

Abstract
Antimicrobial resistance poses a significant threat to our ability to treat infections. Especially concerning is the emergence of carbapenem-resistant Enterobacteriaceae (CRE). In the new 2019 United States Centers for Disease Control and Prevention Antibiotic Resistance Report, CRE remain in the most urgent antimicrobial resistance threat category. There is good reason for this concerning designation. In particular, the combination of several resistance elements in CRE can make these pathogens untreatable or effectively untreatable with our current armamentarium of anti-infective agents. This article reviews recently approved agents with activity against CRE and a range of modalities in the pipeline, from early academic investigation to those in clinical trials, with a focus on structural aspects of new antibiotics. Another article in this series addresses the need to incentive pharmaceutical companies to invest in CRE antimicrobial development and to encourage hospitals to make these agents available in their formularies. This article will also consider the need for change in requirements for antimicrobial susceptibility testing implementation in clinical laboratories to address practical roadblocks that impede our efforts to provide even existing CRE antibiotics to our patients.
Clearly, new therapies are urgently needed for carbapenem-resistant Enterobacteriaceae (CRE). In 2019, the United States Centers for Disease Control and Prevention issued an updated version of its 2013 Antibiotic Resistance Report; once again, CRE are included in highest antimicrobial resistance threat category, “urgent”, along with carbapenem-resistant Acinetobacter baumannii, multidrug-resistant Neisseria gonorrhoeae, Candida auris, and Clostridioides difficile.1 Broad-spectrum agents that provide reliable activity against CRE and other resistant pathogens would have great value in settings where such pathogens are frequent enough to necessitate inclusion in empiric regimens. Directed agents that could be used specifically for CRE when accompanied by appropriate diagnostics to identify such resistance would also address current therapeutic gaps. CRE antibiotics with excellent bioavailability that could be dosed orally would allow earlier and simpler transition to outpatient settings and would fill an important unmet need by reducing the costs and health risks associated with prolonged inpatient hospitalization. Ideally, new therapies would address the three broad classes of carbapenem resistance: serine carbapenemase production, metallo-carbapenemase production, and, either alone or in combination with carbapenemases, altered access to the antimicrobial target through porin mutation and efflux pump activity.2 It would be remiss to consider CRE in isolation. Gram-negative antibiotic resistance is a larger problem and new therapeutics will have greater utility if they can address additional unmet needs coincident with their activity against CRE; concomitant activity against multidrug-resistant Pseudomonas aeruginosa and A. baumannii in particular would provide significant advantage.
Traditional targets.
As has been reviewed elsewhere, most traditional antimicrobials with staying power engage multiple targets, broadly considered, and thereby are probabilistically relatively immune to spontaneous resistance events, and/or engage the active sites of critical enzymes such that target mutational resistance would in most cases undermine critical cellular function and thereby impact fitness.3 Such targets remain of great practical interest in the development of new drugs for CRE. The penicillin binding proteins (PBPs) fulfill both criteria when multiple PBPs are engaged, and, therefore, anti-CRE antibiotic development has mostly centered on modifications to PBP-targeting β-lactam antibiotics to prevent hydrolysis by broad-spectrum β-lactamases and/or through inhibition of the latter class of enzymes.
The ribosome, the largest molecular machine in the cell, also remains a highly rich target. It has multiple potential vulnerabilities exemplified by the large number of different antibiotic classes that engage distinct regions of the complex and interfere with different aspects of translation.
Importantly, the ribosome is an RNA and protein-based machine. Enterobacteriaceae, in general, have 7–8 copies of the rrn operons, which encode the ribosomal 16S, 23S and 5S rRNAs.4 For the aminoglycoside class, mutational resistance in the 16S rRNA antibiotic target is generally a recessive phenotype.5, 6 Emergence of resistance in a clinical setting almost always results from acquisition of modifying enzymes rather than target modification. Circumventing this resistance (similar to strategies used to extend the spectrum of β-lactams) has been successfully performed through chemical modification to weaken modifying enzyme engagement. This approach is exemplified by the addition of a hydroxaminoybutyric acid (HABA) group via an amide bond to the N-1 position of the 2-deoxystreptomycin ring (2-DOS) of kanamycin to form the antibiotic amikacin (Fig. 1A). This modification makes amikacin susceptible to a smaller subset of aminoglycoside modifying enzymes than kanamycin.7 In the more recent conversion of the aminoglycoside sisomicin to plazomicin, this was accomplished through the addition of a HABA group to the N-1 position of the 2-DOS ring and a hydroxyethyl group to the 6’-N position of the A ring, essentially eliminating modification by generally circulating aminoglycoside modifying enzymes (Fig. 1B).8, 9
Figure 1. Traditional Targets.
