

Summary
Identifying oncogenic drivers and tumor suppressors remains a challenge for many forms of cancer, including rhabdomyosarcoma. Anticipating gene expression alterations encrypted by DNA copy-number variants to be particularly important, we developed a computational and experimental strategy incorporating a Bayesian algorithm and CRISPR/Cas9 “mini-pool” screen enabling both genome-scale assessment of disease genes and functional validation. The algorithm, called iExCN, identified 29 rhabdomyosarcoma drivers and suppressors enriched for cell cycle and nucleic acid binding activities. Functional studies showed many iExCN genes to represent rhabdomyosarcoma line-specific or shared vulnerabilities. Complementary experiments addressed modes of action and demonstrated coordinated repression of multiple iExCN genes during skeletal muscle differentiation. Analysis of two separate cohorts revealed that the number of iExCN genes harboring copy-number alterations correlates with survival. Our findings highlight rhabdomyosarcoma as a cancer in which multiple drivers influence disease biology and demonstrate a generalizable capacity for iExCN to unmask previously-unrecognized disease genes in cancer.
Introduction
In the genomics era, technological advancements and sophisticated computational approaches to identify mutated genes have illuminated the molecular basis of many cancer hallmarks (Hanahan and Weinberg, 2011), but only rarely has that insight reached the clinic. The shortcoming is variably due to the paucity or the vast number of somatic single nucleotide variants (SNVs) in a tumor, limited recurrence of specific SNVs within an individual cancer type, lack of ability to target a cancer-driving mutation, and incorrect assumptions about the relevance of individual aberrations (Heuckmann and Thomas, 2015). This problem applies to rhabdomyosarcoma (RMS), an aggressive soft tissue sarcoma composed of skeletal myoblast-like cells (Hettmer and Wagers, 2010; Saab et al., 2011). The cancer is divided into two major histologic subtypes marked by embryonal or alveolar histology features (ERMS and ARMS, respectively), with most ARMS carrying a balanced translocation generating an oncogenic fusion protein consisting of PAX3 or PAX7 and FOXO1 (Saab et al., 2011). Next-generation sequencing approaches reveal the fusion-positive (FP) RMS cases to harbor few additional chromosomal rearrangements and somatic SNVs; in contrast, fusion-negative (FN) cases display many more SNVs, but recurrence of SNVs across RMS specimens is very limited (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014).
In contrast to SNVs, DNA copy-number variations (CNVs) are common and more highly recurrent in FN RMS (Paulson et al., 2011). Therefore, we considered that altered expression of key genes might be “hard-wired” by coordinately increased or decreased copies of those genes. To sift through the vast number of CNVs, we developed a pipeline incorporating Bayesian methodology (Kruschke, 2013; Kruschke and Liddell, 2017) to integrate expression and copy-number (iExCN pipeline). The iExCN analysis of FN RMS revealed candidate oncogenic drivers and tumor suppressor genes (TSGs), most of which were not previously known to be important in this disease. We used a “mini-pool” of lentiviral CRISPR/Cas9 vectors and additional functional analyses to validate the importance of many iExCN-defined genes. We revealed multiple genes with coordinate low-level copy-number and expression changes that contribute to RMS; illuminated mechanisms underlying a corrupted myogenic differentiation program; and identified potentially actionable therapeutic targets and a prognostic biomarker for this disease. Taken together, we provide a general approach to identify oncogenic drivers and TSGs in forms of cancer in which mutation calling and CNV analyses have failed to reveal the full spectrum of disease drivers.
Results
The iExCN pipeline reveals 29 candidate RMS oncogenic drivers and tumor suppressors
Using published (Shern et al., 2014) and unpublished data derived from 290 RMS specimens (Figure 1A, Table S1), our analyses confirmed recent reports showing the paucity of recurrent SNVs in most protein-coding genes (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014). For example, in 134 FN and FP RMS cases with WGS or WES data, we identified somatic protein-altering SNVs in 3827 genes, with 3034 (79%) found in only single cases and 3748 (98%) identified in three or fewer different cases (Figure 1B; Table S2). Mutated genes with recurrence in three or more cases included NF1, KRAS, NRAS, PIK3CA, and FGFR4, again highlighting the importance of RTK/RAS/PIK3CA signaling in RMS (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014).
Figure 1. Integrative genomic analysis identifies few recurrent SNVs and frequent copy-number alterations in RMS.
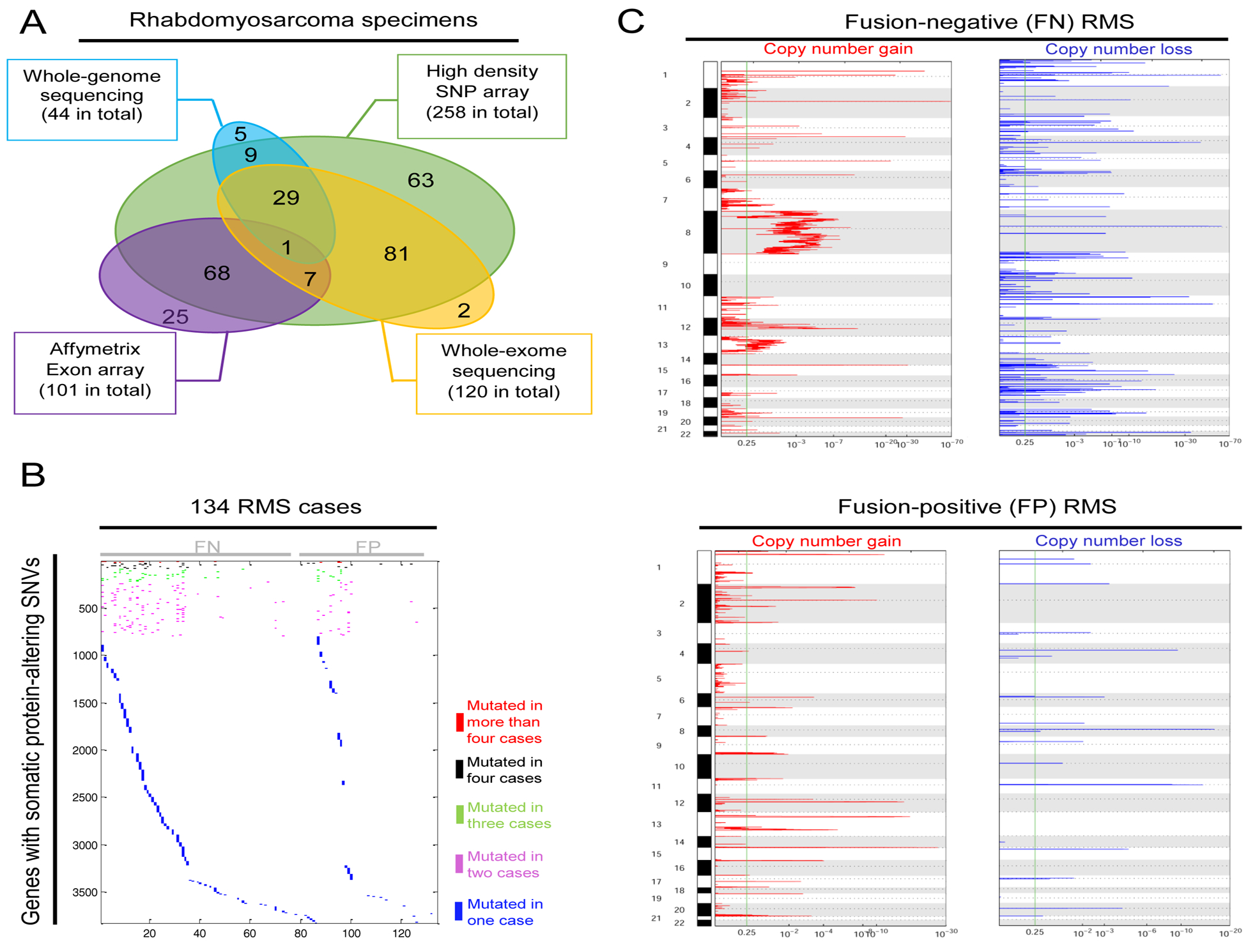
(A) Venn diagrams display the number of RMS cases and type of genomic data used in this analysis.
