

Abstract
Wound infections associated with multidrug-resistant (MDR) bacteria are one of the important threats to public health. Bacteriophage (phage) therapy is a promising alternative or supplementary therapeutic approach to conventional antibiotics for combating MDR bacterial infections. In recent years, significant effort has been put into the development of phage formulations and delivery methods for topical applications, along with preclinical and clinical uses of phages for the treatment of acute and chronic wound infections. This paper reviews the application of phages for wound infections, with focuses on the current status of phage formulations (including liquid, semi-solid and liposome-encapsulated formulations, phage-immobilized wound dressings), safety and efficacy assessment in clinical settings and major challenges to overcome.
Keywords: Phage therapy, wound infections, multidrug-resistant bacteria, phage formulations, safety, efficacy
1. Introduction
Skin is the human body’s largest sensory organ. It also acts as a physical barrier and protects from potential assault by toxic substances or pathogenic organisms. The skin integrity can be compromised by mechanical injuries such as cuts or burns, which exposes the subcutaneous tissue to the surrounding environment. The exposed tissue provides a warm, moist and nutritious environment for pathogenic microorganisms to thrive in [1]. Hence, the wound site becomes vulnerable to microbial colonization and proliferation and any wound is at some risk of becoming infected. Wound infection increases the trauma experienced by patients and causes financial burdens in the healthcare system [1]. For example, post-operative surgical patients often suffer from surgical site infections [2]. This surgical infection is associated with high morbidity and mortality, with 25% of patients developing severe sepsis that requires transfer to intensive care unit [3]. Unfortunately, the severity of the burden has aggravated due to increased prevalence of infections associated with multidrug resistant (MDR) bacteria [4]. World Health Organization reported that more than 2 million illnesses result from MDR bacteria with direct and indirect costs exceeding USD 55 billion annually [4].
The worsening crisis of MDR bacterial infections has heightened the interest in bacteriophage (phage) therapy. Phage therapy utilizes virulent (lytic) phages, which obligately kill their bacterial host whilst self-replicating during the lytic cycle of infection. Although there has been one report using genome engineering of lysogenic phages [5], until recently only lytic phages have been prioritised for therapy and explored for the treatment of wound infections. Advances in techniques used to engineer phages for therapy are reviewed elsewhere [6]. Phage therapy has regained attention due to its ability to kill bacteria regardless of their antibiotic-resistance profile [7]. The first report of phage therapy for surgical and wound infection was during the Finnish Campaign in 1939–1940. The soldiers were treated with a mix of Staphylococcus and Streptococcus phages prepared at the Eliava Institute of Bacteriophages, Microbiology and Virology in Tbilisi, Georgia. Phage therapy saved the lives of 83% of infected soldiers, compared with 58% using other treatment options [8]. Similarly, another report showed 81% survival in phage-treated soldiers and 46% survival in those on other medications [9]. Furthermore, mobile sanitary brigades were in operation to provide prophylactic treatment of wounds, which reduced the number of gas gangrene by 30% in three independent brigades [9]. Despite such an excellent treatment outcome, the use of phages was soon discontinued in the Western counties with the discovery of a broad-spectrum antibiotic, penicillin [10]. Fortunately, phages therapy was continuously practiced and improved in the Eastern European countries, particularly in Russia, Poland and Georgia. Over the past decade, phage therapy has regained traction in response to emergence of MDR bacteria. Now, there are a growing number of studies indicating that phage therapy could be a promising prospect for treating acute and chronic wound infections caused by MDR bacteria.
In this review, we discuss recent progress in phage therapy for wound infections. We will first cover formulation and delivery of liquid and semi-solid phage formulations, incorporation of phages in existing products and liposome-encapsulated phage formulations and immobilized phage preparation on wound dressings. Preclinical and clinical efficacy and safety will be discussed next, and possible concerns and challenges faced will be covered in the last section.
2. Liquid formulation and delivery
Currently, liquid formulations are the vehicle of choice for delivering phages to the wound infection site. Theoretically, the preparation of liquid phage formulations is simpler with relatively minimal formulation development required for phage stability. However, there are very few reports describing the long-term stability (> 1 or 2 years) of phage mixes, particularly after purification processes. Phages are commonly formulated in sterile buffered solutions, such as phosphate-buffered saline (PBS) or Tris-buffered salt-magnesium buffer (SMB). Although the liquid phage formulations are generally considered to be stable under refrigeration, the optimal storage condition seems to be highly phage dependent. Eliava Biopreparations (Tbilisi, Georgia) produces several unpurified liquid phage mix preparations (Pyo Bacteriophage, Intesti Bacteriophage, Staphylococcal Bacteriophage and SES Bacteriophage) with recommended storage at 2–8 °C in a dry place, protected from direct light for 18–24 months [11]. These products contain phages formulated in bacteriological growth medium and sodium saline with chinazolin as a conservator, which seems to sufficiently stabilize the phages over the shelf-life.
Addition of divalent ions, including Mg2+ and Ca2+ (10 mM each) further aids in promoting phage stability during storage [12]. These cations interact with negatively charged moieties on the surface of phages, which helps with phage stabilization in aqueous buffered systems [13]. Even the chemical composition of water can impact phage stability in the liquid formulations. Although pure water with minimal contaminants is often perceived as the gold standard for microbiological laboratory work, a high level of purity does not necessarily correlate with phage stability in liquid formulations. In fact, the chemical composition of water may influence phage stability in liquid formulations. Governal and Gerba (1997) reported greater inactivation of phage MS2 in reverse osmosis water as compared with tap water [14]. Water free of contaminants is thought to have become a more aggressive solvent that has a higher chance of degrading phage genetic materials. Furthermore, ultrapure water can compromise phage bioactivity as a result of direct oxidation, causing capsid degradation, tail fragmentation, and release of phage DNA or RNA [14]. It was also observed that phages are less stable in tap water than distilled water, owing to halogenating agents in tap water inactivating the phages [15]. Conversely, ultrapure water may be required for human applications, including the preparation of products for intravenous delivery, thus adding a layer of complication to the development of liquid formulations.
In the recent PhagoBurn study [16], the instability of the purified phage cocktail PP1131 was noticed during the trial, which may have impacted the therapeutic outcome. The phage cocktail (comprised of 12 different phages) was formulated in PBS at a titer of 109 plaque-forming units (PFU) per mL. Each individual phage was stable at ≥109 PFU/mL for over 24 months. However, once mixed, the total titer of the phage cocktail rapidly dropped to 104–105 PFU/mL over 6 months. The instability may have been due to aggregation by electrostatic interactions, adsorption on the surface of the storage container, oxidation or chemical degradation [17]. Since phages may adversely interact with other phages, phage stability should be monitored as the final product (cocktail of phages) inside the primary container. Furthermore, it is crucial to implement Quality by Design principles to produce robust formulation of liquid preparations, which in turn will save time and resources in the long run.
For wound infection treatment, phage lysate have been prepared at a high titer and then diluted in isotonic saline prior to topical application [16]. Reports on the use of liquid phage preparations have described topical application by dripping the solution into the infected wound cavity [18] and/or by applying a gauze soaked with the preparation [18]. Unfortunately, these commonly used methods of delivery complicate precise control of the phage dose applied to the infection site. Liquid formulations dripped on the skin can easily run off from the infection site, thereby restricting the mobility of patients temporarily. This issue is often overcome by applying a gauze soaked with phage preparation. However, the release of phages from the gauze that reflects the actual dose given has never been reported in the literature. Phages may get stuck in the gauze, hindering the release of phages and subsequent bacteriolysis. Spray devices can potentially be utilized to aerosolize the liquid phage preparation directly on the wound infection site (Figure 1A). Phage spray has been mostly reported for food applications to protect fruit, meat and cattle hides [19, 20], but it can be applied for the treatment of wound infections. Furthermore, phages can be formulated in gels or creams to overcome the limitations of the liquid formulations.
