

Abstract
Microbe-based therapeutics (MBTs) are an emerging therapeutic modality for the treatment of gastrointestinal infections and inflammatory bowel diseases. Current formulations for oral delivery of MBTs use capsules to achieve safe gastric transit, but oral formulations that control the spatiotemporal concentration of MBTs have yet to be developed, despite well-established connections between all therapeutics and their location, concentration, and distribution at sites of action. We report the development a multi-functional polymer-based encapsulation system to formulate MBTs for enhanced storage and delivery through formulation of a model MBT, Lactobacillus casei ATCC393. Our approach enables the additive inclusion of excipients and polymers to grant specific functions, towards the development of a modular MBT platform. For example, through addition of established excipients, our formulation provides long-term storage of the encapsulated MBT. By adding higher molecular weight polymers, the release kinetics of the encapsulated MBTs can be modified. The inclusion of a mucoadhesive polymer significantly increases the adhesion force between our formulation and the intestinal tissue. Together, mucoadhesive and sustained release properties can be used to modulate the spatiotemporal concentration of MBTs. Our formulation is compatible with standard oral capsules, thus maintaining existing clinical advantages of oral capsules while providing new functions from polymer film encapsulation.
Keywords: Drug delivery, probiotics, encapsulation, bacteria, microbes
Graphical Abstract
Microbe-based therapeutics (MBTs) are an emerging therapeutic modality for the treatment of gastrointestinal infections and inflammatory bowel diseases. Current formulations for the oral delivery of MBTs use oral capsules to ensure safe gastric transit. Here, we report a polymeric-film-based approach to encapsulate, provide long-term storage, provide mucoadhesion, and control the release and spatiotemporal distribution of encapsulated MBTs.
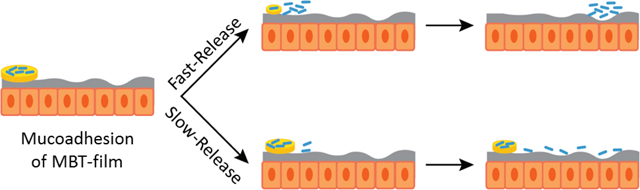
Introduction
Microbe-based therapeutics (MBTs) are an emerging class of FDA-regulated drugs that include microbiota transplants, spore-based therapeutics, probiotics, and genetically engineered microbes that secrete drugs or act as local catalysts to modulate the metabolome.[1] In clinical settings, MBTs are being used as curative treatments for gastrointestinal (GI) diseases such as ulcerative colitis and recurring Clostridium difficile which currently lack therapeutic approaches that cure these diseases in all patients.[2] These diseases are associated with dysbiosis, or an imbalance in protective versus harmful species within the microbiome,[3] and MBTs must initiate microbiome modulation to displace the bacteria implicated in dysbiosis. MBTs can potentially address long-standing limitations of antibiotics used for microbiome modulation since antibiotics can: (i) destroy the commensal microbiome thereby worsening dysbiosis,[4] (ii) create an environment in which pathogens can opportunistically thrive,[5] and (iii) face difficulties in treating antimicrobial resistant infections.[6] A separate class of clinically-tested MBTs are genetically engineered to metabolize non-digestible proteins in the lumen or secrete cytokines as patient-friendly alternatives to injectable dosage forms.[7] These MBTs have potential in reducing dosing frequency by providing a sustained drug source delivered via the oral route, thereby improving patient compliance and acceptance. As such, MBTs represent a new therapeutic modality that provide unique mechanisms to cure diseases through mediating immune system interactions and modulating the microbiome,[2c,8] or improve patient compliance by sustainably producing macromolecules in situ to decrease dosing frequencies.[9]
Broadly, different MBTs are used to treat a variety of diseases through distinct mechanisms of action and this require different delivery considerations. However, the clinical-standard delivery system for MBTs is the oral capsule, a technology predominately used for small molecule delivery.[10] While key parameters such as local concentration, residence time, and target location have yet to be defined for optimizing MBT delivery, their importance in governing toxicity, side effects, and efficacy for all other therapeutic modalities is widely accepted.[11] Development of technologies to control these parameters for small molecule drugs and biologics remains a major focus for academic and industrial research teams;[12] however, these parameters have been largely unexplored in the context of MBT delivery. Some MBTs also require a high dose burden (up to 40 capsules in a single sitting) due to the inclusion of high-volume excipients for both storage and administration purposes.[13] Alongside high dosing regimens are strict storage requirements, often requiring temperatures as low as −80°C.[2c,7a] Altogether, various formulation characteristics limit the potential for MBTs to be a patient-compliant and patient-friendly therapeutic that can be taken at home. There are considerably fewer tools and approaches to modify MBT systems for these formulation and delivery considerations; this is likely the reason why oral capsules have been the dominant formulation strategy. Development and implementation of MBTs as a therapeutic modality, distinct from small molecules and biologics, may be accelerated through the design and engineering of delivery systems specific for MBTs.
Here, we describe a film-based MBT encapsulation platform (MBT-films) that simultaneously provides enhanced storage and new delivery modalities (e.g. mucoadhesion, sustained release) for MBTs. MBT-films were synthesized using biocompatible polymers and scalable solvent casting. Lactobacillus casei ATCC393 (L. casei ATCC393) was chosen as a model MBT since it has been demonstrated to provide health benefits[14] such as maintaining intestinal homeostasis and mitigating inflammatory pathways in Crohn’s disease.[15] Our MBT-film technology was additionally designed to be compatible with the existing clinical standard delivery approach, thereby maintaining existing advantages of oral capsules. For example, enteric-coated capsules are widely used to enable payloads to bypass harsh stomach conditions (e.g. acid) through polymer coatings of pH-responsive enteric polymers that are insoluble in acidic stomach conditions (< 5 pH), but soluble in neutral intestinal conditions (> pH 6) to facilitate payload release downstream of the stomach (Figure 1A).[16] Extending beyond these advantages, our platform provides mucoadhesion to the GI tract and tunable MBT release, which, in combination, can be used to control the spatiotemporal concentration of microbes (Figure 1B). Additionally, our encapsulation system enables viable storage of MBTs up to one year. Altogether, this platform provides novel tools to study and optimize microbe formulation and delivery for advancing MBTs.
Figure 1.
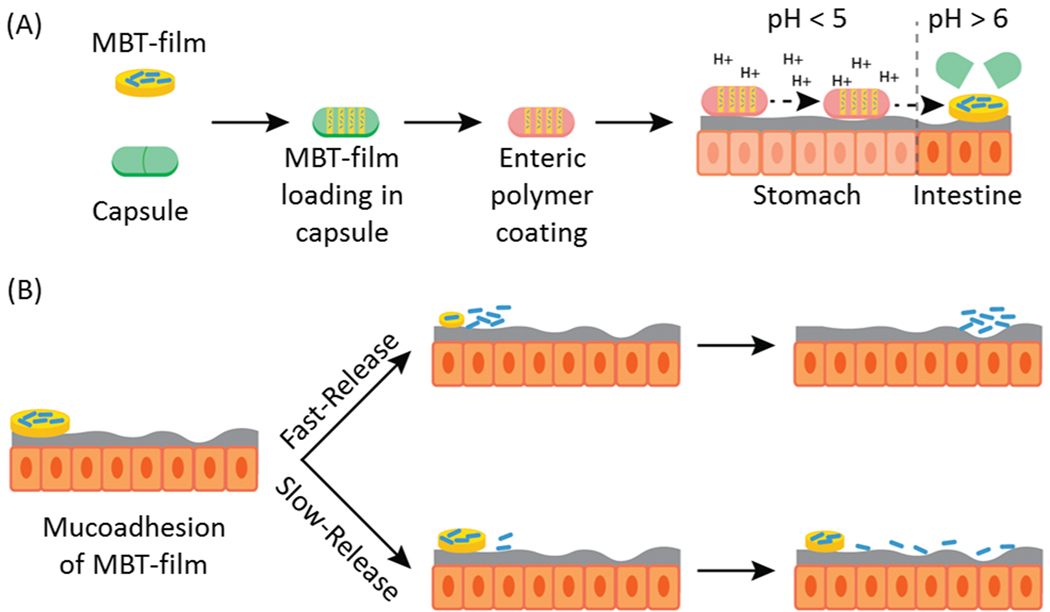
Schematic of the envisioned MBT-film delivery system. (A) Microbes are first encapsulated with storage excipients in MBT-films followed by encapsulation into oral capsules with enteric coatings. After oral administration, the capsules pass through stomach, dissolve, and release MBT-films in the intestines. (B) Building on these existing advantages of capsules, MBT-films can be engineered for mucoadhesion and tunable release for modifying the spatiotemporal distribution of the MBT along the intestines.
