

Abstract
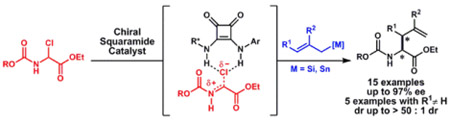
We report a chiral-squaramide-catalyzed enantio- and diastereoselective synthesis of α-allyl amino esters. The optimized protocol provides access to N-carbamoyl-protected amino esters via nucleophilic allylation of readily accessible α-chloro glycinates. A variety of useful α-allyl amino esters were prepared—including crotylated products bearing vicinal stereocenters that are inaccessible through enolate alkylation—with high enantioselectivity (up to 97% ee) and diastereoselectivity (> 10:1). The reactions display first-order kinetic dependence on both the α-chloro glycinate and the nucleophile, consistent with rate-limiting C–C bond formation. Computational analysis of the uncatalyzed reaction predicts an energetically inaccessible iminium intermediate, and a lower energy concerted SN2 mechanism.
Graphical Abstract
Among the many classes of unnatural amino acids devised by synthetic chemists, α-allyl amino acids have proven particularly valuable in a wide variety of contexts.1-6 The alkenyl functional handle in these building blocks can be elaborated for site-selective protein modification,1,2 preparation of glycopeptides,3 or generation of peptide staples4 and macrocycles. Noteworthy applications include construction of all-carbon analogs of disulfide-bridged macrocyclic peptides such as oxytocin,5 and thioether-bridged lantibiotics such as nisin.6
As a result of their broad utility, α-allyl amino acids have been targets of widespread synthetic effort.7-13 Among catalytic approaches, the phase-transfer-catalyzed (PTC) allylation of Schiff base, ester enolates was pioneered by O’Donnell8 (Scheme 1A) and advanced to the current state-of-the-art by Maruoka through the discovery of highly effective, C2 symmetric, quaternary ammonium salt catalysts.9 This methodology provides access to unbranched α-allyl amino esters, but the preparation of branched products via PTC is generally not possible. Approaches involving transition-metal π-allyl intermediates have also been developed, but preparing branched products is hampered by requiring substitution to occur on a hindered electrophilic partner.10 More effective approaches require the preparation of glycine allyl esters or allyl ammonium salts, which undergo enantioselective Claisen11 or [2,3]-sigmatropic rearrangements,12 respectively.
Scheme 1.

Approaches to the Asymmetric Catalytic Synthesis of α-Allyl Amino Esters
The use of nucleophilic allylating agents allows stereocontrolled construction of β-branched α-allyl amino acid derivatives, as elegantly demonstrated by Lectka and Jørgensen in Lewis-acid-catalyzed allylations of N-tosyl α-imino esters using chiral copper complexes (Scheme 1B).13 Inspired by this precedent, we envisioned a new approach involving nucleophilic allylation of α-halo amino esters catalyzed by chiral hydrogen-bond-donor catalysts (Scheme 1C). This strategy benefits from the ready accessibility of N-carbamoyl α-chloro amino esters as α-imino ester equivalents.14,15 The products resulting from the proposed nucleophilic allylation protocol would possess readily cleavable carbamate protecting groups, facilitating further manipulations.16 Herein, we report the highly enantioselective and diastereoselective synthesis of α-allyl amino esters via anion-abstraction catalyzed addition of allylsilane and allylstannane nucleophiles to N-carbamoyl-α-chloro amino esters.
We selected the allylation of α-chloro glycinate 1-Cbz with 2-methallyltrimethylsilane as a model reaction for catalyst optimization (Figure 1). An extensive survey of chiral dual-hydrogen-bond-donors revealed that arylpyrrolidinosquaramides 4a-g17 catalyzed the formation of 2a with promising levels of enantiocontrol. As observed previously in reactions involving stabilized cationic intermediates, enantioselectivity was strongly responsive to the polarizability of the arene substituent on the pyrrolidine.18 Such effects have been ascribed to stabilizing π interactions between the catalyst and positively charged intermediates and/or transition states, in which the diastereomeric transition state leading to the major product is stabilized preferentially. However, arene polarizability alone does not account for the effects on reaction enantioselectivity in the allylation reaction (Figure 1B).19 The point of attachment of the arene to the pyrrolidine is also important, with arenes bearing substitution ortho to the pyrrolidine group affording lower enantioselectivity. Such sensitivity to the spatial disposition of the aryl group is consistent with a highly ordered network of attractive non-covalent interactions in the enantioselectivity-determining transition state assembly.18
Figure 1.

