

Abstract
Although tumorigenesis is dependent on the reprogramming of cellular metabolism, the metabolic pathways engaged in the formation of metastases remain largely unknown. The transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) plays a pleiotropic role in the control of cancer cell metabolism and has been associated with a good prognosis in prostate cancer. Here, we show that PGC1α represses the metastatic properties of prostate cancer cells via modulation of the polyamine biosynthesis pathway. Mechanistically, PGC1α inhibits the expression of c-MYC and ornithine decarboxylase 1 (ODC1), the rate-limiting enzyme for polyamine synthesis. Analysis of in vivo metastases and clinical data from patients with prostate cancer support the proposition that the PGC1α/c-MYC/ODC1 axis regulates polyamine biosynthesis and prostate cancer aggressiveness. In conclusion, downregulation of PGC1α renders prostate cancer cells dependent on polyamine to promote metastasis.
Introduction
Metabolic reprogramming is a hallmark of cancer cells and a consequence of adaptation to a hostile microenvironment with decreased oxygen concentration and nutrients (glucose and glutamine; ref. 1). These metabolic changes are required for rapid proliferation and invasion and are well characterized for cancer cells from primary tumors but poorly described for metastatic cancer cells. Recent advances in the understanding of cancer cell metabolism has allowed for the emergence of new therapeutic approaches that specifically target these adaptations. For example, in cancer cells that rely on oxidative phosphorylation, we have shown that interfering with the mitochondrial respiration could exhibit repression of tumor growth, cancer cell proliferation, and the formation of metastasis (2–4).
One of the main regulators of cellular metabolism is the transcriptional coactivator PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha). PGC1α controls mitochondrial biogenesis, oxidative phosphorylation, and fatty acid oxidation (5). Recently, PGC1α has been shown to facilitate mitochondrial biogenesis in invasive breast cancer cells and to increase their metastatic potential (6). In contrast, overexpression of PGC1α decreased the formation of metastasis in melanoma and prostate, and was associated with poor prognosis and the formation of metastasis in melanoma and prostate cancer (7–9). However, the metabolic and molecular modifications driving the aggressiveness of prostate cancer cells remain poorly understood.
Oncogenes and tumor suppressors regulate metabolic adaptations of cancer cells. Several studies have demonstrated that the MYC gene copy number is upregulated by 30% in human prostate cancer (10, 11). Furthermore, transgenic mice overexpressing c-MYC in the prostate developed prostatic intraepithelial neoplasia followed by invasive adenocarcinoma, demonstrating that c-MYC drives tumorigenesis in the prostate (12). Expression of the proto-oncogene c-MYC increases glycolysis and glutaminolysis (13, 14), by controlling the expression of genes involved in glucose and glutamine metabolism as well as other metabolic pathways, such as polyamine via the ornithine decarboxylase 1 (ODC1), the rate-limiting enzyme of polyamine biosynthesis (15).
In this study, we demonstrate that PGC1α is the regulator of a c-MYC-driven onco-metabolic pathway that promotes prostate cancer aggressiveness through the polyamine pathway. The unravelling of this metabolic circuit represents a new therapeutic target in prostate cancer that may help to curb the advanced form of the disease.
Materials and Methods
Cell culture
PC3, DU145, and LNCaP cells were purchased from the ATCC. Upon reception, cells are thawed at low passages. All cells used in this study were within 20 passages after thawing and tested monthly for Mycoplasma. PC3 and DU145 cells were cultured in DMEM (Gibco). LNCaP cells were cultured in RPMI1640 medium (Gibco). Cell media were supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively; Gibco) and incubated at 37°C and 5% CO2. DU145 and LNCaP cells were infected with lentiviral constructs at different MOI (multiplicity of infection; 2, 5, and 10) in the presence of 8 μg/mL hexadimethrine bromide (Sigma). These cells were infected with a pLKO.1 nontarget control short hairpin RNA (Sigma Mission, shRNA) or individually with five different pLKO.1 Mission shRNA targeting PGC1α (Sigma) to generate cell lines with stable PGC1α knockdown. After 24 hours of infection, the stably shRNA-expressing cells were selected with 1 mg/mL puromycin (Sigma) for 7 days and maintained in the culture medium with 0.5 μg/mL puromycin. Efficiency of PGC1α knockdown was determined by qRT-PCR and immunoblots and stably shRNA-expressing cell lines with the best knockdown of PGC1α, were selected for all the experiments.
Lipofectamine RNAiMAX was used for transfection of siRNAs according to the manufacturer’s instructions (Invitrogen). Transfections of human prostate cell lines were performed using siRNA pools consisting of four siRNA oligos specifically targeting four different sites of the target mRNAs (On Target Plus; Dharmacon). The purpose of the siRNA pools is to use less of each oligo to minimize any off-target effects. The siRNA sequences are: SMART-pool: ON-TARGETplus PPARGC1A siRNA: L-005111, SMART-pool: ON-TARGETplus MYC siRNA: L-003282, SMARTpool: ON-TARGETplus ESRRA siRNA: L-003403, siGENOME Non-Targeting siRNA Pool #1: D-001206-13. Briefly, cells were seeded at 70%–80% confluence in 6-well plates overnight before transfection, and then transfected with 15 nmol/L siRNA sequences or with a nonspecific siRNA using Lipofectamine RNAiMAX. siRNA and Lipofectamine RNAiMAX reagent were diluted in Opti-MEM (Gibco), and the contents were mixed gently. The mixtures were incubated for 10–20 minutes at room temperature. The incubated mixtures of siRNA and RNAiMAX were added to cells and incubated for 48–72 hours.
Lipofectamine 2000 reagent was used for transfections of plasmids according to the manufacturer’s instructions (Invitrogen). All the plasmids were purchased from Addgene: pAdTrack Flag-HA-PGC-1 alpha and an empty vector as control pAd-Track-CMV. For plasmid transfection, cells were seeded to be 70%–90% confluent at the time of transfection. Plasmids and Lipofectamine 2000 reagent were diluted in Opti-MEM (Gibco). The diluted plasmids were mixed with Lipofectamine 2000 and incubated for 5 minutes at room temperature. For transient transfection, the transfected cells were collected after 24–48 hours for experiments. For the stable PC3 cell line generation-overexpressing PGC1α, cells were selected, after transfection, with 500 αg/mL G418 for 30 days and maintained in the culture medium with 100 μg/mL G418. Efficiency of PGC1α overexpression was determined by qRT-PCR and immunoblots.
Steady-state metabolomics
To determine the relative levels of intracellular metabolites, extracts were prepared and analyzed by LC/MS-MS. Quadruplicate 10-cm plates (~80% confluent) were incubated in serum-free medium for 15 hours. Metabolites were extracted on dry ice with 4 mL 80% methanol (−80°C), as described previously (16). Insoluble material was pelleted by centrifugation at 3,000 × g for 5 minutes, followed by two subsequent extractions of the insoluble pellet with 0.5 mL80% methanol, with centrifugation at 16,000 × g for 5 minutes at 4°C. The 5-mL metabolite extract from the pooled supernatants was dried down under nitrogen gas using an N-EVAP (Organomation Associates, Inc). Dried pellets were resuspended using 20 μL HPLC-grade water for mass spectrometry. A 7-μL sample was injected and analyzed using a 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC System (Shimadzu) via selected reaction monitoring of a total of 300 endogenous water-soluble metabolites for steady-state analyses of samples (17). The normalized areas were used as variables for the univariate statistical data analysis. All univariate analyses and modeling on the normalized data were carried out using Metaboanalyst 4.0 (http://www.metaboanalyst.ca). Univariate statistical differences of the metabolites between two groups were analyzed using two-tailed Student t test.
