation sites, and it is necessary for ESCRT‐mediated incorporation of ubiquitinated molecules into MVBs. Ubiquitination sites known for PHF‐tau are K254, K311, and K353, 34 and phosphorylation of tau at S396 may facilitate its MVB sorting through ubiquitination of those lysine residues. In addition, we also observed tau fragments from the mid‐region (156 to 406), which is inclusive of proline‐rich domains (151 to 240), and microtubule binding repeat domains (24 to 369). Tau can be cleaved by various proteases including calpain‐1 and ‐2 (at R230), thrombin, cathepsins, asparagine endopeptidase (at D255 and N368), caspase‐2 (at D314), and caspase‐3 (at D421). 35 , 36 , 37 , 38 A further investigation is needed to determine how tau is truncated, phosphorylated, and enriched in AD brain‐derived EVs.
In the present study, we observed that the size of EVs derived from AD brain samples was smaller than from control EVs. It is possible that cholesterol level might be related to EV size, as small EVs contain significantly higher cholesterol than large EVs 39 , 40 and accumulation of cholesterol is associated with AD. 41 Enhancement of cholesterol levels increases Aβ production in cellular and animal models of AD, and inhibition of cholesterol synthesis reduces Aβ in this model. 39 , 40 , 41 , 42 , 43 Therefore, AD brain cells that accumulate high cholesterol might secrete smaller EVs than controls.
The gene ontology of proteins identified in AD group showed mitochondria metabolism category. It is well known that mitochondrial dysfunction occurs in AD and by Aβ stimulation. 44 , 45 Recombinant tau mutant mimicking S396/S404 phosphorylation can enhance Aβ‐induced mitochondrial damage and neuronal dysfunction, 46 and mitochondrial dysfunction can be an upstream inducer of tau phosphorylation in AD. 44 In addition, mitochondrial dysfunction leads to their sorting to endolysosomal system and MVBs, which can release mitochondria‐derived vesicles into the extracellular space as exosomes. 47 These reports suggest that Aβ accumulation induces mitochondrial dysfunction, increases tau phosphorylation at pS396, and enriches mitochondrial molecule in the EVs. Indeed, accumulation of misfolded proteins and mitochondrial dysfunction induces secretion of large vesicles (exophers) containing dysfunctional organelle and misfolded proteins for neuroprotection in Caenorhabditis elegans. 48 In terms of enriched ANXA5 in AD brain‐derived EVs, the cellular apoptosis induced by mitochondrial dysfunction expresses phosphatidylserine on the cell surface and recruit ANXA5, which is a well‐known phosphatidylserine binding molecule. ANXA5‐containing plasma membrane may be incorporated into the endosome, eventually to MVBs and secreted in EVs. There is, however, no literature to understand the connection between mitochondrial dysfunction and VGF, GPM6A, or ACTZ.
The quantitative proteomic analysis of brain‐derived EV samples isolated from AD and control patients found enriched glia‐specific molecules in AD brain‐derived EVs, and identified ANXA5, VGF, GPM6A, and ACTZ as potential candidate molecules for monitoring the progression of AD. Although the Aβ and tau are tested for the diagnosis or progression of Aβ and tau pathology in AD, it may not cover atypical AD cases in which A+T+N+ phenotype is incomplete. The four candidate molecules that would predict degenerative changes of neuronal and non‐neuronal cells may facilitate the cellular based evaluation of the brain state in AD. Zhang et al. have recently reported that ANXA5 is associated with familial late‐onset AD by whole exome sequencing. 49 Further, Sohma et al. have reported a significantly higher plasma level of ANXA5 in AD cases compared to controls. 50 A number of previous studies in CSF and brain tissue have reported markedly lowered concentration of VGF in AD cases compared to controls. 51 , 52 , 53 , 54 GPM6A expression level was reported to be downregulated in the hippocampus of human AD brain tissues and transgenic AD mouse brains. 55 , 56 Because GPM6A homolog is located in neuropil in Drosophila melanogaster, 57 elevated GPM6A in AD brain EVs may be an indicator of the neuropil loss. Indeed, GPM6A is reported to cluster in lipid rafts upon palmitoylation, which are also enriched in sphingolipids and cholesterol. 58 Thus, reduction in GPM6A in brain tissue may be negatively correlated with GPM6A enrichment in EVs. In our study, ANXA5 was detected in brain‐derived EVs from validation cohort, but VGF, GPM6A, and ACTZ were undetectable by commercial ELISA kits. Further study is necessary to validate these molecules with higher sensitivity ELISA. Finally, the combination of Aβ, tau, and cell type–specific molecules from brain cells, including ANAX5, VGF, GPM6A, or ACTZ may serve as potential biomarker candidate molecules in AD patient body fluid samples.
