

Abstract
The musculoskeletal system is critical for movement and the protection of organs. In addition to abrupt injuries, daily physical demands inflict minor injuries, necessitating a coordinated process of repair referred to as the acute‐phase response (APR). Dysfunctional APRs caused by severe injuries or underlying chronic diseases are implicated in pathologic musculoskeletal repair, resulting in decreased mobility and chronic pain. The molecular mechanisms behind these phenomena are not well understood, hindering the development of clinical solutions. Recent studies indicate that, in addition to regulating intravascular clotting, the coagulation and fibrinolytic systems are also entrenched in tissue repair. Although plasmin and fibrin are considered antithetical to one another in the context of hemostasis, in a proper APR, they complement one another within a coordinated spatiotemporal framework. Once a wound is contained by fibrin, activation of plasmin promotes the removal of fibrin and stimulates angiogenesis, tissue remodeling, and tissue regeneration. Insufficient fibrin deposition or excessive plasmin‐mediated fibrinolysis in early convalescence prevents injury containment, causing bleeding. Alternatively, excess fibrin deposition and/or inefficient plasmin activity later in convalescence impairs musculoskeletal repair, resulting in tissue fibrosis and osteoporosis, while inappropriate fibrin or plasmin activity in a synovial joint can cause arthritis. Together, these pathologic conditions lead to chronic pain, poor mobility, and diminished quality of life. In this review, we discuss both fibrin‐dependent and ‐independent roles of plasminogen activation in the musculoskeletal APR, how dysregulation of these mechanisms promote musculoskeletal degeneration, and the possibility of therapeutically manipulating plasmin or fibrin to treat musculoskeletal disease.
Keywords: acute‐phase reaction, fibrinogen, musculoskeletal diseases, plasminogen, plasminogen activators, rheumatic diseases
Essentials.
Musculoskeletal disease results in pain and poor mobility with limited treatment options.
Beyond clot degredation, plasmin is essential for musculoskeletal health.
Inappropriate plasmin activity in disease can drive musculoskeletal degeneration.
The plasminogen activation system presents new potential targets to treat musculoskeletal disease.
1. INTRODUCTION
The musculoskeletal system serves primarily to counteract gravity, protect vital structures, and allow locomotion. These functions rely on the synergistic interactions of bones and muscles. Muscles attach to bones through tendons and lever them through bone articulations, or joints. 1 , 2 , 3 Additionally, the musculoskeletal system serves as a primary hematopoietic center and a systemic regulator of essential minerals, including calcium and phosphate. 1 , 2 Proper function and maintenance of the musculoskeletal system is therefore essential for these processes.
The musculoskeletal system is repeatedly injured as the physical demands of daily life outweigh the capacity of the tissue to absorb strain. 4 Injuries to the musculoskeletal system differ in nature, severity, and immediate impact but can all lead to chronic complications without timely repair. 3 , 4 , 5 For instance, everyday engagement of the musculoskeletal system results in daily microinjuries, instigating reparative processes imperceptible to the individual. 3 , 6 Alternatively, a significant acute injury, such as a muscle tear or a bone fracture, immediately obviates limb or organ function until function is restored. 6 When musculoskeletal injuries occur in the context of disease, the mechanisms of repair are compromised, and debilitating complications persist. For example, inflammatory diseases, such as diabetes, obesity, and autoimmune conditions, in addition to natural aging, result in the loss of muscle (sarcopenia) and bone (osteopenia) as a result of unresolved daily microinjuries. 5 , 7 , 8 , 9 , 10 Unresolved injuries or degenerative diseases of the musculoskeletal system cause poor range of motion, chronic pain, and loss of limb function, ultimately resulting in significant disability. 11 , 12 , 13 In the United States, musculoskeletal disease or injury affects 1 of every 2 people over the age of 18 and accounts for approximately two thirds of the 68,000 disease conditions listed in the International Classification of Diseases, 10th Revision (ICD‐10). 9 , 14 Therefore, both acute and chronic damage to the musculoskeletal system requires effective and timely repair to preserve function and quality of life.
2. COAGULATION IS CRITICAL FOR MUSCULOSKELETAL INTEGRITY AND REPAIR
The high physical demands of the musculoskeletal system require an abundant supply of energy. Consequently, musculoskeletal tissues are highly vascularized. 1 , 3 Regardless of the severity, injuries to musculoskeletal tissue result in the activation of coagulation and fibrinolytic factors that promote the formation and degradation of fibrin for the primary purpose of hemostasis. 3 , 5 , 15 However, discoveries over the past 2 decades clearly indicate that the roles of the coagulation and fibrinolytic systems in musculoskeletal injury extend beyond their canonical roles in hemostasis to include proper repair and regeneration of tissues. 3 , 4 , 16 This review summarizes current knowledge on the roles of fibrin and the fibrin‐dependent and ‐independent roles of plasmin in musculoskeletal repair and degeneration, highlighting the temporal‐spatial context of their functions and considerations for pharmacologic manipulation of each.
3. PLASMIN AND THE ACUTE‐PHASE RESPONSE
The enzyme plasmin, classically known for its ability to maintain blood flow by cleaving fibrin and breaking down intravascular clots or thrombi, is also essential for tissue repair through both fibrin‐dependent and ‐independent mechanisms. 17 , 18 , 19 , 20 , 21 Since the first observations of plasmin activity by John Hunter and Giovanni Morgagni in the late 1700s 22 , 23 and Albert Dastre in 1893, 22 , 24 the fibrinolytic properties of plasmin have been recognized as both dangerous and beneficial based on context. Consequently, plasmin has been inhibited by antifibrinolytic drugs to prevent blood loss during trauma or harnessed through administration of its activator, tissue‐type plasminogen activator (t‐PA) for thrombolysis in patients with ischemic stroke. 22 , 25 , 26 , 27 In recent decades, additional plasmin functions have been documented that extend beyond fibrinolysis and include promotion of angiogenesis, chemotaxis, hematopoiesis, and cellular differentiation, 28 , 29 , 30 , 31 , 32 revealing an essential role in musculoskeletal repair and maintenance. Importantly, because of these diverse functions, plasminogen activation requires tight regulation based on spatiotemporal context following injury.
Following an acute injury to musculoskeletal tissues, the body must rapidly respond to resolve bleeding, risk of infection, and tissue hypoxia. 3 , 15 , 33 To manage these afflictions, a systemic response known as the acute‐phase response (APR) is initiated. 34 , 35 The APR can be segregated into 2 phenotypically distinct phases: survival and repair. In the survival phase, coagulation system activation prevents bleeding and restores hemostasis. Simultaneously, an acute inflammatory response functions to prevent infection, effectively containing the initial damage 3 , 15 (Figure 1A). During survival APR, fibrin is deposited in both the intra‐ and extravascular spaces. 15 Intravascular fibrin stabilizes clots to prevent bleeding, and extravascular fibrin is deposited within injured tissues to form a temporary matrix that prevents pathogen invasion and absorbs strain. 3 , 15 Once the temporary or provisional fibrin matrix is in place and survival is ensured, the body transitions into the repair APR. Locally generated plasmin is critical during the repair phase to clear fibrin and initiate both the resolution of hypoxia and restoration of tissue function. 17 , 29 , 36 First, plasmin degrades deposited fibrin and stimulates a reparative inflammatory response to promote the removal of damaged and necrotic tissue. 37 , 38 Once the fibrin and damaged tissues are removed, plasmin activity continues in the damaged zone to promote angiogenesis or revascularization to help prevent sustained tissue hypoxia. 29 , 39 Additionally, the plasmin‐dependent release of growth factors contributes to remodeling and cellular regeneration, restoring function to the injured tissue 32 , 40 (Figure 1B). Effective tissue repair is dependent upon a coordinated sequence of fibrin deposition and plasmin activation, with a specific timing, location, and magnitude for each. Conversely, an imbalance of these elements in the musculoskeletal system can prevent repair following injury and drive degeneration of these tissues in disease conditions (Figure 2).