(B) Analysis of somatic protein-altering single nucleotide variants (SNVs) in 134 pairs of RMS tumor-normal specimens with whole-genome sequencing (WGS) and/or whole-exome sequencing (WES) data reveals that the vast majority of more than 3827 mutated genes were found only in single RMS cases.
(C) Charts show statistically significant regions displaying copy number gains (left) and losses (right) identified by applying Genomic Identification of Significant Targets in Cancer 2.0 (GISTIC2) algorithm in RMS cases with PAX3/7-FOXO1 fusion genes (FP cases) or without fusion genes (FN cases).
Given the limited SNV recurrence, we considered whether integrative analyses of CNVs could provide a more complete description of cancer drivers and suppressors. GISTIC analysis demonstrated many CNVs in both RMS forms, with many more (n = 1797) significantly altered genes in FN RMS (Figure 1C, Table S3), consistent with previous reports (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014). Considering that functionally relevant CNVs would likely influence expression of the involved protein-coding genes, we developed iExCN based on Bayesian estimation of the probability that expression of individual genes is correlated with CNV (Figure S1A). [Details of development, testing, and a link to iExCN source code provided in Supplemental Experimental Procedures) (Figure S1)].
Focusing on FN RMS, which harbors many more CNVs, we used iExCN to uncover coordinate protein-coding gene copy-number and expression changes. This analysis revealed 25 candidate oncogenic drivers and four tumor suppressors, assuming that copy-number gain and increased expression signified oncogenes and vice versa for TSGs (Figure 2A). Unlike SNVs that usually applied to only single FN RMS cases (Figure 1B), most iExCN genes were broadly expressed in FN RMS (Figure 2B). Similarly, the copy-number gains and losses in iExCN genes each involved 50 or more FN RMS cases in our cohort (Figure 2C and D).
Figure 2. iExCN analysis identifies RMS oncogenic drivers and tumor suppressors.
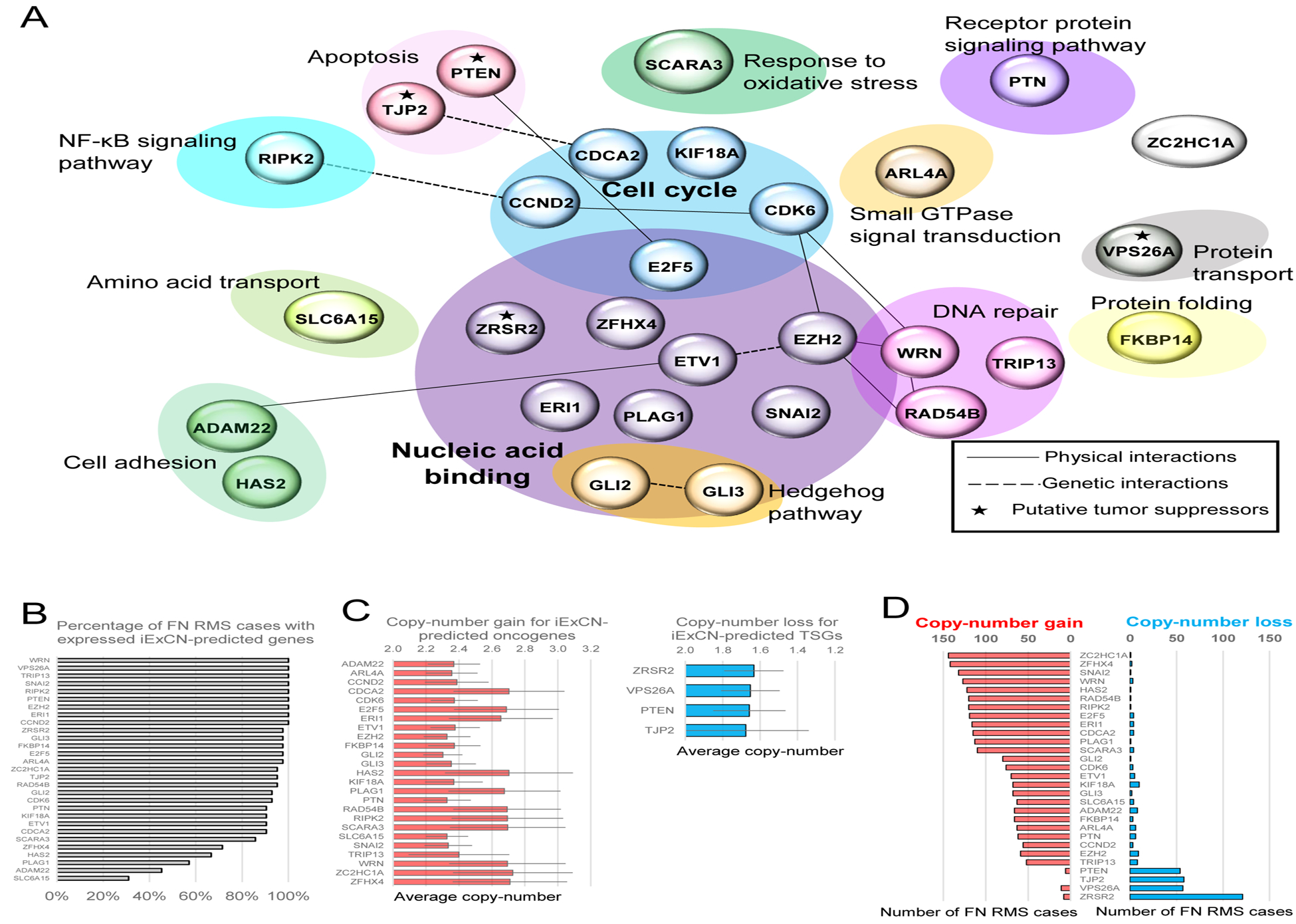
(A) Map shows 29 candidate RMS oncogenic drivers and tumor suppressors (*) within Gene Ontology terms (shaded ovals). Physical and genetic interactions from GeneMANIA database are indicated by solid and dashed lines, respectively. Cell cycle and Nucleic acid binding are the only categories with statistically significant enrichment.
(B) Chart displays the number of FN RMS cases (%) in which each of 29 candidate RMS driver or tumor suppressor has expression level (FPKM) of 1 or greater based on RNA sequencing.
(C) Charts display the average copy-number level for copy-number gain events of iExCN-predicted oncogenes, and copy-number loss events of iExCN-predicted tumor suppressor genes (TSGs), separately.
(D) Charts display the number among 209 FN RMS cases with copy-number gain (marked red) and loss (marked blue) events on 29 iExCN-defined RMS disease genes.
See also Figure S1.
Functional categories of iExCN genes
Eight (28%) of the 29 candidates were previously linked to RMS (CCND2, CDK6, EZH2, GLI2, GLI3, KIF18A, PTEN, RIPK2) (Chen et al., 2013; Ehlers et al., 2008; Hatley et al., 2012; Li et al., 2007; Marchesi et al., 2012; Saab et al., 2006; Shern et al., 2014), and 28% are listed in the COSMIC Cancer Gene Census (CCND2, CDK6, ETV1, EZH2, PLAG1, PTEN, WRN and ZRSR2). This represents a significant enrichment when compared to the 3% found when sets of 29 genes were randomly chosen from a) all protein-coding genes expressed in FN RMS (binomial test; P < 104), or b) from the 1797 genes identified by GISTIC to harbor CNVs (binomial test; P < 104). Of note, 15 of the iExCN genes were among the 1797 GISTIC genes (Table S4). We also note that six iExCN genes (CCND2, CDK6, GLI2, HAS2, SLC6A15, and ZFHX4) harbor PAX3-FOXO1 binding elements identified in previous ChIP-seq studies (Cao et al., 2010; Gryder et al., 2017). Finally, nine of the 29 iExCN genes (CCND2, CDK6, HAS2, KIF18A, RAD54B, SLC6A15, TJP2, TRIP13 and ZFHX4) contained somatic missense SNVs in 134 RMS cases. Each mutation was found in only a single case, except for TJP2 and TRIP13, mutated in two and three cases, respectively. To our knowledge, the significance of these rare SNVs remains to be determined.