AUTHOR CONTRIBUTIONS
Satoshi Muraoka, Annina M. DeLeo, and Tsuneya Ikezu designed research; Satoshi Muraoka, Annina M. DeLeo, Manveen K. Sethi, Kayo Yukawa‐Takamatsu, Zhi Ruan, Yang You, Yuzhi Wang, and Mei Chen performed research; Satoshi Muraoka, Annina M. DeLeo, Manveen K. Sethi, Zijian Yang, Jina Ko, John D. Hogan, Weiming Xia, Joseph Zaia, and Tsuneya Ikezu analyzed data; Santhi Gorantla and Howard E. Gendelman provided brain samples; Satoshi Muraoka and Tsuneya Ikezu wrote the paper; Satoshi Muraoka, Annina M.DeLeo, Manveen K. Sethi, Seiko Ikezu, Jina Ko, Zijian Yang, Maria Medalla, David Issadore, Weiming Xia, Joseph Zaia, and Tsuneya Ikezu edited the paper.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The author thanks Maria Ericsson (Electron Microscopy Facility, Harvard Medical School) for electron microscopic imaging services, participating laboratories of NIH NeuroBioBank for providing frozen human brain tissue specimens, and Li Wu (University of Nebraska Medical Center) for providing human brain tissue specimens.
FUNDING INFORMATION
This work is in part funded by Alzheimer's Association AARF‐9550302678 (AMD & SM), DVT‐14‐320835 (TI), BrightFocus Foundation (A2016551S), Cure Alzheimer's Fund (TI), National Institute of Health (NIH), National Institute in Aging (NIA) and National Institute of Neurological Disorders and Stroke (NINDS) RF1AG054199 (TI), NIH R56AG057469 (TI), NIH R01AG054672 (TI), NIH R21NS104609 (TI).
CONFLICTS OF INTEREST
Tsuneya Ikezu collaborates with Abbvie Inc. (USA), Aethlone Medical, Inc. (USA), Eisai (Japan/USA) and Ono Pharmaceutical (Japan), and consults Takeda (Japan/USA).
Muraoka S, DeLeo AM, Sethi MK, et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer's disease human brain tissues. Alzheimer's Dement. 2020;16:896–907. 10.1002/alz.12089
Satoshi Muraoka and Manveen K. Sethi contributed equally to this study.