Figure 1.
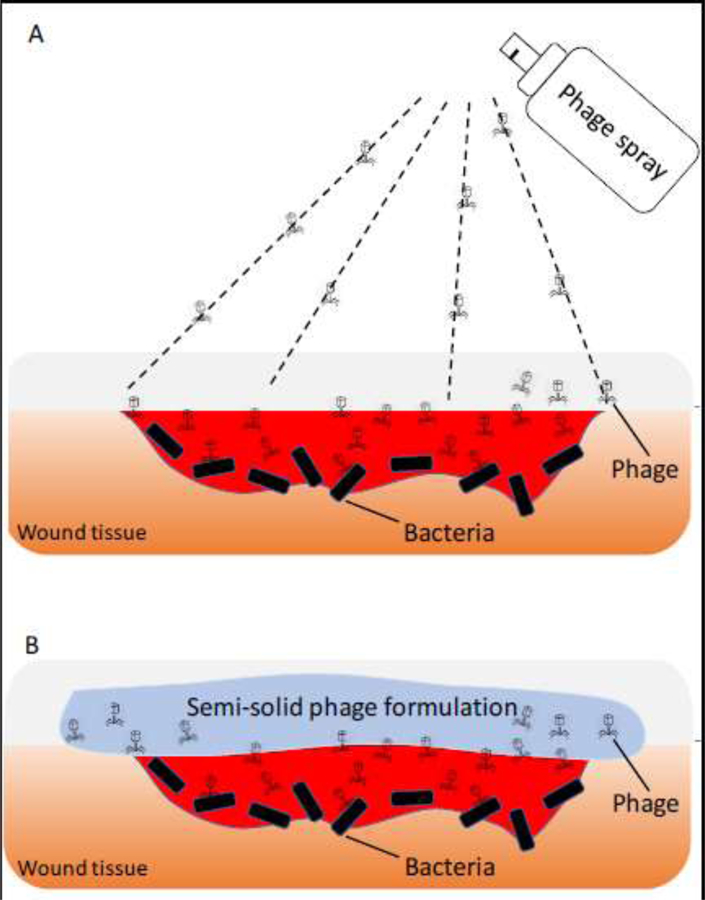
Topical application of phage spray formulation (A) and semi-solid phage formulation (B) to wound infection site.
3. Semi-solid formulation and delivery
Semi-solid formulations such as gels, creams and ointments are intended for topical application to skin or mucus membranes. These formulations not only provide skin protection and hydration, but also act as a delivery vehicle for phages. Semi-solid formulations are easy to apply, minimally irritating on the skin and often easily washable with water. As some phages tend to inactivate in alcohol [21], water-based semi-solid formulations (eg. Hydrogels) are more suitable over organic solvent-based formulations (eg. Organo gels) for delivering phages. Hydrogels are highly absorbent and are capable of retaining a large amount of water. Water-soluble polymers such as hydroxyethyl cellulose (HEC), hydroxypropyl methylcellulose (HPMC), carbomer and agarose have been used to prepare phage hydrogels [22–24]. These polymers are non-allergenic and biodegradable, and help protect the skin against excessive loss of body fluids while absorbing wound excreta [25]. Hydrogel formulation not only improves the balance of hydration of the wound site [26], but also enables hydrogen bond formation between water and phage proteins for phage stabilization. However, not all types of hydrogels confer phage stability in the semi-solid formulation.
Phages well tolerate and remain stable in hydrogels formulated with non-ionic polymers, whereas anionic polymers quickly cause inactivation. Non-ionic polymers promote phage stabilization by minimizing any charge induced phage inactivation. At physiological pH, the phage capsid exhibits an overall net negative charge and the tail has a net positive charge [27]. Anionic polymers can unfavourably interact with positively charged tails via electrostatic interaction and block the receptors in the tail fibers that are responsible for bacteria recognition and binding. When the phage tail becomes inaccessible for bacterial host attachment, it is no longer infective. Carbol et at. (2018) prepared phage hydrogels using a non-ionic and anionic polymers to formulate 5% HEC and 0.75% Carbomer gels, respectively, containing phages active against Propionibacterium acnes (Table 1) [23]. Phages suspended in HEC gel remained biologically stable for the tested period of four weeks, whereas those in Carbomer gel quickly inactivated with 99.95% titer loss by the fourth week. The observed phage inactivation is, perhaps, reversible if the electrostatic interaction is substantially reduced. Another non-ionic polymer, HPMC, has been used to formulate hydrogel containing Klebsiella phage lysate (Table 1) [22]. The final phage hydrogel had a titer of 108 PFU/mL and the phages remained stable over a seven-day storage at 37°C. These two studies highlighted the importance of selecting non-ionic polymers, but the duration of the stability test is too short to draw conclusions on the storage stability of phages in semi-solid formulations. Nonetheless, the use of non-ionic polymers is preferred over charged polymers to promote phage stabilization by minimizing any charge-induced phage inactivation.
Table 1.
List of phage gels, creams and ointments formulated for topical application.
| Phage (host bacteria) | Formulation | Results | Reference |
|---|---|---|---|
| Name not specified (P. acnes) | 5% HEC gel 0.75% Carbomer gel Preservatives: 0.1% methylparaben and 0.02% propylparaben, 1% phenoxyethanol, and 0.2% propylparaben |
Phages were stable in HEC gel for four weeks 99.95% titer loss in Carbomer gel Preservatives had no effect on phage viability | [23] |
| PAC1 (P. acnes) | Cetomacrogol cream, aqueous cream, cetrimide cream, ointments and pastes | < 1 log, 2 log and 3 log titer loss in Cetomacrogol, cetrimide and aqueous creams, respectively, after a 90 day-storage at 4°C in dark No phage lytic activity observed in zinc pastes | [35] |
| Cetomacrogol cream aqueous | <1 log titer loss after storage at 4°C in dark Phage inactivation when stored at 45°C for 14 days Phage inactivation within 21 days of storage at 20–25°C with full light exposure | [34] | |
| Phage Kpn5 (K. pneumoniae) | 3% HPMC | Stable for seven days at 37°C | [22] |
| Phage K (S. aureus) | Poly(N-isopropylacrylamide) co-polymerized with allylamine grafted to fabrics | Stable for four weeks Phages released after incubation at 37°C forming a zone of bacteria clearance | [36] |
| 0.7% agarose gel containing phages with a layer of hyaluronic acid | No stability study conducted 1% of phages released after 6 h | [24] | |
| Oil-based cream | Phage cream inhibited bacterial growth in liquid broth culture and on a petri dish No stability study conducted | [40] | |
| Phages PNM & 14–1 (P. aeruginosa), Acibel004 & Acibel007 (A. baumannii), and ISP (S. aureus) | Bactroban, colistin milk, P.O.H., Sulfamylon cream, Flaminal Forte, Flaminal Hydro, Flammazine 1%, Fucidin, Furacin, Hibidil, Intrasite Gel, Iruxol and iso-Betadine Gel 10% | Stable in Iruxol, Intrasite gel, Fucidin and sulfamylon Phage inactivation in isobetadine gel, solistin milk and P.O.H <2 log titer loss in Flaminal Hydro up to 9 log titer loss in Flaminal Forte | [77] |
In addition to biological stability of phages, the formulation vehicle must remain stable over the shelf-life. Aqueous solutions, emulsions and suspensions provide favourable environment for microbial growth, such as yeast, molds and bacteria [28]. A possible method of producing sterile semi-sold phage formulations (Figure 2) is to firstly sterilize the semi-solid formulations by autoclaving, and then aseptically add purified sterile phage preparation using geometric dilution [22]. As autoclave sterilization exposes the formulation to high temperature and pressure, the viscosity and other physical properties should be assessed to ensure it has not been altered. Preservative systems can also assist to maintain an aseptic condition throughout the shelf-life, although the inclusion of agents with antibacterial activity need to be considered carefully when running clinical studies [29]. A range of preservative systems, including 0.1% methylparaben and 0.02% propylparaben, 1% phenoxyethanol, and 0.2% propylparaben can potentially be used. These preservatives had no effect on the stability of phages formulated in 3% HPMC over the tested period of four weeks [23]. It is expected that these preservative systems will help maintain the sterility of semi-solid formulations containing phages to meet the United States Pharmacopeia [30] and British Pharmacopeia [31] standards. However, whether the phage stability will be compromised during long-term storage is unknown.