Figure 1.
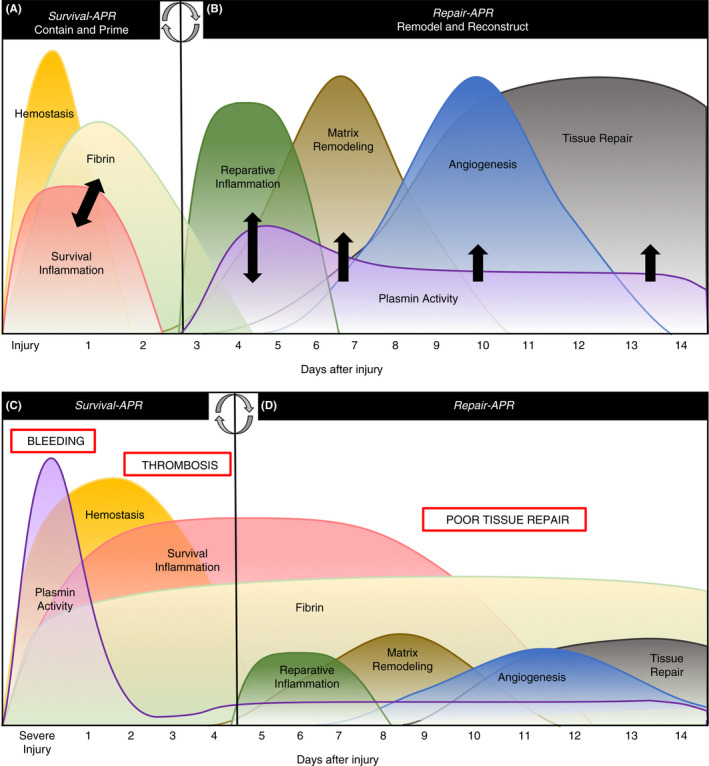
(A) Following an isolated injury to the musculoskeletal system, the survival phase of the acute‐phase response (APR) exists to contain the injury by activating coagulation and the innate immune system to prevent bleeding and infection, respectively. During survival APR, thrombin‐mediated fibrin formation and survival inflammation fuel one another (black arrows) to effectively contain damage and prime the tissue for reparative cells and proteases. (B) Once the injury is contained, the body enters the repair phase of the APR, during which plasmin is activated to remove fibrin deposited during survival, promote macrophage function, and to stimulate matrix remodeling and angiogenesis (black arrows). A normal APR resolves the injury without complications. (C) A severe or traumatic injury provokes a pathologic APR in which plasmin is activated during survival causing bleeding complications, or it is shut down, increasing risk of thrombosis and (D) provoking persistent fibrin deposition and poor tissue repair later in convalescence. Modified from Figure 1 of 3
Figure 2.

(A) Physiologic musculoskeletal health results in effective muscle and fracture repair (yellow arrows). (B) Pathologic musculoskeletal repair may result in poor fracture repair, including fracture nonunion (yellow arrows), and poor muscle repair, including bone formation in muscle (heterotopic ossification‐red arrows). Physiologic maintenance of musculoskeletal organs promotes healthy bone (C) and joints (E), while pathologic musculoskeletal maintenance and inflammation provoke degenerative disease, including significant bone loss (osteoporosis) (D) and arthritis (F). Images shown are from healthy and diseased mouse tissues
The diverse functions of plasmin make it both essential and dangerous, depending on the context. Many diseases pathologically alter plasmin’s functions, causing both acute and chronic complications due to an excess or lack of plasmin activity. 33 , 41 Excess plasmin activity, either locally or systemically, can exacerbate a plethora of disease conditions. For example, traumatic injuries and invasive surgeries provoke systemic plasmin activation during the survival phase of the APR, causing aggressive fibrinolysis at the site of injury and subsequent bleeding complications 25 , 42 , 43 , 44 (Figure 1C). Interestingly, more recent research in trauma has suggested that reduced fibrinolysis based on thromboelastography is associated with poor outcomes, including thrombosis and organ dysfunction. 43 , 44 However, it is unclear if this suggested change in fibrinolysis and possible change in plasmin activity is a causative factor in these outcomes or if it reflects the general dysfunction in inflammation, coagulation, and fibrinolysis that occurs following traumatic injuries (Figure 1D). Furthermore, diseases including infection, diabetes, cancer, and autoimmune disorders have reported pathologic changes in plasmin activity and/or function. 33 , 40 , 41 , 45 Therefore, while plasmin is essential for proper tissue repair, inappropriate plasmin activity can greatly exacerbate pathology in disease conditions.
4. PLASMIN AND FIBRIN IN SKELETAL REPAIR
Following a musculoskeletal injury, such as a fracture, thrombin is activated at the site of injury to form a fibrin and platelet sealant that both prevents blood loss and provides a temporary matrix to absorb the strain of the injury. 15 Neutrophils are recruited to the fracture, where they deposit neutrophil extracellular traps (NETs) that bolster the fibrin matrix against sterile and nonsterile pathology. 46 In addition to protecting against microbial invasion, neutrophils also function to promote to tissue repair by augmenting localized inflammation at the site of injury. 46 , 47 , 48 Following fibrin deposition and neutrophil activation, infiltrating macrophages present heterodimeric αMβ2 integrin on their surface that stimulates an acute inflammatory response upon binding fibrin. 49 αMβ2 integrin activation stimulates the production of inflammatory cytokines, including interleukin (IL)‐1β, IL‐6, and tumor necrosis factor‐α (TNF‐α), promoting further recruitment and activation of proinflammatory, antimicrobial (M1) macrophages at the site of the injury. 49 , 50 , 51 Plasminogen bound to both fibrin at the site of the fracture and immobilized on the cell surface of infiltrating macrophages is activated to plasmin. 3 , 19 Plasmin facilitates the transition to the repair phase by removing provisional fibrin matrices and stimulating reparative, anti‐inflammatory macrophage function. 37 , 52 , 53 While its role in NET removal remains unclear, plasmin is present in NET matrices and degrades proteins in both NETs and protein aggregates presented on the surface of necrotic cells. 48 , 54 , 55 , 56 Plasmin has been shown to reprogram macrophages from a proinflammatory (M1) phenotype, to an anti‐inflammatory, reparative (M2) phenotype 57 , 58 and to stimulate chemotaxis and efferocytosis 37 , 59 in these macrophages. A number of reports have implicated both the interaction of plasmin(ogen) with the PlgRKT receptor and α‐enolase and plasmin‐mediated activation of the protease‐activated receptor‐1 (PAR1) as being critical for plasmin‐mediated macrophage function. 28 , 58 , 59 , 60 Specifically, plasmin activation of PAR1 and coreceptors, including integrin α9β1, on macrophages initiates extracellular signal‐related kinase (ERK1/2) signaling and expression of chemokine (C‐C motif) ligand 2, augmenting phagocytosis and macrophage migration at the site of tissue injury. 28 , 61 Plasminogen binding to PlgRKT facilitates activation of plasmin on the surface of macrophages, leading to activation of pro‐MMP9 to promote macrophage migration. 62 In contrast, plasmin increases phagocytosis but inhibits production of inflammatory cytokines and cell migration in dendritic cells, providing an alternative proreparative, anti‐inflammatory effect of plasmin on phagocytic cells. 63 However, one recent study demonstrated that fibrinolysis itself enhances macrophage ingress and egress by preventing macrophage tethering to fibrin through αMβ2 binding. 64 In addition, the plasminogen activators t‐PA and urokinase plasminogen activator (u‐PA) bind to the annexin A2/S100A10 heterotetrameric complex and the u‐PA receptor (u‐PAR), respectively, to facilitate activation of plasmin on the surface of macrophages and damaged cells. 65 , 66 , 67 Reparative macrophages remove necrotic tissue and fibrin degradation products, 68 , 69 while facilitating the migration of mesenchymal stem cells. 70 As plasmin and macrophages remove the fibrin and dead or necrotic tissue, plasmin activates pro‐MMP9 and vascular endothelial growth factor A (VEGF‐A) at the site of the fracture to stimulate matrix remodeling and angiogenesis, respectively. 29 , 39 With the damaged tissue cleared of fibrin and remodeled, angiogenesis restores the vascularity disrupted by the fracture, allowing bone to form around the fracture site. Within weeks to months, the newly formed bone is remodeled into strong, functional bone capable of withstanding significant force 3 , 5 (Figure 2A). Therefore, plasmin provokes a reparative response from macrophages through fibrin‐dependent and ‐independent mechanisms.