REFERENCES
- 1. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule‐associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051‐4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Primers. 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 4. Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. [DOI] [PubMed] [Google Scholar]
- 5. Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. You Y, Ikezu T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol Dis. 2019;130:104512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Delpech J‐C, Herron S, Botros MB, Ikezu T. Neuroimmune crosstalk through extracellular vesicles in health and disease. Trends Neurosci. 2019;42:361‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell. 2016;30:836‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeLeo AM, Ikezu T. Extracellular vesicle biology in Alzheimer's disease and related tauopathy. J Neuroimmune Pharmacol. 2018;13:292‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo BB, Bellingham SA, Hill AF. Stimulating the release of exosomes increases the intercellular transfer of prions. J Biol Chem. 2016;291:5128‐5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2014;35:1792‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bulloj A, Leal MC, Xu H, Castaño EM, Morelli L. Insulin‐degrading enzyme sorting in exosomes: a secretory pathway for a key brain amyloid‐beta degrading protease. J Alzheimers Dis. 2010;19:79‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid‐modulated exosome secretion promotes clearance of amyloid‐β by microglia. J Biol Chem. 2012;287:10977‐10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dujardin S, Bégard S, Caillierez R, et al. Ectosomes: a new mechanism for non‐exosomal secretion of tau protein. Plos One. 2014;9:e100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Polanco JC, Li C, Durisic N, Sullivan R, Götz J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol Commun. 2018;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y, Balaji V, Kaniyappan S, et al. The release and trans‐synaptic transmission of Tau via exosomes. Mol Neurodegener. 2017;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polanco JC, Scicluna BJ, Hill AF, Götz J. Extracellular vesicles isolated from the brains of rtg4510 mice seed tau protein aggregation in a threshold‐dependent manner. J Biol Chem. 2016;291:12445‐12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crotti A, Sait HR, McAvoy KM, et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci Rep. 2019;9:9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vella LJ, Scicluna BJ, Cheng L, et al. A rigorous method to enrich for exosomes from brain tissue. J Extracell Vesicles. 2017;6:1348885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perez‐Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl‐terminal fragments from the cell into the brain extracellular space. J Biol Chem. 2012;287:43108‐43115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hurwitz SN, Sun L, Cole KY, Ford CR, Olcese JM, Meckes DG. An optimized method for enrichment of whole brain‐derived extracellular vesicles reveals insight into neurodegenerative processes in a mouse model of Alzheimer's disease. J Neurosci Methods. 2018;307:210‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gallart‐Palau X, Serra A, Sze SK. Enrichment of extracellular vesicles from tissues of the central nervous system by PROSPR. Mol Neurodegener. 2016;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adusumilli R, Mallick P. Data conversion with proteowizard msConvert. Methods Mol Biol. 2017;1550:339‐368. [DOI] [PubMed] [Google Scholar]
- 25. Ko J, Baldassano SN, Loh P‐L, Kording K, Litt B, Issadore D. Machine learning to detect signatures of disease in liquid biopsies ‐ a user's guide. Lab on A Chip. 2018;18:395‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ko J, Bhagwat N, Yee SS, Ortiz N, et al. Combining machine learning and nanofluidic technology to diagnose pancreatic cancer using exosomes. ACS Nano. 2017;11:11182‐11193. [DOI] [PubMed] [Google Scholar]
- 27. Silverman JM, Christy D, Shyu CC, et al. CNS‐derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J Biol Chem. 2019;294:3744‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saman S, Lee NCY, Inoyo I, et al. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer's disease. J Alzheimers Dis. 2014;40(Suppl 1):S47‐S70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma K, Schmitt S, Bergner CG, et al. Cell type‐ and brain region‐resolved mouse brain proteome. Nat Neurosci. 2015;18:1819‐1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Statist Soc B. 2005;67:301‐320. [Google Scholar]
- 31. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112‐119. [DOI] [PubMed] [Google Scholar]