Results
Synthesis and characterization of L. casei ATCC393 in MBT-films
Polymer solution casting is a facile and scalable approach for fabricating polymeric films that contain a variety of excipients and payloads.[17] Here, we used this approach to synthesize MBT-films of different polymer compositions with a number of excipients of distinct physicochemical properties for L. casei ATCC393 encapsulation. Briefly, aqueous mixtures of polymer, excipients, and bacteria were mixed and cast onto polystyrene well-plates where the water was then allowed to evaporate at 4°C, leaving behind the solids in the form of a polymeric MBT-film (Figure 2).
Figure 2.
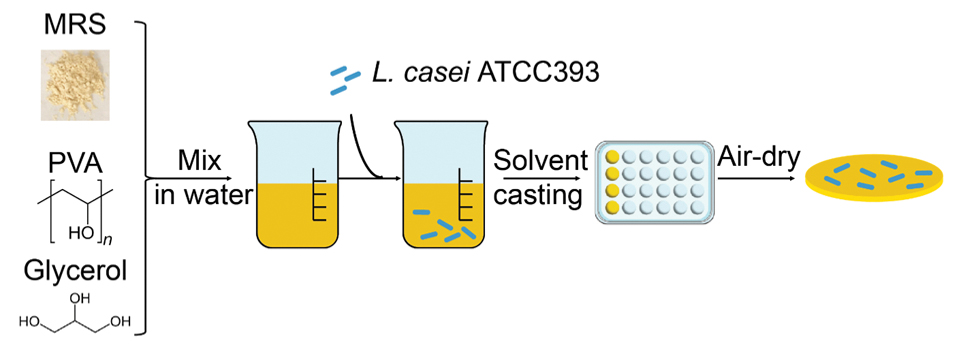
Schematic of the synthesis process for L. casei ATCC393 MBT-films. Poly(vinyl alcohol) (PVA), MRS broth, and glycerol were first dissolved in water and then L. casei ATCC393 was added to the aqueous mixture. The mixture was then cast into 24-well plates followed by a 2-day dehydration process.
Poly(vinyl alcohol) (PVA) was chosen as the main polymer encapsulant for our studies because of its widespread use in FDA-approved oral formulations and minimal cytotoxicity,[18] established use as a film-former,[19] and compatibility with additive blending approaches to make multifunctional composite films.[20] Scanning electronic microscopy (SEM) was used to determine if L. casei ATCC393 (Figure 3A) was encapsulated in PVA. SEM imaging of an intact MBT-film with L. casei ATCC393 from a top-down view demonstrated that L. casei ATCC393 were not visible on the surface or exterior of MBT-films (Figure 3B). However, SEM imaging of MBT-film cross-sections revealed the presence of L. casei ATCC393 inside the MBT-films, confirming encapsulation (Figure 3C). We then studied potential surface interactions between PVA and L. casei ATCC393 with both FTIR and zeta potential measurements. Minor differences of –OH related peak shifts in FTIR were observed when comparing solid formulations with and without L. casei ATCC393 (Figure S1A), which indicate potential interactions between PVA and L. casei ATCC393. Zeta potential measurements were performed to compare L. casei ATCC393: (i) without treatment, and (ii) exposed to MRS, glycerol and 3% PVA, followed by extensive washing. Following exposure to film constituents, zeta potential of L. casei ATCC393 slightly increased (Figure S1B). This slight change indicates that possible interactions between neutrally charged PVA and the bacterial surface may occur at these specific conditions.
Figure 3.

Imaging of L. casei ATCC393 MBT-films. (A) SEM of plain L. casei ATCC393 SEM of L. casei ATCC393 MBT-films from a (B) top-down view and (C) cross-sectional view. (D) Camera image of an L. casei ATCC393-MBT-film. Scale bars = 3 μm unless otherwise noted.
Storage of L. casei ATCC393 in MBT-films
Film formation involves dehydration which can cause loss of microbe viability as shown with other MBT dehydration approaches such as lyophilization.[21] As such, we sought to develop our films for long-term storage of L. casei ATCC393 by including storage excipients within the films. We added DeMan-Rogosa-Sharpe (MRS) broth, a lactobacillus-specific culture formulation, as an excipient component of the film because we hypothesized that the constituents of MRS broth (e.g. sugars, amino acids) could provide storage benefits as they have been shown to protect other sensitive cargos like biologics.[22] Furthermore, we confirmed that MRS broth exhibited minimal toxicity against intestinal epithelial cells (Figure S2). Glycerol was also investigated as an excipient since it is the most commonly used protectant for microbe storage.[23] Additionally, glycerol is a known plasticizer that grants flexibility to the films,[24] a feature that could allow the films to be manipulated into different geometries. These excipients were investigated individually and in combination, both with film-encapsulated L. casei ATCC393 and non-encapsulated (plain) L. casei ATCC393. MBT-films that contained all excipients maintained structural integrity after dehydration and removal from well-plate templates (Figure 3D), and exhibited high consistency in loading (Figure S3).
We first investigated storage of L. casei ATCC393 at 4°C (Figure 4A), to evaluate the role that each of the above excipients play at long-times. Plain L. casei ATCC393 (no excipients or polymers) suffered a nearly 4 log reduction in CFU after 1 week and were undetectable thereafter. The main conclusion was that MRS-containing formulations provided significantly higher maintenance of L. casei ATCC393 as compared to non-MRS formulations at each timepoint. MRS alone, without PVA and glycerol, provided the largest benefit to L. casei ATCC393 storage at 1 year at 4°C. Interestingly, a possible trend at increasing PVA concentration was observed, despite all PVA-containing formulations demonstrating significantly lower CFU maintenance as compared to MRS alone. To investigate the possible trend with PVA further, we performed a separate storage study at an increased temperature of 37°C (Figure 4B), which mimics developing-world conditions, and at PVA amount ranging from 1 to 20%. We observed complete loss of L. casei ATCC393 CFU viability after three days of storage in all groups, except those with 10% and 20% PVA concentration (Figure 4B), indicating that PVA, and higher PVA percentages provide advantages at storage under higher temperature conditions. Altogether, a complex relationship between excipients, PVA concentration, and storage conditions was observed and we describe these findings in more detail in the discussion section.
Figure 4.
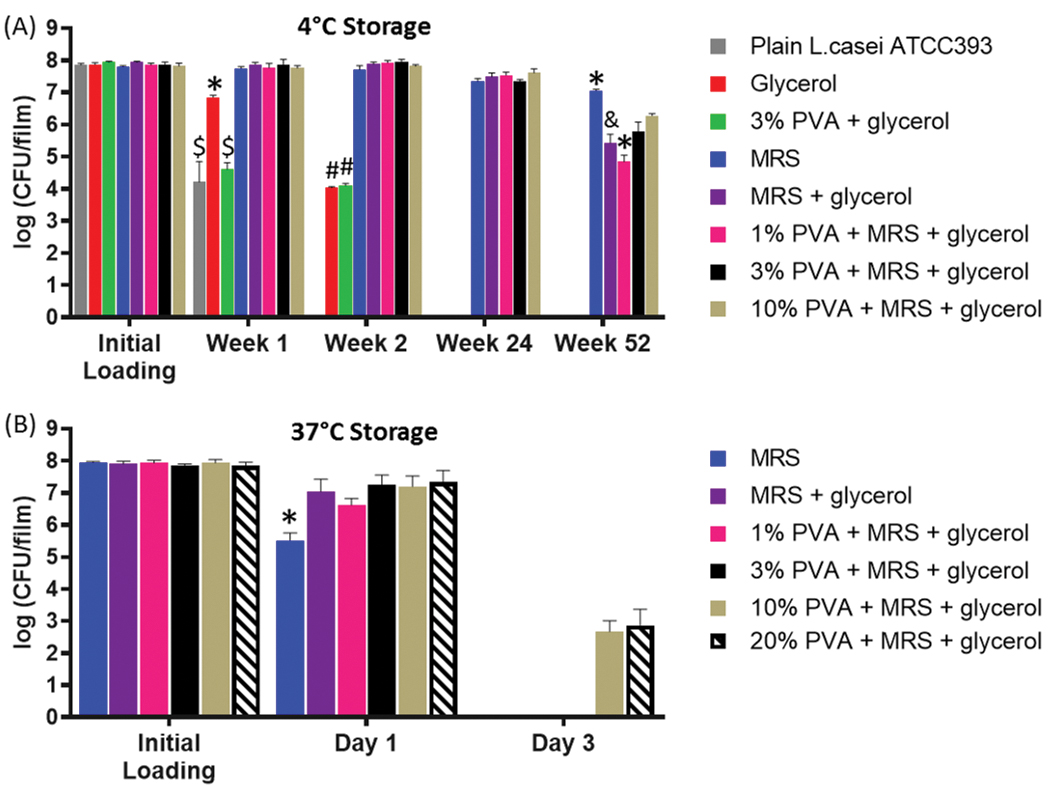
Storage of L. casei ATCC393 in MBT-films. Viability of L. casei ATCC393 with different combinations of excipients at (A) 4°C and (B) 37°C. Each error bar represents standard deviation (n = 3). Statistical analysis was conducted using one-way ANOVA followed by post hoc Tukey’s HSD test for pairwise comparison at each timepoint (statistical significance defined at p < 0.05). *: significantly different from the rest of the groups within the corresponding timepoint. $: significantly different from glycerol, MRS, MRS+glycerol, 1% PVA+MRS+glycerol, 3% PVA+MRS+glycerol and 10% PVA+MRS+glycerol. #: significantly different from MRS, MRS+glycerol, 1% PVA+MRS+glycerol, 3% PVA+MRS+glycerol and 10% PVA+MRS+glycerol. &: significantly different from MRS, 1% PVA+MRS+glycerol and 10% PVA+MRS+glycerol.
