

Summary
Influenza virus M2 and PB1-F2 proteins have been proposed to activate the Nod-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome in macrophages by altering intracellular ionic balance or mitochondrial reactive oxygen species (ROS) production. However, the precise mechanism by which these viral proteins trigger the NLRP3 inflammasome activation remains unclear. Here we show that influenza virus stimulates oxidized DNA release from macrophages. Ion channel activity of the M2 protein or mitochondrial localization of the PB1-F2 protein was required for oxidized DNA release. The oxidized DNA enhanced influenza virus-induced IL-1β secretion, whereas inhibition of mitochondrial ROS production by antioxidant Mito-TEMPO decreased the virus-induced IL-1β secretion. In addition, we show that influenza virus stimulates IL-1β secretion from macrophages in an AIM2-dependent manner. These results provide a missing link between influenza viral proteins and the NLRP3 inflammasome activation and reveal the importance of influenza virus-induced oxidized DNA in inflammasomes activation.
Subject Areas: Immunology, Viral Microbiology
Graphical Abstract
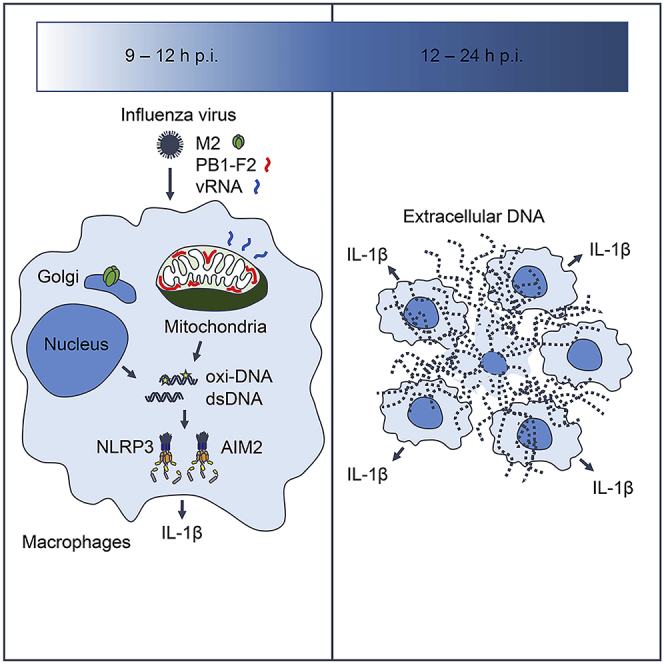
Highlights
-
•
M2 protein triggers oxidized DNA release
-
•
PB1-F2 protein triggers oxidized DNA release in the presence of viral RNA
-
•
Mitochondrial localization of PB1-F2 protein is required for oxidized DNA release
-
•
Influenza virus stimulates AIM2-dependent IL-1β secretion
Immunology; Viral Microbiology
Introduction
Recognition of influenza virus infection by the innate immune system plays a key role not only in limiting viral replication at early stages of infection but also in initiating the virus-specific adaptive immune responses (Iwasaki and Pillai, 2014). Toll-like receptor 7 (TLR7) recognizes influenza virus genomic RNA in endosomes (Diebold et al., 2004; Lund et al., 2004). In cytosol, retinoic acid inducible gene I (RIG-I) detects the 5′ triphosphate end of influenza genomic RNA (Hornung et al., 2006; Pichlmair et al., 2006; Rehwinkel et al., 2010; Weber et al., 2013). These two recognition pathways lead to the induction of type I interferons to restrict influenza virus replication. In contrast to TLRs and RIG-I-like helicases that recognize viral RNA, the NLRP3 senses cellular damage or distress induced by viral infection (Chen and Ichinohe, 2015). Upon activation, NLRP3 is recruited to the mitochondria (Ichinohe et al., 2013; Subramanian et al., 2013) or dispersed trans-Golgi network (Chen and Chen, 2018) to form the multi-molecular protein complex termed the NLRP3 inflammasome. The NLRP3 inflammasome activates caspase-1, which cleaves the precursor forms of proinflammatory cytokines, interleukin 1 beta (IL-1β) and IL-18, and stimulates their secretion (Evavold et al., 2018; Kayagaki et al., 2015; Shi et al., 2015). These inflammasome-dependent cytokines play a key role in the induction of adaptive immune responses and the initiation of tissue healing responses to influenza virus infection (Allen et al., 2009; Ichinohe et al., 2009, 2011; Moriyama and Ichinohe, 2019; Pang et al., 2013; Thomas et al., 2009).