(A) Yield and enantioselectivity of 2a for a series of 2-arylpyrrolidine- and 2-aryl-2-methylpyrrolidine-substituted catalysts. Yields were determined by 1H NMR by integration against an internal standard. (B) Relationship between the enantiomeric ratio of 2a and the polarizability of the arene substituents on the catalysts 4a–4g (R2 = H, blue series) and 4a’–4e’ (R2 = Me, red series).
Further evidence for the importance of the conformational properties of the arylpyrrolidine in enantioinduction was provided through the evaluation of 2-aryl-2-methyl-substituted pyrrolidine-derived catalysts. Dual H-bond-donor catalysts bearing such fully substituted arylpyrrolidine derivatives have been found to exist predominantly as the amide (Z)-rotamer, whereas the unsubstituted analogs exist as rotameric mixtures.20 In the model allylation reaction, catalysts bearing fully substituted arylpyrrolidines (R2 = Me, 4a’–4e’, Figure 1B, red circles) all induce higher enantioselectivities than the corresponding unsubstituted catalysts (R2 = H 4a–4e, Figure 1B, blue squares). A dependence of ee on arene group polarizability and positioning was observed with the fully substituted arylpyrrolidine catalysts similar to that of the unsubstituted catalysts. At this stage it may be concluded that the properties of the arylpyrrolidine have a strong influence on reaction enantioselectivity, but that full elucidation of the bases for these effects and their relative importance will require a sophisticated multiparameter analysis.21 Finally, optimization of the aniline-derived portion of the catalyst revealed that the more electron deficient dicyano derivative 5d’ afforded comparable enantioselectivities but substantially improved rates and product yields than the bis-trifluoromethyl derivative 4d’.
The scope of the α-allyl amino ester synthesis was investigated with optimal catalyst 5d’ (Table 1).22 Consistently high yields and excellent enantioselectivities were obtained with several different 2-substituted allylsilane nucleophiles. 2-Arylallyltrimethylsilanes with electronically neutral or donating substituents furnished the corresponding products 2b–g in 90-96% ee. β-Branched-α-allyl amino esters were produced with high diastereoselectivities and enantioselectivities using 2,3-disubstituted allylsilanes as nucleophilic reacting partners (Table 1, 2i–l). As noted above, these products are generally inaccessible using electrophilic allylation reagents in traditional enolate allylations.9,10 The enantio- and diastereoselectivies of branched products 2i-l and 2n are generally higher than those obtained with previously reported catalytic asymmetric methods.13 Additional benefits of this method include a larger range of reported nucleophiles, as well as products containing easily cleavable N-protecting groups. In contrast, allylsilane nucleophiles lacking 2-substituents were completely unreactive under the catalytic conditions, an observation consistent with the fact that allyltrimethylsilane (N = 1.68)23 is substantially less nucleophilic than 2-methylallylsilane (N = 4.41).24 The more nucleophilic allyltributylstannanes (N = 5.46 for the parent compound)23 were evaluated and found to be effective reacting partners, allowing construction of α-allyl amino esters 2m–o with high yields and enantioselectivities. Reoptimization of the reaction conditions and carbamate protecting group was necessary for reactions employing stannane nucleophiles; in particular the Fmoc-protected analog of 1 was found to provide improved enantioselectivities (e.g., 93 vs 74% ee with prenylstannane nucleophile). Crotylstannanes underwent reaction to furnish the branched product 2n. Both the (E)- and (Z)-crotyltributylstannane afforded the syn diastereomer predominantly (8:1 with the (E)-isomer, 4:1 with the (Z)-isomer). A similar outcome was observed using silane nucleophiles leading to product 2i, indicating that the allylation reactions proceed through open transition states. Reaction of prenyl tributylstannane with 1-Fmoc generated the reverse-prenyl amino ester 2o in 93% ee.
Table 1.