Figure 2.
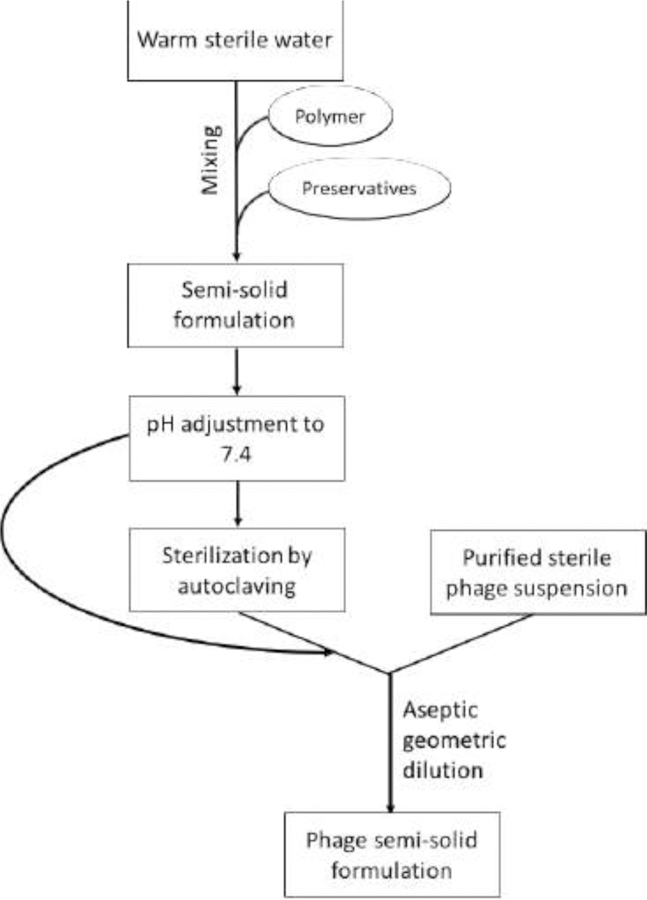
Schematic diagram showing steps involved in preparation of semi-solid formulations containing phages.
For an effective phage therapy, infective phages need to be released from the semi-solid carrier systems (Figure 1B). Drug (phage) release profile assessment is an important part of the formulation study that can impact the reliability of the phage effectiveness. The Franz diffusion cell method commonly used for measuring drug release [32] is potentially suitable for evaluating the in vitro release profiles of phages in topical semi-solid formulations (Figure 3) A receptor solution such as PBS or saline is placed into the receptor compartment which is maintained at 32°C [33]. A suitable membrane with low protein binding properties (such as polyethersulfone membranes) are placed over the diffusion cell opening. The phage semi-solid formulation is loaded on top of the membrane and the cells are continuously stirred using a magnetic stir bar. The receptor solution is sampled at specified timepoints and the sampled volume is replaced with an equal volume of fresh buffer. The titer of infective phages released from the formulation is assayed using a standard overlay plaque assay. This system can be modified to study the phage release profile in the presence of bacteria. As phage concentration increases in the presence of its host pathogen, bacteria can be applied between the membrane and phage formulation to better represent topical bacterial infection in vitro.
Figure 3.
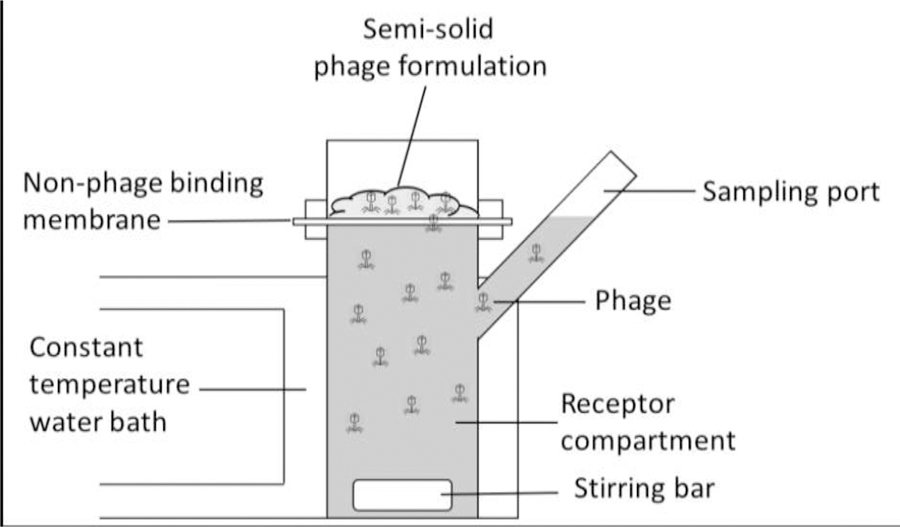
Franz diffusion apparatus which can be adopted to assess the release rate of phages from semi-solid formulations.
At present, majority of the reported studies have not assessed in vitro phage release profiles using Franz diffusion cell or other methods recommended by the Pharmacopeia. There is no guarantee that phages mixed with various polymer vehicles will migrate through the mixture into the infection site. A preclinical study demonstrated that topical application of phage hydrogel can exert therapeutic efficacy [22], implying that the phages are migrating through the gel into the infection site. Since phage release may be matrix and/or phage dependent, it is important to assess the release in vitro. Some studies have qualitatively assessed the release of infective phages from semi-solid formulations by applying the formulation on top of a bacterial overlay plate, followed by incubation at 37°C overnight [34, 35]. Phages are thought to be released from the formulation if a zone of clearance is observed on a bacterial plate. Although this method provides an indication of phage release and subsequent bacteriolysis, the data is qualitative rather than quantitative with only a single time point assessed.
In an effort to better control the release profile of phages, polymeric release systems have been developed. A thermally responsive polymer poly(N-isopropylacrylamide) has been utilized to induce heat-triggered release of Staphylococcus phage K (Table 1) [36]. The polymer was co-polymerized with allylamine to form nanospheres that collapse at 37°C. The nanospheres were grafted to non-woven polypropylene fabric and then soaked with phage K (5 × 108 PFU). Any unbound phages were removed by rinsing with water and left to air-dry. Incubation at 37°C released the phages, which subsequently cleared S. aureus on Petri dish. Although the zone of bacteria clearance implies phage release, the total amount of infective phages released and the stability of these phages in dry polypropylene swath remained unknown. It is indispensable in clinical work to understand the dose released and the total concentration of phage that patients will be exposed to throughout a study.