Thus far, several possible mechanisms could explain how the NLRP3 detects influenza virus infection. First, influenza virus M2 protein, a proton-selective ion channel, stimulates ion flux from the trans-Golgi network and activates the NLRP3 inflammasome (Ichinohe et al., 2010). Second, phagocytosed PB1-F2 aggregates induce mitochondrial reactive oxygen species (ROS) production and stimulate the NLRP3 inflammasome-mediated IL-1β secretion from macrophages (McAuley et al., 2013; Pinar et al., 2017). Third, Z-DNA binding protein 1 (ZBP1) senses the ribonucleoprotein (RNP) complex to facilitate influenza virus-induced NLRP3 inflammasome activation (Kuriakose et al., 2016). However, the precise mechanisms of NLRP3 inflammasome activation by influenza virus M2 and PB1-F2 proteins remain unclear.
Recently, we demonstrated that the ion channel activity of influenza virus M2 protein is essential for mitochondrial DNA (mtDNA) release into the cytosol (Moriyama et al., 2019). In addition, recent studies highlight the importance of oxidized mtDNA in NLRP3 inflammasome activation (Shimada et al., 2012; Zhong et al., 2018). Consistent with these observations, we and others have shown that mtDNA-depleted ρ0 J774A.1 or bone marrow-derived macrophages (BMMs) abrogated caspase-1 activation and IL-1β secretion (Ichinohe et al., 2013; Nakahira et al., 2011; Zhong et al., 2016). These observations prompted us to examine whether influenza virus-induced oxidized mtDNA may trigger the NLRP3 inflammasome activation. Here we examined the mechanisms by which influenza virus stimulates oxidized mtDNA release into the cytosol and role of oxidized mtDNA in influenza virus-induced NLRP3 inflammasome activation.
Results
Influenza A Virus Triggers Oxidized DNA Release from Macrophages
We previously demonstrated that influenza virus stimulates cytosolic mtDNA release in mouse lung fibroblasts, human A549, and human embryonic kidney cell line 293FT (HEK293FT) cells (Moriyama et al., 2019). To determine whether influenza virus stimulates mtDNA release in macrophages, we infected J774A.1 macrophages with influenza virus A/PR8 strain. Although influenza virus replication was abortive in J774A.1 macrophages (Figure S1A), we detected productive translation of the viral proteins (Figure 1A). To detect mtDNA in the cytosol, we first extracted pure cytosolic fraction from mock- or virus-infected J774A.1 macrophages (Figure 1A). Analysis of the pure cytosolic extracts demonstrated that cytosolic release of mtDNA as well as nuclear DNA became apparent starting around 4 h post infection (p.i.) and peaking around 9 h p.i. (Figures 1B, 1C, S1B, and S1C). In addition, we detected significant amounts of mtDNA and nuclear DNA in the supernatants of influenza virus-infected J774A.1 macrophages as early as 8 h p.i. (Figures 1D, 1E, S1D, and S1E). Consistent with this observation, we observed double-stranded DNA (dsDNA) in extracellular web-like structures from influenza virus-infected J774A.1 and BMMs (Figures 1F and S1F). Furthermore, we detected oxidized DNA in the pure cytosolic fraction of influenza virus-infected J774A.1 macrophages (Figure 1G). In addition, a recent A/Narita/1/2009 (H1N1) strain stimulated cytosolic mtDNA release and IL-1β secretion from J774A.1 macrophages (Figures S1G–S1I). Together, these data suggest that influenza virus stimulates oxidized DNA release in macrophages.
Figure 1.

Influenza Virus Triggers Oxidized DNA Release from Macrophages
(A) J774A.1 macrophages were subjected to digitonin fractionation as described in Methods and pellets (P) or cytosolic extracts (C) were analyzed by western blotting using the indicated antibodies.
(B–E) J774A.1 macrophages were infected with PR8 virus. Pure cytosolic extracts and supernatants were collected at indicated time points. Relative levels of mtDNA (B and D) or nuclear DNA (C and E) in the cytosol and supernatants were assessed by quantitative PCR.
(F) J774A.1 macrophages were infected with PR8 virus. At 24 h post infection, cells were stained with anti-dsDNA (AC-30-10) and anti-Tom20 antibodies and analyzed by confocal microscopy. Scale bars, 10 μm.