Reaction Scopea
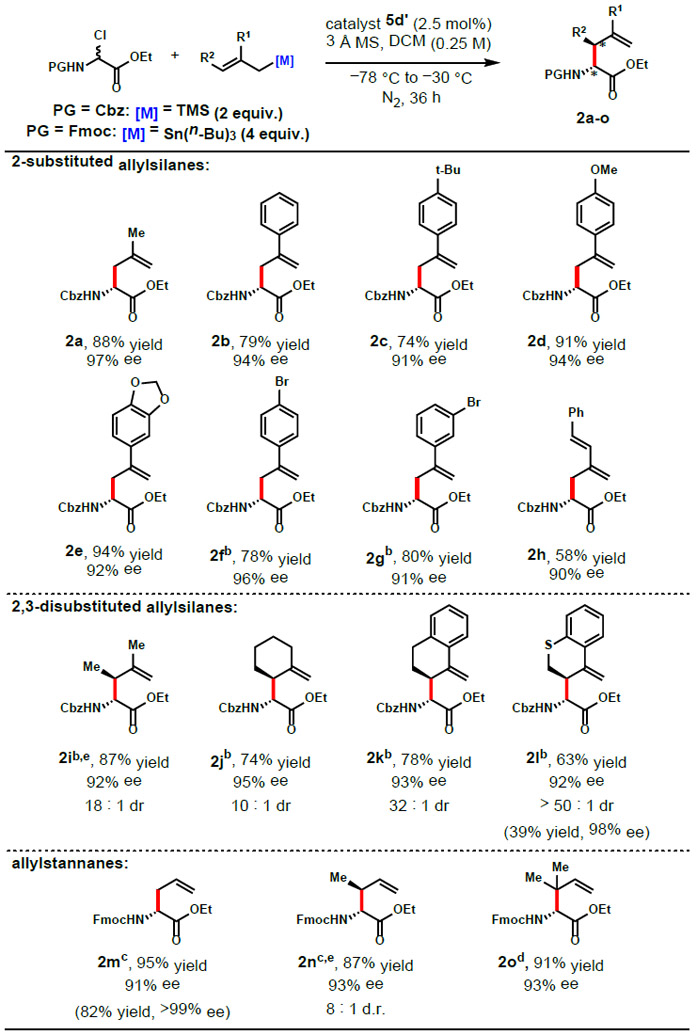 |
Conditions: substrate (0.5 mmol), catalyst (0.0125 mmol), nucleophile (1.0 mmol), 3 Å MS (60 mg), DCM (2 mL), under N2, initially cooled to −78 °C and stirred at −30 °C, 36 h. Enantiomeric excess determined by HPLC. Diastereomeric ratio determined by 1H NMR of the crude product, yield reflects isolation of major diasteromer.
−5 °C.
catalyst (0.05 mmol), nucleophile (2.0 mmol), DCM (5 mL), −50 °C, 72 h.
catalyst (0.05 mmol), nucleophile (1.5 mmol), DCM (5 mL), −30 °C, 72 h.
(E)-trimethyl(2-methylbut-2-en-1-yl)silane and (E)-crotylstannane employed as the nucleophiles. Yields and enantioselectivities in parentheses are of crystallized products.
The new catalytic protocol proved to be readily adaptable to the preparative-scale synthesis of α-allyl amino esters. For example, using either the α-chloro amino ester 1-Cbz or 1-Fmoc and appropriate allylsilane or allylstannane nucleophiles, the β-branched products 2i and 2o were generated on gram scale (Scheme 2). The reaction of 2,3-dimethylallylsilane with 1-Cbz could be conducted at relatively high concentrations (0.5 M in DCM) with 1.5 equiv. of the nucleophile and 1 mol% catalyst with no compromise in enantioselectivity relative to the smaller scale reaction. The addition of prenylstannane to 1-Fmoc could not be conducted at higher concentration due to the limited solubility of 1-Fmoc in DCM, but the catalyst loading could be lowered to 5 mol% while preserving the high enantioselectivity of the reaction.
Scheme 2.