Towards defining a mechanism for MRS-based storage enhancement, we examined various film and pre-film characteristics. We evaluated the pH of various pre-dried film solutions and of the final film formulation to determine if pH differences could be responsible for viability loss, but no significant differences in pH were observed (Table S1). We then evaluated loss on drying (Figure S4) as an indicator of moisture content but no correlation between percent moisture content with storage viability was observed. Finally, we investigated whether glucose, the most abundant protectant contained in MRS, was responsible for conferring the storage benefits in the MRS formulations. MRS was replaced by glucose at the same concentration of glucose in the MRS broth. Within 8 weeks, a continuous viability loss was observed at 4°C (Figure S5), indicating that while glucose is likely an important storage component in our MRS formulation, glucose alone cannot recapitulate the complete storage benefits provided by MRS. Given that MRS improved bacteria viability regardless of the presence of PVA (Figure 4A), we postulated that the MRS-based storage strategy is compatible with other polymers. As such, we investigated the compatibility of our film system with other commonly used polymers for oral delivery such as sodium alginate, PVP K90, HPMC and gelatin type B.[25] Storage studies at room temperature with L. casei ATCC393 demonstrated that high viability was maintained for 1 week in all these biomaterials, with statistically significant reduction in CFUs for alginate and PVP (Figure S6).
Tunable Release of L. casei ATCC393 from MBT-films
To evaluate whether different release profiles could affect spatial distribution of MBTs along the intestines, we used a custom 3D-printed holder (Figure S7A) for mounting sections of intestines (Figure S7B). At one end, a hole was created for infusion of suspensions that recapitulate different release rates of MBTs on the intestinal surface so as to mimic transit time in the intestines. A bioluminescent non-pathogenic E. coli strain was chosen for the study for ease of quantification and to visualize microbe distribution along the intestine via IVIS.[26] To mimic burst release, as is provided by current clinical MBT capsules, 107 CFU were introduced as a bolus at the inlet, followed by immediate infusion of water at 1 mL min−1 for 6 minutes at room temperature to mimic physiological flow rates in the intestines.[27] To mimic slow release, an equivalent amount of bacteria were introduced as an infusion using identical flow conditions and for the same amount of time. At the end of the infusion, the signals of total bioluminescent flux for the two release groups were comparable (Figure S7C), indicating similar total dosing. Effectively, this allowed us to compare how release rate of MBTs affects their intestinal distribution at short timepoints. Our results demonstrate that the slow release groups provided 1.5 fold more intestinal surface area coverage of MBT as compared to the fast release group (Figure 5A and 5B). These differences in MBT distribution motivated the development of film formulations that enable distinct release profiles towards controlling intestinal distribution. However, when attempted to modulate the release of 3% PVA films, we observed that PVA exhibited rapid release (< 15 min), independent of film thickness (Figure S8A), indicating that PVA on its own provides a burst-like release profile. To modify our film system to achieve sustained release for our base PVA films, we blended in a high-molecular-weight (700 kDa) sodium carboxymethylcellulose (NaCMC), since NaCMC has been shown to provide sustained release properties to various formulations.[28] A blend of NaCMC and PVA was chosen because we observed that NaCMC films without PVA exhibited poor pliability and flexibility, thus rendering them difficult to for post-fabrication handling and eventual dosing (Figure S8B). We first confirmed that addition of NaCMC into the PVA films did not compromise the storage of L. casei ATCC393 (Figure 5C), and then systematically investigated how addition of NaCMC affected release profiles. We demonstrated that as NaCMC amount increases in relation to PVA, release of MBTs is slowed (Figure 5D). We then investigated how film thickness can be used to modulate release. Although release was not affected by thickness for PVA-only films (Figure S8A), increasing thickness of PVA-NaCMC films substantially prolonged bacteria release profiles (Figure 5E), providing 3 distinct release profiles that achieve 99% total CFU release in 15 minutes, 2 hours, or 8 hours, depending on film thickness. Finally, we demonstrated that increasing surface area of MBT films containing NaCMC, while maintaining identical volume and CFUs, could also be used to provide sustained release profiles (Figure S8C).
Figure 5.
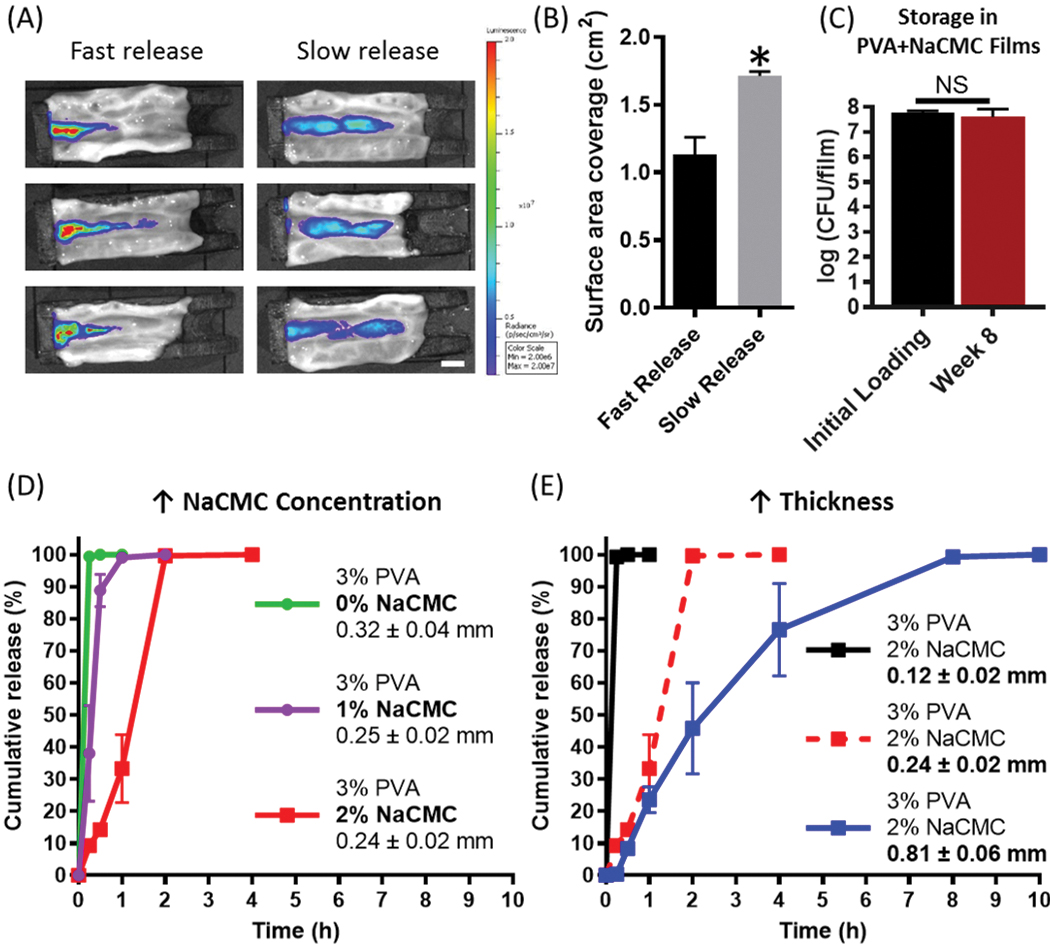
Tunable release of microbes from MBT-films. (A) Microbe distribution on the intestinal surface under fast and slow release conditions. (B) Intestinal surface coverage of microbes under fast and slow release conditions. (C) Storage of L. casei ATCC393 in 3%PVA+2%NaCMC MBT-films at 4°C. (D) Microbe release kinetics from 3%PVA films with 0%, 1%, or 2% NaCMC added. (E) Microbe release kinetics from 3%PVA+2%NaCMC at various film thicknesses. Each error bar represents standard deviation (n = 3). Student’s t-test was used for comparing PVA-NaCMC to positive control and film weight individually (statistical significance defined at p < 0.05). *: significantly different. Statistical analysis was conducted using one-way NS: No significance.