Animal models of fracture have revealed the specific roles for both coagulation and fibrinolysis in fracture repair. 5 , 19 While it was widely presumed that fibrin is essential for fracture repair, 71 fibrinogen‐deficient (FBG‐/‐) mice maintained the ability to heal a fracture normally, despite significantly more fracture‐related blood loss. In contrast, plasminogen‐deficient (PLG−/−) mice, which are unable to clear fibrin from the injury site, had poor fracture repair in both drill hole and femur fracture murine models of bone injury. In the drill hole model, PLG‐/‐ mice had reduced cartilage matrix and bone formation at the site of injury. 21 Similarly, in a transverse femur fracture model with significantly more bone and vascular disruption than the drill hole model, PLG‐/‐ mice had little to no bridging of vascularity across the fracture callus. 19 Without union of vascularity across a fracture callus, the bone does not unite, referred to as a nonunion, or remodel 3 , 5 , 16 , 19 (Figure 2B). Collectively, the phenotypes displayed by fibrin and plasmin‐deficient animals after injury confirm the temporal nature of fibrin and plasmin’s respective roles in healing: Fibrin is critical for the initial containment of the injury through hemostasis, while plasmin is essential later during bone repair. These studies, however, are incapable of resolving the fibrin‐dependent and fibrin‐independent nature of plasmin’s role in skeletal repair. To answer this question, our lab has demonstrated that fibrinogen deficiency partially restores the normal sequence of fracture repair in PLG−/− mice. Specifically, fibrinogen deficiency in PLG−/− mice resulted in increased angiogenesis and the restoration of bone union at the site of fracture. 19 These data are consistent with studies suggesting fibrin‐independent roles for plasmin in fracture repair, but also clearly demonstrate that a principal role of plasmin is the removal of fibrin.
Studies segregating t‐PA– and u‐PA–dependent plasminogen activation suggest that their roles in fracture repair may be nonredundant. Fractures in u‐PA–deficient (u‐PA−/−) mice exhibit poor remodeling and large callus formation due to poor macrophage migration and vascular bridging at the site of the fracture, 53 , 72 similar to PLG−/− animals. t‐PA–deficient (t‐PA−/−) mice, on the other hand, exhibited only a delay in fracture repair. This delay was attributed to reduced proliferation of the bone‐forming cells, osteoblasts, due to a lack of plasmin‐mediated activation of the ERK1/2 pathway. 73 Moreover, t‐PA has been found to induce hypoxia‐inducible factor 1α and VEGF‐A activity at the fracture site, increasing the rate of neovascularization during repair. 73 No such function has been described for u‐PA.
5. PLASMIN AND FIBRIN IN MUSCLE REPAIR
Plasmin is also critical for proper muscle repair and regeneration. 20 , 32 , 74 Consistent with a normal APR, fibrin is deposited during survival to contain the zone of injury, and it is removed during the repair phase for regeneration to occur. 20 , 32 Work from the Muñoz‐Cánoves lab demonstrated that plasmin activity peaks in injured muscle within 3‐5 days of the injury to remove fibrin and necrotic tissue and to regenerate new muscle. 32 , 75 Activated plasmin removes fibrin and signals through M2 macrophages to promote the removal of necrotic tissue. 57 , 58 , 68 Once the provisional fibrin matrix and necrotic debris is removed, plasmin proteolytically activates growth factors and proregenerative factors (ie, VEGF‐A, pro‐MMPs, etc) released from surrounding, regenerating muscle cells to remodel and revascularize the zone of injury. 29 , 39 In the presence of adequate blood supply and an acute, localized inflammatory response, cells surrounding the injury regenerate, and satellite stem cells differentiate into functional myotubes to replace the area of damage. 20 , 32 As in bone repair, t‐PA and u‐PA do not function interchangeably in muscle repair. Studies of plasmin activity in both cardiotoxin and freeze‐crush models of muscle injury have demonstrated that u‐PA activity increases in the muscle following injury, while there is little change in t‐PA activity. 32 , 75 Furthermore, in vivo muscle repair and in vitro myogenesis are dependent on u‐PA– but not t‐PA–mediated plasmin activation. 75
A failure of coordinated repair in muscle results in a persistent state of tissue strain, hypoxia, and inflammation. 12 These chronic complications, including the development of muscle fibrosis, muscle calcification, and sarcopenia, can cause significant pain and permanent loss of muscle function in patients. 12 , 76 , 77 Animal studies have demonstrated that a plasmin deficiency causes ineffective macrophage infiltration and function, persistent fibrin deposition, and chronic inflammation of injured tissues. 57 , 58 , 62 , 64 , 78 In a muscle injury specifically, the absence of plasmin results in fibrosis, skeletal muscle calcification, and bone formation within injured muscle, better known as heterotopic ossification (HO) (Figure 2B). 20 , 75 As little as a 50% deficiency in plasminogen and plasmin activity is sufficient to drive calcification of skeletal muscle in mice following injury and the development of HO. 20 These studies suggest the possibility that deficiencies in plasmin activity routinely encountered in the clinic, such as those observed in trauma patients, may be sufficient to drive pathologic repair of injured muscle. 19 , 20 These data establish a paradox for plasmin’s role in musculoskeletal repair. The role of plasmin in mineralization appears to be tissue specific: Within the context of bone, plasmin is essential for bone formation, 19 , 21 but in skeletal muscle, plasmin activity prevents bone formation (HO). 20 Interestingly, unlike in bone repair, fibrin(ogen) deficiency improves macrophage migration and prevents fibrosis in injured muscle, but it is insufficient to completely restore muscle repair in PLG−/− mice. 20 Therefore, plasmin mediates muscle repair through both fibrin‐dependent and ‐independent mechanisms.
6. MUSCULOSKELETAL DEGENERATION: A “CHRONIC WOUND”
Like the repair of an acute injury, maintenance of musculoskeletal tissue function throughout life requires a delicate balance between fibrin and plasmin. Healthy bones and joints should not contain a significant amount of fibrin, given that the tissue is not damaged and therefore does not require hemostasis. In certain inflammatory diseases, such as diabetes and autoimmune conditions, and during aging, the spatiotemporal regulation of fibrin formation and plasmin activation is often disrupted. 10 , 45 , 50 , 79 In conditions of poor plasmin activity or excess activation of coagulation, fibrin is deposited throughout tissues, provoking localized survival APR inflammation and constant tissue remodeling. 7 , 45 , 50 Consequently, daily microinjuries provoke a persistent cycle of the APR that ultimately leads to musculoskeletal degeneration rather than repair (Figure 3).
Figure 3.