Only two Gene Ontology (GO) terms were significantly enriched in the iExCN-defined genes (Figure 2C). The largest category, Nucleic Acid Binding, encompassed 12 of the 29 genes (FDR adjusted P = 0.01) including transcriptional regulators related to the cell division cycle (E2F5), Hedgehog signaling (GLI2 and GLI3), histone modification (EZH2), epithelial-to-mesenchymal transition (SNAI2), and IGF2 signaling (PLAG1). The next largest GO term, Cell Cycle, contained five genes (FDR adjusted P = 0.04), including those influencing the G1/S phase transition (CCND2, CDK6, and E2F5) and associated with microtubule formation/dynamics (KIF18A). Because transcription factor regulation and cell cycle arrest are intimately linked to skeletal muscle differentiation, enrichment of those pathways underlines how RMS represents a cancer with derailed developmental programs (Hettmer and Wagers, 2010; Saab et al., 2011).
CRISPR/Cas9 “mini-pool” screen validates iExCN-defined genes in RMS
We verified the functional importance of iExCN genes using a competitive assay with a mini-pool of CRISPR/Cas9 lentiviral vectors targeting a) iExCN genes, b) negative control genes chosen randomly, and c) positive controls, previously shown to be essential (Shalem et al., 2014) (Figure 3A, Tables S5 and S6). The iExCN genes and the negative and positive controls were all expressed in most FN RMS cases (Figures 2B, 3B and 3C). Following transduction and serial passage of two authenticated FN RMS lines (JR1 and RD) (Figures S2A and S2B), lentiviral vector DNA was analyzed by targeted deep sequencing to assess relative changes in vector representation (Figures 3A and S2C). In aggregate, the normalized mean read depth for vectors targeting positive control genes decreased by 17% and 20% in JR1 and RD cells (two-tailed Student’s t-test; P = 2 X 10−6 and 2 X10−8, respectively) between days 0 and 14. In contrast, representation of vectors targeting negative control genes did not change significantly [0.4% and 0.1% decrease in JR1 and RD (two-tailed Student’s t-test; P = 0.84 and 0.95, respectively)], confirming the expected assay performance.
Figure 3. iExCN-predicted disease genes are validated by CRISPR/Cas9 “mini-pool” screen.
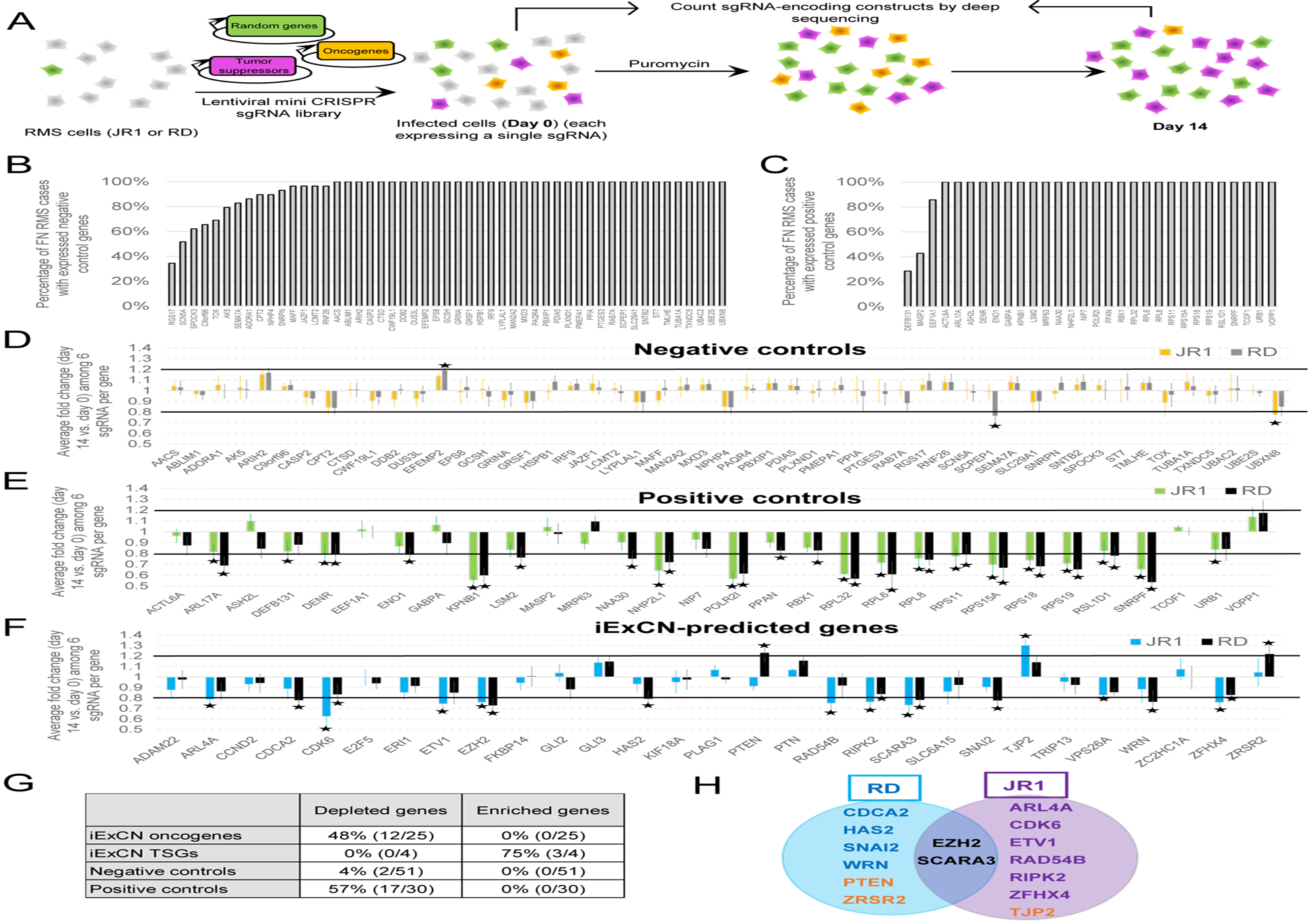
(A) Schematic diagram shows experimental protocol in which RMS cells are transduced with lentiviral vectors effecting CRISPR/Cas9-based targeting of candidate oncogenes and tumors suppressors as well as randomly-chosen genes. Relative changes in lentivirus representation is measured by targeted deep-sequencing at beginning and end of experiment.
(B – C) Charts display the number of FN RMS cases (%) in which each gene has expression level (FPKM) of 1 or greater based on RNA sequencing for negative (B) and positive (C) control genes.
(D – F) Charts display average fold change of read counts of six sgRNA per gene from day 0 to day 14 for negative control (D), positive control (E), and iExCN genes (F). Asterisks represent FDR-corrected P value < 0.05 by two-tailed Student’s t-test. Black lines represent 20% increase or decrease. Data are presented for two RMS cell lines, JR1 and RD, as indicated.
(G) Summary statistics for significantly altered genes targeted by depleted and enriched lentiviral vectors in the screen.
(H) Venn diagram shows genes targeted by vectors that are significantly depleted or enriched (orange text) in the two FN RMS lines (JR1 and RD).
See also Figures S2 and S3, and Tables S5, S6, and S7.
We evaluated changes in the representation of lentiviral vectors targeting individual iExCN-derived and control genes. As in previous reports (Plaisier et al., 2016; Toledo et al., 2015), we defined significantly altered vectors as those meeting both statistical (FDR-corrected P value < 0.05 by two-tailed Student’s t-test) and magnitude of change (≥20%) thresholds (Figure 3D–F, asterisks and black lines, respectively). Only 2 of 51 vectors targeting negative controls were depleted, and none were enriched; in contrast, 17 of 30 vectors targeting positive controls were depleted in one or both of the tested RMS lines (Figure 3D, E, and G). Vectors targeting 15 of 29 iExCN-defined genes changed: 12 of 25 targeting candidate oncogenic drivers were depleted, and 3 of 4 targeting candidate TSGs were enriched (Figure 3F–H).