Stable isotopic tracing analysis
To define the relative abundance of polyamine metabolites by LC/MS-MS analysis, a previously described extraction method optimized for polar metabolite was employed (16, 17). Briefly, cells cultured in 10-cm plates to approximately 90% confluence, were washed with DMEM without arginine prior to addition of labeled [13C6]-arginine (480 μmol/L) for 1 hour. Metabolites were extracted on dry ice with 4 mL of 80% Methanol (LC-MS grade). Cells were scraped and placed at −80°C for 20 minutes before successive centrifugations at 17,000 × g for 5 minutes. Supernatants were collected and dried under nitrogen gas then resuspended in 50% acetonitrile and 25 μL were injected in a Thermo Q-Exactive (LC-MS) in line with an electrospray source and an Ultimate3000 (Thermo Fisher Scientific) series HPLC consisting of a binary pump, degasser, and auto-sampler outfitted with a Xbridge Amide column (Waters; dimensions of 4.6 mm × 100 mm and a 3.5-μm particle size). The mobile phase A contained 95% (vol/vol) water, 5% (vol/vol) acetonitrile, 20 mmol/L ammonium hydroxide, 20 mmol/L ammonium acetate, pH 9.0; B was 100% acetonitrile. The gradient was as follows: 0 minutes, 15% A; 2.5 minutes, 30% A; 7 minutes, 43% A; 16 minutes, 62% A; 16–18 minutes, 75% A; 18–25 minutes, 15% A with a flow rate of 400 μL/minute. The capillary of the ESI source was set to 275°C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, an m/z scan range from 70 to 850 was chosen and MS1 data was collected at a resolution of 70,000. The automatic gain control target was set at 1 × 106 and the maximum injection time was 200 ms. The top 5 precursor ions were subsequently fragmented, in a data-dependent manner, using the higher energy collisional dissociation cell set to 30% normalized collision energy in MS2 at a resolution power of 17,500. The sample volumes of 25 μL were injected. Data acquisition and analysis were carried out by Xcalibur 4.0 software and Tracefinder 2.1 software, respectively (both from Thermo Fisher Scientific).
Immunoblot assays
Cell lines were lysed in RIPA buffer (Tris 50 mmol/L, NaCl 150 mmol/L, SDS 0.1%, sodium deoxycholate 0.5%, NaF 5 mmol/L, NaPY 2.5 mmol/L, NP40 1%, EDTA 10 mmol/L) containing a protease-inhibitor cocktail (Mini-complete, EDTA-free, Roche). Samples were heat denatured with 1 Ü Laemmli buffer. Twenty to 50 μg of protein were loaded per well for the 10% SDS-PAGE. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore). Membranes were blocked for 1 hour in 2% BSA in Tris-buffered saline and 0.1% Tween20 and were probed overnight at 4°C with primary antibodies. After washing, blots were probed for 1 hour at room temperature with secondary anti-mouse or anti-rabbit immunoglobulin G (IgG) antibodies conjugated to horseradish peroxidase. Immunoreactive bands were visualized by exposing membranes to luminescent signals generated after incubating the membrane with ECL (Millipore) using Syngene PXi. ImageJ software was used to quantify band intensity and the ratios of proteins of interest were normalized to a loading control.
Reagents and antibodies
α-difluoromethylornithine (DFMO) was purchased from Toronto Research. Putrescine and spermine were bought from Sigma. SAM486A was purchased from Medkoo Bioscience Inc. The c-Myc inhibitor 10058-F4, antibodies for PGC1α (sc-13067, 1:1,000), c-Myc (sc-40, 1:1,000), HSP90 (sc-13119, 1:10,000), and ERK2 (sc-1647, 1:10,000) were purchased from Santa Cruz Biotechnology. ERRα (C15410230, 1:1,000) was purchased from Diagenode. ODC1 (O1136, 1:100) and anti-α-tubulin (T6199, 1:10,000) were purchased from Sigma. HRP-conjugated Affini-Pure donkey anti-mouse IgG (H+L; 715-035-150, 1:5,000) and HRP-conjugated AffiniPure donkey anti-mouse IgG (H+L) (711-035-152, 1:5000) were purchased from Jackson ImmunoResearch Laboratories.
RNA isolation and RT-PCR
Total RNA from cells was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). The quantity and the quality of the isolated RNA were determined using a NanoDrop One Spectrophotometer (Thermo Fisher Scientific). One microgram of total RNA was reverse transcribed into first strand cDNA using the reverse transcription system (Promega). Real-time quantitative PCR was performed using Fast SYBR Green master mix (Applied Biosystems) on a StepOnePlus System (Applied Biosystems). The gene-specific primer sets were used at a final concentration of 1 μmol/L in a 10 μL final volume. RPLP0 mRNA level was used as an endogenous control to normalize relative expression values of each target gene. The relative expression was calculated by the comparative Ct method. All real-time RT-PCR assays were performed in triplicate with three independent experiments. For RNA isolation and RT-PCR from paraffin-embedded prostate tumor tissues, total RNA was extracted with PureLink FFPE RNA Isolation Kit according to the manufacturer’s instructions (Invitrogen). For each RNA purification, 6 pieces of 10-μm thick sections of paraffin-embedded prostate tissues, with a tissue surface area of 0.5–1 cm2, were used. For each patient, a sample of both normal prostate and tumor tissue were collected. Briefly, the main steps were: first, deparaffinization in melting buffer at 72°C for 10 minutes. Then, the tissue was separated from the melted paraffin by centrifugation and digested at 60°C with Proteinase K for 3 hours. The tissue lysate was further processed by selective binding of RNA to a silica-based membrane in the spin cartridge. Impurities were removed by thorough washing with wash buffer. Total RNA was eluted in RNase-Free Water and the quality and quantity of the purified RNA were determined with a NanoDrop One Spectrophotometer (Thermo Fisher Scientific). One microgram of total RNA was reverse transcribed into first strand cDNA using Maxima First Strand cDNA Synthesis Kit (K1671) according to the manufacturer’s instructions (Thermo Fisher Scientific). Real-time quantitative PCR was performed with TaqMan Fast Universal Master Mix (Applied Biosystems) using the ABI PRISM 7500/Step-One Fast Real Time PCR System following the manufacturer’s protocols (Applied Biosystems). Taq-Man gene expression assays purchased from Applied Biosystems: PPARGC1 (Hs01016719_m1), MYC (Hs00153408_m1), ODC1 (Hs00159739_m1), and AMACR (Hs00204885_m1). Gene expression values were normalized to the value of the housekeeping gene 18S (Hs99999901_s1) and calculated on the basis of the comparative cycle threshold Ct method (ΔΔCt).
Chromatin immunoprecipitation
The Chromatin Immunoprecipitation Kit was purchased from EMD Millipore, and the assay was performed according to the manufacturer’s protocol. Briefly, cells were cross-linked with formaldehyde for 10 minutes at 37°C and washed in ice-cold PBS, unreacted formaldehyde was quenched with glycine, and then the cells were washed with PBS and resuspended in SDS buffer. Samples were sonicated, diluted in dilution buffer with inhibitors, and precleared with agarose G beads. The supernatant was used directly for immunoprecipitation with anti-ERRα or goat IgG (for a negative control). The immunocomplexes were mixed with DNA-coated agarose G beads and incubated overnight. Pellets were washed in a low-salt wash buffer, high-salt wash buffer, LiCl wash buffer, and Tris-EDTA buffer. This was followed by elution of the protein/DNA complex and reversal of cross-linking with 5 mol/L NaCl overnight. The protein was then digested with proteinase K, followed by DNA purification with elution buffer. Quantification of chromatin immunoprecipitation (ChIP) enrichment by quantitative real-time PCR (qPCR) were carried out using the Step One PCR (Applied Biosystems).
Migration and invasion assays in Boyden chambers
Migration and invasion assays were performed using cell culture inserts with 8.0-μm pore transparent PET membrane coated with 10 μg/mL fibronectin. For invasion experiments, the inserts were coated with 25 μg/mL of Matrigel and incubated for 3 hours at 37°C in a CO2 incubator, while for the migration assay, transwells were left uncoated. Briefly, overnight serum-starved cells (1.2 × 105 cells) were seeded into the top chamber in medium without FBS, while medium with 10% FBS was present in the bottom chamber. The cells were incubated for 3–6 hours or 17 hours for migration and invasion, respectively. The media and remaining cells were removed from the top chamber with a cotton swab and washed two times with PBS. The bottom chamber was aspirated and washed two times with PBS. The inserts were fixed with 4% PFA and incubated with a DAPI solution (2 mg/mL) to label cell nuclei. Cell migration and invasion were determined by counting all cells on the total areas at the bottom surface of the membrane with a Nikon A1/A1R Confocal Laser Microscope System. Nuclei were counted and analyzed using ImageJ software and normalized to area.