Mucoadhesion of MBT-films
Mucoadhesive properties facilitate strong interactions between macroscopic delivery systems and the GI tract.[29] These properties enable prolonged GI residence time or even enhanced retention to specific GI areas.[30] NaCMC, used above to provide sustained release properties to MBT-films, is also a mucoadhesive polymer,[28a] and as such PVA-NaCMC films were examined for mucoadhesion. The mucoadhesive force, measured by a pulley system, was defined as the minimal force to detach MBT-films from the lumen side of ex vivo porcine intestine (Figure 6A). A mucoadhesive force of over 40 mN was recorded for the NaCMC containing MBT-films, over 100 times higher than the MBT-film weight (Figure 6B).
Figure 6.
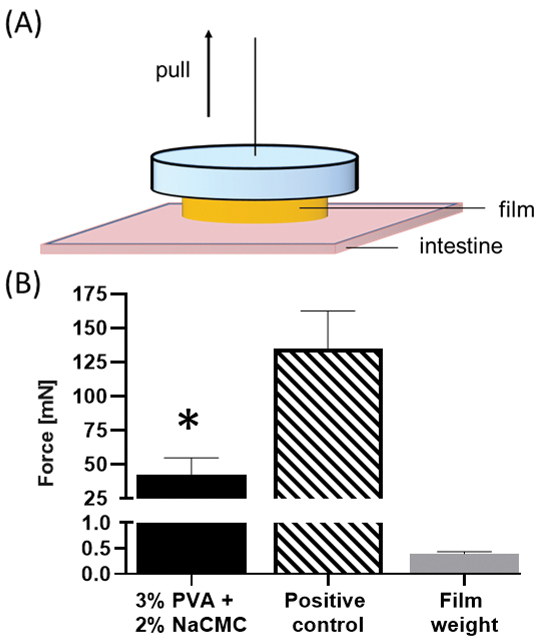
Mucoadhesion of 3% PVA+2% NaCMC MBT-films. Using a standard (A) pulley system, (B) the minimal force to detach a 3% PVA+2% NaCMC MBT-film from porcine intestine was measured at 37°C after incubation of film on ex vivo porcine intestine for 2 minutes at 37°C. Positive control consists of 3% PVA+2% NaCMC MBT-film super-glued onto the intestinal surface. Error bars represent standard deviation (n = 5 for experimental and control groups; n = 10 for film weight group). Student’s t-test was used for comparing PVA-NaCMC to positive control and film weight individually (statistical significance defined at p < 0.05). *: significantly different from positive control and film weight.
Compatibility of MBT-films with current clinical capsules
We then sought to demonstrate that the MBT-film platform is compatible with existing oral delivery modalities that are used in the clinic for MBT delivery. To better visualize the compatibility of MBT-films and capsules, we utilized rhodamine B to dye the PVA MBT-film. The flexibility of PVA MBT-films, granted by plasticizing effects of glycerol, enabled folding and subsequent insertion into a standard, 00-sized oral capsule (Figure 7A–D and Figure S9A). Slow-releasing PVA-NaCMC-MBT films of the highest NaCMC concentration tested could also be inserted into a standard 00-sized oral capsule (Figure S9B). Furthermore, a single 00-sized capsule could be used to deliver at least ten MBT-films without affecting the integrity of the capsule system (Figure 7E–G). Storage compatibility with capsules was also evaluated with ten PVA MBT-films stored at 4°C inside capsules for five days, demonstrating statistically insignificant viability loss (Figure 7H). Furthermore, capsule dissolution time comparing empty capsules and capsules with ten films inside demonstrated no significant differences in release (Figure 7I). Through increasing encapsulation of L. casei ATCC393 up to 109 CFU per MBT-film whilst maintaining long-term storage benefits (Figure 7J), we demonstrate that our MBT-film system could deliver 1010 CFU of L. casei ATCC393 via a single capsule, a dose that would meet the requirements of many clinically used MBTs.[2c,13a]
Figure 7.
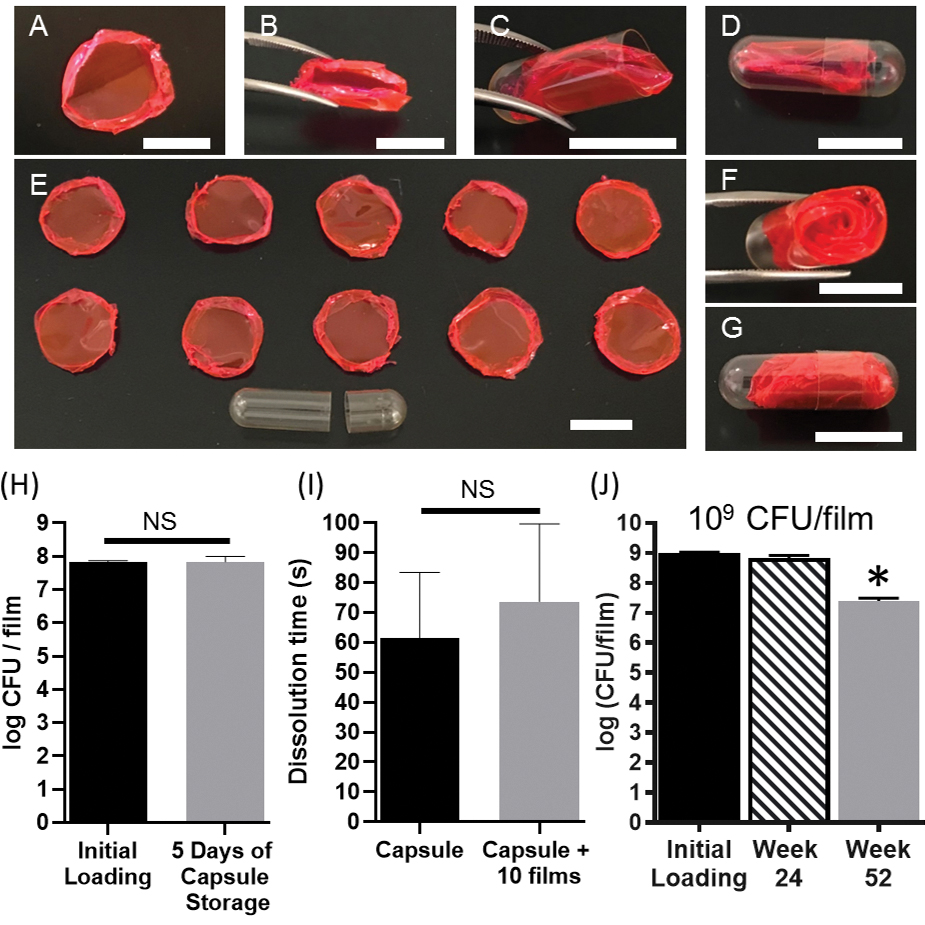
Compatibility of the MBT-film with standard oral capsules. (A) A rhodamine-dyed MBT-film. (B) Folding of the rhodamine-dyed MBT-film. (C) Insertion and (D) capping of the rhodamine-dyed MBT-film into a 00-sized oral capsule. (E) Ten rhodamine-dyed MBT-films and an empty 00-sized capsule. (F) Insertion and (G) capping of ten rhodamine-dyed MBT-films into a 00-sized capsule. Scale bars = 1 cm. (H) Storage of MBT films (3%PVA+MRS+glycerol with 108 L. casei ATCC393) in 00-sized capsules. (I) Capsule dissolution time with or without film inclusion. (J) Storage of L. casei ATCC393 in 3%PVA+MRS+glycerol at 4°C at 109 CFU film−1 loading. Each error bar represents standard deviation (n = 3). (H-I) Statistical analysis was conducted using Student’s t-test (statistical significance defined at p < 0.05). NS: not significant. (J) Statistical analysis was conducted using one-way ANOVA followed by post hoc Dunnett’s test (statistical significance defined at p < 0.05). *: significantly different from initial loading.