Non-specific toxicity mediated by CRISPR/Cas9 targeting genes with copy-number amplification has been reported by several groups (Aguirre et al., 2016; Munoz et al., 2016; Wang et al., 2015), and this raises the concern that certain of the 12 oncogenes identified as line-specific vulnerabilities in the mini-pool screen (Figure 3H) might include false-positives due to gene copy-number gain. To address this, we first examined copy-number of the iExCN and the positive control genes in the two rhabdomyosarcoma models (JR1 and RD). We found that 75% (JR1) and 67% (RD) of CRISPR/Cas9 hits in iExCN genes and over 80% of the hits in positive control genes lacked measurable copy-number amplification in these particular cell lines (Table S7). Furthermore, siRNA knockdown of each of the iExCN-defined oncogenic drivers impaired cell accumulation in the relevant lines (Figures 5A–F and S3). Taken together, our data indicate that over half of the iExCN genes represent true shared or line-specific vulnerabilities in the tested FN RMS models.
Figure 5. siRNA and pharmacological inhibition of iExCN-defined oncogenes decreases RMS cell accumulation.
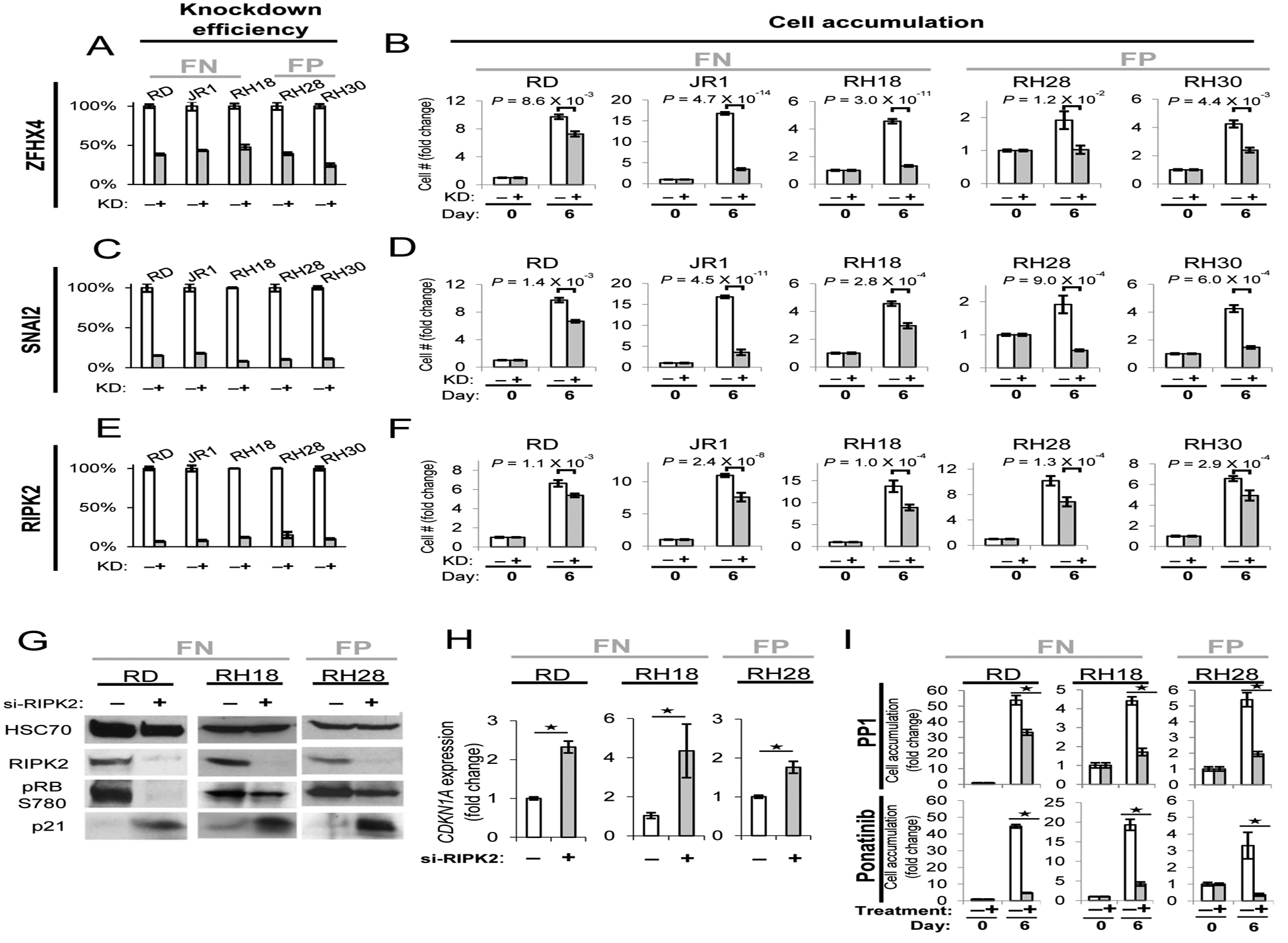
(A – F) siRNA knockdown in three iExCN-predicted oncogenes (ZFHX4, SNAI2 and RIPK2) in three FN (JR1, RD and RH18) and two FP (RH28 and RH30) cell lines. Charts display knockdown efficiency (A, C and E) and inhibition of cell accumulation (B, D and F) for siRNA knockdown. “KD” is short for knockdown. P values were calculated based on the two-tailed Student’s t-test.
(G, H) Representative western blots (G) and qRT-PCR data (H) demonstrate that RIPK2 suppression by transient transfection of targeting (+) or scrambled (−) siRNA represses CDK4-dependent phosphorylation of RB and enhances expression of CDKN1A (p21Cip1) in the indicated fusion positive (FP) and negative (FN) RMS lines.
(I)Charts demonstrate that exposure of FP and FN RMS cells to RIPK2 inhibitors PP1 and ponatinib decreases RMS cell accumulation. Asterisks represent P value < 0.05 by two-tailed Student’s t-test.
See also Figures S3, S6, and S7.
Functional interplay between iExCN genes and muscle differentiation
RMS is composed of myoblast-like cells in which the capacity for terminal differentiation is corrupted, but mechanisms underlying that differentiation defect are incompletely understood (Saab et al., 2011). Hypothesizing that iExCN-predicted drivers might impair differentiation, we investigated whether they were differentially expressed in myoblasts versus differentiated myocytes. Analysis of human skeletal myoblasts propagated in vitro showed enhanced expression of muscle differentiation markers in myoblasts cultivated in differentiation-inducing medium (Figure 4A, red curves), while the expression of 14 (48%) of all 29 iExCN-predicted genes (56% of 25 candidate oncogenic drivers) negatively correlated with differentiation (Pearson correlation coefficients < −0.5) (Figure 4A, blue curves). In contrast, expression of the four TSGs did not significantly associate with differentiation (Figure 4A, green curves). Iteratively testing sets of 29 genes randomly selected from a) all genes expressed in FN RMS (gray bars, Figure 4B) or b) the 1797 genes identified by GISTIC (gray bars, Figure S4A) revealed most genes to display little expression change with muscle differentiation (Pearson correlation coefficients −0.4 – 0.4). In both cases, finding expression of 48% of iExCN genes to fall with differentiation (Pearson correlation coefficient −0.5 or less) significantly exceeded the 15% found by chance (binomial test; P < 10−6) (blue bars, Figure 4B and S4A). Because no part of the iExCN pipeline filtered candidates based on expression changes during muscle differentiation, this finding further illustrates the potential for iExCN to identify biologically-relevant genes.
Figure 4. Expression of iExCN genes falls with skeletal muscle differentiation.