Gram-scale reactions
While the direct generation of carbamate-protected amino esters provides amino acid derivatives in conveniently protected form, we were naturally concerned about the likely incompatibility of the alkenyl group in the allylated products with typical protocols for Cbz group removal. Indeed, under standard hydrogenolysis conditions, compound 2i underwent competitive reduction of the alkene. However, reduction with triethylsilane under Pd(OAc)2 catalysis left the alkenyl group intact and afforded the corresponding free amine in 87% yield with no measurable racemization.25
A simplified catalytic cycle for the new allylation reaction is outlined in Figure 2A. The catalytic mechanism is proposed to involve three fundamental steps: 1) complexation of the electrophile 1-Cbz to catalyst 5d’, 2) substitution involving C–Cl bond breaking and C–C bond formation, and 3) trimethylsilyl elimination from the β-silyl cation to form product 2a and TMSCl. The detailed mechanism of the substitution step and the role of the catalyst in promoting it are of greatest interest, since that process results in formation of the new C–C bond and is enantiodetermining. Although enantioselective anion-binding catalysis has been applied successfully to many reactions proceeding via N-acyliminium ion intermediates,18b,26 several considerations led us to question whether such species are involved in the system under consideration here. α-Chloro carbonyl compounds such as 1 are generally activated toward SN2 substitution pathways and deactivated toward dissociative SN1 mechanisms.27 Optimal enantioselectivities are obtained using DCM as solvent in the allylations of 1 (Figure S7). In contrast, nonpolar ethereal or aromatic solvents are generally required to achieve high enantioselectivities in ion-pairing catalysis promoted by chiral H-bond donors. Indeed, DCM has been found to promote solvent separation of ion pairs in these systems, resulting in severe diminution of enantioinduction.28
Figure 2:

(A) Proposed catalytic cycle. Observed first order dependence on 1-Cbz and Nuc indicate that nucleophilic addition is the rate-determining step. (B) Concerted SN2 and stepwise SN1 mechanisms require different mechanisms for enantioinduction. (C) Computational evaluation of the uncatalyzed reaction predicts that the SN1 pathway is much higher in energy than the SN2 pathway. All DFT calculations were performed at the B3LYP-D3(BJ)/6-311+G(d,p) level of theory including solvent corrections PCM(DCM). For a fuller discussion of the computational study, see SI.
Two plausible mechanisms for the substitution step are outlined in Figure 2B. If a stepwise SN1 is operant, anion abstraction would precede C–C bond formation with the intermediacy of a discrete, highly electrophilic iminium ion intermediate. The unexpected solvent effect noted above might be reconciled with a secondary attractive interaction holding the ion-pair together, such as the iminium–Cl H-bond depicted in Figure 2B (right). If the reaction proceeds through a concerted, stereospecific nucleophilic displacement, a dynamic kinetic resolution with rapid racemization of the α-chloro glycinate would be required to account for the observed high yields and enantioselectivies. Such a racemization could occur via chloride dissociation, bimolecular chloride displacement, or sequential tautomerization pathways. The catalytic reaction was determined to obey a second-order rate law, with first-order dependence on both the α-chloro glycinate and allylsilane concentrations, consistent with either a concerted SN2 mechanism or rate-determining allylation of an iminium intermediate in an SN1 pathway.
Clear differentiation of the two mechanisms and determination of the basis for enantioinduction will require considerable further study and are under active investigation. However, a preliminary computational analysis of the uncatalyzed transformation predicts that the concerted SN2 substitution proceeds through a substantially lower-energy pathway than the stepwise SN1 mechanism.29,30 The global ground state was identified as a dipole-dipole interaction complex between 1 and Nuc. Six staggered SN2 transition states were located with activation barriers < 20 kcal/mol, with the lowest at 12.7 kcal/mol (Figure 2C). In contrast, the iminium–chloride ion-pair implicated in the SN1 mechanism was 31.6 kcal/mol above the ground state. Recognizing that nucleophile addition is rate-determining and therefore must involve a discrete activation barrier, the concerted SN2 pathway is thus computed to be more than 18.9 kcal/mol lower in energy than the SN1 pathway.31,32
In summary, an enantio- and diastereoselective synthesis of α-allyl amino esters was accomplished via allylation of α-chloro glycine esters with a chiral squaramide hydrogen-bond donor as an anion-abstraction catalyst. Using either allylsilane or allylstannane nucleophiles, 15 representative α-allyl amino esters were constructed in high enantio- (up to 97% ee) and diastereoselectivities (> 10:1 dr). The silane addition reaction was carried out on gram-scale at 0.5 M concentration using 1 mol% catalyst. Experimental observations, kinetic data, and DFT calculations all point to an energetically inaccessible acyliminium ion intermediate and a relatively favorable, concerted SN2 mechanism for the substitution step. Our current efforts are directed toward elucidating the racemization mechanism, the origin of enantioselectivity, and the specific non-covalent interactions involved in the selectivity-determining transition state. We anticipate that a full mechanistic understanding of the origin of selectivity in the allylation of 1 could enable extension of the methodology to other classes of nucleophiles, providing a broadly applicable approach to the enantioselective synthesis of α-substituted amino esters.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (GM043214), by an NSF predoctoral fellowship to A.J.B., and by a Samsung Scholarship Foundation predoctoral fellowship to S.C.K. We thank Dr. Adam Trotta (Harvard University) for helpful discussions, Mr. Richard Liu for valuable experimental assistance, and Dr. Shao-Liang Zheng (Harvard University) for determination of the X-ray crystal structures.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05556.