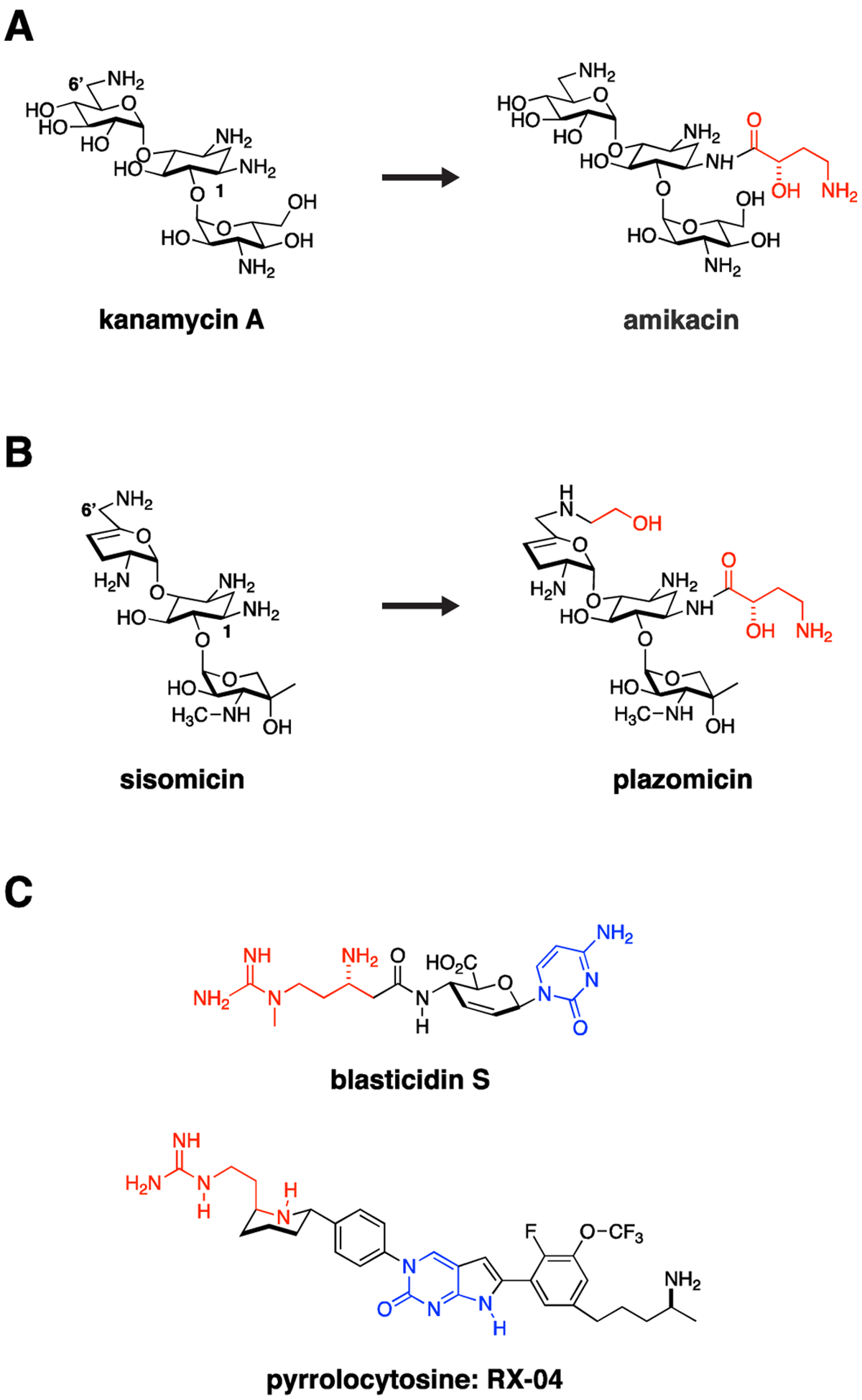
The ribosome and bacterial cell wall remain tried and true targets for Gram-negative antimicrobials. Resistance often occurs through acquisition of antibiotic modifying enzymes or hydrolases. Medicinal chemistry strategies can be used to overcome these liabilities by altering antibiotics to impede action of these resistance enzymes. For example, (A) kanamycin was modified through addition of a hydroxyaminobutyric (HABA) acid (colored red) to the deoxystreptamine ring to yield amikacin. The HABA group sterically blocks several aminoglycoside-modifying enzymes, significantly broadening the activity spectrum of amikacin, which in doing so may preserve activity against CRE strains. (B) This strategy was taken one step further in development of plazomicin from sisomicin with both hydroxyethyl and HABA modifications to the parent sisomicin shown in red. This constellation of changes makes plazomicin immune to essentially all aminoglycoside modifying enzymes circulating in CRE. Unfortunately, plazomicin is not able to overcome emerging 16S ribosomal RNA methylasetransferase-based aminoglycoside resistance. (C) The pyrrolocytosine series is an example of a new class of antimicrobials modeled in part on the interaction of blasticidin S with the ribosome but extending into unique ligand binding space. These molecules share a 1,3-aminoguanidine (red) and a cytosine ring system (blue). The presumption from this strategy is that pre-existing resistance enzymes that modify either drug or the binding target in the ribosome should not have previously evolved, thereby extending the useful lifetime of the antimicrobial.