Animal experiments
To conduct pulmonary extravasation assays, 1 × 106 DU145, LNCaP, or PC3 cells were labeled for 1 hour with 15 μmol/L CellTracker Green and injected via the tail vein of 6-week-old nude mice. After 24 hours, mice were sacrificed and the lungs were harvested, fixed with 4% PFA, stained with DAPI, and analyzed with a Nikon A1/A1R Confocal Laser Microscope System. Two mice per group were sacrificed after 30 minutes to confirm that an equal number of cells reached the lung. Quantification of the lung micrometastasis were performed as follows with ImageJ. First, the user sets a large threshold based on the DAPI labeling, and the macro measures the tissue area. This value is used at the end of the analysis to normalize cell numbers relative to the imaged tissue area. Second, the number of positive cells is counted using the Find Maxima function. This function searches for the local maximum of intensity in a defined area. We adapted this area size depending on the noise in the image, which was different depending on the phenotype. All mice were maintained in specific pathogen-free conditions and all experimental procedures were approved by the national Animal Care and Use Committee and the regional ethics committee.
Bioinformatic analysis
The Cancer Genome Atlas (TCGA) data for Prostate Adenovarcinoma (PRAD) were downloaded from the TCGA data portal (https://portal.gdc.cancer.gov). RNA-seq data were normalized using the Bioconductor package DESeq2. Expression levels of the following genes were evaluated in these patient datasets: PPARGC1A (PGC1α), MYC (c-MYC), AMACR, and ODC1. We compared the expression levels of each individual gene in primary tumors and in a reference population (patients without prostate tumors or metastasis). For TCGA data, we compared the PPARGC1A gene expression levels in tumor samples, distinguished by the Gleason grade. P values were calculated by the Wilcoxon rank sum test.
Results
PGC1α inhibits aggressiveness and polyamine synthesis in prostate cancer cells
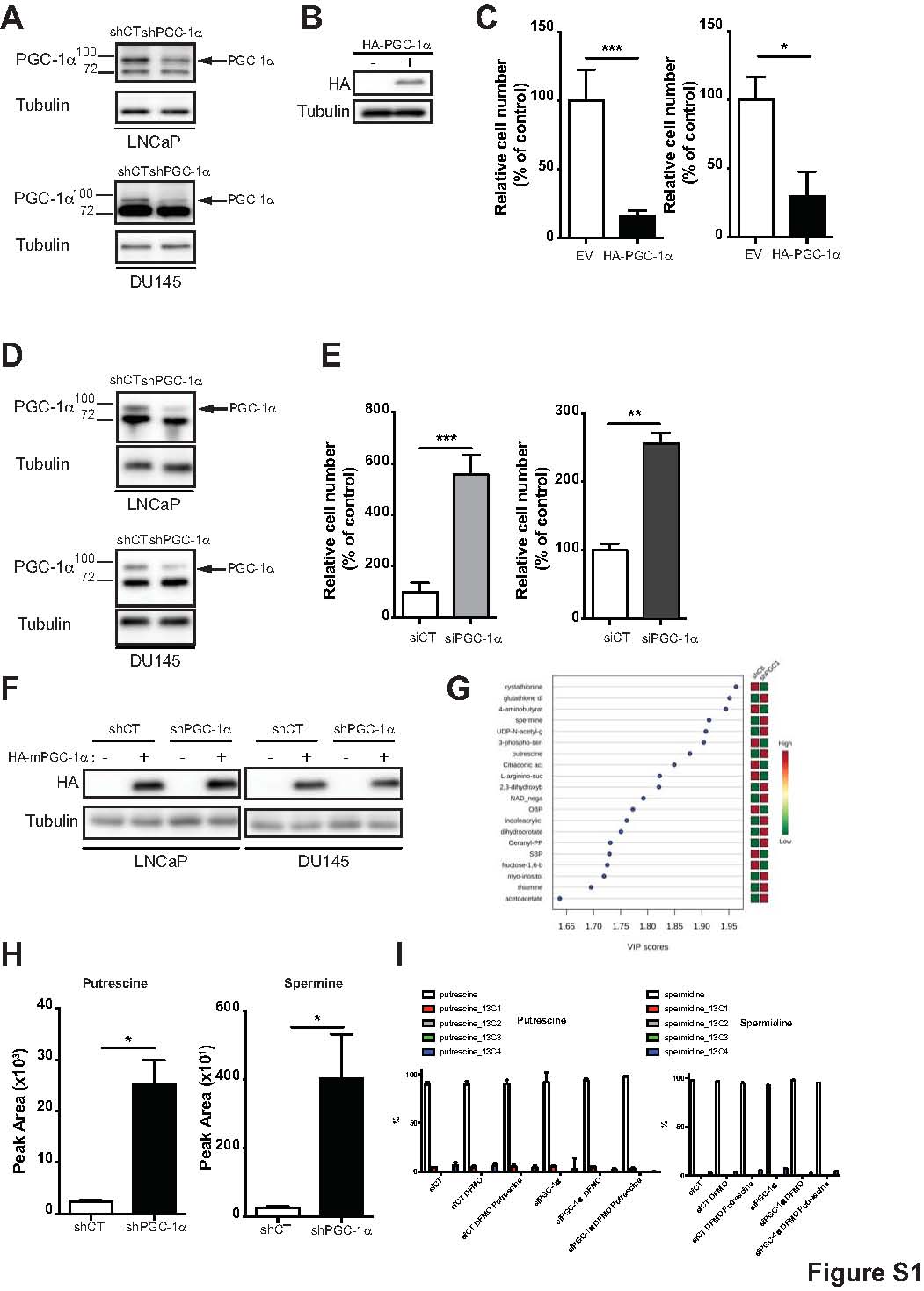
To understand the metabolic modifications associated with PGC1α in prostate cancer, we modulated the expression of PGC1α in human prostate cancer cell lines. Given the tumor suppressor activity of PGC1α (8), we performed migration and invasion assays in two poorly aggressive prostate cancer cell lines, DU145 and LNCaP, which stably express PGC1α shRNA (Supplementary Fig. S1A). The same assays were performed in PC3, the most aggressive prostate cancer cells, overexpressing PGC1α (Supplementary Fig. S1B). The loss of PGC1α significantly increased the migration and the invasion of the LNCaP and DU145 shPGC1α cell lines (Fig. 1A and B), whereas the overexpression of PGC1α inhibited the aggressive phenotype of PC3 cells (Supplementary Fig. S1C).
Figure 1.
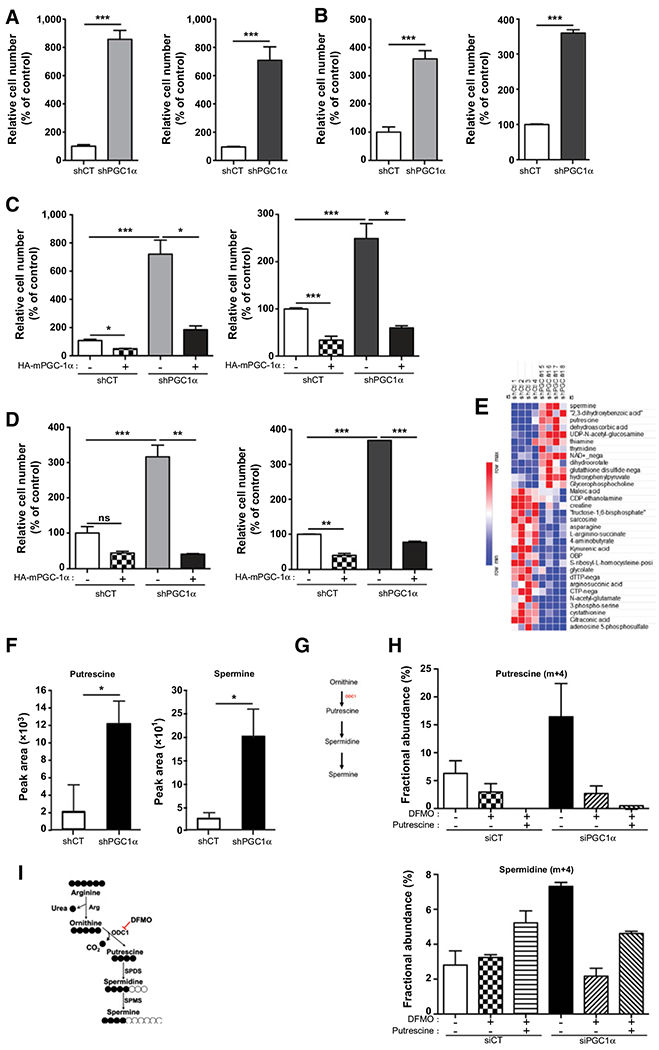
PGC1-α silencing results in cellular polyamine build-up and increased cell migration and invasion. A, Migration assay of LNCaP (orange) and DU145 (blue) shCT and shPGC1α cell lines. B, Invasion assays of LNCaP (orange) and DU145 (blue) in shCT and shPGC1α cell lines after the restoration of PGC1α expression. C, Migration assay of LNCaP and DU145 in shCT and shPGC1α cell lines after the restoration of PGC1α expression. D, Invasion assay of LNCaP and DU145 in shCT and shPGC1α cell lines after the restoration of PGC1α expression. E, Steady-state metabolite profile of shCT and shPGC1-α DU145 cells. Intracellular metabolites from four independent samples per condition were profiled by LC/MS-MS, and those significantly altered in PGC1-α knockdown cells, relative to control cells, are shown as row-normalized heatmaps ranked according to fold-change log2 (shPGC1-α/shCT). F, Normalized peak area of putrescine and spermine in DU145 shCT and shPGC1α cancer cell line, P < 0.02. G, Schematic representation of the polyamine biosynthesis pathway. H and I, Abundance of putrescine and spermidine in DU145 cells transfected with PGC1α siRNA, treated with DFMO (5 mmol/L) and/or putrescine (100 mmol/L) after a pulse of 13C arginine. *, P < 0.05; **, P < 0.01; ***, P < 0.005.