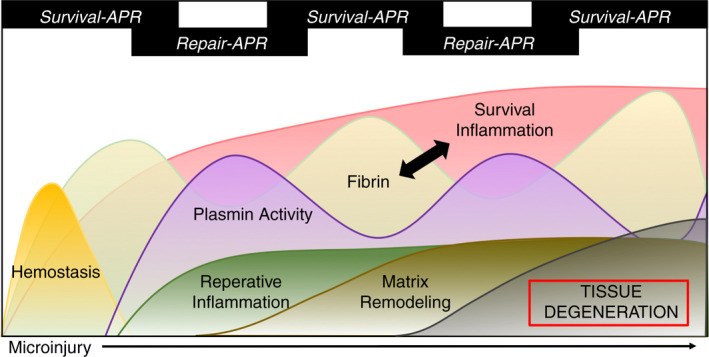
In chronic inflammatory conditions and aging, microinjuries sustained during daily movement trigger a persistent APR cycle in which fibrin deposition and plasmin activation are dysregulated in musculoskeletal tissues. The consequence of this cyclical acute‐phase response (APR) is chronic inflammation, inappropriate tissue remodeling, and ultimately, degeneration of the musculoskeletal tissues. Recurring fibrin deposition and inflammation positively feedback upon one another (black arrow), furthering tissue degeneration
7. FIBRIN ACCUMULATION IN BONE DEGENERATION
Osteoporosis is the debilitating loss of bone resulting in significant costs in both health care expenses and quality of life (Figure 2D). In the United States, osteoporosis afflicts over 10 million individuals, increasing their risk of fractures and loss of mobility. 7 , 14 Age, sex, and body mass index are all factors in the risk of developing osteoporosis. 7 , 9 Inflammation and uncontrolled bone resorption are current therapeutic targets for this disease, but recent data suggest that fibrin may be an initiator of these pathologies, making it a more efficacious target. 50 Studies in PLG‐/‐‐deficient mice have demonstrated that the absence of plasmin activity drives the deposition of fibrin in bone. 50 Further, fibrin in bone activates resident macrophages through binding the αMβ2 integrin, stimulating local production of IL‐6, IL‐1β, and TNF‐α. These inflammatory cytokines work synergistically with one remodeling protein,receptor activator of nuclear factor κB ligand (RANKL) to stimulate the proliferation and differentiation of bone‐resorbing osteoclasts. 49 , 50 The net effect of this is increased bone resorption, resulting in decreased bone mass (osteopenia) and bone degeneration (osteoporosis). Consequently, the genetic ablation of fibrinogen (FBG−/−) or the αMβ2 binding motif on fibrinogen (Fib‐γ390‐396A) effectively prevented osteoporosis in PLG−/− mice, demonstrating a direct role for persistent fibrin deposition in bone disease progression. Mechanistically, these studies determined that engagement of macrophage‐1 antigen on fibrin potentiates the fusion of monocytes to form osteoclasts in the presence of RANKL. 50 These studies indicate that persistent fibrin in bone may potentiate osteoporosis by driving osteoclastogenesis, thereby uncoupling the bone remodeling unit favoring a catabolic phenotype. Although early clinical studies have identified a relationship to circulating fibrinogen and osteoporosis, 80 further studies are required to determine if targeting fibrin(ogen) is a viable therapeutic approach to prevent osteoporosis.
8. COAGULATION AND FIBRINOLYSIS IN DEGENERATIVE JOINT DISEASE
In addition to loss of bone, chronic inflammatory conditions cause degeneration in joints by hijacking thrombin and plasmin activity. Since the 1960s, inappropriate fibrin formation and plasmin activity have been detected in the synovial fluid of arthritic joints. 79 , 81 , 82 , 83 Affecting approximately 1.3 million individuals in the United States, rheumatoid arthritis is an autoimmune disease marked by a persistent APR: localized inflammation, fibrin formation, cellular infiltration, and plasmin activation within the synovium (Figure 3), which eventually leads to cartilage and bone degradation 8 , 84 (Figure 2F). Similar to osteoporosis, IL‐6, IL‐1β, and TNF‐α have all been implicated in rheumatoid arthritis (RA), indicating anticytokine monoclonal antibodies for treatment. 84 , 85 In a collagen‐induced arthritis (CIA) murine model, fibrinogen deficiency reduced the local inflammatory response to the collagen injection and conferred resistance to RA in the paw joints, suggesting that fibrin is a pathologic driver in this disease. In the same model, Fib‐γ390‐396A mice expressing fibrinogen unable to bind to αMβ2 on the macrophage were resistant to CIA. 51 Furthermore, the prevention of fibrin crosslinking by removing or inhibiting transglutaminase factor XIII is sufficient to reduce local inflammation and bone erosion in CIA. 86 Interestingly, another study using the CIA model demonstrated the seemingly contradictory finding that a plasminogen deficiency was found to protect mice from the development of RA in the paw joints. 87 This was further investigated in a more clinically relevant model of RA: transgenic mouse overexpressing human TNF‐α (Tg197), in which mice spontaneously develop the disease. Work in Tg197 mice demonstrated that while plasminogen deficiency exacerbated disease progression in the paw joints, it reduced disease progression in the knee joints, demonstrating that plasmin has both pro‐ and antiarthritic functions based on the specific joint tissue and the model of disease. 88 It was found that in Tg197 mice, fibrin drives the disease progression of RA in the paw joints, while in the knee joints, plasmin activation of MMP9 plays a significant role in joint degeneration. Furthermore, combined plasmin(ogen) and fibrin(ogen) deficiency negated both the pro‐ and antiarthritic roles of plasmin in RA. 88 Collectively, these data suggest that plasmin and fibrin may work cooperatively to promote disease progression through different mechanisms.
More recent research implicates a specific plasminogen activator, receptor, and cell type in RA disease progression. Previous studies in synovial samples from RA patients have demonstrated that cells present in synovial fluid, including fibroblasts and macrophages, overexpress u‐PAR. 89 Further studies in a CIA model of RA demonstrated that a deficiency or inhibition of macrophages, u‐PA, or u‐PAR conferred resistance to CIA. 90 , 91 To investigate if the pathologic effect of plasmin in RA is mediated by u‐PAR on the surface of macrophages, bone marrow transplants from u‐PAR–deficient mice into wild‐type mice were employed. Transplant of u‐PAR–deficient hematopoietic cells into wild‐type mice prevented the development of arthritis in a CIA model. 91 Therefore, u‐PA–u‐PAR–mediated activation of plasmin on the surface of hematopoietic cells, including macrophages, is a possible mechanism for inappropriate plasmin activation in arthritic joints. In contrast, a t‐PA deficiency exacerbated arthritis in the same murine model. 92 These findings of plasminogen activator specificity in arthritis suggest that t‐PA drives fibrinolysis in the joints to reduce inflammation, while u‐PA activates plasmin on the surface of macrophages and fibroblasts via u‐PAR, propagating inflammation and tissue remodeling. Interestingly, patients with hereditary hemophilia A and B often develop bleeding within the joints, which leads to persistent inflammation, cartilage remodeling, and joint degeneration. 93 , 94 Recent research from the Mosnier lab has demonstrated that insufficient thrombin‐activated fibrinolytic inhibitor (TAFI) activation in factor VIII−/− mice results in unchecked plasmin activation in hemophilic joints. Similarly to the joint pathology observed in RA, unbridled u‐PA–mediated plasmin generation, secondary to defective TAFI activation within joints, was found to drive hemophilic joint bleeds and ensure arthropathy. 94 New research is investigating the possible therapeutic use of u‐PA inhibitors to treat joint degeneration, 95 but it should be applied with caution, as u‐PA also plays a critical role in musculoskeletal repair. Research from the Hamilton laboratory has demonstrated that while u‐PA deficiency protects against CIA, if there is a soft‐tissue injury adjacent to the joint, a u‐PA deficiency worsens local inflammation, and arthritis develops. 96 , 97 While the fibrinolytic function of plasmin prevents fibrin‐mediated inflammation in joints, chronic plasmin‐mediated macrophage activation and matrix remodeling in joints drives cartilage degeneration and bone erosion.