Bean et al. (2014) utilized enzyme-driven polymer degradation for targeted release of phages from agarose gels (Table 1) [24]. Phage K was formulated in a gel containing 0.2, 0.4 and 0.7% of agarose at a final concentration of 108 PFU/mL. Within 30 min, less than 2% of phages were released from these agarose gels, while after 6 h, 20%, 1% and 0.6% of phages were released from the 0.2%, 0.4% and 0.7% agarose gel. Phage K was thought to be partially entangled in the presence of higher polymer concentration, causing inefficient phage release. In spite of this, 0.7% agarose gel was selected for further formulation work due to ease of handling, highlighting the challenges of balancing the practicalities of use and the stability and availability of the phages in the various carriers. Although the general belief is that phage titer would increase in the presence of its host in the wound infection site, the chances of a successful therapeutic outcome will be greatly reduced if the availability of the active ingredient is compromised from the start and should be avoided. The trigger-release system consisted of a bilayer hydrogel architecture, where phage K reservoir (0.7 % agarose gel) was covered by a top layer of hyaluronic acid. Phages were released (106 PFU/mL; equivalent of approximately 1% release) from the bilayer hydrogel system only in the presence of hyaluronidase, an enzyme released by S. aureus. Such release rate is very low and may not produce the adequate phage dose required for phage therapy, according to the therapeutic phage concentration of 108–109 PFU/mL recommended for biocontrol of bacteria [37]. Based on the low release rate, the phage titre in the reservoir would need to be at least 100-fold higher for this trigger-release system to achieve minimal effective phage density.
Interestingly, the Eliava phage cocktail products available for topical application contains phages at a titer of ~107 PFU/mL [38], which is 10–100 times lower than the recommended PFU/mL by Abedon et al [37]. Further clinical work is still required to determine the minimum effective dose of any phage product in a particular clinical indication and the contribution of the human immune response in the eradication of bacteria.
4. Utilizing commercial products
Several topical burn wound care products are commercially available with some containing antibiotics. Phage preparations may be used in combination with these products to maximize the therapeutic outcome with minimal formulation development. However, commercial products have complex formulation compositions and additives which may compromise the phage biostability. Thus, it is crucial to test the viability of individual phages in these formulations. Merabishvili et al. (2017) studied the biostability of five phages active against Pseudomonas aeruginosa (PNM and 14–1), Acinetobacter baumannii (Acibel004 and Acibel007) and Staphylococcus aureus (ISP) in commercial burn wound products (Table 1). The wound care products were mixed with the phage suspension formulated in 0.9% NaCl (109 PFU/mL) at a ratio of 1:1. All five phages remained stable in Iruxol, Intrasite gel, Fucidin and sulfamylon over 24 h. Products with low pH (iso-betadine gel, solistin milk and P.O.H.) quickly inactivated the phages. The phage most resistant to pH-induced titer loss was myovirus 14–4, which belongs to the genus Pbunavirus characterized by acid-resistant capsids [39]. All five phages remained relatively stable with only 1.3–1.9 log titer loss in hydrogel Flaminal Hydro which contains 3.5% alginate, whereas 1.7–9.0 log titer loss was seen in hydrogel Flaminal Forte (containing 5.5% alginate). Negatively charged alginate may adversely impact phage infectivity, similar to that observed in the presence of negatively charged carbomer gel (see section 3).
Commercial creams have also been utilized as a semi-solid carrier system of phages for topical application [34, 35, 40, 41]. These creams are emollients or moisturisers for treating dry skin conditions but have been used for delivery of phages. O’Flaherty et al. (2005) incorporated phage K into an oil-based cream containing bismuth subnitrate (Cross Vetpharm Group Ltd, Ireland) at a final phage concretion of 108 PFU/g. The phage cream inhibited the growth of S. aureus in liquid broth culture and on petri dish. Although the antibacterial activity of the phage cream was demonstrated, the phage release profile is unknown. Brown et al. (2016) formulated Propionibacterium acnes phages into a non-ionic cream, cetomacrogol cream aqueous (Biotech Pharmaceuticals, Australia) (Table 1) [34]. Phage stability was achieved when the formulation was stored at 4°C in a light protected bottle (~1 log titer loss). Storage at high temperature (45°C) or exposure to full light at room temperature (20–25°C) caused phage inactivation within 14 and 21 days, respectively.
In a separate study, P. acnes phage PAC1 was formulated in various semi-solid formulations, including creams, ointments and pastes [35] according to published formularies [42]. Phage PAC1 formulated in cetomacrogol cream aqueous, aqueous cream and cetrimide cream aqueous all remained biologically active for 90 days when stored at 4°C protected from light. Storage at 45°C in the dark resulted in loss of antibacterial activity within 14 days. Exposure to constant light resulted in complete inactivation of phage by day 21 of storage at 25°C. Of the three formulations, a non-ionic base cetomacrogol provided the best phage stability with ~1 log titer reduction over 90 days as compared with 2 log and 3 log losses in cetrimide (cationic base) and aqueous creams (anionic base), respectively. Thus, creams with non-ionic base should be selected when formulating phages for topical application.
Additives such as sodium lauryl sulphate can rapidly inactivate phages due to its virucidal properties [43]. Phage PAC1 formulated in zinc pastes did not exhibit lytic activities, likely due to the ionic binding of phages to bivalent zinc cations, prohibiting phage release. Alternatively, the phages may have been inactivated in the presence of zinc oxide [44], resulting in the absence of phage lytic activity. Furthermore, the thickness of ointments may contribute to hindered release of phages in the formulation [35]. The release rate of phages depends on the diffusion rate through the matrix of semi-solid formulations. Compared with antibiotics, phages have a much lower diffusion rate due to their significantly larger (> 105 times) molecular weight. The diffusion coefficient of T4 phages was 2.2 × 10−11 m2/s through 0.2% agarose gel and the diffusion velocity was decreased with the thickness of the gel [45]. Once a phage semi-solid formulation is applied to the infection site, the phages that are in direct contact with the targeted site will be available for bacteriolysis. Subsequent phage replication is expected to increase the phage titer and promote active phage therapy at the infection site.
Though the use of commercial gels, creams and other wound care products for phage preparation may simplify the formulation development process, it is crucial to test the viability of individual and mixed phages in these formulations. In particular, wound care products have complex formulation compositions. While these additives may confer stabilizing effects to the active components, phage biostability may be compromised long-term. Products with potential phage-inactivating properties, such as a low pH, antiseptic activity and anionic polymer should be avoided. Light-induced phage inactivation during storage can be minimized by packaging or storing the phage formulations in opaque or light-protected containers. Furthermore, storage at 4°C is recommended to preserve the bioactivity of phages in the formulations.
5. Liposome-encapsulated phages
Liposome encapsulation can help improve the phage viability, stability and retention time at wound infection site [46, 47]. Liposomes are composed of natural lipids and mimics biological membranes, enabling epidermal layer penetration. The non-immunogenic, biodegradable and biocompatible properties of liposomes [48] make it a suitable lipid-based delivery system for phages. Furthermore, liposomes can protect phages from pH-induced inactivation, an important attribute particularly in an acidic environment such as wound infection sites. Cationic liposomal formulations have been used to encapsulate phages [47, 48]. Cationic liposomes are also used to enhance the interaction with negatively charged bacteria. Bacteria flocculation is observed in the presence of positively charged liposomes and larger liposomes have higher affinity for the bacteria [49]. Furthermore, cationic liposomes exhibit mucoadhesive properties which enables prolonged drug retention at the application site, thereby providing a controlled release and improved therapeutic outcome [50].
Colom et al. (2015) prepared a cationic liposomal formulation containing Salmonella phages. Rotary evaporation was used to produce a thin lipid film comprising phosphatidylcholine:cholesteryl polyethylene glycol 600:cholesterol:cholesteryl 3β-N-(dimethylaminoethyl)carbamate hydrochloride (1:0.1:0.2:0.7) [48]. The lipid film was hydrated using phage lysate in PBS and then extruded through 400 nm pore-sized membranes. The phage-encapsulated liposomes were 309–356 nm in size, positively charged and had an encapsulation efficiency of 47–49%. These physical properties and encapsulation yield remained unchanged during a three-month storage at 4°C.