(G) J774A.1 macrophages were infected with PR8 virus. Pure cytosolic extracts were collected at indicated time points. Oxidized DNA in the cytosol was detected by dot blot analysis using anti-8OH-dG antibody. These data are from three independent experiments (B–E; mean ± SEM).
∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001 versus mock-infected cells (one-way ANOVA and Tukey's test). See also Figure S1.
Influenza Virus M2 or PB1-F2 Proteins Trigger Oxidized DNA Release
We previously demonstrated that ion channel activity of the influenza virus M2 protein is essential for NLRP3 inflammasome activation (Ichinohe et al., 2010) or mtDNA release into the cytosol (Moriyama et al., 2019). Consistent with our previous observations (Moriyama et al., 2019), overexpression of the M2 protein was sufficient to stimulate mtDNA release into the cytosol (Figure 2A). Since influenza virus stimulates cytosolic mtDNA release in an MAVS-dependent manner (Moriyama et al., 2019), we next examined whether intracellular delivery of polyinosinic-polycytidylic acid (poly(I:C)), a synthetic analog of double-stranded RNA, into HEK293FT cells by transfection stimulates mtDNA release into the cytosol. Interestingly, we found that, although intracellular delivery of poly(I:C) was insufficient to stimulate mtDNA release into the cytosol, the PB1-F2 protein stimulated cytosolic mtDNA release in the presence of poly(I:C) or viral RNA (Figures 2A–2C). In addition, we found that ion channel activity of the M2 protein or PB1-F2 together with poly(I:C) protein stimulated oxidized DNA release into the cytosol (Figures 2D and 2E). Furthermore, the recombinant influenza virus lacking amino acids 29–31 of the M2 protein (termed rgPR8/M2del29-31), which has lost its ion channel activity and failed to stimulate the NLRP3 inflammasome-mediated IL-1β secretion (Ichinohe et al., 2010; Moriyama et al., 2019), or the PB1-F2 protein (termed rgPR8/ΔPB1-F2) reduced its ability to stimulate mtDNA release into the cytosol (Figures 2F and 2G). These data indicate that the ion channel activity of M2 protein or PB1-F2 protein together with poly(I:C) stimulates mtDNA and oxidized DNA release into the cytosol.
Figure 2.

Influenza Virus M2 and PB1-F2 Proteins Stimulate Oxidized DNA Release
(A–C) HEK293FT cells were transfected with the expression plasmid encoding EGFP or influenza virus proteins in the presence or absence of poly(I:C) (A, B) or influenza viral RNA (C). Cytosolic mtDNA was assessed by quantitative PCR at 24 h post transfection.
(D and E) HEK293FT cells were transfected with the expression plasmid encoding EGFP or influenza virus proteins in the presence or absence of poly(I:C). Pure cytosolic extracts were collected at 24 h post transfection. Oxidized DNA in the cytosol was detected by dot blot analysis using anti-8OH-dG antibody.
(F and G) HEK293FT cells were infected with WT (rgPR8), rgPR8/M2del29–31 (F), or rgPR8/ΔPB1-F2 virus (G). Cytosolic mtDNA was assessed by quantitative PCR at 24 h post infection. These data are from three independent experiments (A–C, F, G; mean ± SEM).
∗∗p < 0.01 and ∗∗∗p < 0.001 versus EGFP-transfected or rgPR8-infected cells (one-way ANOVA and Tukey's test).
We further examined the cellular mechanism by which PB1-F2 protein stimulates mtDNA and oxidized DNA release. We previously demonstrated that the PB1-F2 protein translocates into mitochondria via translocase of the outer membrane 40 (Tom40) channels (Yoshizumi et al., 2014). Consistent with our previous observation (Yoshizumi et al., 2014), knockdown of Tom40 significantly blocked cytosolic mtDNA release induced by PB1-F2 protein together with poly(I:C) without affecting the expression levels of the PB1-F2 protein (Figures 3A and 3B). In addition, a C-terminal truncated mutant of PB1-F2 protein, which failed to translocate into the mitochondrial inner membrane space (Figure 3C) (Yoshizumi et al., 2014), lost the ability to stimulate mtDNA and oxidized DNA release in the presence of poly(I:C) (Figures 3D and 3E). Collectively, these data indicate that mitochondrial localization of PB1-F2 protein is required for mtDNA and oxidized DNA release in poly(I:C)-stimulated cells.
Figure 3.