Discussion
Here, we report a polymer-based encapsulation system that simultaneously enables the long-term storage of L. casei ATCC393 and facilitates new delivery modalities for MBTs such as mucoadhesion and tunable release of MBTs. The MBT-film system employed here exhibits additive properties as different excipients and polymers are included at the synthesis stage; thus, our MBT-films have the potential to form the basis of a modular platform for MBT delivery (Figure 2). Through the addition NaCMC, the MBT-film system was modified for sustained release and mucoadhesive functions. Furthermore, we show compatibility of the MBT-film system with other polymers such as alginate and gelatin which are commonly used in many MBT formulations.[31] We believe that due to the tunable and additive nature of this system, our MBT-films can be engineered for storage and delivery of a variety of clinically-relevant MBTs, beyond the L. casei ATCC393 example highlighted here. Future development of these systems may lead to approaches to modulate GI residence time and the spatial distribution of microbes in oral delivery.
We first confirmed that L. casei ATCC393 was completely encapsulated in the MBT-films following dehydration by visualizing the surface and cross-sections of MBT-films. SEM imaging of the films highlighted how L. casei ATCC393 was only observed in the films interior following cross-sectioning (Figure 3). We then demonstrated that MRS, both alone and when formulated with PVA, preserves L. casei ATCC393 at 4°C (Figure 4A) and 37°C (Figure 4B). Compared with 4°C, storage at high temperatures resulted in faster loss of viability, which is consistent with the trend reported in other studies.[32] However, we observed conflicting results at 4°C (Figure 4A) and 37°C (Figure 4B). We saw that MRS alone provided enhanced storage at 52 weeks compared to PVA+MRS formulations, but the opposite trend was observed at 37°C. Towards describing this, we closely analyzed the components of MRS that likely contribute to enhanced storage (e.g. glucose (Figure S5), amino acids). Both sugar and amino acid excipients have been demonstrated to require hydrophobic, ion-dipole, and hydrogen-bonding interactions to stabilize surface proteins to grant storage benefits.[22c,33] Since MBTs are protein-rich, the benefits that glucose and amino acids provide in stabilizing proteins may underlie the storage benefits that the MRS components contribute to MBTs. Because of this, we hypothesize that as PVA is introduced into the formulation, the physical interactions between storage excipients and the MBTs that occur at the excipient-microbe interface are changed, since excipient mass remained constant for each group. Altering the interactions between excipients and MBTs may be responsible for the demonstrated differences in storage. This hypothesis is supported by our characterization work that suggests PVA and excipients may interact with the MBT surface (Figure S1). Interestingly, at both temperatures we observed a trend that at higher PVA concentrations, storage was improved. Since oxidation-mediated death of dehydrated microbes has been shown for many other strains as they are exposed to storage conditions in oxygen-rich environments and the fact that PVA is widely-known and used as an oxygen barrier,[34] we postulate that increasing PVA amount can potentially ameliorate oxidation-mediated viability loss via PVA limiting oxygen diffusion. Considering these results and our hypotheses, it is clear a complex relationship between PVA, excipients, and storage conditions (e.g. temperature, humidity) exists and that further study is needed to define optimal formulations for specific storage conditions. We then demonstrated compatibility of MRS with polymers other than PVA (Figure S6) and believe this potentiates the application of MRS in the context of a wide range of biomaterials, further supporting the MBT-film system and MRS as a potential platform for MBT storage and delivery. However, inclusion of MRS and potentially other components that are widely used with bacteria for a variety of reasons (i.e. as growth or storage media) should be considered for their toxicity against mammalian cells. Since the specific combination of MRS ingredients, such as peptone, yeast extracts and a variety of salts could pose toxicity risks against mammalian host cells, we assessed its cytotoxicity on an intestinal epithelial cell line. No statistical significance of cell viability was identified among the MRS treated groups and control, implicating MRS has minimal, if any, impact on Caco-2 cells in vitro (Figure S2).
We then 3D-printed a holder to mount sections of intestines so we could investigate how different MBT release profiles alters their distribution along the intestinal surface. We aimed to recreate and subsequently compare two extreme MBT release profiles, bolus and sustained release. We conducted this experiment at room temperature to minimize bacteria growth, which could potentially confound our observations. In the bolus case, which mimics current delivery of MBTs via capsule, 107 CFU were exposed directly to the surface of the intestines followed by water flow to mimic intestinal transport. In the sustained release case, which could potentially be mimicked by our tunable MBT-film system, an identical number of microbes (Figure S7C) were exposed to the same area of the intestines, but over a longer period of time. Effectively, we compared how microbes exposed to the intestines over a short period of time at high concentration differs in intestinal distribution as compared to a longer period of time at lower concentration. Both cases were exposed to identical physiologically relevant flow rates during the duration of the experiment.[27] Our quantitative imaging analysis showed that slower release rate correlated with higher surface area coverage as compared to the bolus release (Figure 5A). While it has not been shown in any previous work that we could identify, it is likely that altered distributions of MBTs along the GI tract play a role in the ability of the MBT to colonize. This is likely the case because the first step in MBT colonization is adhesion, and adhering to a larger surface area may increase the number of individual MBT-intestinal interactions, potentially increasing the likelihood of colonization. Considering another main therapeutic function of MBTs, secretion of drugs, increasing surface area coverage would lead to higher absorption of drugs across the epithelium. As such, modifying the spatial distribution of MBTs through formulation design has the potential to alter how MBTs interact with target tissues. However, this experimental system relied on a static depot of MBTs as the source of MBTs, a situation that may be more difficult to achieve in a dynamic in vivo situation. Taken together, we were motivated to create a sustained release and mucoadhesive delivery system towards tailoring microbe distribution profiles in the GI tract.
As such, we focused on the development of MBT-films with distinct release profiles. Our initial result showed that NaCMC alone exhibited poor flexibility and pliability as compared to PVA+NaCMC films (Figure S8B) which would render NaCMC films incompatible with post-fabrication handling for incorporation into standard oral capsules. Flexibility and pliability maintains structural integrity of the films throughout the fabrication and capsule-loading process, and potentially under the dynamic gastrointestinal conditions in vivo, which is important as geometry (Figure 5E) and surface area (Figure S8C) are shown to be key structural parameters in ensuring reproducible and predictable release profiles. As a result, we chose the strategy of polymer blending with PVA and slow-releasing NaCMC to slow film dissolution. Through the additive blending of NaCMC, our MBT-films provided sustained release of MBTs over a period of up to 10 hours or over a period of as short as 15 minutes (Figure 5 and S8) We further demonstrated that tuning the NaCMC content (Figure 5D), thickness (Figure 5E), and surface area (Figure S8C) are all viable strategies to grant sustained release properties. Many other potential strategies exist to slow dissolution of PVA films, such as inducing crystallization of PVA by treating the film with freeze-thaw cycles and chemical crosslinking of PVA chains.[35] However, these methods introduce additional physical or chemical challenges to the encapsulated microbes which may reduce viability during fabrication.