To exclude any off-target effects of the shRNA, we transfected LNCaP and DU145 cells with a pool of siRNA targeting PGC1α (Supplementary Fig. S1D). As with the shPGC1α cells, the siRNA knockdown of PGC1α increased the migration of the cells (Supplementary Fig. S1E). To further validate the role of PGC1α, we restored its expression in LNCaP and DU145 shPGC1α cells using a cDNA encoding for a HA-tagged mouse PGC1α (HA-mPGC-1α; Supplementary Fig. S1F). The over-expression of PGC1α hampered the migration and the invasion of the shPGC1α cell lines, but it also significantly decreased the aggressive phenotype of the shCT cells (Fig. 1C and D). Thus, our results demonstrate that PGC1α inhibits prostate cancer cells’ aggressiveness.
To characterize the metabolic changes associated with this phenotype, we performed an unbiased metabolomic profiling of cells knockdown for PGC1α. Of the 262 small metabolites identified by LC/MS-MS), the steady-state levels of 32 metabolites were significantly altered in the PGC1α knockdown cells relative to cells transfected with control shRNA (P < 0.02). Eventually, we found 12 metabolites that were significantly upregulated upon PGC1α silencing, among them, spermine and putrescine, two metabolites that are representative of the polyamine pathway (Fig. 1E–G), were among the most upregulated with a Variable Importance in Projection score above 1.85 (Supplementary Fig. S1G). Consistently, the increase of putrescine and spermine was confirmed in another shPGC1α-stable cell line transfected with a shRNA targeting a different region of PGC1α (Supplementary Fig. S1H). To confirm the regulation of the polyamine pathway upon PGC1α loss, we employed a stable isotope-tracing LC/MS-MS experiment with [13C6]-arginine a precursor for polyamine synthesis in cells depleted or not for PGC1α. As expected, thedownregulation of PGC1α, although not significant, increased the abundance of 13C putrescine and 13C spermidine (Fig. 1H; Supplementary Fig. S1I). We also treated the cells with DFMO, a specific and well-characterized inhibitor of the ODC1 the rate-limiting enzyme of polyamine synthesis (Fig. 1I; ref. 18). As expected, DFMO abolished M+4 labeling of putrescine and spermidine induced by the downregulation of PGC1α. Addition of exogenous putrescine restored partially the flux of polyamine synthesis as illustrated by the increase of spermidine (Fig. 1H).
These results demonstrate that the increase in polyamine synthesis is one of the major metabolic alteration induced by the downregulation of PGC1α.
PGC1α inhibits ODC1 expression via the downregulation of c-MYC
The rate-limiting enzyme of polyamine synthesis is the ODC1. We found, in accordance with the upregulation of polyamine synthesis, that the expression of ODC1 mRNA and protein was significantly increased in shPGC1α cells (Fig. 2A; Supplementary Fig. S2A). To validate that PGC1α inhibits the expression of ODC1, we overexpressed PGC1α in shPGC1α cells and found that it hampered the expression of ODC1 (Fig. 2A).
Figure 2.
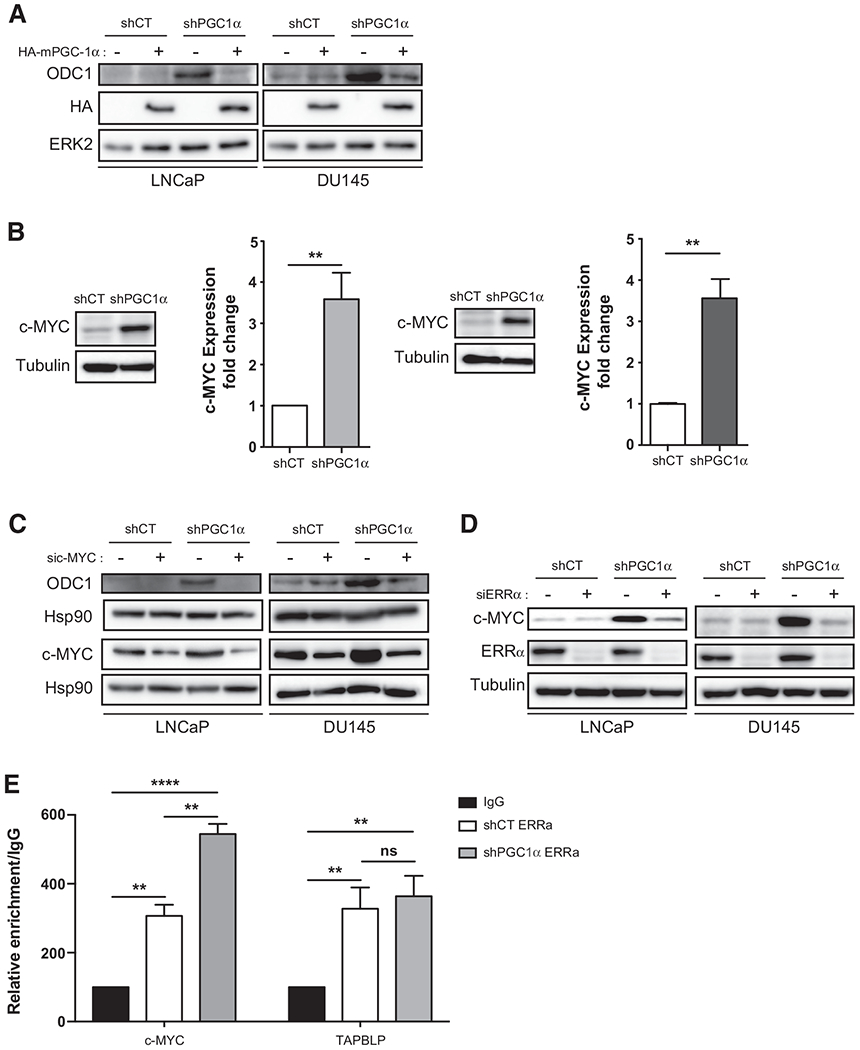
PGC1α represses c-MYC and ODC1 expression. A, Expression of ODC1 in LNCaP and DU145 shPGC1α cell lines overexpressing or not HA-mPGC-1α. B, Expression of c-MYC in the shPGC1α cell lines. The graph represents the quantification of three independent experiments. C, Expression of ODC1 in LNCaP and DU145 shPGC1α cell lines transfected with sic-MYC. D, Expression of c-MYC in LNCaP and DU145 shPGC1α cell lines transfected with siERRα. E, CHIP analysis of ERRα binding in the promoter region of c-MYC and TAPBLP in shCT and shPGC1α DU145 cells. Data are shown as mean ± SEM. **, P < 0.01; ***, P < 0.0001. P values are calculated by Student t test. ns, nonsignificant.
ODC1 is one of the first metabolic genes shown to be transcriptionally regulated by the proto-oncogene c-MYC (19), and c-MYC increases polyamine synthesis (20). Thus, we monitored the expression of c-MYC in shPGC1α cells. We showed that c-MYC mRNA and protein are significantly increased in shPGC1α cells (Fig. 2B; Supplementary Fig. S2B), suggesting, that like ODC1, PGC1α represses c-MYC expression. In accordance with this result, we found a decrease in the expression of c-MYC mRNA and protein in PC3 cells overexpressing PGC1α (Supplementary Fig. S2C). To validate that c-MYC upregulates ODC1 expression, we silenced c-MYC with siRNA in shPGC1α cells and found that the downregulation of c-MYC abrogates the expression of ODC1 (Fig. 2C).