Mitochondrial Localization of PB1-F2 Protein Is Required for Oxidized DNA Release
(A and B) HEK293FT cells were transfected with siRNA targeting Tom40, Tom22, or control siRNA. Two days later, cells were transfected with the expression plasmid encoding EGFP or PB1-F2 protein in the presence of poly(I:C). Cell lysates were collected at 24 h post transfection and blotted using the indicated antibodies (A). Cytosolic mtDNA was assessed by quantitative PCR at 24 h post transfection (B).
(C) HeLa cells were transfected with expression plasmids encoding PB1-F2 or its C-terminal truncated mutant. At 24 h post transfection, cells were stained with anti-PB1-F2 and anti-Tom20 antibodies to visualize the PB1-F2 protein and mitochondria, respectively, and analyzed by confocal microscopy. Scale bars, 10 μm.
(D and E) HEK293FT cells were transfected with the expression plasmid encoding EGFP, PB1-F2, or its C-terminal truncated mutant in the presence of poly(I:C). Pure cytosolic extracts were collected at 24 h post transfection. Cytosolic mtDNA was assessed by quantitative PCR (D). Oxidized DNA in the cytosol was detected by dot blot analysis using anti-8OH-dG antibody (E). These data are from three independent experiments (B, E; mean ± SEM).
∗∗∗p < 0.001 (one-way ANOVA and Tukey's test).
Oxidized DNA Enhances Influenza Virus-Induced IL-1β Secretion
Given that oxidized mtDNA triggers NLRP3 inflammasome activation (Shimada et al., 2012; Zhong et al., 2018), oxidized DNA could enhance influenza virus-induced IL-1β secretion. To test this idea, we generated oxidized mtDNA fragment by standard PCR using 8-oxo-dGTP (Figure 4A). Since influenza virus alone failed to induce detectable levels of pro-IL-1β and stimulated marginal amounts of IL-1β secretion (Figures S2A–S2C), we stimulated macrophages with LPS after influenza virus infection. In LPS-stimulated J774A.1 or BMMs infected with influenza virus, expression levels of PB1-F2, M2, and NS1 proteins became apparent starting around 6 h p.i. and peaking around 9 h p.i. (Figures S2B and S2C). In addition, IL-1β secretion from influenza virus-infected J774A.1 or BMMs became apparent starting around 9 h p.i. and peaking around 18–24 h p.i. (Figures S2B and S2C). Consistent with our previous report (Ichinohe et al., 2010), we detected active caspase-1 in the supernatants of the virus-infected macrophages at 24 h p.i. (Figures S2B and S2C). To test whether oxidized mtDNA might enhance influenza virus-induced IL-1β secretion, we infected J774A.1 or BMMs with influenza virus in the presence of 8-oxo-dG- or 8-oxo-dG+ DNA. Notably, oxidized mtDNA (8-oxo-dG+ DNA) significantly enhanced influenza virus-induced IL-1β secretion from LPS-stimulated J774A.1 and BMMs compared with control 8-oxo-dG- DNA (Figures 4B and S2D). Similarly, 8-oxo-2′-deoxyguanosine (8-oxo-dG), an oxidized derivative of deoxyguanosine, significantly increased influenza virus-induced IL-1β secretion from LPS-stimulated J774A.1 and BMMs compared with deoxyguanosine (dG) control (Figures 4C and S2E). In addition, increased levels of influenza virus-induced IL-1β secretion by 8-oxo-dG treatment was significantly inhibited by Mcc950, a specific inhibitor of the NLRP3 inflammasome, or Ac-YVAD-cmk, a specific peptide inhibitor of caspase-1 (Figures 4C and S2E). Furthermore, 8-oxo-dG restored IL-1β secretion from rgPR8/M2del29-31 virus-infected J774A.1 and BMMs (Figures 4D and S2F). Conversely, treatment with antioxidant Mito-TEMPO, a scavenger specific for mitochondrial reactive oxygen species, inhibited IL-1β secretion or oxidized DNA release from influenza virus-infected J774A.1 and BMMs (Figures 4E, S2G, and S2H). Together, these data suggest oxidized DNA may be important for the NLRP3 inflammasome-mediated IL-1β secretion after influenza virus infection.
Figure 4.

Oxidized DNA Enhances Influenza Virus-Induced IL-1β Secretion
(A) Oxidized mtDNA fragment was prepared as described in Methods. dsDNA and oxidized DNA were detected by dot blot analysis using anti-dsDNA (35I9 DNA) or anti-8OH-dG antibodies.