9. FUTURE PERSPECTIVES
Plasminogen activation has been implicated in many disease states, including musculoskeletal disease, and future studies in this field may identify therapeutic targets within the coagulation and fibrinolytic systems to diagnose and treat diseases beyond the vascular system. Specifically, there is a great clinical need for improved therapeutics to treat musculoskeletal repair problems and degenerative diseases, and the pharmacologic manipulations of plasmin and/or fibrin may present effective treatment options. Potent amiloride‐derived u‐PA inhibitors developed in the Ranson and Kelso laboratories show promising potential for the treatment of u‐PA–mediated pathologies, including joint degeneration, 95 , 98 but further studies are required to determine optimal use without affecting u‐PA–mediated tissue repair. Additionally, novel inhibitors of plasminogen activator inhibitor‐1 (PAI‐1) may also provide therapeutic benefit if dosed within the appropriate spatiotemporal context to enhance plasmin activation. 99 Likewise, the pathologic effects of persistent fibrin deposition in musculoskeletal repair and degeneration are clear, but anticoagulant or fibrin‐targeting drugs carry the risk of bleeding side effects 100 and may not have efficacy in extravascular compartments.
Because macrophages can have both reparative and pathologic roles within the musculoskeletal system, they may represent another target for musculoskeletal disease. Recent research in cancer, cardiovascular disease, and inflammatory conditions have identified macrophage polarization as a possible therapeutic target for treatment of these diseases. 101 , 102 Because much of plasmin and fibrin’s respective roles within the musculoskeletal system are mediated or amplified by macrophages, and alternative approaches may target receptors for plasmin(ogen), plasminogen activators (ie, u‐PAR), or fibrin (ie, αMβ2) on the surface of macrophages without dramatically affecting global plasmin(ogen) activation or hemostasis. While macrophages have long been associated with tissue repair mechanisms, new research has elucidated diverse functions for neutrophils in tissue repair and degeneration. 46 , 47 Prolonged tissue inflammation mediated by neutrophils can have deleterious effects on repair, but acute inflammation instigated by neutrophil cytokines and NETs provoke a reparative response. 47 Investigations on the roles of neutrophils in thrombosis, inflammation, and tissue repair suggest that NETs within injured tissue may also stimulate tissue repair through macrophage function. 46 , 48 Collectively, the plasminogen activation system and downstream effector cells present promising therapeutic targets for a range of conditions beyond vascular disease, including the treatment of musculoskeletal disease.
10. ISTH 2019 MELBOURNE REPORT
State‐of‐the‐art abstracts presented at ISTH 2019 Congress presented both new insights and techniques in the study of coagulation and fibrinolysis in physiology and disease. Two separate reports were presented that focused on the contribution of platelet‐localized activation of TAFI to the regulation of fibrinolysis. Suzuki and colleagues presented data indicating that supplementation of normal plasma with platelets prolonged in vitro clot lysis times in a concentration‐dependent manner. 103 Plasminogen accumulation at the lysis front was accelerated with addition of either a thrombomodulin inhibitory antibody or a direct TAFI inhibitor (TAFIaI). Finally, these authors showed that addition of TAFIaI to whole blood clots formed under flow conditions in a capillary chamber prolonged occlusion time. 103 Kim and colleagues 104 documented that activation of human or mouse platelets with thrombin resulted in plasminogen and t‐PA binding to the platelet surface in fibrin‐dependent manner. The authors presented additional data indicating TAFIa activated significantly attenuated t‐PA and plasminogen binding to platelets and subsequent platelet‐associated plasmin generation. Finally, these authors had data supporting the innovative hypothesis that a thrombin‐dependent modification of fibrin other than fibrinopeptide release was required for plasminogen binding. Mass spectrometry analyses further suggested lysine 556 on the α‐chain of fibrinogen as a thrombin‐generated C‐terminal lysine, which is a target for plasminogen binding and TAFIa‐mediated release. 104 Collectively, these studies provided novel insights into the platelet‐fibrinolysis axis.
The molecular basis of compromised fibrinolytic function in obesity was also a topic of multiple abstracts presented. Zheng and colleagues 105 provided data identifying a novel regulatory network in hepatocytes that controls the expression and release of t‐PA and PAI‐1. These authors illustrated that mRNA levels of both t‐PA and PAI‐1 increase in obese compared to lean mice, but that the increase of PAI‐1 was significantly greater. The authors identified what they described as a compensatory pathway controlling fibrinolysis in obesity by which lowering PAI‐1 in obesity reduced t‐PA and PAI‐1 treatment induced hepatocyte t‐PA mRNA production. Using small interfering RNA gene‐silencing strategies, the authors identified that the regulatory pathway was controlled by PAI‐1 activation of the transcription factor CAMP responsive element‐binding protein 1 that drives t‐PA expression, whereas expression is counterbalanced by the transcriptional repressor dachshund homolog 1. 105 Miszta and coworkers 106 developed and validated a novel plasmin generation (PG) assay. The premise of the assay is akin to that of the established thrombin generation (TG) assay whereby plasmin activity is measured by conversion of a fluorogenic substrate to calculate a plasmin generation curve over time. Parameters may be quantified including generation rate, lagtime, peak, time to peak, and endogenous plasmin potential. The assay was shown to be dependent on fibrin formation and sensitive to t‐PA, PAI‐1, and α2‐antiplasmin. Using the assay, the authors documented that plasma from mice fed a high‐fat diet displayed delayed plasmin generation lagtime, time to peak, and reduced rate relative to control diet–fed mice. The authors documented that contrary to expectations, the delayed plasmin generation was not due to elevated PAI‐1 levels. The authors performed analyses integrating PG and TG to show that the effects of obesity on TG and PG resulted in normal fibrin formation kinetics but delayed clot lysis. 106
A novel targeting strategy to drive plasmin generation and thus thrombolysis in patients with thrombotic thrombocytopenic purpura was presented by de Maat and colleagues. 107 The basis of the approach was generation of a “mini”‐urokinase catalytic domain fused to a nanobodies targeting either von Willebrand factor (VWF) or glycoprotein 1b. The fusion proteins were able to bind to the specified target with high affinity. The VWF‐targeted fusion protein was found to enhance the destruction of in vitro platelet microthrombi at a 7‐fold faster rate than nontargeting constructs. Further, using flow‐based assays, these authors showed that the targeting constructs accelerated the removal of VWF‐platelet strings from endothelial cell surfaces. 107
11. CONCLUSION
Proper musculoskeletal repair is dependent upon coordinated activation of coagulation and fibrinolysis as part of the survival and repair phases of the APR, respectively. When a specific tissue or entire system gets stuck in an unresolved, perpetual APR, it drives chronic inflammation and tissue degeneration. The delicate interplay between coagulation and fibrinolysis in musculoskeletal health is dependent upon context, which explains why different models have found seemingly conflicting data on their roles in pathology and repair. Because plasmin is essential for repair of muscle and bone following injury, plasmin‐enhancing therapeutics may be an effective strategy to improve musculoskeletal healing in patients with repair problems, but timing, location, and level of plasmin(ogen) activation remain key factors that differentiate plasmin’s roles in repair from its pathologic roles in degeneration. As such, therapeutically targeting plasmin or u‐PA may prevent its proarthritic roles, but it should be considered that inhibition of this system might have deleterious effects on tissue repair after injury. The difference between the beneficial and the pathologic effects of plasmin and fibrin in musculoskeletal health is the spatiotemporal context of each, and therefore therapeutic efforts to target either plasmin or fibrin must take into consideration the precise etiology of disease, stage of disease, and anatomic location of disease to minimize off‐target effects.