Another general strategy worthy of pursuit, with increasingly available structural information, is to exploit new structural space in ways different from known natural product scaffolds, and thereby potentially circumvent pre-existing resistance mechanisms, such as antibiotic modifying enzymes. A beautiful example of this is the development of the pyrrolocytosine series (Fig. 1C), based on and extending beyond existing interactions of blasticidin with the 50S ribosome.10 These small molecule blasticidin analogues possess excellent broad-spectrum activity against carbapenemase-expressing Enterobacteriaceae, P. aeruginosa and A. baumannii, presumably based on absence of existing resistance mechanisms to these “unnatural” products. The recently developed ability of cryogenic electron microscopy (cryo-EM) studies to rapidly solve drug-target structures in solution – for example, the delineation of the 70S ribosome at high resolution in its several dynamic functional states11–13 – will most certainly catalyze similar structure-guided design going forward.
In addition to these examples, there are several new drugs either recently approved or in trials with promising activity against CRE that will be discussed along with consideration of methods to diversify existing antibiotic classes to expand their utility against CRE. The development and potential of novel β-lactamase inhibitor co-formulations will also be considered as a promising therapeutic strategy against this recalcitrant group of organisms. We expect several new options for CRE and other Gram-negatives to become available in the coming years (summarized in Table I), and decisions about their use in patients will have to incorporate considerations including desired spectrum of activity, effectiveness for specific clinical indications, and side effect profiles.
Table I.
Examples of Approved Antimicrobials and Antimicrobials in Development for Treatment of CRE.