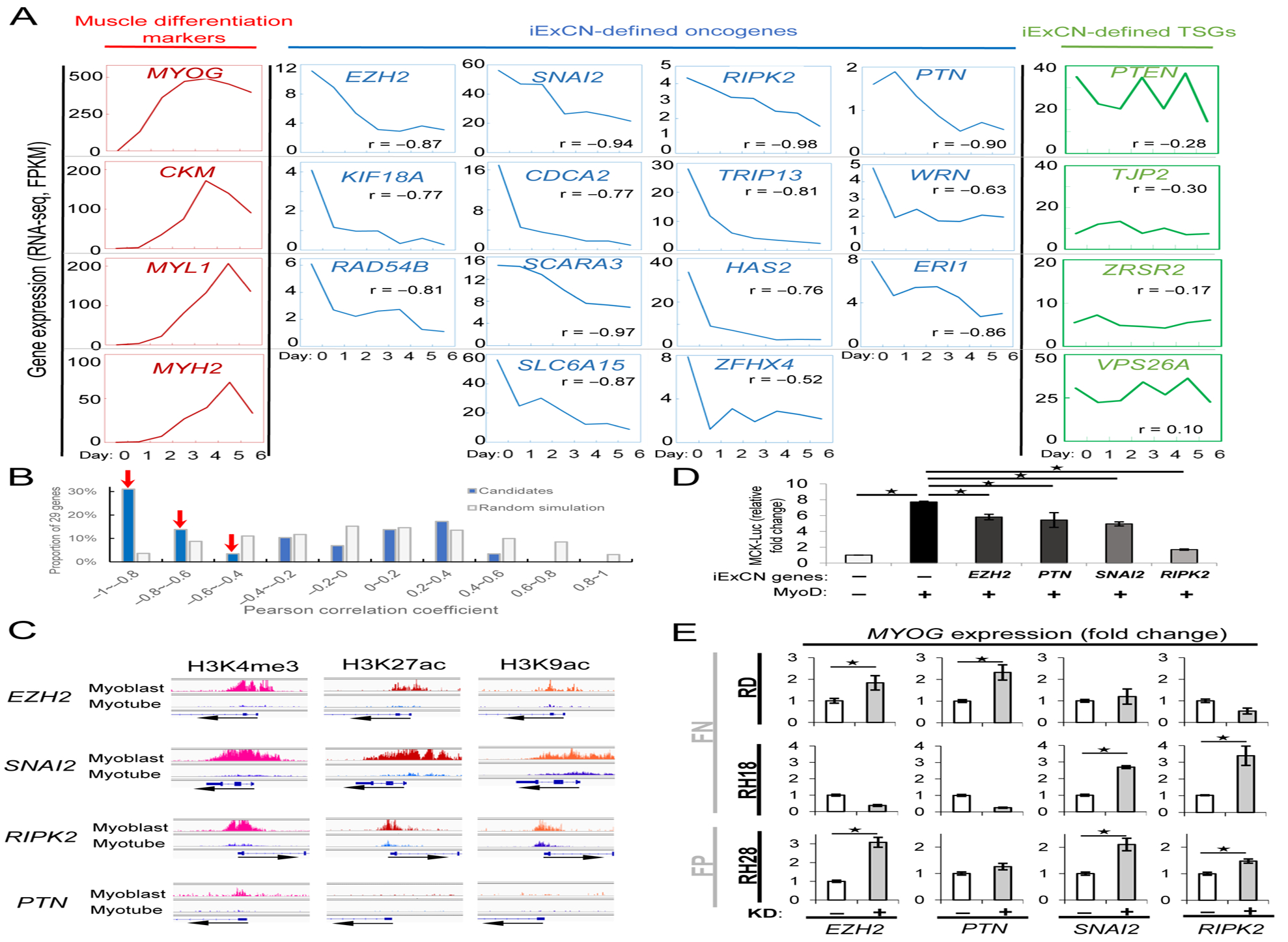
(A) Charts show gene expression for muscle differentiation markers (red), iExCN-derived oncogenic drivers (blue) and iExCN-derived TSGs (green) in human myoblasts cultured in growth medium (day 0) or differentiation medium for the indicated number of days (1–6).
(B) Chart displays the number of iExCN genes (blue bars) or genes randomly chosen from all those expressed in FN RMS (gray bars) with the indicated correlation to the degree of skeletal muscle differentiation. Pearson correlation coefficient near 0 indicates no correlation, whereas correlation coefficients of −1 or 1 indicate repression or induction of individual gene with muscle differentiation.
(C) Charts display ENCODE ChIP-seq data of H3K4me1 and H3K9ac, both of which are histone markers of active promoters, and H3K27ac, which is histone marker for active promoters and enhancers, in human myoblast and myotube on promoter regions of EZH2, PTN, SNAI2, or RIPK2.
(D) Chart demonstrates that ectopically expressed MyoD augments the expression of a muscle creatine kinase (MCK) enhancer/promoter reporter in 10T1/2 fibroblasts, whereas co-transfection of EZH2, PTN, SNAI2, or RIPK2 blunts MyoD activity. Data are average values from replicate samples, normalized to a co-transfected Renilla luciferase. Asterisks represent P value < 0.05 by two-tailed Student’s t-test.
(E) Charts show transient transfection of siRNA targeting EZH2, PTN, SNAI2, or RIPK2 or a scrambled control siRNA, (+ or −, respectively) influences the expression of MYOG in the indicated fusion positive (FP) and fusion negative (FN) cell lines. Asterisks represent P value < 0.05 by two-tailed Student’s t-test.
See also Figures S4 and S5.
To extend our findings beyond a mere correlation, we addressed mechanisms by which the differentially regulated iExCN genes fall with differentiation and how those genes can blunt muscle gene expression. Because a number of miRNA species have been recognized to play a role coordinating the myoblast to myocyte transition (Ge and Chen, 2011), we investigated whether the decreased expression could be explained by miRNA-dependent targeting. After predicting miRNA binding sites in 3’UTR regions, we found 633 miRNAs that could target at least one iExCN gene, whereas each iExCN gene had four to 85 predicted miRNA partners (Figure S4B). The majority (492 of 633) of these miRNAs, though, were predicted to only regulate one iExCN gene, and no miRNA was predicted to regulate more than five (Figure S4C). These findings indicate that coordinated effects of just a few miRNAs known to influence myogenesis is not likely to account for the differential expression of the iExCN genes.
We then focused on several histone marks that are associated with active transcription (ENCODE, 2012). Analyzing DNA located within 5kb of the transcription start sites for the 14 iExCN genes of interest, we observed that nearly all showed significant enrichment for H3K4me3, H3K27ac, and H3K9ac (Figures 4A, 4C and S5). In all, the histone methylation and acetylation fell with differentiation, even for genes like PTN, with relatively low expression and histone marks at baseline (Figures 4A and 4C). Hence, histone remodeling coordinates repression of these oncogenic drivers in muscle differentiation.
High expression of certain iExCN drivers in myoblasts suggested that they might limit the transcriptional activity of MyoD, a lineage-defining myogenic transcription factor (Saab et al., 2011) in myoblasts and RMS cells. Utilizing a system in which MyoD activity is monitored by a reporter driven by enhancer/promoter elements from the CKM gene (Wilson et al., 2016), we found MyoD activity to be significantly blunted by co-expression of EZH2, PTN, SNAI2 and RIPK2 (Figure 4D). To directly test whether those drivers suppressed muscle gene expression in RMS, we studied how their transient knockdown influenced expression of early and later markers of muscle differentiation in two FN (RD and RH18), and also one FP (RH28) line (Figures 4E, 5C, 5E and S6). Focusing on the expression of MYOG, a direct transcriptional target of MyoD (Saab et al., 2011) and early step in the myoblast-to-myocyte transition (Andres and Walsh, 1996), we observed that knockdown of each gene enhanced MYOG expression in one or more of the cell lines, revealing cell line-specific effects of individual iExCN genes (Figure 4E). The induction of later markers of differentiation was less consistent across the lines (Figure S6), highlighting the need for additional feed-forward pathways beyond MyoD activation for terminal differentiation (Penn et al., 2004). Viewed together, our findings indicate that deregulated expression of multiple genes with low-level copy-number gains may act cooperatively to impose a differentiation block in RMS.
iExCN oncogene drivers as potential therapeutic targets in RMS
We used siRNA knockdown to confirm the importance of several of the iExCN drivers depleted in the CRISPR/Cas9 mini-pool: ZFHX4, SNAI2 and RIPK2. Transiently targeting each decreased the native transcript and significantly diminished cell accumulation in three FN RMS lines and two FP RMS lines (RH28 and RH30) (two-tailed Student’s t-test; P < 0.05) (Figure 5A–F). Hence, although FP and FN RMS are initiated by different mechanisms (Hettmer and Wagers, 2010; Saab et al., 2011), their shared myogenic lineage may lead to shared cooperating drivers or tumor suppressors.