AUTHOR CONTRIBUTIONS
This review article was drafted and revised by BHYG with critical revisions and additions made by MTD, and critical revisions made by SNML, MJF, and JGS. All authors approved the final version of this paper.
ACKNOWLEDGMENTS
The authors thank Zack Backstrom, Satoru Egawa, Court Reese, and previous members of the Schoenecker laboratory for their ongoing support.
Gibson BHY, Duvernay MT, Moore‐Lotridge SN, Flick MJ, Schoenecker JG. Plasminogen activation in the musculoskeletal acute phase response: Injury, repair, and disease. Res Pract Thromb Haemost. 2020;4:469–480. 10.1002/rth2.12355
Handling Editor: Yotis Senis
Funding information
This work was supported by the Vanderbilt University Medical Center Department of Orthopaedics and Rehabilitation (JGS), NIGMS R01GM126062 (JGS), Department of Defense W81XWH1810536 (JGS), The Caitlin Lovejoy Fund (JGS) NHLBI predoctoral fellowship F31HL149340 (BHYG), NIAMS training fellowship T32AR059030‐06A1 (BHYG), and NIDDK R01DK112778 (MJF).
Contributor Information
Breanne H.Y. Gibson, @breannegibson10
Matthew T. Duvernay, @bigplatelets.
Stephanie N. Moore‐Lotridge, @Smoorelotridge.
Matthew J. Flick, @fib390_396A.
Jonathan G. Schoenecker, Email: jon.schoenecker@vumc.org, @jonshenmd.
REFERENCES
- 1. Murphy AC, Muldoon SF, Baker D, Lastowka A, Bennett B, Yang M, et al. Structure, function, and control of the human musculoskeletal network. PloS Biol [Internet]. 2018;16(1):1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lowe JS, Anderson PG. Musculoskeletal system In: Stevens and Lowe’s Human Histology, 4th ed, 2015. Mosby Ltd, Maryland Heights, MO, USA. p. 239–62. [Google Scholar]
- 3. Baker CE, Moore‐Lotridge SN, Hysong AA, Posey SL, Robinette JP, Blum DM, et al. Bone fracture acute phase response—a unifying theory of fracture repair: clinical and scientific implications. Clin Rev Bone Miner Metab. 2018;16(4):142–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stocum DL. Regeneration of musculoskeletal tissues In: Regenerative Biology and Medicine, 2nd ed, 2012. Academic Press, Waltham, MA, USA; p. 127–60. [Google Scholar]
- 5. Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol. 2015;11(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ellis A, Taylor T. Trauma and the musculoskeletal system In: The Musculoskeletal System, 2nd ed, 2010. Churchill Livingston, London, UK; p. 137–49. [Google Scholar]
- 7. Wright N, Looker A, Saag K, Curtis J, Delzel E, Randall S, et al. The recent prevalence of osteoporosis and low bone mass in the United States. J Bone Miner Res. 2014;29(11):2520–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hunter TM, Boytsov NN, Zhang X, Schroeder K, Michaud K, Araujo AB. Prevalence of rheumatoid arthritis in the United States adult population in healthcare claims databases, 2004–2014. Rheumatol Int. 2017;37(9):1551–7. [DOI] [PubMed] [Google Scholar]
- 9. Andersson G, AAOO S. United States Bone and Joint Initiative. Burden of Musculoskeletal Diseases in the United States (BMUS). Rosemont, IL: 2014. [Google Scholar]
- 10. Sundararaghavan V, Mazur MM, Evans B, Liu J, Ebraheim NA. Diabetes and bone health: latest evidence and clinical implications. Ther Adv Musculoskelet Dis. 2017;9(3):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson LC, Pacchiana PD. Common complications of fracture repair. Clin Tech Small Anim Pract. 2004;19(3):168–79. [DOI] [PubMed] [Google Scholar]
- 12. Alessandrino F, Balconi G. Complications of muscle injuries. J Ultrasound. 2013;16(4):215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herrmann LG, Caldwell JA. Diagnosis and treatment of post‐traumatic osteoporosis. Am J Surg. 1941;51(3):630–40. [Google Scholar]
- 14. American Academy of Orthopaedic Surgeons . “One in two Americans have a musculoskeletal condition: New report outlines the prevalence, scope, cost and projected growth of musculoskeletal disorders in the U.S..” ScienceDaily. ScienceDaily, 1 March 2016. http://www.sciencedaily.com/releases/2016/03/160301114116.htm
- 15. Reikeras O, Borgen P. Activation of markers of inflammation, coagulation and fibrinolysis in musculoskeletal trauma. PLoS ONE. 2014;9(11):9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Keefe RO. Fibrinolysis as a Target to Enhance Fracture Healing. N Engl J Med. 2015;373(18):1776–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost. 2005;93(4):647–54. [DOI] [PubMed] [Google Scholar]
- 18. Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2(3):287–92. [DOI] [PubMed] [Google Scholar]
- 19. Yuasa M, Mignemi NA, Nyman JS, Duvall CL, Schwartz HS, Okawa A, et al. Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. J Clin Invest. 2015;125(8):3117–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mignemi NA, Yuasa M, Baker CE, Moore SN, Ihejirika RC, Oelsner WK, et al. Plasmin prevents dystrophic calcification after muscle injury. J Bone Miner Res. 2017;32(2):294–308. [DOI] [PubMed] [Google Scholar]
- 21. Kawao N, Tamura Y, Okumoto K, Yano M, Okada K, Matsuo O, et al. Plasminogen plays a crucial role in bone repair. J Bone Miner Res. 2013;28(7):1561–74. [DOI] [PubMed] [Google Scholar]
- 22. Cliffton EE. Physiological mechanisms of fibrinolysis. Acta Haematol. 1958;20:76–85. [DOI] [PubMed] [Google Scholar]
- 23. Flute PT. Coagulation and fibrinolysis after injury. J Clin Pathol. 1970;S3–4(1):102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Misirlioglu YI, Lillehei CW. The physiologic enzymes activating profibrinolysin to fibrinolysin, and the substances which inhibit activation: localizations. Clinical importance and pathogenesis. Angiology. 1962;13(5):185–207. [DOI] [PubMed] [Google Scholar]
- 25. Innes D, Sevitt S. Coagulation and fibrinolysis in injured patients. J Clin Pathol [Internet]. 1964;17:1–13. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=480663&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chapin J, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015;29(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macfarlane R, Biggs R. Fibrinolysis: its mechanism and significance. Blood. 1948;3(10):1167–87. [PubMed] [Google Scholar]
- 28. Carmo AAF, Costa BRC, Vago JP, de Oliveira LC, Tavares LP, Nogueira CRC, et al. Plasmin induces in vivo monocyte recruitment through protease‐activated receptor‐1–, MEK/ERK‐, and CCR2‐mediated signaling. J Immunol. 2014;193(7):3654–63. [DOI] [PubMed] [Google Scholar]
- 29. Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, et al. Plasmin modulates vascular endothelial growth factor‐A‐mediated angiogenesis during wound repair. Am J Pathol [Internet]. 2006;168(2):670–84. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002944010621268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, et al. Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood. 2012;119(24):5879–87. [DOI] [PubMed] [Google Scholar]
- 31. Carmeliet P, Collen D. Development and disease in proteinase‐deficient mice: role of the plasminogen, matrix metalloproteinase and coagulation system. Thromb Res. 1998;91(6):255–85. [DOI] [PubMed] [Google Scholar]
- 32. Suelves M, López‐Alemany R, Lluís F, Aniorte G, Serrano E, Parra M, et al. Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood. 2002;99(8):2835–44. [DOI] [PubMed] [Google Scholar]
- 33. Benvenuti M, An T, Amaro E, Lovejoy S, Mencio G, Martus J, et al. Musculoskeletal infection provoked acute phase response in children. Orthop Clin North Am. 2017;48(2):181–97. [DOI] [PubMed] [Google Scholar]