Chhibber et al. (2019) also used the thin-film hydration method with phosphatidylcholine:cholesterol:tween 80:stearylamine (7:3:1:0.5) to formulate cationic liposomal formulations containing a cocktail of phages [47]. The lipid film was hydrated using a phage cocktail suspension (phages MR-5 and MR-10, 1:1) in PBS, followed by sonication for 30 min. The resulting liposomal formulation exhibited uniform shape and lamellarity with liposome size of 212 nm (polydispersity index below 0.3). Phages were encapsulated at a high efficiency of 87% and had a final titer of 2 × 1010 PFU/mL. The liposomal formulation remained physically stable at 4°C over 9-week storage without reduction in number of encapsulated phages.
Chadha et al. (2017) used the same excipients but at a different ratio of phosphatidylcholine:cholesterol:tween 80:stearylamine (8:2:1:0.5) to prepare a cationic liposomal formulation containing a five phages in the cocktail [46]. Thin-film hydration method produced 230 nm phage-encapsulated liposomes with an encapsulation efficiency of 79% (107 PFU/mL). Positively charged surfactant stearylamine may have helped with phage encapsulation by increasing the overall entrapped volume. Phages were released at a constant and steady pace over 96 h, but with 3% of the encapsulated phages released in total. However, it is uncertain whether all five phages in the cocktail were equally released from the liposomes.
Although all these studies have used cationic liposomes to promote interaction with the target pathogen, the charge may unfavourably interact with phages and compromise phage biostability during storage. Longer storage stability testing would be necessary to study liposome formulations containing phages.
More recently, microfluidic devices have been used to produce phage-encapsulated liposomes [51, 52]. Microfuidic approaches enable precise control over the size of liposomes – narrow particle size distribution compared to the conventional thin-film hydration method. The size distribution is one of the critical factors that influence drug release profile, loading capacity and liposome stability as well as pharmacokinetics and pharmacodynamics. Leung et al. (2018) used soy phosphatidylcholine:cholesterol (4:1) to encapsulate podovirus and myovirus Pseudomonas phages in liposomes. The lipid mixture was dissolved in ethanol (17.5 mg/mL) and then injected through a cross mixer. The phage suspensions were injected from the side channels intersecting the central channel. Phage encapsulation efficiency of 50–59% was achieved at a total flow rate of 160 µL/min and organic/aqueous flow rate ratio of 2:3. Interestingly, the size of phage-encapsulated liposomes was dependent on the size of the phages. Those encapsulating long-tailed myovirus was bigger (261–448 nm) than short-tailed podovirus-containing liposomes (135–218 nm).
Cinquerrui et al. (2018) also used a micro-capillary micro-mixing setup to produce phage K-encapsulated liposomes [52]. A thin-film containing different ratios of DSPC and cholesterol was produced, followed by solubilization in isopropanol (10 mg/mL). The organic and aqueous (phage K in buffer) solutions were delivered using a microfluidics setup with a total flow rate of ~17 µL/min and organic/aqueous flow rate ratio of 1:2. Higher cholesterol content increased the size of liposomes. Encapsulated phages were predominately found in large liposomes (>500 nm), likely due to the relatively large head and long tail features of phage K. Some phages were found interacting with the outer surface of the liposomal membrane, possibly due to electrostatic interaction between the negatively charged phospholipid heads and positively charged tail fibers. The encapsulation yield of tailed phages reported in the literature would have likely included externally bound phages, hence, an overestimation. Microfluics approach allows continuous and automated production of highly uniform liposomes, but the feasibility of a large scale-up production is yet to be established.
6. Immobilized phages on wound dressings
Phage immobilization strategies have been explored for wound dressings and surface disinfection applications [53]. Immobilization of phages on inert polymeric surfaces allow direct application of infective phages on various surfaces, including wounds. In principle, when the phage capsid attaches to polymeric surfaces, the tail end is exposed to the environment for bacteria recognition and subsequent infection. Thus, the orientation of immobilized phages will impact the antimicrobial efficacy [53]. In particular, tailed phages rely on bacteria-binding receptors on their tail fibers and these fibers must be freely available for bacteria interaction. Phages can be immobilized via physisorption (physical absorption), electrostatic interaction or covalent binding. Although physisorption is a very simple approach, phages can easily detach from the substrate in response to changes in pH or temperature. This makes it troublesome for use at the wound infection site where the pH shifts with wound-healing [54]. Another approach is to immobilize the phages via electrostatic interaction between the phage and the substrate. Phages are negatively charged [27]: the capsid is thought to be responsible for the overall negative charge and the tail fibers possess an overall net positive charge [55]. This charge difference between the capsid and the tail was utilized by Anany et al. (2011) for oriented immobilization of Escherichia coli phages on a positively charged cellulose membrane. A cocktail of E. coli phages were immobilized on a cellulose film coated with cationic polymer polyvinylamine [27]. More phages were captured on the modified positively charged cellulose film (95%) as compared with the unmodified membrane (71%). Phage-treated unmodified membrane formed considerably less plaques. Charge modification greatly improved the number of infective phages by immobilizing the phages through their capsid head so that the tail fibers are freely available to target and kill the bacteria. The storage stability of phages immobilized on dressings have not been tested so far. Yet, the stabilization of phages immobilized on a dry substrate is likely to impose challenges.
Semi-solid formulations, including hydrogels, creams and ointments are suitable vehicles for topical phage delivery provided that phage-inactivating excipients are avoided. Hydrophilic creams and ointments are preferred over hydrophobic bases due to improved phage stability during storage as well as ease of miscibility in aqueous solvent, which is a prerequisite for assessment of bioactivity using plaque assay. Thick semi-solid formulations with high solid contents such as pastes may not be suitable as it hinders the release and subsequent bacteriolysis. Although immobilized phages on wound dressings is an attractive concept, the shelf-life of such dry formulations need to be tested and optimized to become a viable product. Alternatively, the dressings can be prepared in pre-soaked wet wipe materials.
7. Safety & Efficacy
Studies on the safety and efficacy of phage treatments delivered topically, both in animals and humans, started shortly after their discovery. Various reviews have been written summarizing the early potential of phage preparations for the treatment of skin infections, surgical and purulent wounds as well as acquired postsurgical infections caused by E. coli, P. aeruginosa, S. aureus and other well-established and prevalent pathogens to this date [56–58]. More recently, Morozova et al. (2018) reported on serial case reports published in the Russian literature [59]. These reviews comprehensively highlight the widespread use of phage therapy prior and even after the discovery of antibiotics, including during World War II, with reports of clinical success ranging from 50% to 69% depending on the target pathogen. Phage treatments were administered both as single phage preparations (monophages) or cocktails containing multiple phage species (polyphages). The use of polyphages applied >5 to ≤8 days was reported as providing faster clinical outcomes even in comparison with antibiotic treatments [59]. The early use of phage treatments was not just limited to the former Soviet Union countries. Abedon et al. (2011) reviewed the early use of topical phage therapy to treat skin infections in France, mainly thanks to Felix d’Herelle presence and work. For the treatment of furunculosis, pads moistened with the phage preparations were applied over the affected areas followed by compresses moistened with phages that were used as the dressings. The treatments were reapplied every 2 days with a total of 8–10 applications reported as required to observe clinical benefits. Clinical benefit was also observed for the treatment of surgical infections with polyphage solutions [60]. Treatment of furunculosis was also reported in the United States as early as in 1929 [61]. Despite all these early promising results in the West, the use of phage therapy after the discovery of antibiotics continues to be rather limited to Russia and the Republic of Georgia [62], where phages are approved for clinical use and sold over the counter, and to Poland where unapproved phage treatments can still be administered following the Declaration of Helsinki guidelines [63, 64]. In Belgium, a new regulatory framework – The Magistral Phage-has been proposed to facilitate the used of tailor-made phage products for treatment of complicated bacterial infections [65], while in other countries administration of phage treatments is only allowed for patients who run out of options under compassionate use ethical codes [66].