| Antibiotic | Category | Stage of Development | Target; Mechanism of Action | Carbapenemase Spectrumg | Major Resistance Mechanism |
|---|---|---|---|---|---|
| Apramycina | Aminoglycoside | Phase 1 | 30S ribosomal subunit; Protein synthesis inhibition | N.A. | Aminoglycoside 3-N-acetyltransferase subtype IV |
| Plazomicin | Aminoglycoside | FDA-approved | 30S ribosomal subunit; Protein synthesis inhibition | N.A. | 16S rRNA ribos om al methyltransferases |
| Meropenem-vaborbactam | Carbapenem + boronic acid β-lactamase-inhibitor | FDA-approved | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, C | Not yet determined |
| Cefepime-taniborbactam | Cephalosporin + cyclic boronate β-lactamase-inhibitor | Phase 3 | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, B, C, D | Not yet determined |
| Ceftazidime-avibactam | Cephalosporin + diazabicyclooctane β-lactamase-inhibitor | FDA-approved | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, B, C, D | Carbapenemase mutations129 |
| Aztreonam-avibactamb | Monobactam/diazabicyclooctane β-lactamase-inhibitor | Phase 2 | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, B, C, D | β-lactamase mutations130 |
| Imipenem-relebactam | Carbapenem + diazabicyclooctane β-lactamase-inhibitor | FDA-approved | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, C | Not yet determined |
| Meropenem-nacubactam | Carbapenem + diazabicyclooctane β-lactamase-inhibitor | Phase 1 | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, C, (B, D)i | Not yet determined |
| Cefepime-zidebactam | Cephalosporin + diazabicyclooctane β-lactamase- inhibitor | Phase 1 | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, C, (B, D)i | Not yet determined |
| Cefpodoxime-ETX0282c | Cephalosporin + diazabicyclooctane β-lactamase-inhibitor | Phase 1 | PBP/β-lactamase enzyme; Cell wall synthesis inhibition | Class A, C | Not yet determined |
| BOS-228 (LYS228) | Monobactam derivative | Phase 2 | PBP; Cell wall synthesis inhibition | Class A, B, Ch, D | Efflux pumps |
| Cefiderocol | Siderophore cephalosporin | FDA-approved | PBP; Cell wall synthesis inhibition | Class A, B, C, D | Mutations in iron uptake genes |
| Eravacyclined | Fluorinated tetracycline analogue | FDA-approved | 30S ribosomal subunit; Protein synthesis inhibition | N.A. | Efflux pumps |
| Fosfomycine | Phosphoenolpyruvate analogue | FDA-approved | Pyruvyl transferase (MurA); Cell wall synthesis inhibition | N.A. | Mutations in fosfomycin uptake systems; fosfomycin-modifying enzymes |
| SPR206 | Polymyxin | Phase 1 | Lipopolysaccharide; Outer membrane disruption | N.A. | Lipid A modification |
| SPR741f | Polymyxin B derivative (antibiotic potentiator) | Phase 1 | Lipopolysaccharide; Outer membrane permeabilization | N.A. | Not yet determined |
PBP: Penicillin-binding protein
Currently used in veterinary medicine.
Combination not yet available; can be given as aztreonam plus ceftazidime-avibactam.
Orally bioavailable.
Example of application and power of total de novo synthesis of natural product analogues
For systemic infections, IV form usually used with other antibiotics; only oral form available in the US.
Not active alone; used as an outer membrane permeabilizing agent.
Indicates activity against strains expressing the indicated molecular classes of carbapenemases and β-lactamases. Class A includes ESBL serine β-lactamases of SHV, TEM, CTX-M types, and the Klebsiella pneumoniae carbapenemase (KPC); class B includes metallo-carbapenemases such as NDM, VIM, and IMP; class C includes chromosomal AmpC and plasmid-borne CMY serine cephalosporinases; and class D includes serine oxacillinases such as OXA-48.131 Non-β-lactam agents are marked as not applicable (N.A.) in this column. These drugs are often active against CRE based on mechanisms unaffected by carbapenemase expression.
Only active against some members of indicated class. More detail is provided in the text.
β-lactamase inhibitor does not inactivate Class B, metallo-carbapenemases and Class D, OXA-carbapenemases; however, β-lactam/β-lactamase inhibitor combination may inhibit strains expressing these carbapenemases based on the intrinsic antimicrobial activity and enhancer effects of the β-lactamase inhibitor.