Experimental and Characterization Data of Catalyst and Substrate Syntheses (PDF)
Procedures and Analytical Data for the Enantioselective Reactions (PDF)
Details of Kinetic Studies (PDF)
Computational Studies (PDF)
Crystallographic Data for 2l (CIF)
The authors declare no competing financial interests.
REFERENCES
- (1).For recent reviews see: Lin YA; Chalker JM; Davis BG Olefin Metathesis for Site-Selective Protein Modification. ChemBioChem 2009, 10, 959–969. [DOI] [PubMed] [Google Scholar]; (b) Lang K; Chin JW Bioorthogonal Reactions for Labeling Proteins. ACS Chem Biol. 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]; (c) deGruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lin YA; Chalker JM; Floyd N; Bernardes G. a. J. L.; Davis BG Allyl Sulfides Are Privileged Substrates in Aqueous Cross-Metathesis: Application to Site-Selective Protein Modification. J. Am. Chem. Soc 2008, 130, 9642–9643. [DOI] [PubMed] [Google Scholar]
- (3).McGarvey GJ; Benedum TE; Schmidtmann FW Development of Co- and Post-Translational Synthetic Strategies to C-Neoglycopeptides. Org. Lett 2002, 4, 3591–3594. [DOI] [PubMed] [Google Scholar]
- (4) (a).Kim Y-W; Grossmann TN; Verdine GL Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protocols 2011, 6, 761–771. [DOI] [PubMed] [Google Scholar]; (b) Verdine GL; Hilinski GJ Stapled peptides for intracellular drug targets. Methods Enzymol. 2012, 503, 3–33. [DOI] [PubMed] [Google Scholar]
- (5) (a).Stymiest JL; Mitchell BF; Wong S; Vederas JC Synthesis of Biologically Active Dicarba Analogues of the Peptide Hormone Oxytocin Using Ring-Closing Metathesis. Org. Lett 2003, 5, 47–49. [DOI] [PubMed] [Google Scholar]; (b) Stymiest JL; Mitchell BF; Wong S; Vederas JC Synthesis of Oxytocin Analogues with Replacement of Sulfur by Carbon Gives Potent Antagonists with Increased Stability. J. Org. Chem 2005, 70, 7799–7809. [DOI] [PubMed] [Google Scholar]
- (6).Ghalit N; Reichwein JF; Hilbers HW; Breukink E; Rijkers DTS; Liskamp RMJ Synthesis of Bicyclic Alkene-/Alkane-Bridged Nisin Mimics by Ring-Closing Metathesis and their Biochemical Evaluation as Lipid II Binders: toward the Design of Potential Novel Antibiotics. ChemBioChem 2007, 8, 1540–1554. [DOI] [PubMed] [Google Scholar]
- (7).(a) Select non-catalytic methods for the preparation of α-allyl amino acids: Myers AG; Gleason JL; Yoon T Practical method for the synthesis of D- or L-.alpha.-amino acids by the alkylation of (+)- or (−)-pseudoephedrine glycinamide. J. Am. Chem. Soc 1995, 117, 8488–8489. [Google Scholar]; Myers AG; Gleason JL Asymmetric Synthesis of α-Amino Acids by the Alkylation of Pseudoephedrine Glycinamide: L-Allylglycine and N-Boc-L-allylglycine Organic Syntheses; Wiley & Sons: New York, 1999; 76, 57. [Google Scholar]; (c) Grigg R; McCaffrey S; Sridharan V; Fishwick CWG; Kilner C; Korn S; Bailey K; Blacker J Enantioselective synthesis of non-proteinogenic 2-arylallyl-α-amino acids via Pd/In catalytic cascades. Tetrahedron 2006, 62, 12159–12171. [Google Scholar]; (d) Sun X-W; Liu M; Xu M-H; Lin G-Q Remarkable Salt Effect on In-Mediated Allylation of N-tert-Butanesulfinyl Imines in Aqueous Media: Highly Practical Asymmetric Synthesis of Chiral Homoallylic Amines and Isoindolinones. Org. Lett 2008, 10, 1259–1262. [DOI] [PubMed] [Google Scholar]