Despite this, a number of preclinical and clinical cases, and clinical trials using phage products delivered topically have been more recently reported. Preclinical studies reported therapeutic utility of topically applied phages in treating acute [67] and chronic [68] infections. Kumari et al. (2011) demonstrated the efficacy of Klebsiella phage Kpn5 formulated in hydrogel in acute mouse burn wound infection model [67]. Mendes et al. (2013) showed that treatment with polyvalent phage cocktail (S. aureus, P. aeruginosa and A. baumannii) can reduce the bacterial numbers and improved wound healing in diabetes mellitus model in rats [68]. Rhoads et el. (2009] reported on the first modern phase I trial in the United States looking to evaluate the safety of phages on difficult to treat wounds [69]. Thirty-nine patients participating in the trial had their chronic venous leg ulcers treated for 12 weeks with either a phage product or placebo. No adverse events or safety concerns were reported. Another clinical trial was conducted to determine at the safety, tolerability and preliminary efficacy of an anti-S. aureus phage product (AB-SA01). It involved healthy volunteers in the United States under an Investigational New Drug application (NCT02757755). The phage product (1 mL at either 108 or 109 PFU/mL) or a placebo was applied daily for 3 consecutive days to the subject’s volar forearm via gauze pads saturated and covered with an occlusive dressing [70]. The potential of a monophage preparation (Sb-1) to resolve diabetic toe ulcers infected with S. aureus under the compassionate use guidelines was also reported by Fish and colleagues (2016) [18]. Phage therapy without the inclusion of antibiotic treatment resulted in wound healing and prevented amputation in all treated patients. The topical clinical protocol used in this case series has been reported as the basis for future randomized trials that appeared to be in planning phase by commercial companies like Pherecydes Pharma in France (https://www.pherecydes-pharma.com/phosa-collaborative-project.html). Technophage, a private company based in Portugal, has also received FDA clearance to begin human clinical trials of its phage product. The product has already been evaluated in vivo in rodent and porcine models showing its potential to resolve chronic infections in conjunction with wound debridement [68]. The accumulated data prior and post the antibiotic era suggest phages can successfully help resolve infections when delivered topically.
The most recent study was the PhagoBurn phase 1/2 trial reported in 2019 [16], using a cocktail of 12 natural lytic phages against P. aeruginosa (1 × 106 PFU/mL applied via an alginate dressing) compared with standard of care (1% sulfadiazine silver emulsion cream) for seven days of treatment of burn wound infection in 25 patients. Insufficient efficacy was demonstrated in the phage treatment which was attributed to a number of causes such as differences in the maximal bacterial burden between the two treatment groups, a much lower than expected phage dose (1 × 102 PFU/mL per daily dose) received by the patients which was due to titre drop after manufacturing. Nonetheless, the study highlighted some important lessons for future design of studies. These include monitoring the shelf-life of the phage cocktail, close collaboration with regulatory agencies, improvement of analytical methods required for good manufacturing practices, the benefits of reducing the number of phages in a product and working towards enhancing phage stability in a product, ensuring the clinical protocol can realistically recruit the number of participants required during the planned period of time and considerations in patient selection (e.g., complication of poly-infection, phage-susceptibility testing prior to commencement of phage treatment).
8. Challenges
The perceived general biological and business development challenges for the introduction of phage therapy as mainstream treatment have been highlighted and discussed by others previously [33, 71–73]. It is the authors’ opinion that many of the perceived biological challenges can now be addressed by developing strategic isolation, characterization and phage selection research plans. There seems to be an agreement on the basic biological characteristics that ought to be considered and what quality deficiencies are not permissible in therapeutic phage products [70, 74]. The molecular tools and in vitro models available for phage characterization are recently much more sophisticated and the costs of performing such analysis have also reduced significantly overtime, alleviating one of the primary issues in early research stages.
The production of purified phages that meet Good Manufacturing Practices (GMP) standards was initially seen as a major obstacle in the field not only because of the cost, but because of the uninformed belief that phages could not be manufactured under controlled, reproducible conditions without introducing substantial changes in their genome and biological activity. AmpliPhi Biosciences was the first company to receive cGMP certification to manufacture phage products for human use in Europe and its GMP products used in clinical studies and compassionate use cases. Pherecydes Pharma, in collaboration with Clean Cells, also produced GMP products for its clinical trial, although as mentioned before, there were product stability issues that impacted the study [16]. Other companies like BiomX and Technophage have now announced the construction of their own GMP facilities, in addition to specialized phage contract manufacturing organizations like Jafral. The increase in the number of GMP facilities dedicated to manufacture phage products suggests an increased level of confidence on the processes and regulatory paperwork required to manufacture high quality products and thus, the initial production challenge in being actively addressed.
Regulatory agencies should provide oversight and make companies accountable for the quality of the products generated to avoid poor clinical outcomes as occurred with the PhagoBurn study. Perhaps of higher interest and concern is the absence of appropriate clinical development plans specific to the intended clinical indication for the phage product in development. The next wave of modern clinical trials will require the development of robust clinical protocols with well-established clinical and microbiological endpoints as well as the development of the specific analytical tools to measure those accurately. A lesson the field has learned after the publication of the study results evaluating the efficacy of two coliphage preparations in a randomized trial in children from Bangladesh [75]. The selection of a clinical indication where the effect of phage therapy delivered topically can be measured against standard of care treatment is not a minor proposition and one that requires careful deliberation. For example, despite the promising reports on the use of phages for the treatment of diabetic foot ulcers and the desperate clinical need, clinical trial protocols designed to test the efficacy of phages must give consideration to the innate variation in clinical diabetic foot ulcers outcomes due to the complexity of the pathogenesis and the local variability in size and grading of the wounds despite the existence of national and international guidelines for the standard care of wounds [76]. The development of robust clinical protocols that maximize the potential of phages in vivo while still keeping within the practical logistics of disease management is not trivial. Improvement on the various delivery methods for topical applications that accurately deliver the expected dose requires further research as reviewed here. Exact dose control is likely to be challenging for topical phage therapy. However, it is imperative to find out the expected dose delivered (phage release from formulations) to ensure sufficient number of infective phages would be released and then delivered to the infection site and observe a therapeutic effect. However, progress should not be halted waiting for a “perfect protocol”. The integration of highly multidisciplinary teams with phage biology, microbiology, formulation, clinical development and regulatory expertise is fundamental to develop logical, balanced clinical protocols that permit the implementation of step grading plans for the sustainable development of phage-based products.
acknowledgements
The authors acknowledge the financial support from the National Health and Medical Research Council (Project Grant APP1140617). H.-K. Chan is supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R33AI121627. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest.
Abbreviations
- MDR
Multidrug resistant
- phage
bacteriophage
- PBS
phosphate-buffered saline
- SMB
salt-magnesium buffer
- PFU
plaque-forming units
- HEC
hydroxyethyl cellulose
- HPMC
hydroxypropyl methylcellulose
- GMP
good manufacturing practices
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no competing financial or otherwise conflicts of interest with respect to the content of this review.