We focused further mechanistic and pharmacological studies on RIPK2 because it represents a targetable vulnerability in RMS (Canning et al., 2015). RIPK2 was previously implicated in myogenesis, but mechanisms by which it acted were not defined (Munz et al., 2002). We tested whether it influenced the Cyclin D/CDK/RB axis because deregulated Cyclin D1 blunts MyoD activity (Skapek et al., 1995); RIPK2 expression enhances NFkB signaling (McCarthy et al., 1998); and NFkB induces Cyclin D1 (Guttridge et al., 1999). Indeed, RIPK2 knockdown in three RMS lines diminished CDK4/6 dependent phosphorylation of RB serine 780 (Figure 5G) and increased expression of the CDKN1A gene product, p21CIP1, a CDK inhibitor induced in differentiating myocytes (Figure 5G and H). Ectopic RIPK2 also blocked the expression of an artificial enhancer/reporter driven by reiterated MyoD-specific E-boxes (Skapek et al., 1995) (Figure S7A), demonstrating that RIPK2 expression interferes with transcriptional activation by MyoD or related transcription factors.
We next considered whether pharmacological strategies to manipulate RIPK2 similarly control RMS accumulation and foster muscle gene expression. We focused on two compounds: PP1, previously shown to inhibit RIPK2 (Bain et al., 2007), and ponatinib, which blocks RIPK2 and other kinases (Canning et al., 2015). RIPK2 knockdown and exposure to PP1 or ponatinib consistently decreased cell accumulation in all tested RMS lines (Figure 5E, F, and I), while RIPK2 knockdown also augmented MYOG expression in RH18 and RH28 cells (Figure 4E). These findings align with another report in a panel of RMS models in vitro and in vivo (Li et al., 2013).
Copy-number variations in iExCN genes correlate with survival
Because our findings support the concept that multiple iExCN drivers can act within a single tumor, we tested whether the number of iExCN genes harboring CNVs influenced outcome in children with FN RMS. We defined CNVs of iExCN genes as copy-number gain in any of the 25 oncogenes or loss in any of the four TSGs. Using two separate FN RMS cohorts, we found that cases with CNVs in 10 or fewer iExCN genes had significantly better failure-free and overall survival than those with more than 10 involved (log-rank test; P < 0.05 for each) (Figure 6A–D). The finding was notable because the iExCN pipeline did not consider outcome or clinical features like tumor stage or clinical group, both of which correlate with outcome for children with RMS (Malempati and Hawkins, 2012). The observation that children with a higher number of iExCN genes harboring copy-number changes had poorer survival supported the existence of gene-gene interactions among the iExCN genes. In that way, the effect of copy-number changes in a small number of iExCN genes may not be evident without cooperative or synergistic effects of other genes.
Figure 6. Increased number of iExCN genes with CNVs correlates with survival in two independent FN RMS cohorts.

(A - D) Kaplan-Meier plots display (A, B) failure-free and (C, D) overall survival in two independent cohorts with different numbers of iExCN genes with CNVs, as indicated. Cohort I and II are described in Experimental Procedures.
(E, F) Chart shows that chromosomal instability index score in FN RMS cases with CNVs involving no more than 10 iExCN genes is not lower than ones involving more than 10 iExCN genes in two cohorts, separately.
(G, H) Chart shows that average relative expression of 25 iExCN oncogenes is higher in FN RMS cases with CNVs in more than 10 iExCN genes in two cohorts, separately.
See also Figure S7.
Three pieces of data indicated that poorer survival in cases with CNVs in a greater number of iExCN genes was not merely related to genomic instability. First, employing an established chromosomal instability (CIN) index score, which positively correlates with genomic instability (Wang et al., 2013), we found that RMS cases with CNVs in a greater number of iExCN genes actually had a slightly lower CIN score in one cohort (Figure 6E). Because that association was not confirmed in a second, independent cohort (Figure 6F), it should be interpreted cautiously. Second, simulations calculating the association between survival and CNVs in randomly-selected, 29-gene sets revealed that less than 1% of 10,000 iterations had a significant association (log-rank test, P < 0.05) in both cohorts. Finally, cases with CNVs involving more than 10 iExCN genes had significantly higher average expression of the 25 oncogenes (Figure 6G and H) (two-tailed Student’s t-test; P < 0.05 for each) and a trend toward lower expression of the four candidate TSGs (Figure S7B). Hence, the prognostic value of CNVs involving iExCN genes did not stem from genome-wide copy number change, per se; it depended on the identity of the involved genes and correlated with higher expression of the encoded drivers.
Discussion
The potential for next-generation sequencing to illuminate oncogenic drivers and deliver better therapy has yet to be fully realize for most cancers, including FN RMS. Working from the premise that gene expression changes drive the cancer phenotype, we developed a computational pipeline to identify gene expression changes encrypted by copy-number changes – anticipating them to be particularly important. This prediction is relevant for FN RMS because a recent DNA methylation analysis revealed that just over 12% of gene expression differences between FP and FN RMS are explained by DNA methylation (Sun et al., 2015). The iExCN analysis identified 29 FN RMS drivers and TSGs, most of which were not uncovered in previous genomics analyses (Chen et al., 2013; Paulson et al., 2011; Seki et al., 2015; Shern et al., 2014). While not discounting the importance of those approaches, we emphasize the value added by the complementary iExCN analysis. For example, the RAS/PIK3CA and Cyclin D/CDK/RB pathways were previously implicated in RMS by SNVs and CDKN2A deletion (Chen et al., 2013; Shern et al., 2014). That iExCN identified PTEN, a negative regulator of RAS/PI3K signaling, and CCND2 and CDK6, negative regulators of RB, cements their importance in FN RMS and highlights coordinated copy-number and expression changes as mechanisms to corrupt those pathways.
Our analysis extends previous reports highlighting somatic SNVs (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014) because, by focusing on coordinate changes between gene expression and copy-number, we identify previously unknown genes that contribute to FN RMS pathogenesis. This observation is supported by another report showing that other types of soft tissue sarcoma are also driven by CNV-coupled gene expression changes (TCGA, 2017). That iExCN genes are expressed and harbor CNVs across the majority of FN RMS cases also contrasts the narrow distribution of somatic SNVs, the majority of which we and others show to be only found in single FN RMS cases (Chen et al., 2013; Seki et al., 2015; Shern et al., 2014). In contrast, many individual FN RMS cases harbor CNVs in each of the iExCN genes, demonstrating their potential to represent commonly shared vulnerabilities. Indeed, that was borne out in our CRISPR/Cas9 screen showing vectors targeting two iExCN genes, EZH2 and SCARA3, to be depleted in both of the tested FN RMS lines. Both genes are expressed in more than 80% of RMS specimens and harbor copy-number gains in over 50 of the approximately 200 cases that we analyzed. While EZH2 has recently been implicated in RMS and cancer (Ciarapica et al., 2014; Kim and Roberts, 2016), SCARA3 encodes a reactive oxygen species (ROS) scavenger receptor family protein with only limited connection to cancer. Interestingly, a previous genomics analysis concluded that genes controlling oxidative stress are important in RMS but did not actually identify lesions in direct ROS regulators (Chen et al., 2013). SCARA3 represents such an ROS regulator, nominated in our study by iExCN and further highlighted in the CRISPR/Cas9 mini-pool screen.
Our findings help to explain mechanisms underlying the well-known terminal differentiation defect in RMS. Perhaps most importantly, finding that the expression of 14 iExCN-defined drivers falls with differentiation in normal skeletal myoblasts highlights the potential for multiple genes with low-level copy-number gains to contribute to a differentiation arrest in RMS. A number of those are already implicated in myogenic differentiation control. For example, D-type cyclins and CDK4/6 are recognized to impair normal muscle gene expression and contribute to RMS (Saab et al., 2011). Our candidates include CCND2 and CDK6, and we provide evidence that RIPK2, previously implicated as a regulator of myogenesis (Munz et al., 2002), contributes to muscle gene repression by influencing CDK4/6 activity. Aberrations in myogenic trans activators, like MYOD1 mutation (Szuhai et al., 2014) and E2A splicing (Yang et al., 2009) align with our identification of EZH2 and SNAI2. The former encodes a histone methyltransferase in the Polycomb repressor complex 2 (PRC2) that can help preserve a non-differentiated state of FN RMS (Ciarapica et al., 2014). SNAI2 encodes Slug, a zinc-finger transcription factor with a consensus DNA binding element (CAGGTG), analogous to the CANNTG “E-box” bound by MYOD-family proteins (Cobaleda et al., 2007; Saab et al., 2011). Indeed, proteins encoded by mouse Snai1 and Snai2 genes block MyoD1 binding and muscle differentiation in myoblasts (Soleimani et al., 2012). Further studies of the iExCN genes may reveal additional mechanisms to either impair cell cycle arrest or the activity of myogenic transcription factors, both of which are needed for terminal differentiation.