- (8).(a) For a review see: O’Donnell MJ The Enantioselective Synthesis of α-Amino Acids by Phase-Transfer Catalysis with Achiral Schiff Base Esters. Acc. Chem. Res 2004, 37, 506–517. [DOI] [PubMed] [Google Scholar]; (b) O’Donnell MJ; Eckrich TM The synthesis of amino acid derivatives by catalytic phase-transfer alkylations. Tetrahedron Lett. 1978, 19, 4625–4628. [Google Scholar]; (c) O’Donnell MJ; Bennett WD; Wu S The stereoselective synthesis of .alpha.-amino acids by phase-transfer catalysis. J. Am. Chem. Soc 1989, 111, 2353–2355. [Google Scholar]
- (9).(a) For a review see: Maruoka K; Ooi T Enantioselective Amino Acid Synthesis by Chiral Phase-Transfer Catalysis. Chem. Rev 2003, 103, 3013–3028. [DOI] [PubMed] [Google Scholar]; (b) Ooi T; Kameda M; Maruoka K Molecular Design of a C2-Symmetric Chiral Phase-Transfer Catalyst for Practical Asymmetric Synthesis of α-Amino Acids. J. Am. Chem. Soc 1999, 121, 6519–6520. [DOI] [PubMed] [Google Scholar]; (c) Kitamura M; Shirakawa S; Maruoka K Powerful Chiral Phase-Transfer Catalysts for the Asymmetric Synthesis of α-Alkyl- and α,α-Dialkyl-α-amino Acids. Angew. Chem. Int. Ed 2005, 44, 1549–1551. [DOI] [PubMed] [Google Scholar]
- (10) (a).Trost BM; Azira X Catalytic Asymmetric Alkylation of Nucleophiles: Asymmetric Synthesis of α-Alkylated Amino Acids. Angew. Chem. Int. Ed 1997, 36, 2635–2637. [Google Scholar]; (b) Trost BM; Dogra K Synthesis of Novel Quaternary Amino Acids Using Molybdenum-Catalyzed Asymmetric Allylic Alkylation. J. Am. Chem. Soc 2002, 124, 7256–7257. [DOI] [PubMed] [Google Scholar]; (c) Huo X; He Rui.; Fu J; Zhang J; Yang G; Zhang W Stereoselective and Site-Specific Allylic Alkylation of Amino Acids and Small Peptides via a Pd/Cu Dual Catalysis. J. Am. Chem. Soc 2017, 139, 9819–9822. [DOI] [PubMed] [Google Scholar]
- (11) (a).Kazmaier U; Krebs A Synthesis of Chiral γ,δ-Unsaturated Amino Acids by Asymmetric Ester Enolate Claisen Rearrangement. Angew. Chem. Int. Ed 1995, 34, 2012–2014. [Google Scholar]; (b) Krebs A; Kazmaier U The asymmetric ester enolate Claisen rearrangement as a suitable method for the synthesis of sterically highly demanding amino acids. Tetrahedron Lett. 1996, 37, 7945–7946. [Google Scholar]; (c) Kazmaier U Application of the Chelate-Enolate Claisen Rearrangement to The Synthesis of γ,δ-Unsaturated Amino Acids. Liebigs Annalen 1997, 1997, 285–295. [Google Scholar]
- (12) (a).Soheili A; Tambar UK Tandem Catalytic Allylic Amination and [2,3]-Stevens Rearrangement of Tertiary Amines. J. Am. Chem. Soc 2011, 133, 12956–12959. [DOI] [PubMed] [Google Scholar]; (b) West TH; Daniels DSB; Slawin AMZ; Smith AD An Isothiourea-Catalyzed Asymmetric [2,3]-Rearrangement of Allylic Ammonium Ylides. J. Am. Chem. Soc 2014, 136, 4476–4479. [DOI] [PubMed] [Google Scholar]; (c) Spoehrle SSM; West TH; Taylor JE; Slawin AMZ; Smith AD Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of α-Amino Acid Derivatives via Allylic Amination and [2,3]-Sigmatropic Rearrangement. J. Am. Chem. Soc 2017, 139, 11895–11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13) (a).Fang X; Johannsen M; Yao S; Gathergood N; Hazell RG; Jørgensen KA Catalytic Approach for the Formation of Optically Active Allyl α-Amino Acids by Addition of Allylic Metal Compounds