References
- [1].Bowler PG, Duerden BI, Armstrong DG. Wound microbiology and associated approaches to wound management. Clin Microbiol Rev 2001;14:244–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Owens CD, Stoessel K. Surgical site infections: epidemiology, microbiology and prevention. J Hosp Infect 2008;70 Suppl 2:3–10. [DOI] [PubMed] [Google Scholar]
- [3].Atilla A, Doganay Z, Kefeli Celik H, Demirag MD, S SK. Central line-associated blood stream infections: characteristics and risk factors for mortality over a 5.5-year period. Turk J Med Sci 2017;47:646–52. [DOI] [PubMed] [Google Scholar]
- [4].World Health Organization. Antimicrobial resistance: global report on surveillance 2014 2014.
- [5].Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med 2019;25:730–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pires DP, Cleto S, Sillankorva S, Azeredo J, Lu TK. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol R 2016;80:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chang RYK, Wallin M, Lin Y, Leung SSY, Wang H, Morales S, et al. Phage therapy for respiratory infections. Adv Drug Deliv Rev 2018;133:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kogin GA. Use of bacteriophages in surgery. Sov Med 1941;9:15–8. [Google Scholar]
- [9].Krestovnikova VA. Phage treatment and phage prophylactics and their approval in the works of the Soviet researchers. J Microb Epidemiol Immun 1947;3:56–65. [Google Scholar]
- [10].Landecker H Antibiotic resistance and the biology of history. Body Soc 2016;22:19–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Eliava Biopreparations. Products Tbilisi, Georgia. [Google Scholar]
- [12].Bourdin G, Schmitt B, Marvin Guy L, Germond JE, Zuber S, Michot L, et al. Amplification and purification of T4-like escherichia coli phages for phage therapy: from laboratory to pilot scale. Appl Environ Microbiol 2014;80:1469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mylon SE, Rinciog CI, Schmidt N, Gutierrez L, Wong GC, Nguyen TH. Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir 2009;26:1035–42. [DOI] [PubMed] [Google Scholar]
- [14].Governal R, Gerba CP. Persistence of MS-2 and PRD-1 bacteriophages in an ultrapure water system. J Ind Microbiol Biot 1997;18:297–301. [DOI] [PubMed] [Google Scholar]
- [15].Jepson CD, March JB. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine 2004;22:2413–9. [DOI] [PubMed] [Google Scholar]
- [16].Jault P, Leclerc T, Jennes S, Pirnay JP, Que YA, Resch G, et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect Dis 2019;19:35–45. [DOI] [PubMed] [Google Scholar]
- [17].Mutti M, Corsini L. Robust approaches for the production of active ingredient and drug product for human phage therapy. Front Microbiol 2019;10:2289–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fish R, Kutter E, Wheat G, Blasdel B, Kutateladze M, Kuhl S. Bacteriophage treatment of intransigent diabetic toe ulcers: a case series. J Wound Care 2016;25:S27–S33.26949862 [Google Scholar]
- [19].Arthur TM, Kalchayanand N, Agga GE, Wheeler TL, Koohmaraie M. Evaluation of bacteriophage application to cattle in Lairage at beef processing plants to reduce Escherichia coli O157:H7 prevalence on hides and carcasses. Foodborne Pathog Dis 2017;14:17–22. [DOI] [PubMed] [Google Scholar]
- [20].Leverentz B, Conway WS, Janisiewicz W, Camp MJ. Optimizing concentration and timing of a phage spray application to reduce Listeria monocytogenes on honeydew melon tissue. J Food Prot 2004;67:1682–6. [DOI] [PubMed] [Google Scholar]
- [21].Olofsson L, Ankarloo J, Andersson PO, Nicholls IA. Filamentous bacteriophage stability in non-aqueous media. Chem Biol 2001;8:661–71. [DOI] [PubMed] [Google Scholar]
- [22].Kumari S, Harjai K, Chhibber S. Topical treatment of Klebsiella pneumoniae B5055 induced burn wound infection in mice using natural products. J Infect Dev Ctries 2010;4:367–77. [PubMed] [Google Scholar]
- [23].Carbol J, Tan PI, Varma Y, Osborne DW. Formulating topical products containing live microorganisms as the active ingredient. Pharm Technol 2018;42:32–6. [Google Scholar]
- [24].Bean JE, Alves DR, Laabei M, Esteban PP, Thet NT, Enright MC, et al. Triggered release of bacteriophage K from agarose/hyaluronan hydrogel matrixes by Staphylococcus aureus virulence factors. Chem Mater 2014;26:7201–8. [Google Scholar]
- [25].Thomas S, Hay P. Fluid handling properties of hydrogel dressings. Ostomy Wound manag 1995;41:54–6, 8–9. [PubMed] [Google Scholar]
- [26].Jones A, Vaughan D. Hydrogel dressings in the management of a variety of wound types: A review. J Orthop Nurs 2005;9:S1–S11. [Google Scholar]
- [27].Anany H, Chen W, Pelton R, Griffiths MW. Biocontrol of Listeria monocytogenes and Escherichia coli O157:H7 in meat by using phages immobilized on modified cellulose membranes. Appl Environ Microbiol 2011;77:6379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Allen LV. Remington -an introduction to pharmacy London and Chicago: Pharmaceutical Press; 2013. [Google Scholar]
- [29].Novack GD, Evans R. Commercially available ocular hypotensive products: preservative concentration, stability, storage, and in-life utilization. J Glaucoma 2001;10:483–6. [DOI] [PubMed] [Google Scholar]
- [30].United States Pharmacopeia Antimicrobial Effectiveness Testing 2018.
- [31].Appendix XVI C Efficacy of Antimicrobial Preservation. British Pharmacopoeia 2019.
- [32].Shah VP, Elkins J, Lam S-Y, Skelly JP. Determination of in vitro drug release from hydrocortisone creams. Int J Pharm 1989;53:53–9. [Google Scholar]
- [33].Jonczyk-Matysiak E, Lodej N, Kula D, Owczarek B, Orwat F, Miedzybrodzki R, et al. Factors determining phage stability/activity: challenges in practical phage application. Expert Rev Anti Infect Ther 2019;17:583–606. [DOI] [PubMed] [Google Scholar]
- [34].Brown TL, Petrovski S, Dyson ZA, Seviour R, Tucci J. The formulation of bacteriophage in a semi solid preparation for control of Propionibacterium acnes growth. PloS one 2016;11:e0151184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brown TL, Thomas T, Odgers J, Petrovski S, Spark MJ, Tucci J. Bacteriophage formulated into a range of semisolid and solid dosage forms maintain lytic capacity against isolated cutaneous and opportunistic oral bacteria. J Pharm Pharmacol 2017;69:244–53. [DOI] [PubMed] [Google Scholar]
- [36].Hathaway H, Alves DR, Bean J, Esteban PP, Ouadi K, Sutton JM, et al. Poly(N-isopropylacrylamide-co-allylamine) (PNIPAM-co-ALA) nanospheres for the thermally triggered release of Bacteriophage K. Eur J Pharm Biopharm 2015;96:437–41. [DOI] [PubMed] [Google Scholar]
- [37].Abedon S Phage therapy pharmacology: calculating phage dosing. Adv Appl Microbiol 2011;77:1–40. [DOI] [PubMed] [Google Scholar]
- [38].Zhvania P, Hoyle NS, Nadareishvili L, Nizharadze D, Kutateladze M. Phage therapy in a 16-year-old boy with Netherton syndrome. Front Med 2017;4:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ceyssens PJ, Miroshnikov K, Mattheus W, Krylov V, Robben J, Noben JP, et al. Comparative analysis of the widespread and conserved PB1-like viruses infecting Pseudomonas aeruginosa. Environ Microbiol 2009;11:2874–83. [DOI] [PubMed] [Google Scholar]