(B) LPS-stimulated BMMs were infected with PR8 in the presence or absence of oxidized (8-oxo-dG+) or control (8-oxo-dG-) DNA.
(C and D) LPS-stimulated BMMs were infected with PR8 (C), rgPR8, or rgPR8/M2del29–31 virus (D) in the presence or absence of dG (246 μM), 8-oxo-dG (246 μM), Mcc950 (20 μM), or Ac-YVAD-cmk (20 μM).
(E) LPS-stimulated BMMs were infected with PR8 virus in the presence or absence of Mito-TEMPO (500 μM). Cell-free supernatants were collected and analyzed for IL-1β by ELISA (B–E). These data are from three independent experiments (B–E; mean ± SEM).
∗∗∗p < 0.001 (one-way ANOVA and Tukey's test). See also Figure S2.
Influenza Virus Stimulates AIM2 Inflammasome-Dependent IL-1β Secretion
Thus far, our data indicated influenza virus-induced oxidized DNA triggers NLRP3 inflammasome activation. Since cytosolic mtDNA triggers the absent in melanoma 2 (AIM2), a cytosolic dsDNA sensor, -dependent inflammasome activation (Dang et al., 2017), the released mtDNA from influenza virus-infected macrophages would elicit the AIM2 inflammasome activity. To test this directly, we infected WT or AIM2-deficient BMMs with influenza virus and measured IL-1β secretion. Consistent with our previous report (Maruzuru et al., 2018), the recombinant herpes simplex virus 1 (HSV-1) lacking VP22 protein stimulated IL-1β secretion in an AIM2-dependent manner (Figure 5A). Notably, IL-1β secretion was significantly reduced in AIM2-deficient cells upon influenza virus infection but not ATP, an NLRP3 agonist (Figures 5B and 5C). Furthermore, a 1,277 bp of mtDNA fragment significantly enhanced influenza virus-induced IL-1β secretion from J774A.1 and BMMs (Figures 5D and S3A). Conversely, addition of DNase I in the extracellular medium significantly blocked IL-1β secretion from influenza virus-infected J774A.1 and BMMs (Figures 5E and S3B). These results indicated that cytosolic or extracellular mtDNA may stimulate AIM2 inflammasome-mediated IL-1β secretion from influenza virus-infected macrophages.
Figure 5.

Influenza Virus Stimulates the AIM2 Inflammasome-Mediated IL-1β Secretion
(A–C) LPS-stimulated BMMs prepared from WT or AIM2-deficient mice were infected with HSV-1ΔVP22 (A) or PR8 virus (B) for 24 h. For ATP stimulation, BMMs were stimulated with LPS for 6 h and pulsed with 2 mM ATP for 30 min (C).
(D and E) LPS-stimulated BMMs were infected with PR8 virus for 24 h in the presence or absence of mtDNA fragment prepared as described in Methods (D) or DNase I (E). Cell-free supernatants were collected and analyzed for IL-1β by ELISA. These data are from three independent experiments (mean ± SEM).
∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA and Tukey's test). See also Figure S3.
Discussion
Influenza virus stimulates the NLRP3 inflammasome-mediated IL-1β secretion from macrophage through its M2 ion channel activity (Ichinohe et al., 2010) or the PB1-F2 aggregates (McAuley et al., 2013; Pinar et al., 2017). However, the precise mechanism by which these viral proteins trigger NLRP3 inflammasome activation remains unclear. In this study, we demonstrated that ion channel activity of the M2 protein or mitochondrial localization of the PB1-F2 through Tom40 channel was essential for oxidized DNA release. We found that these cytosolic or extracellular DNA stimulated both NLRP3 and AIM2 inflammasome-mediated IL-1β secretion from macrophages.