Pipeline Drugs
Cefiderocol is a siderophore-based cephalosporin with enhanced stability to serine and metallo-carbapenemases.14 Based on its iron-chelating catechol moiety (Fig. 2A), the drug is actively taken up through bacterial siderophore uptake mechanisms. As such, cefiderocol can be viewed as a Trojan horse that capitalizes on the pathogen’s need for iron acquisition at sites of infection.15 As a side benefit, this efficient uptake mechanism circumvents porin-based resistance mechanisms, which would otherwise prevent antibiotic diffusion through the outer membrane. A theoretical concern for mutational resistance leading to inactivation of such uptake mechanisms, as occurred with prior investigated siderophore-based antibiotics, appears to be low.16 Cefiderocol also demonstrated potent activity against carbapenem-resistant Enterobacteriaceae, A. baumannii, P. aeruginosa, Burkholderia cepecia, and Stenotrophomonas maltophilia, placing it as a welcome empiric or targeted Gram-negative therapy in multiple infectious conditions. The current options for especially the latter two organisms are particularly limited.14, 17 The anaerobic and Gram-positive activity of cefiderocol is poor and would need to be supplemented with additional agents depending on clinical suspicion. The dynamics of siderophore based uptake and optimal use of this new therapy will need to be considered with unfolding clinical information, as follow up to the unexplained trend towards increased mortality rates for cefiderocol observed in the CREDIBLE-CR study compared to the best available therapy.18
Figure 2. Pipeline Drugs.
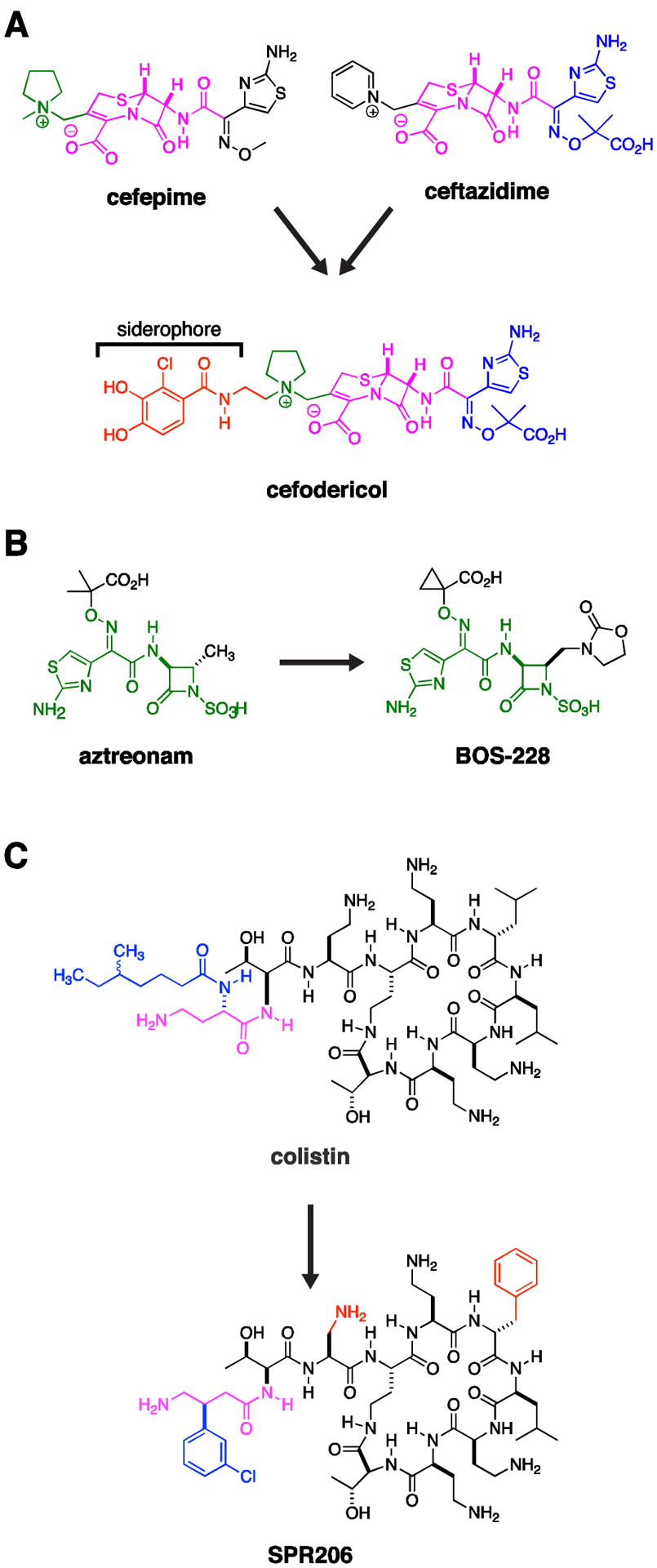
(A) Cefodericol. The first approved siderophore β-lactam, cefiderocol contains an aminothiazole substituent (blue) found in ceftazidime, a pyrrolidine substituent found in cefepime (green), a cephalosporin core (purple), and a catechol siderophore (red), related to those found in naturally occurring bacterial siderophores. The siderophore enhances uptake while the constellation of bulky sidegroups attached to the cephalosporin core prevents hydrolysis by serine and metallo-β-lactamases. (B) Aztreonam is a monolactam. It intrinsically resists hydrolysis by metallo-carbapenemases, but remains susceptible to hydrolysis by serine β-lactamases. Aztreonam serves as the core (green) of BOS-228. Substituents on this latter molecule also prevent hydrolysis by serine carbapenemases. Therefore BOS-228 offers broad-spectrum activity against CRE expressing serine and/or metallo-carbapenemases. (C) SPR206 is a derivative of colistin with reduced renal toxicity in animal models. Differences from colistin are highlighted in red and blue, where the blue β-(m-chlorophenyl) ring on the purple gamma amino butyric amide mimics the blue α-(Δ-methyl-hexanoic amide) residue of colistin.