We subsequently studied how PGC1α regulates c-MYC expression. We took advantage of transcriptomic analyses showing that PGC1α regulates the estrogen-related receptor alpha (ERRα) transcriptional program in prostate cancer cells (8). To determine whether ERRα is involved in the regulation of c-MYC expression by PGC1α, we undertook a silencing approach with a pool of ERRα-targeting siRNA. The silencing of ERRα had no effect on c-MYC expression in shCT cells, but it inhibited the expression of c-MYC induced by the knockdown of PGC1α in LNCaP and DU145 (Fig. 2D). To confirm the involvement of ERRα in the regulation of c-MYC, we performed a ChIP with ERRα antibodies and a quantitative PCR with the putative binding site of ERRα in the promoter region of c-MYC and TAPBPL as a control (21). As expected, ERRα occupied the promoter region of TAPBPL, but the downregulation of PGC1α did not increase its recruitment (Fig. 2E). On contrary, the downregulation of PGC1α significantly increased the recruitment of ERRα on the promoter region of c-Myc. In addition, analysis of the publicly available ChIP-seq data revealed that ERRα is one the regulatory transcription factor of c-MYC in several databases (Supplementary Fig. S2D). Altogether, our data provide a molecular mechanism by which PGC1α represses a c-MYC/ODC1 axis and demonstrate that PGC1α inhibits c-MYC expression through an ERRα-dependent mechanism.
The PGC1α/c-MYC/ODC1 axis controls the aggressive phenotype of prostate cancer cells via polyamine synthesis
To determine the contributing role of c-MYC in the aggressive phenotype of the shPGC1α cells, we inhibited c-MYC with siRNA or the 10058-F4 compound (a specific chemical inhibitor of c-MYC) and performed migration and invasion assays. Under both conditions, the inhibition of c-MYC abolished the promigrative and proinvasive effects of the shPGC1α prostate cancer cells (Fig. 3A–D; Supplementary Fig. S3A). This result clearly demonstrates that c-MYC mediates the effects of PGC1α on prostate cancer cell aggressiveness.
Figure 3.
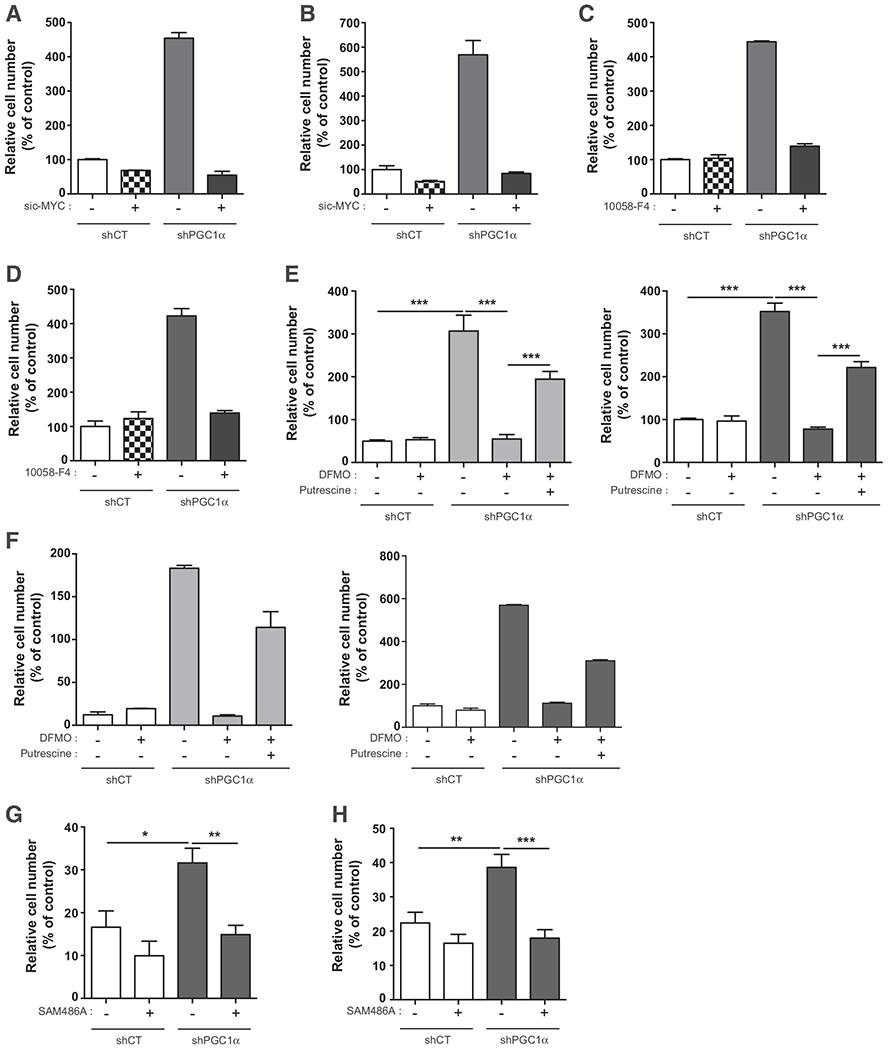
The PGC1α/c-MYC/ODC1 axis controls the aggressive phenotype in prostate cancer cells via polyamine synthesis. A, Migration assay in DU145 shPGC1α cells transfected with sic-MYC. B, Invasion assay in DU145 shPGC1α cells transfected with sic-MYC. C, Migration assay in DU145 shPGC1α cells treated with the c-MYC inhibitor, 10058-F4 (100 μmol/L). D, Invasion assay in DU145 shPGC1α cells treated with the c-MYC inhibitor, 10058-F4 (100 μmol/L). E, Migration assay in LNCaP and DU145 shPGC1α cells treated with DFMO (5 mol/L) and putrescine (10 μmol/L). F, Invasion assay in LNCaP and DU145 shPGC1α cells treated with DFMO (5 mmol/L) and putrescine (10 μmol/L). G, Migration assay in shCT and shPGC1α cells treated with the inhibitor of AMD1, SAM486A (1 μmol/L). H, Invasion assay in shCT and shPGC1α cells treated with the inhibitor of AMD1, SAM486A (1 μmol/L). Data are shown as mean ° SEM. **, P < 0.01; ***, P < 0.001. P values are calculated by Student t test. The graphs in A–D and F are the mean of two independent experiments.
To establish whether polyamine synthesis is the metabolic pathway implicated in the aggressive phenotype of shPGC1α cells, we treated cells with DFMO. DFMO did not inhibit the migration and the invasion of the control cells, but it hampered the aggressive phenotype of the shPGC1α cells (Fig. 3E and F). The contribution of polyamine synthesis to the aggressiveness of shPGC1α cells was validated by the addition of exogenous putrescine for 24 hours in the culture media after the DFMO treatment. Indeed, putrescine partially restored the aggressiveness of the shPGC1α cell lines (Fig. 3E and F). To further confirm the role of ODC1, we used a pool of siRNA (Supplementary Fig. S3B). The decrease of ODC1 inhibited cell migration and invasion induced by the loss of PGC1α in LNCaP and DU145 cells. While the addition of putrescine alone had no effect on control cells (siCT in Supplementary Fig. S3C and S3D), it decreased the invasive properties of shPGC1α cells (shPGC1α cells in Supplementary Fig. S3D) but, it rescued partially the aggressiveness (migration and invasion) of ODC1-depleted cells (Supplementary Fig. S3C and S3D).
Recently, AMD1 (S-adenosylmethionine decarboxylase 1) that regulates the synthesis of decarboxy S-adenosyl-l-methionine required for spermine synthesis has been shown to control polyamine synthesis in prostate cancer (22). We thus asked whether this enzyme plays a role in the upregulation of polyamine synthesis in PGC1α-depleted cells. We treated the cells with an inhibitor of AMD1, SAM486A, validated in preclinical and clinical experiments (23, 24). SAM486A did not significantly affect the migration and the invasion of the ShCT cells but led to a significant decrease in the aggressiveness of shPGC1α cells (Fig. 3G and H). All together, these results support the conclusion that the downregulation of PGC1α reprograms cancer cell metabolism toward polyamine synthesis to promote an aggressive phenotype through an ODC1- and AMD1-dependent pathway.