Mitochondria play a central role in NLRP3 inflammasome activation (Swanson et al., 2019). ROS-producing damaged mitochondria was first implicated in NLRP3 inflammasome activation (Zhou et al., 2011). Consistent with this observation, cytosolic oxidized mtDNA was found to be important for NLRP3 inflammasome activation (Shimada et al., 2012; Zhong et al., 2018). Translocation of mtDNA into the cytosol required activation of the NLRP3 or MAVS, mitochondrial ROS production, or formation of mitochondrial permeability transition (MPT) pores or BAK/BAX macropores (McArthur et al., 2018; Moriyama et al., 2019; Nakahira et al., 2011). In addition to their role in NLRP3 inflammasome activation, mitochondria also act as platforms for the NLRP3 inflammasome activation. MAVS interacts with the NLRP3 and promotes its localization to the mitochondria for full activation of the NLRP3 inflammasome (Subramanian et al., 2013). Following influenza virus infection, the NLRP3 associates with mitofusin 2 to facilitate its localization to the mitochondria (Ichinohe et al., 2013). In this study, we found that the M2 protein of influenza virus was sufficient to trigger oxidized DNA release. Since Ca2+ overload is sufficient to induce the MPT (Lemasters et al., 2009), we speculate that the M2-mediated perturbation of intracellular ionic concentrations may be sufficient to stimulate formation of Ca2+ influx-dependent MPT pores and cytosolic release of oxidized mtDNA. Interestingly, a recent study has demonstrated that disassembly of trans-Golgi network into vesicles serves as a scaffold for NLRP3 aggregation and activation (Chen and Chen, 2018). Since the M2-induced activation of inflammasomes required its localization to the Golgi apparatus (Ichinohe et al., 2010), it is possible that the M2 protein promotes trans-Golgi network disassembly into vesicles and NLRP3 aggregation. In contrast to the M2 protein, the PB1-F2 protein required intracellular delivery of poly(I:C) or viral RNA to stimulate oxidized mtDNA release. Although the PB1-F2-mediated reduction of mitochondrial membrane potential may cause mitochondrial ROS production (Yoshizumi et al., 2014; Zhou et al., 2011), it was insufficient to stimulate oxidized mtDNA release. Thus, mitochondria-localized PB1-F2 protein and MAVS-dependent signals may act synergistically to stimulate oxidized mtDNA release into the cytosol.
Not surprisingly, influenza virus has evolved strategies to counteract host innate antiviral immune responses. We and others previously demonstrated that the NS1 protein of influenza virus suppressed the NLPR3 inflammasome-mediated IL-1β secretion from macrophages (Chung et al., 2018; Moriyama et al., 2016; Stasakova et al., 2005). Interestingly, the RNA-binding domain of the NS1 protein was essential for the NLRP3 inflammasome-mediated IL-1β secretion (Moriyama et al., 2016). More recently, we showed that the RNA-binding domain of the NS1 protein was important for association with cytosolic mtDNA to suppress the stimulator of interferon genes (STING)-dependent interferon responses (Moriyama et al., 2019). These observations suggest that the NS1 protein of influenza virus inhibits the NLRP3 inflammasome-mediated IL-1β secretion by inhibiting the MAVS-dependent mtDNA release or sequestering cytosolic mtDNA.
Inflammasome-dependent cytokine production in the lung of influenza virus-infected mice plays an essential role in the induction of virus-specific adaptive immune responses and in the initiation of tissue repair responses following infectious damage (Allen et al., 2009; Ichinohe et al., 2009, 2011; Moriyama and Ichinohe, 2019; Pang et al., 2013). Somewhat surprisingly, induction of the influenza virus-specific adaptive immune responses was dependent of ASC and caspase-1 but not NLRP3 (Ichinohe et al., 2009). Recent reports indicated that the AIM2 plays an important role in IL-1β secretion in the lung of influenza virus-infected mice (Schattgen et al., 2016; Zhang et al., 2017). We and others detected mitochondrial or nuclear DNA in the bronchoalveolar (BAL) fluid of influenza virus-infected mice (Moriyama et al., 2019; Schattgen et al., 2016). In this study, we detected dsDNA in extracellular web-like structures of influenza virus-infected macrophages. In addition, we showed that influenza virus stimulated the AIM2 inflammasome-mediated IL-1β secretion from macrophages. These observations suggest that activation of both NLRP3 and AIM2 inflammasomes in the lung of influenza virus-infected mice may play an important role in the induction of virus-specific adaptive immune responses or influenza-associated mortality. In addition to influenza viral RNA (Kuriakose et al., 2016), mitochondrial or nuclear DNA could act as ligands for these inflammasomes in the lung tissue.
In summary, our data reveal a missing link between influenza virus M2 or PB1-F2 proteins and the NLRP3 inflammasome activation. Both M2 and PB1-F2 proteins stimulated oxidized DNA release to trigger the NLRP3 and AIM2 inflammasomes activation. Because both M2 and PB1-F2 proteins contribute virulence of influenza A viruses (Chen et al., 2001; Conenello et al., 2007; McAuley et al., 2007, 2010, 2013), our findings suggest a possible effect of influenza virus-induced oxidized DNA in pathogenesis of influenza A virus infections. Better understanding of the role of the M2 and PB1-F2 proteins in regulating inflammatory responses in vivo and pathogenesis of influenza virus infections will aid the development of more effective interventions for the treatment of influenza virus-associated diseases.