Barriers to differentiation in RMS represent opportunities for therapeutics. In mammals, terminal skeletal muscle differentiation has long been known to impose an irreversible cell proliferation arrest (Saab et al., 2011). Even if complete differentiation cannot be achieved, higher expression of certain muscle genes portends better survival, implying more favorable RMS biology (Davicioni et al., 2006). That multiple RMS drivers influence differentiation would explain why targeting an individual node – like CDK4/6, which blocks differentiation in myoblasts – has limited impact (Saab et al., 2006; Skapek et al., 1995). However, the aforementioned coordinate repression of 14 iExCN genes provides a new entry point for “differentiation therapy”. The changes in histone methylation and acetylation at those genes might be choreographed in normal muscle by a small number of chromatin or histone modifying proteins, many of which are already targetable (Helin and Dhanak, 2013).
While increased number of low-level copy-number changes in iExCN genes drives higher expression of those genes and less favorable outcome, we do not yet know how to best identify any single iExCN gene as a targetable vulnerability in an individual RMS specimen. As we show, the propagation of two RMS lines is impaired by targeting individual genes even though some do not harbor measurable copy-number changes. To help explain an apparent lack of specificity for iExCN prediction, we emphasize that iExCN represents a statistical tool that nominates disease genes by correlating gene expression and copy-number changes across many specimens. Obviously, the expression of any one of those genes in a cell line or individual tumor might also be driven by epigenetic mechanisms that are divorced from gene copy-number change. That can also explain how iExCN analysis of FN RMS can also identify genes important in fusion-positive disease because the shared skeletal muscle lineage likely enables both RMS forms (or individual cancers within the same subtype) to co-opt ancestral ontogenetic themes to impair differentiation and drive cell proliferation, migration, and survival as targetable cancer hallmarks (Hanahan and Weinberg, 2011). The application of molecularly-targeted therapies in childhood cancer is more likely to succeed if predictive biomarkers are identified, typically as a protein-altering mutation in a disease driver as in the Pediatric MATCH “basket” trial (Dolgin, 2017). Additional pre-clinical and clinical translational studies are needed to clarify how well gene copy-number, expression, or both predicts any single iExCN gene to be vulnerability in an individual tumor.
Experimental Procedures
RMS cases and genomic data sets
Genomic data from 290 specimens, collected from 290 patients and de-identified before use, were from three sources referred to as NCI, Children’s Oncology Group (COG) and UTSW datasets (Table S1 and Supplemental Experimental Procedures). Genomic data used in this study has been deposited to dbGaP database under accession number phs000720 and Gene Expression Omnibus (GEO) under accession number GSE114621. Genomics analyses of archived patient samples were conducted at UT Southwestern Medical Center with approval of its Institutional Review Board (STU 102011–034).
Genomic sequencing, copy-number and gene expression data analysis
Details are provided in Supplemental Experimental Procedures. In brief, whole-genome and -exome sequencing reads were aligned to human reference genome (hg19) and somatic protein-altering mutations were identified by GATK pipeline. SNP arrays were processed by SNP-FASST segmentation algorithm implemented in Nexus BioDiscovery software (BioDiscovery, El Segundo, CA). Significantly altered CNVs were examined by the Genomic Identification of Significant Targets in Cancer (GISTIC) method using a default q-value of 0.25 to define statistical significance as previously described (Mermel et al., 2011). For gene expression data, RNA was extracted in the Triche lab and processed by Affymetrix Exon 1.0 ST array platform according to manufacturer’s recommendations (Affymetrix, CA). CEL files were analyzed using R/BioConductor with Robust Multiarray Average (RMA) normalization and custom PERL scripts.
Molecular experiments
Human myoblasts (Lonza) and rhabdomyosarcoma cell lines (JR1, RD, RH28, RH18 and RH30), either provided by P. Houghton (St. Jude Children’s Research Hospital) or obtained from American Tissue Type Culture Collection, were cultivated as previously described (Wilson et al., 2016). RMS cells were authenticated by RT-PCR for PAX3/FOXO1 fusion expression and by short tandem repeat (STR) testing to assure distinct cell lines and compatible with known standards (Figure S2A). Transient gene knockdown was achieved using siRNA duplexes (Life Technologies) and Lipofectamine RNAiMAX (Thermo Fisher Scientific). 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo-d-3,4-pyrimidine) (PP1)(10 μM) (EMD chemicals) and ponatinib (Selleck Chemicals) were used to inactivate RIPK2. Expression plasmids encoding a subset of iExCN-defined drivers (obtained from GeneCopoeia or from D. W. Abbott) (Tigno-Aranjuez et al., 2010), with authentication by PCR or sequencing. MyoD activity was assessed using transient transfection and reporter assays as previously described (Wilson et al., 2016). More experimental details are provided in Supplemental Experimental Procedures.
CRISPR/Cas9 “mini-pool” screen and data analysis
A CRISPR/Cas9 lentiviral “mini-pool” screen was carried out in RD and JR1 cells (see Supplemental Experimental Procedures for full details). In brief, lentiviral vectors targeting iExCN-predicted genes and controls (Table S5 and S6) were prepared and used as described (Shalem et al., 2014). Following transduction at low MOI, puromycin selection and cultivation ex vivo for 14 days, genomic DNA was prepared for analysis in the UTSW McDermott Next Generation Sequencing Core using the Illumina Nextseq 500. The supporting reads were mapped to guide RNA sequences (Table S5) by a custom PERL script, and then median-normalized to adjust for the effect of library size and read count distribution. Average read counts for six guide RNAs per gene were used to represent gene-level lentiviral vector reads in each sample. The average gene-level read counts of three replicates from days 0 and 14 were used to calculate fold change for each gene. Two-tailed Student’s t-test with Benjamini-Hochberg FDR correction was used to adjust P values for multiple comparisons.
Statistical analysis
Statistical analyses were conducted using R programming packages (http://www.R-project.org). The comparison of two independent samples was assessed using the two-tailed Student’s t-test (normal distribution) or non-parametric two-tailed Mann–Whitney U-test (non-normal distribution). P value less than 0.05 was considered statistically significant. For survival analysis (Figure 6A–D), P values were calculated based on log-rank test. Cohort I is based on published datasets (Paulson et al., 2011; Shern et al., 2014), while Cohort II is from a unpublished dataset used in this study (COG dataset in Table S1). Note that study subject age and sex, available only on a subset of the data, were not incorporated into the survival analyses because those features are not generally accepted to influence survival. Functional annotation of enriched gene sets was based on Gene Ontology (GO) database (http://www.geneontology.org), and the hypergeometric test was used to determine statistically significant enrichment. P values were adjusted for multiple comparisons using the Benjamini-Hochberg procedure. Random sampling was performed using Statistics::R and List::Util modules in PERL and the ‘sample’ function in R to randomly choose elements from a population based on a user-defined number of elements and 10,000 iterations.
Supplementary Material
Table S1. Genomic data from 290 RMS tumors used in this study. Related to Figure 1.
Table S2. Mutation frequency of genes with somatic protein-altering SNVs in 134 RMS cases. Related to Figure 1.
Table S3. GISTIC-defined significantly altered genes in FN RMS. Related to Figure 1.
Table S6. Negative control and positive control genes. Related to Figure 3.
Table S5. gRNA list for iExCN-predicted, negative control, and positive control genes. Related to Figure 3.