- 34. Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15(2):74–80. Available from: http://www.sciencedirect.com/science/article/pii/0167569994901376%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/7512342%5Cnhttp://www.sciencedirect.com/science/article/pii/0167569994901376%255Cnhttp://www.ncbi.nlm.nih.gov/pubmed/7512342. [DOI] [PubMed] [Google Scholar]
- 35. Kushner I. The phenomenon of the acute phase response. Ann N Y Acad Sci [Internet]. 1982;389:39–48. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=Pub-Med&dopt=Citation&list_uids=7046585. [DOI] [PubMed] [Google Scholar]
- 36. Sulniute R, Shen Y, Guo Y, Fallah M, Ahlskog N, Ny L, et al. Plasminogen is a critical regulator of cutaneous wound healing. Thromb Haemost. 2016;115(05):1001–9. [DOI] [PubMed] [Google Scholar]
- 37. Das R, Ganapathy S, Settle M, Plow EF. Plasminogen promotes macrophage phagocytosis in mice. Blood. 2014;124(5):679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Syrovets T, Lunov O, Simmet T. Plasmin as a proinflammatory cell activator. J Leukoc Biol [Internet]. 2012;92(3):509–19. [DOI] [PubMed] [Google Scholar]
- 39. Pepper MS. Role of the matrix metalloproteinase and plasminogen activator‐plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21(7):1104–17. [DOI] [PubMed] [Google Scholar]
- 40. Deryugina EI. Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol; 2012. p. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77(6):811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor JR, Fox EE, Holcomb JB, Rizoli S, Inaba K, Schreiber MA, et al. The hyperfibrinolytic phenotype is the most lethal and resource intense presentation of fibrinolysis in massive transfusion patients. J Trauma Acute Care Surg. 2018;84(1):25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oelsner WK, Engstrom SM, Benvenuti MA, An TJ, Jacobson RA, Polkowski GG, et al. Characterizing the acute phase response in healthy patients following total joint arthroplasty: predictable and consistent. J Arthroplasty. 2017;32(1):309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schöchl H, Voelckel W, Maegele M, Solomon C. Trauma‐associated hyperfibrinolysis. Hamostaseologie. 2012;32(1):22–7. [DOI] [PubMed] [Google Scholar]
- 45. Kearney K, Tomlinson D, Smith K, Ajjan R. Hypofibrinolysis in diabetes: a therapeutic target for the reduction of cardiovascular risk. Cardiovasc Diabetol. 2017;16(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, et al. Neutrophils orchestrate post‐myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38(3):187–97. [DOI] [PubMed] [Google Scholar]
- 47. Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371(3):531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alpha M beta 2/Mac‐1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113(11):1596–606. Available from: http://www.jci.org/articles/view/20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cole H, Ohba T, Nyman JS, Hirotaka H, Cates JMM, Flick MJ, et al. Fibrin accumulation secondary to loss of plasmin‐mediated fibrinolysis drives inflammatory osteoporosis in mice. Arthritis Rheumatol. 2014;66(8):2222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin αMβ2 binding motif. J Clin Invest. 2007;117(11):3224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li Q, Laumonnier Y, Syrovets T, Simmet T. Plasmin triggers cytokine induction in human monocyte‐derived macrophages. Arterioscler Thromb Vasc Biol. 2007;27(6):1383–9. [DOI] [PubMed] [Google Scholar]
- 53. Kawao N, Tamura Y, Horiuchi Y, Okumoto K, Yano M, Okada K, et al. The tissue fibrinolytic system contributes to the induction of macrophage function and CCL3 during bone repair in mice. PLoS ONE. 2015;10(4):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samson AL, Knaupp AS, Sashindranath M, Borg RJ, Au AEL, Cops EJ, et al. Nucleocytoplasmic coagulation: an injury‐induced aggregation event that disulfide crosslinks proteins and facilitates their removal by plasmin. Cell Rep. 2012;2(4):889–901. [DOI] [PubMed] [Google Scholar]
- 55. Lim CH, Adav SS, Sze SK, Choong YK, Saravanan R, Schmidtchen A. Thrombin and plasmin alter the proteome of neutrophil extracellular traps. Front Immunol. 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Samson AL, Borg RJ, Niego B, Wong CHY, Crack PJ, Tang Y, et al. A nonfibrin macromolecular cofactor for tPA‐mediated plasmin generation following cellular injury. Blood. 2009;114(9):1937–46. [DOI] [PubMed] [Google Scholar]
- 57. Sugimoto MA, Lu A, Ribeiro C, Costa BRC, Vago JP, Lima M, et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood. 2017;129(21):2896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vago JP, Sugimoto MA, Lima KM, Negreiros‐Lima GL, Baik N, Teixeira MM, et al. Plasminogen and the plasminogen receptor, PLG‐RKT, regulate macrophage phenotypic, and functional changes. Front Immunol. 2019;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA. Regulation of macrophage migration by a novel plasminogen receptor Plg‐R KT. Blood. 2011;118(20):5622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Díaz‐Ramos À, Roig‐Borrellas A, García‐Melero A, López‐Alemany R. α‐enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Majumdar M, Tarui T, Shi B, Akakura N, Ruf W, Takada Y. Plasmin‐induced migration requires signaling through protease‐activated receptor 1 and integrin α9β1. J Biol Chem. 2004;279(36):37528–34. [DOI] [PubMed] [Google Scholar]
- 62. Lighvani S, Baik N, Diggs JE, Khaldoyanidi S, Parmer RJ, Miles LA. Regulation of macrophage migration by a novel plasminogen receptor Plg‐RKT. Blood. 2011;118(20):5622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Borg RJ, Samson AL, Au AEL, Scholzen A, Fuchsberger M, Kong YY, et al. Dendritic cell‐mediated phagocytosis but not immune activation is enhanced by plasmin. PLoS ONE. 2015;10(7):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Silva LM, Lum AG, Tran C, Shaw MW, Gao Z, Flick MJ, et al. Plasmin‐mediated fibrinolysis enables macrophage migration in a murine model of inflammation. Blood. 2019;134(3):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hajjar KA. The biology of annexin A2: from vascular fibrinolysis to innate immunity. Trans Am Clin Climatol Assoc. 2015;126:144–55. [PMC free article] [PubMed] [Google Scholar]
- 66. Kim J, Hajjar KA. Annexin II: a plasminogen‐plasminogen activator co‐receptor. Front Biosci [Internet]. 2002;1:341–8. Available from: http://www.bioscience.org/u37153137/gaDTRQo7632rgysaGWQYT64356/2002/v7/d/kim/kim.pdf. [DOI] [PubMed] [Google Scholar]
- 67. Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the urokinase‐type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front. Oncol. 2018;8:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Motley MP, Madsen DH, Jurgensen HJ, Spencer DE, Szabo R, Holmbeck K, et al. A CCR2 macrophage endocytic pathway mediates extravascular fibrin clearance in vivo. Blood. 2016;127(9):1085–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pajarinen J, Lin T, Gibon E, Kohno Y, Maruyama M, Nathan K, et al. Mesenchymal stem cell‐macrophage crosstalk and bone healing. Biomaterials 2019; 196:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berg RM, Kugelmass IN. Calcification in callus formation and fracture repair. Ann Surg. 1931;93(5):1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Popa NL, Wergedal JE, Lau KHW, Mohan S, Rundle CH. Urokinase plasminogen activator gene deficiency inhibits fracture cartilage remodeling. J Bone Miner Metab. 2014;32(2):124–35. [DOI] [PubMed] [Google Scholar]