PGC1α suppresses the formation of micrometastasis through the downregulation of polyamine synthesis
To establish the role of PGC1α in aggressiveness in vivo, we performed an extravasation experiment in mice. DU145 and LNCaP shPGC1α cells and their respective shCT control cells, treated or not treated with DFMO, were injected into the caudal vein of 6-week-old male athymic nude mice. After 24 hours, to allow for the formation of micrometastasis, we analyzed the ability of cells to extravasate through the pulmonary parenchyma and quantified the micrometastasis formation. The downregulation of PGC1α significantly increased the invasive potential of DU145 and LNCaP cells (Fig. 4A and B; Supplementary Fig. S4A). The treatment with DFMO did not affect the aggressiveness of the shCT cells, but it significantly hampered the invasive properties of the shPGC1α cells demonstrating that inhibition of polyamine synthesis reduces micrometastasis in cells with elevated polyamine anabolism. To validate the anti-invasive activity of PGC1α, we tested the aggressiveness of PC3 cells overexpressing PGC1α. The expression of PGC1α strongly inhibited extravasation and the treatment with exogenous putrescine restored the ability to form micrometastases (Fig. 4C). We showed previously that the addition of putrescine on control cells did not affect the migration and invasion (Supplementary Fig. S3C and S3D). To determine whether putrescine affects the formation of micrometastasis, we treated DU145 shCT with putrescine prior to their injection into mice. Putrescine led to a slight but significant decrease of micrometastasis (Supplementary Fig. S4B).
Figure 4.
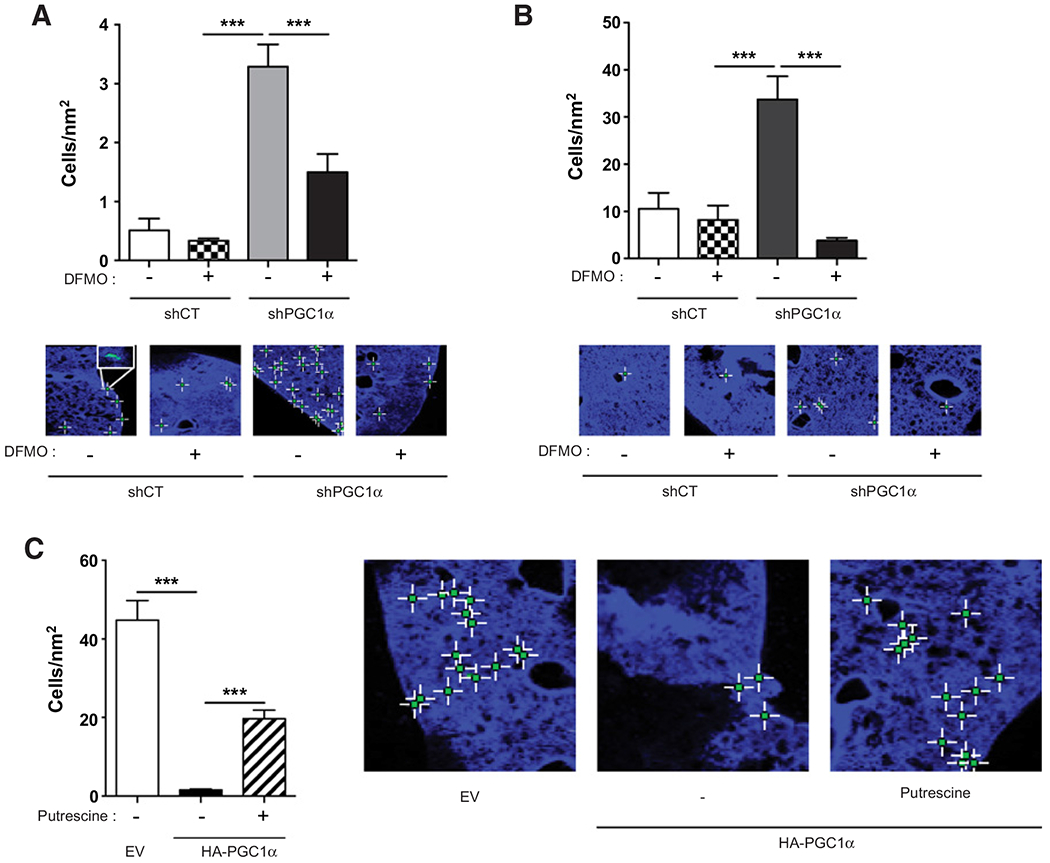
PGC1α inhibits the formation ofmicrometastasis, and putrescine restores the aggressive phenotype. A, Quantification and representative images of lung micrometastasis in mice injected with LNCaP shPGC1α cells and treated with DFMO (5 mmol/L). B, Quantification and representative images of lung micrometastasis in mice injected with DU145 shPGC1α cells and treated with DFMO (5 mmol/L). C, Quantification and representative images of lung micrometastasis in mice injected with PC3 cells expressing PGC1α and treated with putrescine (10 μmol/L). n = 5 mice per group. Data are shown as mean ° SEM. ***, P < 0.001. P values are calculated by Student t test.
Taken together, our data show that putrescine alone does not increase metastasis but PGC1α suppresses the formation of micrometastasis and polyamines are required for this process.
The downregulation of PGC1α is associated with increased c-MYC and ODC1 in human tumor biopsies
To determine whether the PGC1α/c-MYC/ODC1 axis has a clinical relevance, we analyzed mRNA expression of these genes in normal and tumor matched tissues from 73 patients with advanced prostate cancer (Table 1). We extracted mRNA from paraffin-embedded tissues and performed a qRT-PCR analysis on paired tumor and normal adjacent tissue. The expression of AMACR (a well-characterized biomarker of prostate cancer), PGC1α, c-MYC, and ODC1 are presented in Fig. 5A. As expected, the expression of AMACR was significantly higher in tumors compared with the healthy gland. Importantly, we found that the expression of PGC1α mRNA was significantly lower in tumors compared with the adjacent normal tissue and in the same tumor tissue, the expression of c-MYC and ODC1 was increased.
Table 1.
Characteristics of the 73 patients with advanced prostate cancer
| n (%) | |
|---|---|
| Patients | 73 |
| Age, y | |
| Median | 67 (51–81) |
| Mean | 66.3 |
| PSA (ng/mL) | |
| Median | 8 (3.2–39) |
| Mean | 11.5 |
| Tumor stage | |
| pT2a | 4 (5.4) |
| pT2b | 1 (1.4) |
| pT2c | 33 (44.6) |
| pT3a | 28 (37.8) |
| pT3b | 8 (10.8) |
| Final Gleason | |
| Gleason 6 | 4 (5.5) |
| Gleason 7 | 53 (72.6) |
| Gleason ≥ 8 | 16 (21.9) |
Figure 5.

The downregulation of PGC1α is associated with increased c-MYC and ODC1 in tumor biopsies. A, Expression of AMACR, PGC1α, MYC, and ODC1 mRNA in prostate tumor and adjacent normal tissue from 73 patients. B, Heatmap representation of the expression of PGC1α, MYC, and ODC1 from the TCGA database. C, Correlation analysis of the TCGA database for the expression of c-MYC and PPARGC1A (PGC1α) and for the expression of ODC1 and PPARGC1A (PGC1α). D, Association of ODC1 expression with progression-free survival (PFS) in the TCGA database. Data are shown as mean ± SEM. **, P < 0.01; ***, P < 0.001. P values are calculated by Student t test.
On the basis of the TCGA-PRAD database (healthy tissue: n = 52 and primary tumors: n = 498), we constructed a heatmap to visualize the coexpression pattern of the PGC1α/c-MYC/ODC1 axis genes in normal and tumoral tissues with increasing Gleason grades. PGC1α expression is high in normal tissues and gradually decreases with elevated Gleason scores. In contrast, the expression of ODC1 and c-MYC is low in normal tissue and increased with Gleason grade (Fig. 5B). To confirm this observation, we performed a correlative analysis using the same database. We observed a weak but significant correlation between PGC1α and ODC1 expression as well as PGC1α and c-MYC in prostate cancer tumors and normal tissue with r = 0.30 (P = 2.9 × 10−13) and r = 0.24 (P = 6.48 × 10−9), respectively (Fig. 5C).