Acknowledgements
We are grateful to M. DePlaza, J. Singh, and other members of the Skapek laboratory for technical assistance and helpful comments, and to C. Liu and J. Mendell (UT Southwestern) for advice and early technical assistance with CRISPR/Cas9 targeting. We acknowledge support from the Hyundai Hope on Wheels Foundation, the Wipe Out Kids’ Cancer Foundation, the Children’s Cancer Fund, and the Patrick and Beatrice Haggerty Foundation, all of which were secured with assistance from the Children’s Health Children’s Medical Center Foundation; and the Andrew McDonough B+ Foundation. Collection of expression and copy-number data in the Children’s Oncology Group (COG) dataset was supported by grants from the National Cancer Institute (U10CA098543 and U10CA098413). Work in the Skapek laboratory was also aided by grants from the Cancer Prevention and Research Institute of Texas (RP120685-P2) and to the UTSW Harold C. Simmons Comprehensive Cancer Center from the National Cancer Institute (CA142543).
Footnotes
Accession Numbers
Genomic data used in this study has been deposited to dbGaP database under accession number phs000720 and Gene Expression Omnibus (GEO) under accession number GSE114621.
Declaration of Interests
The authors declare no competing interests.
References
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres V, and Walsh K (1996). Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J. Cell Biol 132, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, and Cohen P (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P, Ruan Q, Schwerd T, Hrdinka M, Maki JL, Saleh D, Suebsuwong C, Ray S, Brennan PE, Cuny GD, et al. (2015). Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem. Biol 22, 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ, and Meltzer PS (2010). Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 70, 6497–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, et al. (2013). Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24, 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarapica R, Carcarino E, Adesso L, De Salvo M, Bracaglia G, Leoncini PP, Dall’agnese A, Verginelli F, Milano GM, Boldrini R, et al. (2014). Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC cancer 14, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Perez-Caro M, Vicente-Duenas C, and Sanchez-Garcia I (2007). Function of the zinc-finger transcription factor SNAI2 in cancer and development. Annu. Rev. Genet 41, 41–61. [DOI] [PubMed] [Google Scholar]
- Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, and Anderson MJ (2006). Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 66, 6936–6946. [DOI] [PubMed] [Google Scholar]
- Dolgin E (2017). Pediatric MATCH Trial Opens Enrollment. Cancer Discov. 7, 1054. [DOI] [PubMed] [Google Scholar]
- Ehlers S, Mueck T, Adams S, Landuzzi L, Lollini PL, and Munz B (2008). RIP2 regulates growth and differentiation of normal myoblasts and of rhabdomyosarcoma cells. Eur. J. Cell Biol 87, 163–172. [DOI] [PubMed] [Google Scholar]
- ENCODE (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, and Chen J (2011). MicroRNAs in skeletal myogenesis. Cell cycle 10, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, et al. (2017). PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 7, 884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, and Baldwin AS Jr. (1999). NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell Biol 19, 5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hatley ME, Tang W, Garcia MR, Finkelstein D, Millay DP, Liu N, Graff J, Galindo RL, and Olson EN (2012). A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell 22, 536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, and Dhanak D (2013). Chromatin proteins and modifications as drug targets. Nature 502, 480–488. [DOI] [PubMed] [Google Scholar]
- Hettmer S, and Wagers AJ (2010). Muscling in: Uncovering the origins of rhabdomyosarcoma. Nat. Med 16, 171–173. [DOI] [PubMed] [Google Scholar]
- Heuckmann JM, and Thomas RK (2015). A new generation of cancer genome diagnostics for routine clinical use: overcoming the roadblocks to personalized cancer medicine. Ann. Oncol 26, 1830–1837. [DOI] [PubMed] [Google Scholar]
- Kim KH, and Roberts CW (2016). Targeting EZH2 in cancer. Nat. Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruschke JK (2013). Bayesian estimation supersedes the t test. J. Exp. Psychol. Gen 142, 573–603. [DOI] [PubMed] [Google Scholar]
- Kruschke JK, and Liddell TM (2018). The Bayesian New Statistics: Hypothesis testing, estimation, meta-analysis, and power analysis from a Bayesian perspective. Psychon. Bull. Rev 25, 178–206. [DOI] [PubMed] [Google Scholar]
- Li HG, Wang Q, Li HM, Kumar S, Parker C, Slevin M, and Kumar P (2007). PAX3 and PAX3-FKHR promote rhabdomyosarcoma cell survival through downregulation of PTEN. Cancer Lett. 253, 215–223. [DOI] [PubMed] [Google Scholar]
- Li SQ, Cheuk AT, Shern JF, Song YK, Hurd L, Liao H, Wei JS, and Khan J (2013). Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS ONE 8, e76551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malempati S, and Hawkins DS (2012). Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 59, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi I, Fiorentino FP, Rizzolio F, Giordano A, and Bagella L (2012). The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle 11, 3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JV, Ni J, and Dixit VM (1998). RIP2 is a novel NF-kappaB-activating and cell death-inducing kinase. J. Biol. Chem 273, 16968–16975. [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, et al. (2016). CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 6, 900–913. [DOI] [PubMed] [Google Scholar]
- Munz B, Hildt E, Springer ML, and Blau HM (2002). RIP2, a checkpoint in myogenic differentiation. Mol. Cell Biol 22, 5879–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF, and Cameron S (2011). High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chrom. Cancer 50, 397–408. [DOI] [PubMed] [Google Scholar]
- Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, and Tapscott SJ (2004). A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 18, 2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier CL, O’Brien S, Bernard B, Reynolds S, Simon Z, Toledo CM, Ding Y, Reiss DJ, Paddison PJ, and Baliga NS (2016). Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst. 3, 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab R, Bills JL, Miceli AP, Anderson CM, Khoury JD, Fry DW, Navid F, Houghton PJ, and Skapek SX (2006). Pharmacological inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol. Cancer Ther 5, 1299–1308. [DOI] [PubMed] [Google Scholar]
- Saab R, Spunt SL, and Skapek SX (2011). Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr. Top. Dev. Biol 94, 197–234. [DOI] [PubMed] [Google Scholar]
- Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, Kato M, Chiba K, Tanaka H, Hoshino N, et al. (2015). Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun 6, 7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. (2014). Comprehensive Genomic Analysis of Rhabdomyosarcoma Reveals a Landscape of Alterations Affecting a Common Genetic Axis in Fusion-Positive and Fusion-Negative Tumors. Cancer Discov. 4, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, and Lassar AB (1995). Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science 267, 1022–1024. [DOI] [PubMed] [Google Scholar]
- Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CE, van Ijcken WF, Grosveld F, and Rudnicki MA (2012). Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell 47, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Chatterjee B, Wang Y, Stevenson HS, Edelman DC, Meltzer PS, and Barr FG (2015). Distinct methylation profiles characterize fusion-positive and fusion-negative rhabdomyosarcoma. Mod. Pathol 28, 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuhai K, de Jong D, Leung WY, Fletcher CD, and Hogendoorn PC (2014). Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J. Pathol 232, 300–307. [DOI] [PubMed] [Google Scholar]
- TCGA (2017). Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 171, 950–965 e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigno-Aranjuez JT, Asara JM, and Abbott DW (2010). Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 24, 2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo CM, Ding Y, Hoellerbauer P, Davis RJ, Basom R, Girard EJ, Lee E, Corrin P, Hart T, Bolouri H, et al. (2015). Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 13, 2425–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, Zhu Z, Wang Y, Pocalyko D, Yang WJ, et al. (2013). Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 58, 706–717. [DOI] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RA, Liu J, Xu L, Annis J, Helmig S, Moore G, Timmerman C, Grandori C, Zheng Y, and Skapek SX (2016). Negative regulation of initial steps in skeletal myogenesis by mTOR and other kinases. Sci. Rep 6, 20376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, MacQuarrie KL, Analau E, Tyler AE, Dilworth FJ, Cao Y, Diede SJ, and Tapscott SJ (2009). MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes Dev. 23, 694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genomic data from 290 RMS tumors used in this study. Related to Figure 1.
Table S2. Mutation frequency of genes with somatic protein-altering SNVs in 134 RMS cases. Related to Figure 1.
Table S3. GISTIC-defined significantly altered genes in FN RMS. Related to Figure 1.
Table S6. Negative control and positive control genes. Related to Figure 3.
Table S5. gRNA list for iExCN-predicted, negative control, and positive control genes. Related to Figure 3.