- 32. Takeda S, Wegmann S, Cho H, et al. Neuronal uptake and propagation of a rare phosphorylated high‐molecular‐weight tau derived from Alzheimer's disease brain. Nat Commun. 2015;6:8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case‐control study. Alzheimers Dement. 2015;11:600‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease‐specific conformation of hyperphosphorylated paired helical filament‐Tau is polyubiquitinated through Lys‐48, Lys‐11, and Lys‐6 ubiquitin conjugation. J Biol Chem. 2006;281:10825‐10838. [DOI] [PubMed] [Google Scholar]
- 35. Rissman RA, Poon WW, Blurton‐Jones M, et al. Caspase‐cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Z, Song M, Liu X, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med. 2014;20:1254‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurbatskaya K, Phillips EC, Croft CL, et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathol Commun. 2016;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao X, Kotilinek LA, Smith B, et al. Caspase‐2 cleavage of tau reversibly impairs memory. Nat Med. 2016;22:1268‐1276. [DOI] [PubMed] [Google Scholar]
- 39. Durcin M, Fleury A, Taillebois E, et al. Characterisation of adipocyte‐derived extracellular vesicle subtypes identifies distinct protein and lipid signatures for large and small extracellular vesicles. J Extracell Vesicles. 2017;6:1305677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pfrieger FW, Vitale N. Cholesterol and the journey of extracellular vesicles. J Lipid Res. 2018;59:2255‐2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Torres M, Busquets X, Escribá PV. Brain lipids in the pathophysiology and treatment of Alzheimer's disease. update on dementia. InTech. 2016. 10.5772/64757. [DOI] [Google Scholar]
- 42. Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345‐351. [DOI] [PubMed] [Google Scholar]
- 43. Evangelisti E, Cecchi C, Cascella R, et al. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J Cell Sci. 2012;125:2416‐2427. [DOI] [PubMed] [Google Scholar]
- 44. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer's Disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40:151‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cadonic C, Sabbir MG, Albensi BC. Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol Neurobiol. 2016;53:6078‐6090. [DOI] [PubMed] [Google Scholar]
- 46. Quintanilla RA, Bernhardivon R, Godoy JA, Inestrosa NC, Johnson GVW. Phosphorylated tau potentiates Aβ‐induced mitochondrial damage in mature neurons. Neurobiol Dis. 2014;71:260‐269. [DOI] [PubMed] [Google Scholar]
- 47. Picca A, Guerra F, Calvani R, et al. Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int J Mol Sci. 2019;20:E805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Melentijevic I, Toth ML, Arnold ML, et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang X, Zhu C, Beecham G, et al. A rare missense variant of CASP7 is associated with familial late‐onset Alzheimer's disease. Alzheimers Dement. 2019;15:441‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sohma H, Imai S‐I, Takei N, et al. Evaluation of annexin A5 as a biomarker for Alzheimer's disease and dementia with lewy bodies. Front Aging Neurosci. 2013;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cocco C, D'Amato F, Noli B, et al. Distribution of VGF peptides in the human cortex and their selective changes in Parkinson's and Alzheimer's diseases. J Anat. 2010;217:683‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Llano DA, Devanarayan P, Devanarayan V. Alzheimer's disease neuroimaging initiative (adni). vgf in cerebrospinal fluid combined with conventional biomarkers enhances prediction of conversion from mci to aD. Alzheimer Dis Assoc Disord. 2019;33:307‐314. [DOI] [PubMed] [Google Scholar]
- 53. Hölttä M, Minthon L, Hansson O, et al. An integrated workflow for multiplex CSF proteomics and peptidomics‐identification of candidate cerebrospinal fluid biomarkers of Alzheimer's disease. J Proteome Res. 2015;14:654‐663. [DOI] [PubMed] [Google Scholar]
- 54. Brinkmalm G, Sjödin S, Simonsen AH, et al. A parallel reaction monitoring mass spectrometric method for analysis of potential csf biomarkers for Alzheimer's disease. Proteomics Clin Appl. 2018;12:1700131. [DOI] [PubMed] [Google Scholar]
- 55. Xu P‐T, Li Y‐J, Qin X‐J, et al. Differences in apolipoprotein E3/3 and E4/4 allele‐specific gene expression in hippocampus in Alzheimer disease. Neurobiol Dis. 2006;21:256‐275. [DOI] [PubMed] [Google Scholar]
- 56. Woo J‐M, Park SJ, Kang HI, et al. Characterization of changes in global gene expression in the brain of neuron‐specific enolase/human Tau23 transgenic mice in response to overexpression of Tau protein. Int J Mol Med. 2010;25:667‐675. [DOI] [PubMed] [Google Scholar]
- 57. Zappia MP, Bernabo G, Billi SC, Frasch AC, Ceriani MF, Brocco MA. A role for the membrane protein M6 in the drosophila visual system. Bmc Neurosci. 2012;13:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ito Y, Honda A, Igarashi M. Glycoprotein M6a as a signaling transducer in neuronal lipid rafts. Neurosci Res. 2018;128:19‐24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information