- [40].O’Flaherty S, Ross RP, Meaney W, Fitzgerald GF, Elbreki MF, Coffey A. Potential of the polyvalent anti-Staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl Environ Microbiol 2005;71:1836–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen L-K, Liu Y-L, Hu A, Chang K-C, Lin N-T, Lai M-J, et al. Potential of bacteriophage ΦAB2 as an environmental biocontrol agent for the control of multidrug-resistant Acinetobacter baumannii. BMC Microbiol 2013;13:154–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sansom L Australian pharmaceutical formulary and handbook, 23rd edn (in press). Canberra: Pharmaceutical Society of Australia; 2014. [Google Scholar]
- [43].Piret J, Desormeaux A, Bergeron MG. Sodium lauryl sulfate, a microbicide effective against enveloped and nonenveloped viruses. Curr Drug Targets 2002;3:17–30. [DOI] [PubMed] [Google Scholar]
- [44].You J, Zhang Y, Hu Z. Bacteria and bacteriophage inactivation by silver and zinc oxide nanoparticles. Colloid Surface B 2011;85:161–7. [DOI] [PubMed] [Google Scholar]
- [45].Hu J, Miyanaga K, Tanji Y. Diffusion properties of bacteriophages through agarose gel membrane. Biotechnol Prog 2010;26:1213–21. [DOI] [PubMed] [Google Scholar]
- [46].Chadha P, Katare OP, Chhibber S. Liposome loaded phage cocktail: Enhanced therapeutic potential in resolving Klebsiella pneumoniae mediated burn wound infections. Burns 2017;43:1532–43. [DOI] [PubMed] [Google Scholar]
- [47].Chhibber S, Kaur J, Kaur S. Liposome entrapment of bacteriophages improves wound healing in a diabetic mouse MRSA infection. Front Microbiol 2018;9:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Colom J, Cano-Sarabia M, Otero J, Cortés P, Maspoch D, Llagostera M. Liposome-encapsulated bacteriophages for enhanced oral phage therapy against Salmonella spp. Appl Environ Microbiol 2015;81:4841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Campanhã MTN, Mamizuka EM, Carmona-Ribeiro AM. Interactions between cationic liposomes and bacteria: the physical-chemistry of the bactericidal action. J Lipid Res 1999;40:1495–500. [PubMed] [Google Scholar]
- [50].Shaikh R, Raj Singh TR, Garland MJ, Woolfson AD, Donnelly RF. Mucoadhesive drug delivery systems. J Pharm Bioallied Sci 2011;3:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Leung SSY, Morales S, Britton W, Kutter E, Chan HK. Microfluidic-assisted bacteriophage encapsulation into liposomes. Int J Pharm 2018;545:176–82. [DOI] [PubMed] [Google Scholar]
- [52].Cinquerrui S, Mancuso F, Vladisavljevic GT, Bakker SE, Malik DJ. Nanoencapsulation of bacteriophages in liposomes prepared using microfluidic hydrodynamic flow focusing. Front Microbiol 2018;9:2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hosseinidoust Z, Van de Ven TG, Tufenkji N. Bacterial capture efficiency and antimicrobial activity of phage-functionalized model surfaces. Langmuir 2011;27:5472–80. [DOI] [PubMed] [Google Scholar]
- [54].Schneider LA, Korber A, Grabbe S, Dissemond J. Influence of pH on wound-healing: a new perspective for wound-therapy? Arch Dermatol Res 2007;298:413–20. [DOI] [PubMed] [Google Scholar]
- [55].Serwer P, Hayes SJ. Agarose gel electrophoresis of bacteriophages and related particles. I. Avoidance of binding to the gel and recognizing of particles with packaged DNA. Electrophoresis 1982;3:76–80. [Google Scholar]
- [56].Summers WC. Felix d’Herelle and the origins of molecular biology New Haven, CT: Yale University Press; 1999. [Google Scholar]
- [57].Chanishvili N A literature review of the practical application of bacteriophage research New York: Nova Science Publishers; 2009. [Google Scholar]
- [58].Chanishvili N Bacteriophages as therapeutic and prophylactic means: summary of the Soviet and post Soviet experiences. Curr Drug Deliv 2016;13:309–23. [DOI] [PubMed] [Google Scholar]
- [59].Morozova VV, Vlassov VV, Tikunova NV. Applications of bacteriophages in the treatment of localized infections in humans. Front Microbiol 2018;9:1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. Phage treatment of human infections. Bacteriophage 2011;1:66–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Larkum NW. Bacteriophage treatment of Staphylococcus infections. J Infect Dis 1929;45:34–41. [Google Scholar]
- [62].Parfitt T Georgia: an unlikely stronghold for bacteriophage therapy. Lancet 2005;365:2166–7. [DOI] [PubMed] [Google Scholar]
- [63].Slopek S, Weber-Dabrowska B, Dabrowski M, Kucharewicz-Krukowska A. Results of bacteriophage treatment of suppurative bacterial infections in the years 1981–1986. Arch Immunol Ther Exp (Warsz) 1987;35:569–83. [PubMed] [Google Scholar]
- [64].Slopek S, Kucharewicz-Krukowska A, Weber-Dabrowska B, Dabrowski M. Results of bacteriophage treatment of suppurative bacterial infections. IV. Evaluation of the results obtained in 370 cases. Arch Immunol Ther Exp (Warsz) 1985;33:219–40. [PubMed] [Google Scholar]
- [65].Pirnay JP, Verbeken G, Ceyssens PJ, Huys I, De Vos D, Ameloot C, et al. The Magistral Phage. Viruses 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Borysowski J, Górski A. Compassionate use of unauthorized drugs: Legal regulations and ethical challenges. Eur J Intern Med 2019;65:12–6. [DOI] [PubMed] [Google Scholar]
- [67].Kumari S, Harjai K, Chhibber S. Bacteriophage versus antimicrobial agents for the treatment of murine burn wound infection caused by Klebsiella pneumoniae B5055. J Med Microbiol 2011;60:205–10. [DOI] [PubMed] [Google Scholar]
- [68].Mendes JJ, Leandro C, Corte-Real S, Barbosa R, Cavaco-Silva P, Melo-Cristino J, et al. Wound healing potential of topical bacteriophage therapy on diabetic cutaneous wounds. Wound Repair Regen 2013;21:595–603. [DOI] [PubMed] [Google Scholar]
- [69].Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care 2009;18:237–8, 40–3. [DOI] [PubMed] [Google Scholar]
- [70].Lehman SM, Mearns G, Rankin D, Cole RA, Smrekar F, Branston SD, et al. Design and preclinical development of a phage product for the treatment of antibiotic-resistant Staphylococcus aureus infections. Viruses 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Torres-Barcelo C Phage therapy faces evolutionary challenges. Viruses 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Caflisch KM, Suh GA, Patel R. Biological challenges of phage therapy and proposed solutions: a literature review. Expert Rev Anti Infect Ther 2019;17:1011–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Brussow H Hurdles for phage therapy to become a reality-an editorial comment. Viruses 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Reindel R, Fiore CR. Phage therapy: considerations and challenges for development. Clin Infect Dis 2017;64:1589–90. [DOI] [PubMed] [Google Scholar]
- [75].Sarker SA, Sultana S, Reuteler G, Moine D, Descombes P, Charton F, et al. Oral phage therapy of acute bacterial Ddarrhea with two coliphage preparations: A randomized trial in children from Bangladesh. EBioMedicine 2016;4:124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jeffcoate WJ, Vileikyte L, Boyko EJ, Armstrong DG, Boulton AJM. Current challenges and opportunities in the prevention and management of diabetic foot ulcers. Diabetes care 2018;41:645–52. [DOI] [PubMed] [Google Scholar]
- [77].Merabishvili M, Monserez R, van Belleghem J, Rose T, Jennes S, De Vos D, et al. Stability of bacteriophages in burn wound care products. PloS one 2017;12:e0182121. [DOI] [PMC free article] [PubMed] [Google Scholar]