- 73. Kawao N, Tamura Y, Okumoto K, Yano M, Okada K, Matsuo O, et al. Tissue‐type plasminogen activator deficiency delays bone repair: Roles of osteoblastic proliferation and vascular endothelial growth factor. Am J Physiol – Endocrinol Metab. 2014;307(3):278–88. [DOI] [PubMed] [Google Scholar]
- 74. López‐Alemany R, Suelves M, Muñoz‐Cánoves P. Plasmin generation dependent on a‐enolase‐type plasminogen receptor is required for myogenesis. Thromb Haemost. 2003;90(04):724–33. [DOI] [PubMed] [Google Scholar]
- 75. Lluís F, Roma J, Suelves M, Parra M, Aniorte G, Gallardo E, et al. Urokinase‐dependent plasminogen activation is required for efficient skeletal muscle regeneration in vivo. Blood. 2001;97(6):1703–11. [DOI] [PubMed] [Google Scholar]
- 76. Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cholok D, Lee E, Lisiecki J, Agarwal S, Loder S, Ranganathan K, et al. Traumatic muscle fibrosis: from pathway to prevention. J Trauma Acute Care Surg. 2017;82(1):174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rømer J, Bugge T, Pyke C, Lund LR, Flick MJ, Degen JL, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Fibrinolysis. 1996;10(Suppl. 3):78. [DOI] [PubMed] [Google Scholar]
- 79. Barnhart MI, Riddle JM, Bluhm GB, Quintana C. Fibrin promotion and lysis in arthritic joints. Ann Rheum Dis. 1967;26(3):206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen JT, Kotani K. Inverse correlation between fibrinogen and bone mineral density in women: Preliminary findings. J Formos Med Assoc. 2016;115(1):54–6. [DOI] [PubMed] [Google Scholar]
- 81. Carmassi F, De Negri F, Morale M, Pucetti R, Chung S. Elastase‐ and plasmin‐mediated fibrinolysis in rheumatoid arthritis. Int J Tissue React. 1994;16(2):89–93. [PubMed] [Google Scholar]
- 82. Busso N, Péclat V, So A, Sappino AP. Plasminogen activation in synovial tissues: Differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis. 1997;56(9):550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoppe B, Dörner T. Coagulation and the fibrin network in rheumatic disease: a role beyond haemostasis. Nat Rev Rheumatol. 2012;8(12):738–46. [DOI] [PubMed] [Google Scholar]
- 84. Barnhart MI, Riddle JM, Bluhm GB. Immunocytology in arthritic joints. Ann Rheum Dis. 1967;26(4):281–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Brennan FM, Mcinnes IB. Evidence that cytokines play a role in rheumatoid arthritis. Journal of Clinical Investigation. 2008;118(11):3537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Raghu H, Cruz C, Rewerts CL, Frederick MD, Thornton S, Mullins ES, et al. Transglutaminase factor XIII promotes arthritis through mechanisms linked to inflammation and bone erosion. Blood. 2015;125(3):427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li J, Ny A, Leonardsson G, Nandakumar KS, Holmdahl R, Ny T. The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II‐induced arthritis. Am J Pathol. 2005;166(3):783–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Raghu H, Jone A, Cruz C, Rewerts CL, Frederick MD, Thornton S, et al. Plasminogen is a joint‐specific positive or negative determinant of arthritis pathogenesis in mice. Arthritis Rheumatol. 2014;66(6):1504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Serrataĩ S, Margheri F, Chillà A, Neumann E, Müller‐Ladner U, Benucci M, et al. Reduction of in vitro invasion and in vivo cartilage degradation in a SCID mouse model by loss of function of the fibrinolytic system of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2011;63(9):2584–94. [DOI] [PubMed] [Google Scholar]
- 90. Almholt K, Hebsgaard JB, Nansen A, Andersson C, Pass J, Rønø B, et al. Antibody‐mediated neutralization of uPA proteolytic function reduces disease progression in mouse arthritis models. J Immunol. 2018;200(3):957–65. [DOI] [PubMed] [Google Scholar]
- 91. Thornton S, Raghu H, Cruz C, Frederick MD, Palumbo JS, Mullins ES, et al. Urokinase plasminogen activator and receptor promote collagen‐induced arthritis through expression in hematopoietic cells. Blood Adv. 2017;1(9):545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yang YH, Carmeliet P, Hamilton JA. Tissue‐type plasminogen activator deficiency exacerbates arthritis. J Immunol. 2001;167(2):1047–52. [DOI] [PubMed] [Google Scholar]
- 93. Nieuwenhuizen L, Schutgens REG, Coeleveld K, Mastbergen SC, Schiffelers RM, Roosendaal G, et al. Silencing of protease‐activated receptors attenuates synovitis and cartilage damage following a joint bleed in haemophilic mice. Haemophilia. 2016;22(1):152–9. [DOI] [PubMed] [Google Scholar]
- 94. Wyseure T, Cooke EJ, Declerck PJ, Behrendt N, Meijers JCM, von Drygalski A, et al. Defective TAFI activation in hemophilia A mice is a major contributor to joint bleeding. Blood. 2018;132(15):1593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Buckley BJ, Ali U, Kelso MJ, Ranson M. The urokinase plasminogen activation system in rheumatoid arthritis: pathophysiological roles and prospective therapeutic targets. Curr Drug Targets. 2018;20(9):970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. De Nardo CM, Lenzo JC, Pobjoy J, Hamilton JA, Cook AD. Urokinase‐type plasminogen activator and arthritis progression: contrasting roles in systemic and monoarticular arthritis models. Arthritis Res Ther. 2010;12(5):R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cook AD, De Nardo CM, Braine EL, Turner AL, Vlahos R, Way KJ, et al. Urokinase‐type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther. 2010;12(2):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Buckley BJ, Aboelela A, Minaei E, Jiang LX, Xu Z, Ali U, et al. 6‐Substituted hexamethylene amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer. J Med Chem. 2018;61(18):8299–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li SH, Reinke AA, Sanders KL, Emal CD, Whisstock JC, Stuckey JA, et al. Mechanistic characterization and crystal structure of a small molecule inactivator bound to plasminogen activator inhibitor‐1. Proc Natl Acad Sci U S A. 2013;110(51):E4941–E4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shoeb M, Fang MC. Assessing bleeding risk in patients taking anticoagulants. J Thromb Thrombolysis. 2013;35(3):312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ponzoni M, Pastorino F, Di Paolo D, Perri P. Brignole C. Targeting macrophages as a potential therapeutic intervention: impact on inflammatory diseases and cancer. Int J Mol Sci. 2018;19(7):2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cheng Y, Rong J. Macrophage polarization as a therapeutic target in myocardial infarction. Curr Drug Targets. 2018;19(6):651–62. [DOI] [PubMed] [Google Scholar]
- 103. Suzuki Y, Sano H, Maess M, Honkura N, Urano T. Platelet‐dependent activation of thrombin‐activatable fibrinolysis inhibitor (TAFI). ISTH 2019 Congress. 2019;74.
- 104. Kim P, Ni R, Neves MAD, Wu C, Cerroni SE, Gross PL, et al. Activated thrombin activable fibrinolysis inhibitor (TAFIa) regulates plasmin generation on thrombin‐activated platelets. ISTH 2019 Congress. 2019;76–77.
- 105. Zheng Z, Tabas I. The imbalanced hepatic production of PAI1 and tPA contributes to decreased fibrinolysis in obesity. ISTH 2019 Congress. 2019;75.
- 106. Miszta A, Kopec A, Pant A, Holle L, Byrnes J, Lawrence DA, et al. Novel plasmin generation assay reveals a plasminogen activator inhibitor‐1 independent mechanism that contributes to the hemostatic imbalance in experimental obesity. ISTH 2019 Congress. 2019;75–6.
- 107. de Maat S, Clark C, Waning M, van Moorsel M, Barendrecht A, Lenting P, et al. Targeting thrombolytics for thrombotic thrombocytopenic purpura. ISTH2019 Congress. 2019;76.