We then analyzed the Taylor and colleagues dataset that includes healthy tissues (n = 29), primary tumors (n = 131), and metastasis (n = 19; ref. 25). ODC1 and C-MYC are significantly increased in metastasis and the primary tumor compared with the healthy tissue and PGC1α is dowregulated in the tumor and the metastasis (Supplementary Fig. S5A).
Finally, in accordance with our results and other studies, high expression of ODC1 (P = 0.0146; Fig. 5D) and low expression or PGC1α (8) are significantly correlated with progression-free survival.
Taken together, our data demonstrate that the low expression of PGC1α is associated with high expression of c-MYC and ODC1 in patients with prostate cancer. It provides evidence that the PGC1α/c-MYC/ODC1 axis defines a gene signature that may have a prognostic value for aggressive prostate cancer and may also be the target of future therapies.
Discussion
We demonstrate here that the transcriptional coactivator, PGC1α, is one of the regulators of an onco-metabolic program regulating prostate cancer cell aggressiveness. The downregulation of PGC1α makes prostate cancer cells dependent on the polyamine pathway to promote metastasis. Indeed, the decrease of PGC1α induces the upregulation of c-MYC and ODC1, the rate-limiting enzyme of polyamine synthesis. Both the bioinformatic analysis of a prostate cancer dataset and our results on human biopsies demonstrate that low expression of PGC1α correlates with higher levels of c-MYC and ODC1 in tumors. In addition, we demonstrate, based on the analysis of public databases, that high expression of ODC1 is associated with a lower PFS. Thus, our results highlight the importance of the PGC1α/c-MYC/ODC1 axis as a potential prognostic marker of prostate cancer aggressiveness.
Recent studies have demonstrated that the downregulation of PGC1α promotes cancer cell aggressiveness and metastasis in prostate cancer, melanoma, and hepatocarcinoma (8, 26, 27). However, the role of PGC1α is different in breast cancer, where the expression of PGC1α promotes breast cancer metastasis (28) and circulating (metastatic) breast cancer cells express high levels of PGC1α (6). The elevated expression of PGC1α correlates with increased mitochondrial respiration and glucose consumption as well as greater fuel flexibility in breast cancer cells. This opposite role of PGC1α in different cancer is puzzling. PGC1α interacts with a wide variety of transcription factors and it is regulated by several signaling pathways (29), thus, depending on the organs the target genes may differ. Hormones may also interfere with PGC1α normal function especially in prostate and breast. Indeed, PGC1α interacts with the androgen receptor (30) and the estrogen receptor (31). Finally, because of the different metabolic status of tumors (glycolytic vs. oxidative phosphorylation), modifications of PGC1α expression may induce different biological responses.
Even though PGC1α plays a central role in mitochondrial biogenesis, we did not observe significant differences in the energetic status of the cell lines with modified expression of PGC1α. The ATP concentration as well as the mitochondrial mass were not altered. This not surprising because PGC-1β (another member of the PGC-1 family) has overlapping and cooperative actions in vivo, and may therefore compensate the dowregulation of PGC1α (32, 33).
The importance of polyamines in prostate cancer proliferation is well established (34, 35). It was recently demonstrated that this pathway drives prostate cancer aggressiveness through the activation of the pro-oncogenic complex mTORC1. mTORC1 controls the stability of S-adenosylmethionine decarboxylase 1 (AMD1), the enzyme that regulates the production of decarboxylated S-adenosylmethionine, a substrate for polyamine synthesis (22). In our study, the inhibition of AMD1 reversed the aggressiveness of shPGC1α cells, reinforcing the role of polyamine metabolism in the formation of metastasis induced by PGC1α loss. Thus, we have characterized a new regulatory pathway orchestrated by PGC1α, which drives prostate cancer cancer aggressiveness through polyamine synthesis. Our data demonstrate that addition of exogenous putrescine alone does not increase aggressiveness of prostate cancer cells, suggesting that the phenotype observed is solely linked to the loss of PGC1α. Indeed, we demonstrate that the downregulation of PGC1α triggers a metabolic switch that makes prostate cancer cells depend on polyamine synthesis to promote aggressiveness. This conclusion is strengthened by the fact that putrescine rescues the inhibitory effects of DFMO. Targeting polyamine synthesis may be interesting for cancer therapy in prostate cancer with low PGC1α levels. Many studies have shown that DFMO (the inhibitor of ODC1) inhibits the proliferation and the aggressiveness of gastric, prostate, nonmelanoma skin cancer, and neuroblastoma cell lines. DFMO is also an inhibitor of tumor growth in animal models, where it reduced pancreatic intraepithelial neoplasm by 40% (36). Similarly, addition of DFMO in drinking water decreased significantly tumor growth in TRAMP mice, a transgenic model of prostate cancer (35). Preclinical evidence strongly supports the use of DFMO in cancer therapy and many clinical trials have already been performed, while others are ongoing for the treatment of different cancers such as gliomas, prostate, and gastric cancer (37). So far, the results of these clinical trials have shown moderate effects (38–40). Even though DFMO very efficiently inhibits polyamine synthesis in patients (41), dietary sources of polyamines compensate for the inhibitory effects of DFMO on endogenous polyamine synthesis. Thus, strategies combining a low polyamine diet and DFMO may be potentially interesting. A recent study that combines DFMO and the inhibitor of the solute carrier family 3 member 2 (AMXT 1501), a transporter of polyamines, demonstrated the efficiency of this strategy in neuroblastoma (42).
We found that PGC1α knockdown led to the increase of c-MYC expression in prostate cancer cells and, conversely, that the overexpression of PGC1α decreases c-MYC protein levels. The c-MYC/ PGC1α balance has been shown to control not only the metabolic phenotype of pancreatic cancer stem cells, but also their sensitivity to metabolic drugs targeting the mitochondria, such as metformin. Low expression of c-MYC allows high expression of PGC1α, resulting in higher bioenergetic capacities in cancer stem cells (43). Here, we found that low expression of PGC1α allows for high expression of c-MYC in prostate cancer cells, thus triggering the increase of polyamine synthesis and making prostate cancer more aggressive. The link between c-MYC and polyamine synthesis is not new because ODC1 expression is known to be regulated by c-MYC, and glutaminolysis has been shown to be directly linked to polyamine synthesis in a c-MYC-dependent manner during T-lymphocyte activation (44). Our findings demonstrate that PGC1α regulates a pathway integrating c-MYC and ODC1. Mechanistically, we also show that PGC1α and ERRα act together to regulate c-MYC expression. When PGC1α is downregulated, ERRα is required for the increase of c-MYC. This interesting result shed light on a complex interaction existing between ERRα and PGC1α. PGC1α is not the only coactivator of ERRα, studies have shown that members of the p160 steroid receptor coactivator (SRC) family, namely SRC-1 and SRC-2 increase ERRα transcriptional activity (45, 46). The loss of PGC1α may promote the recruitment of other coactivators such as SRC-1/2 that may induce a stronger activity of ERRα and thus increase c-MYC expression. Analogous interplay between ERRα, PGC1α, and SRC-1 has already been described by Zhang and colleagues (47). These results are in agreement with recent studies demonstrating that ERRα is involved in prostate cancer progression and the formation of bone metastasis (48, 49).
In summary, our study reinforces the central role of the polyamine pathway in prostate cancer progression, but it also reveals a link between PGC1α and an onco-metabolic pathway. Our findings not only describe new signatures of cancer progression but also provide tools to identify new therapeutic options.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Significance:
These findings show that a major regulator of mitochondrial metabolism controls polyamine synthesis and prostate cancer aggressiveness, with potential applications in therapy and identification of new biomarkers.
Acknowledgments
We thank Charlotte Hinault for carefully reading the manuscript. This work was supported by a grant from the Fondation ARC pour la recherche sur le Cancer, l’Association pour la Recherche sur les Tumeurs de la Prostate (ARTP), ITMO-Cancer. This work has been supported by the French government, through the UCAJEDI Investments in the Future project managed by the National Research Agency (ANR) with the reference number ANR-15-IDEX-01. L. Kaminski is supported by the French Ministry of Research. S. Torrino has a grant from the Fondation de France. R. Haider and E. Jaune were supported by the foundation ARC and F.-R. Roustan by the CHU of Nice. I. Ben-Sahra is supported by the NIH R00-CA194192 and LAM Foundation grants. F. Bost, N. M. Mazure, and J.F. Tanti are CNRS investigators. Metabolomics services were performed by the Metabolomics Core Facility at Robert H. Lurie Comprehensive Cancer Center of Northwestern University and the Beth Israel Deaconess Medical Center Mass Spectrometry Facility of Harvard Medical School.