Limitations of the Study
In the present study, we demonstrated that influenza virus stimulates oxidized DNA release from macrophages. Although we detected significant amounts of both mitochondrial and nuclear DNA in cytosol and supernatants of influenza virus-infected macrophages, the current study does not determine the relative contributions of mtDNA versus nuclear DNA in activation of the NLRP3 and AIM2 inflammasomes. In addition, we showed that ion channel activity of the M2 protein or mitochondrial localization of the PB1-F2 protein is required for oxidized DNA release. We generated recombinant influenza viruses lacking amino acids 29–31 of the M2 protein or PB1-F2 protein to examine the effect of deficiency of the M2 ion channel activity or PB1-F2 protein in influenza virus-induced IL-1β secretion. However, we found that the expression levels of the M2 and PB1-F2 proteins were different between WT and the mutant virus-infected macrophages. Thus, we cannot simply compare the ability to stimulate inflammasome-dependent IL-1β secretion from macrophages infected with rgPR8 (WT), rgPR8/M2del29-31, or rgPR8/ΔPB1-F2 virus. Since virus-encoded ion channels (viroporins) of other RNA viruses such as encephalomyocarditis virus and severe acute respiratory syndrome coronavirus activate the NLRP3 inflammasome (Chen et al., 2019; Ito et al., 2012; Nieto-Torres et al., 2015), future studies to dissect the importance of the viroporins in oxidized DNA release and activation of the NLRP3 and AIM2 inflammasomes will be important extensions of this work.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Takeshi Ichinohe (ichinohe@ims.u-tokyo.ac.jp).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
No custom code, software, or algorithm were used in this research.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Y. Kawaoka (University of Wisconsin and University of Tokyo) for providing the plasmids for reverse genetics. This work was supported by the Tokyo Biochemical Research Foundation and the SENSHIN Medical Research Foundation. M.M. is the Research Fellow of the Japan Society for the Promotion of Science.
Author Contributions
M.M. and T.I. designed research; M.M., M.N., Y.M., and T.I. performed experiments and analyzed data; T.K. and Y.K. provided critical reagents and advice; T.I. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101270.
Supplemental Information
References
- Allen I.C., Scull M.A., Moore C.B., Holl E.K., McElvania-TeKippe E., Taxman D.J., Guthrie E.H., Pickles R.J., Ting J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.Y., Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015;23:55–63. doi: 10.1016/j.tim.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Chen I.Y., Moriyama M., Chang M.F., Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:50. doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Chen Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564:71–76. doi: 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Calvo P.A., Malide D., Gibbs J., Schubert U., Bacik I., Basta S., O'Neill R., Schickli J., Palese P. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- Chung W.C., Kang H.R., Yoon H., Kang S.J., Ting J.P., Song M.J. Correction: influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS One. 2018;13:e0200624. doi: 10.1371/journal.pone.0200624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conenello G.M., Zamarin D., Perrone L.A., Tumpey T., Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–1421. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang E.V., McDonald J.G., Russell D.W., Cyster J.G. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171:1057–1071.e11. doi: 10.1016/j.cell.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S.S., Kaisho T., Hemmi H., Akira S., Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Evavold C.L., Ruan J., Tan Y., Xia S., Wu H., Kagan J.C. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44.e36. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M. 5'-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Ichinohe T., Lee H.K., Ogura Y., Flavell R., Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T., Pang I.K., Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010;11:404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T., Pang I.K., Kumamoto Y., Peaper D.R., Ho J.H., Murray T.S., Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. U S A. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T., Yamazaki T., Koshiba T., Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. U S A. 2013;110:17963–17968. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Yanagi Y., Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 2012;8:e1002857. doi: 10.1371/journal.ppat.1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A., Pillai P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Stowe I.B., Lee B.L., O'Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kuriakose T., Man S.M., Malireddi R.K., Karki R., Kesavardhana S., Place D.E., Neale G., Vogel P., Kanneganti T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016;1 doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters J.J., Theruvath T.P., Zhong Z., Nieminen A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J.M., Alexopoulou L., Sato A., Karow M., Adams N.C., Gale N.W., Iwasaki A., Flavell R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U S A. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruzuru Y., Ichinohe T., Sato R., Miyake K., Okano T., Suzuki T., Koshiba T., Koyanagi N., Tsuda S., Watanabe M. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe. 2018;23:254–265.e7. doi: 10.1016/j.chom.2017.12.014. [DOI] [PubMed] [Google Scholar]