to α-Imino Esters. J. Org. Chem 1999, 64, 4844–4849. [DOI] [PubMed] [Google Scholar]; (b) Ferraris D; Young B; Cox C; Dudding T; Drury WJ; Ryzhkov L; Taggi AE; Lectka T Catalytic, Enantioselective Alkylation of α-Imino Esters: The Synthesis of Nonnatural α-Amino Acid Derivatives. J. Am. Chem. Soc 2002, 124, 67–77. [DOI] [PubMed] [Google Scholar]; (c) Ogawa C; Sugiura M; Kobayashi S Stereospecific, Enantioselective Allylation of α-Hydrazono Esters by Using Allyltrichlorosilanes with BINAP Dioxides as Neutral-Coordinate Organocatalysts. Angew. Chem. Int. Ed 2004, 43, 6491–6493. [DOI] [PubMed] [Google Scholar]; (d) Jonker SJT; Diner C; Schulz G; Iwamoto H; Eriksson L; Szabó KJ Catalytic asymmetric propargyl- and allylboration of hydrazonoesters: a metal-free approach to sterically encumbered chiral α-amino acid derivatives. Chem. Commun 2018, 54, 12852–12855. [DOI] [PubMed] [Google Scholar]
- (14) (a).Roche SP; Samanta SS Autocatalytic one pot orchestration for the synthesis of α-arylated, α-amino esters. Chem. Commun 2014, 50, 2632–2634. [DOI] [PubMed] [Google Scholar]; (b) Samanta SS; Roche SP In Situ-Generated Glycinyl Chloroaminals for a One-Pot Synthesis of Non-proteinogenic α-Amino Esters. J. Org. Chem 2017, 82, 8514–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wasa M; Liu RY; Roche SP; Jacobsen EN Asymmetric Mannich Synthesis of α-Amino Esters by Anion-Binding Catalysis. J. Am. Chem. Soc 2014, 136, 12872–12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wuts PGM; Greene TW Chapter 7: Protection for the Amino Group Greene’s Protective Groups in Organic Synthesis, Ed. 4;John Wiley and Sons, Inc.: Hoboken, N.J. 2006; 696–926. [Google Scholar]
- (17).Zhang H; Lin S; Jacobsen EN Enantioselective Selenocyclization via Dynamic Kinetic Resolution of Seleniranium Ions by Hydrogen-Bond Donor Catalysts. J. Am. Chem. Soc 2014, 136, 16485–16488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) For a review see: Kennedy CR; Lin S; Jacobsen EN The Cation–π Interaction in Small-Molecule Catalysis. Angew. Chem. Int. Ed 2016, 55, 12596–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knowles RR; Lin S; Jacobsen EN Enantioselective Thiourea-Catalyzed Cationic Polycyclizations. J. Am. Chem. Soc 2010, 132, 5030–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lin S; Jacobsen EN Thiourea-catalysed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nat. Chem 2012, 4, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Arene polarizability values taken from: Waite J; Papadopoulos MG; Nicolaides CA Calculations of induced moments in large molecules. III. Polarizabilities and second hyperpolarizabilities of some aromatics. J. Chem. Phys 1982, 77, 2536–2539. [Google Scholar]
- (20).Lehnherr D; Ford DD; Bendelsmith AJ; Kennedy CR; Jacobsen EN Conformational Control of Chiral Amido-Thiourea Catalysts Enables Improved Activity and Enantioselectivity. Org. Lett 2016, 18, 3214–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fully substituted α-chloro amino esters such as α-chloro phenylglycine and α-chloro alanine proved not to be useful substrates in the allylation reactions, with the former undergoing decomposition to unidentified products and the latter undergoing elimination to the dehydroalanine derivative.