Footnotes
The costs of publication of this article were defrayed in part bythe payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008;27: 3576–86. [DOI] [PubMed] [Google Scholar]
- 3.Bost F,Decoux-Poullot AG, Tanti JF, Clavel S. Energy disruptors: rising stars in anticancer therapy? Oncogenesis 2016;5:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dirat B, Ader I, Golzio M, Massa F, Mettouchi A, Laurent K, et al. Inhibition of the GTPase Rac1 mediates the antimigratory effects of metformin in prostate cancer cells. Mol Cancer Ther 2015;14:586–96. [DOI] [PubMed] [Google Scholar]
- 5.Villena JA.Newinsights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 2015;282: 647–72. [DOI] [PubMed] [Google Scholar]
- 6.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, et al. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016;537:422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic co-regulator PGC1alpha suppresses prostate cancer metastasis. Nat Cell Biol 2016;18:645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bost F, Kaminski L. The metabolic modulator PGC-1α in cancer. Am J Cancer Res 2019;9:198–211. [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins RB, Qian J, Lieber MM, Bostwick DG. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res 1997;57:524–31. [PubMed] [Google Scholar]
- 11.Qian J, Jenkins RB, Bostwick DG. Detection of chromosomal anomalies and c-myc gene amplification in the cribriform pattern of prostatic intraepithelial neoplasia and carcinoma by fluorescence in situ hybridization. Mod Pathol 1997;10:1113–9. [PubMed] [Google Scholar]
- 12.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003;4:223–38. [DOI] [PubMed] [Google Scholar]
- 13.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucoseindependent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012;15:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 1997;94:6658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A 1993;90: 7804–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013;339:1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ionswitching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 2012;7: 872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bey P, Danzin C, Van Dorsselaer V, Mamont P, Jung M, Tardif C. Analogues of ornithine as inhibitors of ornithine decarboxylase. New deductions concerning the topography of the enzyme’s active site. J Med Chem 1978; 21:50–5. [DOI] [PubMed] [Google Scholar]
- 19.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogarty MD, Norris MD, Davis K, Liu X, Evageliou NF, Hayes CS, et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res 2008;68:9735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deblois G, Hall JA, Perry MC, Laganiere J, Ghahremani M, Park M, et al. Genome-wide identification of direct target genes implicates estrogenrelated receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res 2009;69:6149–57. [DOI] [PubMed] [Google Scholar]
- 22.Zabala-Letona A, Arruabarrena-Aristorena A, Martin-Martin N, Fernandez-Ruiz S, Sutherland JD, Clasquin M, et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017;547:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eskens FA, Greim GA, van Zuylen C, Wolff I, Denis LJ, Planting AS, et al. Phase I and pharmacological study of weekly administration of the polyamine synthesis inhibitor SAM 486A (CGP 48 664) in patients with solid tumors. European Organization for Research and Treatment of Cancer Early Clinical Studies Group. Clin Cancer Res 2000;6:1736–43. [PubMed] [Google Scholar]
- 24.Regenass U, Mett H, Stanek J,Mueller M, Kramer D, Porter CW.CGP48664, a new S-adenosylmethionine decarboxylase inhibitorwith broad spectrum antiproliferative and antitumor activity. Cancer Res 1994;54:3210–7. [PubMed] [Google Scholar]
- 25.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo C, Widlund HR, Puigserver P. PGC-1 Coactivators: shepherding the mitochondrial biogenesis of tumors. Trends Cancer 2016;2:619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu R, Zhang H, Zhang Y, Li S, Wang X, Wang C, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha acts as a tumor suppressor in hepatocellular carcinoma. Tumour Biol 2017;39:1010428317695031. [DOI] [PubMed] [Google Scholar]
- 28.Andrzejewski S, Klimcakova E, Johnson RM, Tabaries S, Annis MG, McGuirk S, et al. PGC-1alpha promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab 2017;26:778–87. [DOI] [PubMed] [Google Scholar]
- 29.Deblois G, St-Pierre J, Giguère V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013;32:3483–90. [DOI] [PubMed] [Google Scholar]
- 30.Shiota M, Yokomizo A, Tada Y, Inokuchi J, Tatsugami K, Kuroiwa K, et al. Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor (AR) and promotes prostate cancer cell growth by activating the AR. Mol Endocrinol 2010;24:114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP. Modulation of estrogen receptor-alpha transcriptional activity by the coactivator PGC-1. J Biol Chem 2000;275:16302–8. [DOI] [PubMed] [Google Scholar]
- 32.Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, et al. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 2010;12: 633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev 2008;22:1948–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettuzzi S, Davalli P, Astancolle S, Carani C, Madeo B, Tampieri A, et al. Tumor progression is accompanied by significant changes in the levels of expression of polyamine metabolism regulatory genes and clusterin (sulfated glycoprotein 2) in human prostate cancer specimens. Cancer Res 2000;60:28–34. [PubMed] [Google Scholar]
- 35.Gupta S, Ahmad N, Marengo SR, MacLennan GT, Greenberg NM, Mukhtar H. Chemoprevention of prostate carcinogenesis by alpha-difluoromethylornithine in TRAMP mice. Cancer Res 2000;60:5125–33. [PubMed] [Google Scholar]
- 36.Mohammed A, Janakiram NB, Madka V, Ritchie RL, Brewer M, Biddick L, et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev Res 2014;7: 1198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexiou GA, Lianos GD, Ragos V, Galani V, Kyritsis AP. Difluoromethylornithine in cancer: new advances. Future Oncol 2017;13:809–19. [DOI] [PubMed] [Google Scholar]
- 38.Horn Y, Schechter PJ, Marton LJ. Phase I-II clinical trial with alpha-difluoromethylornithine–an inhibitor of polyamine biosynthesis. Eur J Cancer Clin Oncol 1987;23:1103–7. [DOI] [PubMed] [Google Scholar]
- 39.Levin VA, Hess KR, Choucair A, Flynn PJ, Jaeckle KA, Kyritsis AP, et al. Phase III randomized study of postradiotherapy chemotherapy with combination alpha-difluoromethylornithine-PCV versus PCV for anaplastic gliomas. Clin Cancer Res 2003;9:981–90. [PubMed] [Google Scholar]
- 40.Lynch PM, Burke CA, Phillips R, Morris JS, Slack R, Wang X, et al. An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut 2016;65:286–95. [DOI] [PubMed] [Google Scholar]
- 41.Simoneau AR, Gerner EW, Nagle R, Ziogas A, Fujikawa-Brooks S, Yerushalmi H, et al. The effect of difluoromethylornithine on decreasing prostate size and polyamines in men: results of a year-long phase IIb randomized placebo-controlled chemoprevention trial. Cancer Epidemiol Biomarkers Prev 2008;17:292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gamble LD, Purgato S, Murray J, Xiao L, Yu DMT, Hanssen KM, et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci Transl Med 2019;11. doi: 10.1126/scitranslmed.aau1099. [DOI] [PubMed] [Google Scholar]
- 43.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 2015;22:590–605. [DOI] [PubMed] [Google Scholar]
- 44.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011;35:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu D, Benlhabib H, Mendelson CR. cAMP enhances estrogen-related receptor alpha (ERRalpha) transcriptional activity at the SP-A promoter by increasing its interaction with protein kinase A and steroid receptor coactivator 2 (SRC-2). Mol Endocrinol 2009;23:772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie W, Hong H, Yang NN, Lin RJ, Simon CM, Stallcup MR, et al. Constitutive activation of transcription and binding of coactivator by estrogenrelated receptors 1 and 2. Mol Endocrinol 1999;13:2151–62. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, Teng CT. Interplay between estrogen-related receptor alpha (ERRalpha) and gamma (ERRgamma) on the regulation of ERRalpha gene expression. Mol Cell Endocrinol 2007;264:128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fradet A, Bouchet M, Delliaux C, Gervais M, Kan C, Benetollo C, et al. Estrogen related receptor alpha in castration-resistant prostate cancer cells promotes tumor progression in bone. Oncotarget 2016;7: 77071–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misawa A, Inoue S. Estrogen-related receptors in breast cancer and prostate cancer. Front Endocrinol 2015;6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.