Mucoadhesive properties allow formulations to reside in the dynamic environment of GI tract for extended periods of time.[30] We showed the addition of NaCMC into PVA films enables mucoadhesion on intestines ex vivo, where the magnitude of the adhesive force is comparable to that of other intestinal devices reported in the literature.[36] We envision that in the context of drug-secreting MBTs,[9b] prolonged residence of MBTs enabled by mucoadhesion could extend the time for which MBTs secrete drugs at sites of absorption (e.g. the small intestine), which would potentially lead to higher therapeutic doses or longer duration of therapy. Also, mucoadhesion interfaces MBTs close to host tissues and thus could alter or accelerate interactions between MBTs and the mucus, since colonization requires adherence to mucus. Translation of these advantages towards in vivo performance of the films to manipulate GI residence time requires further evaluation. Generally, microbiome manipulation with MBTs in treating recurrent Clostridium difficile infections and inflammatory bowel diseases is assumed to require sustainable colonization for boosting therapeutic efficacy.[37] However, modulating colonization of MBTs through controlling their interactions at sites of desirable colonization represents an under-explored area. By providing a mucoadhesive anchor of MBT-formulations upstream of colonization sites and subsequently providing tunable release kinetics, MBTs may interact with colonization sites in varied and controllable spatiotemporal profiles (Figure 2), which may influence whether and how MBTs colonize. As such, mucoadhesive and tunable release properties of PVA-NaCMC films may serve as an enabling tool to systematically study the impacts of spatiotemporal factors of MBT delivery on the colonization and therapeutic outcome in vivo.
Finally, we demonstrated compatibility of our MBT-films with standard capsules (Figure 7) to highlight that our MBT-films can additionally use the existing advantages of capsules. For example, enteric coatings can be used to bypass the stomach in an effort to limit interactions between MBTs and the acid environment. Moreover, multiple MBT-films, possibly of different MBT composition or even release rates, can be included in a single capsule with up to 1010 CFU capsule−1 (Figure 7J), which satisfies the dosing requirements of clinical studies that require the highest dose of MBTs.[2c,13a] Given that each film can include unique bacteria, we speculate that the film-capsule combinatorial system can also be used for delivering a microbial consortia. Overall, we report a multifunctional polymeric film system for storage and delivery of L. casei ATCC393. We found MRS enhanced storage of L. casei ATCC393 at two different temperatures. Mucoadhesion and tunable release properties of the film system may serve as a powerful tool to control parameters of oral microbe delivery including residence time and spatial distribution. The additive approach we used to endow additional functions such as improved storage, tunable release, or mucoadhesion highlight the potential of using this system as a future platform for MBT delivery.
Methods
Materials
Poly(vinyl alcohol) (PVA, 87%−90% hydrolyzed, 30–70 kDa), sodium carboxymethyl cellulose (NaCMC, 700 kDa), polyvinylpyrrolidone (PVP) K90, sodium alginate and sodium borohydride were purchased from Sigma-Aldrich (Missouri, USA). Gelatin type B was purchased from Acros Organics. Methocel (hydroxypropyl methylcellulose, HPMC) K4M was a kind gift from Dow Chemical Company (Michigan, USA). Glycerol, ethanol, acetone, glutaraldehyde and rhodamine B were purchased from Fisher Scientific (Massachusetts, USA). DeMan-Rogosa-Sharpe (MRS) broth and Lactobacillus casei ATCC393 were purchased from Thermo Scientific (California, USA). MRS agar was purchased from Becton, Dickinson and Company. pGEN-luxCDABE was a gift from Harry Mobley (Addgene plasmid # 44918; http://n2t.net/addgene:44918 ; RRID:Addgene_44918). Porcine intestine was purchased from Lampire Biological Laboratories (Pennsylvania, USA). The intestine holder was 3D printed with XT-BLACK 2.85/750 co-polyester (colorFabb, Netherlands) by Ultimaker 3 (Ultimaker, Netherlands). Enzyme-free simulated intestinal fluid (SIF), LB broth, glucose and Loctite super glue were purchased from VWR (Pennsylvania, USA). Capsules were a kind gift from Torpac Inc (New Jersey, USA). Caco-2 (ATCC #HTB 37) cells were purchased from the University of North Carolina at Chapel Hill Tissue Culture Facility. All the water used in this paper was sterilized Milli-Q water.
Bacteria Growth
L. casei ATCC393 was inoculated into autoclaved MRS broth and grew overnight statically at 37°C in sealed 50 mL conical tubes. The overnight culture was diluted down with MRS broth to OD600 < 0.3 and harvested when OD600 reached 0.50 ± 0.05. Bioluminescent and ampicillin-resistant E. coli was inoculated into autoclaved LB broth containing 100 μg mL−1 ampicillin and grew in vented tubes overnight at 200 rpm until harvest, 37°C.[26] Before use, bacteria culture was centrifuged at 4000 rpm for 10 minutes at room temperature and the pellet was washed once in sterile water. OD600 values were measured by GENESYS 30 visible spectrophotometer (Thermo Scientific, California, USA) with bacteria-free media subtracted.
Preparation of Polymer Solutions
3% wt vol−1 PVA, sodium alginate, PVP K90 or gelatin type B was dissolved in 80°C sterile water. 2% wt vol−1 HPMC was first dispersed in 80°C water under stirring and then dissolved at room temperature. To prepare polymer solution containing both PVA and NaCMC, 1% or 2% wt vol−1 NaCMC was added into 3% wt vol−1 PVA in water and stirred overnight at room temperature until complete dissolution. 1% vol vol−1 glycerol was added into all the polymer solutions as the film plasticizer. In the formulations with MRS, 5.2% wt vol−1 MRS formula was dissolved in water and autoclaved before polymer dissolution.
Film preparation
107 to ~109 CFU mL−1 L. casei ATCC393 was added and stirred in polymer solutions at room temperature. In the formulations without polymer, L. casei ATCC393 was directly introduced into the solution accordingly. For storage studies, 300 μL of the prefabricated film solution was cast into individual wells of 24-well plates, followed by air-dry at 4 °C for two days (Figure 2). For SEM analysis, L. casei ATCC393 was first crosslinked with 2.5% vol vol−1 glutaraldehyde under shaking for 1 hour at room temperature, followed by addition of sodium borohydride (10 mg mL−1) to quench the crosslinking process. Then bacteria were pelleted, washed once in water and introduced into the PVA solution (with no MRS, syringe-filtered with 0.22 μm filter) to a concentration of ~109 CFU mL−1. For mucoadhesion studies, the 3% PVA-2% NaCMC film was fabricated by four rounds of solvent casting. For bacteria release studies, one, two and four rounds of solvent casting were done separately to make three films with different thickness for 3% PVA and 3% PVA-2% NaCMC films. Four rounds of solvent casting were used for making 3% PVA-1% NaCMC films. One round of solvent casting for PVA films with NaCMC was defined as casting 4g of the prefabricated film solution (~108 CFU mL−1 L. casei ATCC393) into a sterile, 60 mm x15 mm, round petri dish followed by solvent evaporation at 4°C for 2 days Some 3% PVA-1% NaCMC films were sliced obtain halves of films at half thickness. For making rhodamine B-loaded films, rhodamine B (0.03 M) was added into PVA solution (with no MRS) followed by casting 1 mL into individual wells of 24-well plates.
SEM analysis
Films were sectioned using a razor blade and mounted onto a SEM stub with adhesive carton tape for imaging. Plain L. casei ATCC393 was prepared by resuspending crosslinked L. casei ATCC393 in acetone and loading an aliquot on a stub. Plain L. casei ATCC393 was imaged after acetone evaporation.
FTIR analysis
The interaction between PVA and L. casei ATCC393 in solid films containing MRS and glycerol was evaluated using Fourier Transform Infrared (FTIR) Spectroscopy (Nicolet 380 FT-IR Spectrometer, Thermo Scientific, California, USA). Data processing was performed with OMNIC Spectroscopy Software.
Zeta Potential Measurement
Bacteria culture was centrifuged as described above and the pellet was washed three times in water. Then pellets were resuspended with 3% wt vol−1 PVA solution (including 1% vol vol−1 glycerol and 5.2% wt vol−1 MRS) and incubated at room temperature for 30 minutes under rotation. Then the suspensions were centrifuged and washed three times in water as described above. Final pellets were resuspended with water for zeta potential measurement with zeta sizer (Malvern, PA, USA). Bacteria without incubation with polymer solutions was used as control.
pH and Moisture Analysis
After L. casei ATCC393 was introduced into the polymer solutions, pH was measured at room temperature. For pH measurement of dried films, 5 films of 3% PVA-MRS-glycerol at day 11 were dissolved in 1.5 mL water and pH was measured. Moisture analysis was conducted by weighing samples (10–80 mg) in glass vials with the Mettler Toledo XP26 DeltaRange Microbalance and heating films at 105°C for 1 hour. Weight loss-on-drying was presented as percentage of pre-drying film mass.