- (22).Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]
- (23).Ammer J; Nolte C; Mayr H Free Energy Relationships for Reactions of Substituted Benzhydrylium Ions: From Enthalpy over Entropy to Diffusion Control. J. Am. Chem. Soc 2012, 134, 13902–13911. [DOI] [PubMed] [Google Scholar]
- (24).Mayr H; Bug T; Gotta MF; Hering N; Irrgang B; Janker B; Kempf B; Loos R; Ofial AR; Remennikov G; Schimmel H Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles. J. Am. Chem. Soc 2001, 123, 9500–9512. [DOI] [PubMed] [Google Scholar]
- (25) (a).Sakaitani M; Kurokawa N; Ohfune Y N-carboxylate ion equivalent. II. Novel transformations of N-benzyloxycarbonyl (Z) group and N-allyloxycarbonyl group into N-t-butyldimethylsilyloxycarbonyl intermediate. Tetrahedron Lett. 1986, 27, 3753–3754. [Google Scholar]; (b) Coleman RS Chemoselective Cleavage of Benzyl Ethers, Esters, and Carbamates in the Presence of Other Easily Reducible Groups. Synthesis 1999, S1, 1399–1400. [Google Scholar]; (c) Wipf P; Uto Y Total Synthesis and Revision of Stereochemistry of the Marine Metabolite Trunkamide A. J. Org. Chem 2000, 65, 1037–1049. [DOI] [PubMed] [Google Scholar]
- (26) (a).Raheem IT; Thiara PV; Peterson EA; Jacobsen EN Enantioselective Pictet–Spengler-Type Cyclizations of Hydroxylactams: H-Bond Donor Catalysis by Anion Binding. J. Am. Chem. Soc 2007, 129, 13405–13406. [DOI] [PubMed] [Google Scholar]; (b) Raheem IT; Thiara PV; Jacobsen EN Regio- and Enantioselective Cyclization of Pyrroles onto N-Acyliminium Ions. Org. Lett 2008, 10, 1577–1580. [DOI] [PubMed] [Google Scholar]; (c) Peterson EA; Jacobsen EN Enantioselective, Thiourea-Catalyzed Intermolecular Addition of Indoles to Cyclic N-Acyl Iminium Ions. Angew. Chem. Int. Ed 2009, 48, 6328–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Park Y; Schindler CS; Jacobsen EN Enantioselective Aza-Sakurai Cyclizations: Dual Role of Thiourea as H-bond Donor and Lewis Base. J. Am. Chem. Soc 2016, 138, 14848–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27) (a).Conant JB; Kirner WR; Hussey RE The Relation Between the Structure of Organic Halides and the Speeds of Their Reaction with Inorganic Iodides. III. The Influence of Unsaturated Groups. J. Am. Chem. Soc 1925, 47, 488–501. [Google Scholar]; (b) Paddon-Row MN; Santiago C; Houk KN Possibility of .pi.-electron donation by the electron-withdrawing substituents CN, CHO, CF3, and +NH3. J. Am. Chem. Soc 1980, 102, 6561–6563. [Google Scholar]; (c) Kost D; Aviram K SN2 transition state. 4. Effect of .alpha.-substituents on SN2 reactivity and the SN2-SN1 borderline problem. A molecular orbital approach. J. Am. Chem. Soc 1986, 108, 2006–2013. [Google Scholar]; (d) Bach RD; Coddens BA; Wolber GJ Origin of the reactivity of allyl chloride and .alpha.-chloroacetaldehyde in SN2 nucleophilic substitution reactions: a theoretical comparison. J. Org. Chem 1989, 51, 1030–1033. [Google Scholar]
- (28).Rötheli AR A Mechanistic Approach Towards Highly Efficient Anion-Binding Catalysts. Ph. D. Thesis. Harvard University, 2016. [Google Scholar]
- (29).(a) Computations were carried using DFT at the B3LYP-D3BJ/6-311+G(d,p) level of theory: Ditchfield R; Hehre WJ; Pople JA Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys 1971, 54, 724–728. [Google Scholar]; (b) Hehre WJ; Ditchfield R; Pople JA Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]; (c) Hariharan PC; Pople JA The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- (30).(a) Given the observed solvent sensitivity and likely importance of attractive non-covalent interactions, we used the polarization continuum model (PCM) with a DCM dielectric and Grimme’s dispersion correction with Becke-Johnson damping (D3BJ). Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132 154104. [DOI] [PubMed] [Google Scholar]; (b) Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem 2010, 32 1456–1465. [DOI] [PubMed] [Google Scholar]; (c) Scalmani G; Frisch MJ Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys 2010, 132, 114110. [DOI] [PubMed] [Google Scholar]
- (31).Given that the rate-limiting step is known to be 1st-order in nucleophile and therefore associative based on the kinetic analysis, electronic energies (ΔE) rather than free energies (ΔG) are provided. Inappropriate entropic contributions from comparing computed associative (SN2) and dissociative (SN1) pathways are thereby avoided. As expected, the energy difference between the two pathways is smaller if free energies are compared, but the SN2 pathway is still substantially lower in energy: ΔG‡ for SN2 = 13.7 kcal/mol, ΔG‡ for SN1 >24.1 kcal/mol, ΔGrxn = –33.0 kcal/mol.
- (32).Using a water PCM, we found the SN2 pathway to be favored by an even greater margin, 22.8 kcal/mol, providing a strong indication that the high energy predicted for the iminium ion intermediate was not simply an artifact of comparing unbenchmarked covalent and ionized energies in a solvent PCM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.