- McArthur K., Whitehead L.W., Heddleston J.M., Li L., Padman B.S., Oorschot V., Geoghegan N.D., Chappaz S., Davidson S., San Chin H. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359 doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- McAuley J.L., Chipuk J.E., Boyd K.L., Van De Velde N., Green D.R., McCullers J.A. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010;6:e1001014. doi: 10.1371/journal.ppat.1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley J.L., Hornung F., Boyd K.L., Smith A.M., McKeon R., Bennink J., Yewdell J.W., McCullers J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249. doi: 10.1016/j.chom.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley J.L., Tate M.D., MacKenzie-Kludas C.J., Pinar A., Zeng W., Stutz A., Latz E., Brown L.E., Mansell A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013;9:e1003392. doi: 10.1371/journal.ppat.1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M., Chen I.Y., Kawaguchi A., Koshiba T., Nagata K., Takeyama H., Hasegawa H., Ichinohe T. The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1beta secretion. J. Virol. 2016;90:4105–4114. doi: 10.1128/JVI.00120-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M., Ichinohe T. High ambient temperature dampens adaptive immune responses to influenza A virus infection. Proc. Natl. Acad. Sci. U S A. 2019;116:3118–3125. doi: 10.1073/pnas.1815029116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M., Koshiba T., Ichinohe T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 2019;10:4624. doi: 10.1038/s41467-019-12632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Torres J.L., Verdia-Baguena C., Jimenez-Guardeno J.M., Regla-Nava J.A., Castano-Rodriguez C., Fernandez-Delgado R., Torres J., Aguilella V.M., Enjuanes L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang I.K., Ichinohe T., Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat. Immunol. 2013;14:246–253. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Pinar A., Dowling J.K., Bitto N.J., Robertson A.A., Latz E., Stewart C.R., Drummond G.R., Cooper M.A., McAuley J.L., Tate M.D. PB1-F2 peptide derived from avian influenza A virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J. Biol. Chem. 2017;292:826–836. doi: 10.1074/jbc.M116.756379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J., Tan C.P., Goubau D., Schulz O., Pichlmair A., Bier K., Robb N., Vreede F., Barclay W., Fodor E. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Schattgen S.A., Gao G., Kurt-Jones E.A., Fitzgerald K.A. Cutting edge: DNA in the lung microenvironment during influenza virus infection tempers inflammation by engaging the DNA sensor AIM2. J. Immunol. 2016;196:29–33. doi: 10.4049/jimmunol.1501048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasakova J., Ferko B., Kittel C., Sereinig S., Romanova J., Katinger H., Egorov A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005;86:185–195. doi: 10.1099/vir.0.80422-0. [DOI] [PubMed] [Google Scholar]
- Subramanian N., Natarajan K., Clatworthy M.R., Wang Z., Germain R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson K.V., Deng M., Ting J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.G., Dash P., Aldridge J.R., Jr., Ellebedy A.H., Reynolds C., Funk A.J., Martin W.J., Lamkanfi M., Webby R.J., Boyd K.L. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M., Gawanbacht A., Habjan M., Rang A., Borner C., Schmidt A.M., Veitinger S., Jacob R., Devignot S., Kochs G. Incoming RNA virus nucleocapsids containing a 5'-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013;13:336–346. doi: 10.1016/j.chom.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizumi T., Ichinohe T., Sasaki O., Otera H., Kawabata S., Mihara K., Koshiba T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014;5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- Zhang H., Luo J., Alcorn J.F., Chen K., Fan S., Pilewski J., Liu A., Chen W., Kolls J.K., Wang J. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J. Immunol. 2017;198:4383–4393. doi: 10.4049/jimmunol.1600714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Liang S., Sanchez-Lopez E., He F., Shalapour S., Lin X.J., Wong J., Ding S., Seki E., Schnabl B. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Umemura A., Sanchez-Lopez E., Liang S., Shalapour S., Wong J., He F., Boassa D., Perkins G., Ali S.R. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No custom code, software, or algorithm were used in this research.