Cell Viability Assay
1×104 Caco-2 cells were seeded into tissue culture-treated 96-well plates and grown at 37°C for 72 hours. Then supernatants were replaced with 5.2% and 0.52% wt vol−1 MRS dissolved in cell culture medium consisting of DMEM, 20% Fetal Bovine Serum (FBS), and 100 U mL−1 penicillin/100 μg mL−1 streptomycin and incubated at 37°C for 4 hours. Cell viability was evaluated with Vybrant® MTT Cell Proliferation Assay Kit (Thermo Scientific, California, USA) according to manufacture instructions.
Quantification of Viability of Bacteria for Storage Study
Films were dissolved by adding 1ml of sterile water, followed by shaking for 10–20 minutes until the films dissolved. Dissolved films solutions were serially diluted and drop-plated (10 μL) on MRS agar, incubated at 37°C for 48–60 hours, and enumerated for CFU. Prefabricated film formulation was used as control (day 0).
Mucoadhesion Measurement
An 8-mm diameter film was punched and incubated with the luminal side of a piece of porcine intestine (~3 cm x 5 cm) for 2 minutes at 37°C before force measurement. The adhesive force between the film and the intestine was measured with a standard pulley system. Specifically, a plastic cylinder (~3 cm in diameter and ~1 cm in height) was super glued to the top side of the film on the intestine. The opposite side of the cylinder was attached to a string and loaded onto a pulley system. The opposite end of the string attached to a conical plastic container (volume, 50 mL). Water was gradually added into the container until the film detached from the intestine. The weights of the cap, the holder and the added water were recorded. As a positive control, the film was super glued to the intestinal surface. The adhesive force was calculated as: Adhesive Force = Weight (holder + water) - Weight (cylinder).
Bacteria Distribution on Intestinal Surface
The intestine holder was 3D-printed by Ultimaker 3 with XT-BLACK 2.85/750 co-polyester. One piece of intestine (~3 cm x 5 cm) was sectioned from bulk porcine intestine and fitted into the holder with the luminal side facing upwards. Silicone tubing (#89501–666, Thermo Scientific, California, USA) was inserted into the hole at the left end of the holder for bacteria suspension or pure water infusion over the intestine. Fast release was mimicked by loading TOP10 E. coli (5 μL, 1.1±0.3×107 CFU in total) onto the inlet and subsequently infusing water (100 μL) at 1 mL h−1 for 6 min at room temperature, where the flow rate was determined based on porcine large intestine transit time.[26–27] Slow release was mimicked by infusing 100 μL E. coli suspension with equivalent amount of total CFU at the same flow conditions. Infusion was controlled by Pump 11 Elite Syringe Pump (Harvard Apparatus, Massachusetts, USA). Bioluminescent images were taken and analyzed with In vivo Imaging System (Perkin Elmer, Massachusetts, USA).
Bacteria Quantification for Sustained Release Study
An 8-mm diameter film was punched out and incubated with 5 mL filter-sterilized SIF in a 50 mL conical tube at 200 rpm, 37°C. 5 mL supernatant was removed and replaced with 5 mL fresh SIF at pre-determined timepoints. CFU in the supernatant was enumerated as described above. Film thickness was measured by a digital caliper (World Precision Instruments, Florida, USA).
Compatibility of Films with Capsules
Ten 3% PVA+MRS+glycerol films were loaded into a 00-sized oral capsule after 2 day air-dry process. At day 7, films were taken out of the capsule and dissolved in 10 mL sterile water. CFU was enumerated as described above. Capsule dissolution time was evaluated by incubating a single capsule with or without ten films included in 50 mL water at 37°C under rotation. Time was recorded when capsules started to dissolve.
Statistical Analysis
All CFU data were presented after log transformation unless otherwise noted. Cumulative release data were normalized to total released CFU counts. Experiments were conducted in triplicate unless otherwise noted. The data were presented as mean ± SD. Significant differences were assessed with parametric two-sided Student’s t-test (two groups) or one-way ANOVA (more than two groups). Tukey’s HSD test was chosen for post hoc comparison unless otherwise noted. α = 0.05. P- value less than 0.05 was considered significantly different. Statistical analysis was conducted with RStudio Version 1.1.456.
Supplementary Material
Acknowledgements
We thank UNC CHANL facility for guidance of SEM analysis. We thank Dr. Daniel C. Sweeney and the UNC BEAM facility for assistance in fabricating the 3D-printed intestine holder. I.Y. was funded through the UNC Solar Program. Research reported in this publication was supported by the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number R21DK123583. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is the peer reviewed version of the following article: Qiu, K., Young, I., Woodburn, B. M., Huang, Y., Anselmo, A. C., Polymeric Films for the Encapsulation, Storage, and Tunable Release of Therapeutic Microbes. Adv. Healthcare Mater. 2020, 9, 1901643., which has been published in final form at https://doi.org/10.1002/adhm.201901643. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Use of Self-Archived Versions.
References
- [1].a) Young VB, Clinical Microbiology and Infection 2016, 22, 905; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vargason AM, Anselmo AC, Bioengineering and Translational Medicine 2018, 3, 124; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Riglar DT, Silver PA, Nature Reviews Microbiology 2018, 16, 214. [DOI] [PubMed] [Google Scholar]
- [2].a) Diao L, Nnamani MC, Obrien E, Desjardins C, Martinez A, Simmons S, Litcofsky K, McGovern B, Weigand S, Cook D, Wortman JR, Henn MR, Gastroenterology 2019, 156, S;31462210 [Google Scholar]; b) Orenstein R, Dubberke E, Hardi R, Ray A, Mullane K, Pardi DS, Ramesh MS, Investigators PC, Clin Infect Dis 2016, 62, 596; [DOI] [PubMed] [Google Scholar]; Khanna S, Pardi DS, Kelly CR, Kraft CS, Dhere T, Henn MR, Lombardo MJ, Vulic M, Ohsumi T, Winkler J, Pindar C, McGovern BH, Pomerantz RJ, Aunins JG, Cook DN, Hohmann EL, J Infect Dis 2016, 214, 173. [DOI] [PubMed] [Google Scholar]
- [3].a) Tamboli CP, Neut C, Desreumaux P, Colombel JF, Gut 2004, 53, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ, Microbial Ecology in Health and Disease 2015, 26, 26191; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E, Nat Rev Immunol 2017, 17, 219. [DOI] [PubMed] [Google Scholar]
- [4].Petersen C, Round JL, Cellular Microbiology 2014, 16, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lewis BB, Buffie CG, Carter RA, Leiner I, Toussaint NC, Miller LC, Gobourne A, Ling L, Pamer EG, The Journal of Infectious Diseases 2015, 212, 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV, Nature Reviews Microbiology 2014, 13, 42. [DOI] [PubMed] [Google Scholar]
- [7].a) Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, Nature biotechnology 2018; [DOI] [PubMed] [Google Scholar]; b) Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, van Deventer SJH, Neirynck S, Peppelenbosch MP, Steidler L, Clinical Gastroenterology and Hepatology 2006, 4, 754. [DOI] [PubMed] [Google Scholar]
- [8].Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, Shiota A, Takeshita K, Yasuma-Mitobe K, Riethmacher D, Kaisho T, Norman JM, Mucida D, Suematsu M, Yaguchi T, Bucci V, Inoue T, Kawakami Y, Olle B, Roberts B, Hattori M, Xavier RJ, Atarashi K, Honda K, Nature 2019, 565, 600. [DOI] [PubMed] [Google Scholar]
- [9].a) Kurtz CB, Millet YA, Puurunen MK, Perreault M, Charbonneau MR, Isabella VM, Kotula JW, Antipov E, Dagon Y, Denney WS, Wagner DA, West KA, Degar AJ, Brennan AM, Miller PF, Science Translational Medicine 2019, 11, eaau7975; [DOI] [PubMed] [Google Scholar]; b) Steidler L, Hans W, Schotte L, Neirynck S, Obermeier F, Falk W, Fiers W, Remaut E, Science 2000, 289, 1352. [DOI] [PubMed] [Google Scholar]
- [10].Helfand WH, Cowen DL, Pharmacy in history 1983, 25, 3. [PubMed] [Google Scholar]
- [11].Langer R, Science 1990, 249, 1527. [DOI] [PubMed] [Google Scholar]
- [12].a) Langer R, NATURE-LONDON- 1998, 5; [PubMed] [Google Scholar]; b) Anselmo AC, Mitragotri S, Journal of Controlled Release 2014, 190, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Anselmo AC, Gokarn Y, Mitragotri S, Nature Reviews Drug Discovery 2018. [DOI] [PubMed] [Google Scholar]
- [13].a) Kao D, Roach B, Silva M, Beck P, Rioux K, Kaplan GG, Chang HJ, Coward S, Goodman KJ, Xu H, Madsen K, Mason A, Wong GK, Jovel J, Patterson J, Louie T, JAMA 2017, 318, 1985; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Orenstein R, Investigators f. t. P. C, Dubberke E, Investigators f. t. P. C., Hardi R, Investigators f. t. P. C., Ray A, Investigators f. t. P. C., Mullane K, Investigators f. t. P. C., Pardi DS, Investigators f. t. P. C., Ramesh MS, Investigators f. t. P. C., Investigators f. t. P. C., Dubberke ER, Hardi R, Kelly C, Mariani P, Misra B, Mullane K, Orenstein R, Pardi DS, Price CS, Ramesh MS, Ray A, Drekonja D, Clinical Infectious Diseases 2015, 62, 596.26565008 [Google Scholar]
- [14].Goldin BR, British Journal of Nutrition 1998, 80, S203. [PubMed] [Google Scholar]
- [15].a) Eun CS, Kim YS, Han DS, Choi JH, Lee AR, Park YK, Apmis 2011, 119, 49; [DOI] [PubMed] [Google Scholar]; b) Tien M-T, Girardin SE, Regnault B, Le Bourhis L, Dillies M-A, Coppée J-Y, Bourdet-Sicard R, Sansonetti PJ, Pédron T, The Journal of Immunology 2006, 176, 1228; [DOI] [PubMed] [Google Scholar]; c) Llopis M, Antolin M, Carol M, Borruel N, Casellas F, Martinez C, Espín-Basany E, Guarner F, Malagelada JR, Inflammatory Bowel Diseases 2008, 15, 275. [DOI] [PubMed] [Google Scholar]
- [16].a) Brogmann B, Beckert TE, Drugs and the Pharmaceutical Sciences 2001, 115, 1; [Google Scholar]; b) Thoma K, Bechtold K, Enteric coated hard gelatin capsules, Capsugel Library, 1992. [Google Scholar]
- [17].a) Prabhu P, Malli R, Koland M, Vijaynarayana K, D’Souza U, Harish N, Shastry C, Charyulu R, International journal of pharmaceutical investigation 2011, 1, 99; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Semalty M, Semalty A, Kumar G, Indian journal of pharmaceutical sciences 2008, 70, 43; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) El-Setouhy DA, El-Malak NSA, Aaps Pharmscitech 2010, 11, 1018; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Moon HT, Y.-k. Lee, J. K. Han, Y. Byun, Biomaterials 2001, 22, 281. [DOI] [PubMed] [Google Scholar]
- [18].DeMerlis CC, Schoneker DR, Food and Chemical Toxicology 2003, 41, 319. [DOI] [PubMed] [Google Scholar]
- [19].Jayasekara R, Harding I, Bowater I, Christie G, Lonergan GT, Polymer testing 2004, 23, 17. [Google Scholar]
- [20].a) Pereira VA Jr, de Arruda INQ, Stefani R, Food Hydrocolloids 2015, 43, 180; [Google Scholar]; b) Caykara T, Demirci S, Journal of Macromolecular Science, Part A 2006, 43, 1113. [Google Scholar]
- [21].Coulibaly I, Kouassi EK, N’guessan E, Destain J, Béra F, Thonart P, Annual Research & Review in Biology 2018, 1. [Google Scholar]
- [22].a) Alcock R, Cottingham MG, Rollier CS, Furze J, De Costa SD, Hanlon M, Spencer AJ, Honeycutt JD, Wyllie DH, Gilbert SC, Bregu M, Hill AVS, Science Translational Medicine 2010, 2, 19ra12; [DOI] [PubMed] [Google Scholar]; b) Leung V, Szewczyk A, Chau J, Hosseinidoust Z, Groves L, Hawsawi H, Anany H, Griffiths MW, Ali MM, Filipe CDM, ACS Biomaterials Science & Engineering 2018, 4, 3802; [DOI] [PubMed] [Google Scholar]; c) Ajmera A, Scherließ R, International Journal of Pharmaceutics 2014, 463, 98. [DOI] [PubMed] [Google Scholar]
- [23].a) Guerin-Danan C, Microbial Ecology in Health and Disease 1999, 11, 180; [Google Scholar]; b) Feltham R, POWER AK, PELL PA, Sneath P, Journal of Applied Bacteriology 1978, 44, 313. [DOI] [PubMed] [Google Scholar]
- [24].Kolhar P, Anselmo AC, Gupta V, Pant K, Prabhakarpandian B, Ruoslahti E, Mitragotri S, Proceedings of the National Academy of Sciences 2013, 110, 10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sonia TA, Sharma CP, Drug discovery today 2012, 17, 784. [DOI] [PubMed] [Google Scholar]
- [26].Lane MC, Alteri CJ, Smith SN, Mobley HL, Proceedings of the National Academy of Sciences 2007, 104, 16669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wilfart A, Montagne L, Simmins H, Noblet J, van Milgen J, British Journal of Nutrition 2007, 98, 54. [DOI] [PubMed] [Google Scholar]
- [28].a) Boddupalli BM, Mohammed ZN, Nath RA, Banji D, Journal of advanced pharmaceutical technology & research 2010, 1, 381; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kamel S, Ali N, Jahangir K, Shah S, El-Gendy A, Express Polym Lett 2008, 2, 758. [Google Scholar]
- [29].Mitragotri S, Burke PA, Langer R, Nature Reviews Drug Discovery 2014, 13, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Laffleur F, Expert opinion on therapeutic patents 2016, 26, 377. [DOI] [PubMed] [Google Scholar]
- [31].Cook MT, Tzortzis G, Charalampopoulos D, Khutoryanskiy VV, Journal of controlled release 2012, 162, 56. [DOI] [PubMed] [Google Scholar]
- [32].a) Soukoulis C, Behboudi-Jobbehdar S, Yonekura L, Parmenter C, Fisk ID, Food Chemistry 2014, 159, 302; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Soukoulis C, Singh P, Macnaughtan W, Parmenter C, Fisk ID, Food Hydrocolloids 2016, 52, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Selmer‐Olsen E, Birkeland SE, Sørhaug T, Journal of Applied Microbiology 1999, 87, 429; [DOI] [PubMed] [Google Scholar]; b) Santivarangkna C, Naumann D, Kulozik U, Foerst P, Annals of microbiology 2010, 60, 235. [Google Scholar]
- [34].a) Naylor H, Smith P, Journal of bacteriology 1946, 52, 565; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lion M, Microbiology 1963, 32, 321; [DOI] [PubMed] [Google Scholar]; c) Castro H, Teixeira P, Kirby R, Applied Microbiology and Biotechnology 1995, 44, 172; [Google Scholar]; d) Lim M, Kwon H, Kim D, Seo J, Han H, Khan SB, Progress in Organic Coatings 2015, 85, 68. [Google Scholar]
- [35].a) Peppas NA, Scott JE, Journal of controlled release 1992, 18, 95; [Google Scholar]; b) Kamoun EA, Chen X, Eldin MSM, Kenawy E-RS, Arabian Journal of chemistry 2015, 8, 1. [Google Scholar]
- [36].a) Gupta V, Hwang BH, Lee J, Anselmo AC, Doshi N, Mitragotri S, Journal of controlled release 2013, 172, 753; [DOI] [PubMed] [Google Scholar]; b) Duraj‐Thatte AM, Courchesne NMD, Praveschotinunt P, Rutledge J, Lee Y, Karp JM, Joshi NS, Advanced Materials 2019, 1901826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Chu ND, Crothers JW, Nguyen LT, Kearney SM, Smith MB, Kassam Z, Collins C, Xavier R, Moses PL, Alm EJ, bioRxiv 2019, 649384; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smillie CS, Sauk J, Gevers D, Friedman J, Sung J, Youngster I, Hohmann EL, Staley C, Khoruts A, Sadowsky MJ, Allegretti JR, Smith MB, Xavier RJ, Alm EJ, Cell Host & Microbe 2018, 23, 229; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mimee M, Citorik RJ, Lu TK, Advanced Drug Delivery Reviews 2016, 105, 44; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sartor RB, Wu GD, Gastroenterology 2017, 152, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.