BOS-228 (formerly LYS228) is a monobactam derivative now in phase II clinical trials, which similar to aztreonam is resistant to metallo-β-lactamases and which was additionally engineered for stability to serine β-lactamases (Fig. 2B). Accordingly, it demonstrated high efficacy when tested against Enterobacteriaceae expressing serine- and metallo-carbapenemases and extended spectrum β-lactamase (ESBL) enzymes.19–21 There are rare reports of a particular inducible plasmid ampC β-lactamase that confers resistance, resolved by combination of BOS-228 with β-lactamase inhibitors such as avibactam.22
SPR206, a new polymyxin drug (Fig. 2C), is currently in Phase 1 trials. This drug has similar in vitro activity to the clinically available polymyxin B,23 but demonstrated less nephrotoxicity in a mouse model.24 If this reduction in nephrotoxicity is borne out in human trials it could have significant clinical implications, as polymyxins are one of the few classes of drugs with activity against a majority of CRE isolates, yet their utility is limited by their nephrotoxicity (along with concerns about clinical efficacy).25
The majority of CRE drugs that have been recently approved or are in the pipeline are β-lactam-β-lactamase inhibitor agents, which are discussed in the following section.
Combination Therapy
β-lactam-β-lactamase inhibitor combinations
The ability of bacteria to develop resistance to antibiotics soon after their development is notorious, and nowhere is the arms race between microbes and the drugs developed to control them more clearly illustrated than in the case of β-lactam antibiotics and β-lactamase enzymes. β-lactam antibiotics, which bind to penicillin-binding proteins (PBPs) and inhibit peptidoglycan synthesis, are one of the oldest classes of antibiotics. By the time ampicillin and amoxicillin, the first β-lactam drugs with significant activity against Enterobacteriaceae, were introduced in the 1970s,26 plasmid-mediated β-lactamase enzymes capable of hydrolyzing them had already been recognized for a decade;27 such naturally-occurring resistance to antibiotics that are derived from compounds produced by microbes themselves is not uncommon. These narrow-spectrum β-lactamases, which include TEM-1, TEM-2, and SHV-1, can be inhibited by the β-lactamase inhibitor clavulanic acid, which was approved in 1984.26 Inevitably, however, β-lactamases that are stable against clavulanic acid and similar β-lactamase inhibitors emerged, necessitating the development of the broader-spectrum cephalosporin antibiotics.
Following the predictable appearance of cephalosporinase enzymes, an even broader class of β-lactam antibiotics, the carbapenems, were developed, but the evolution of carbapenemase β-lactamase enzymes has threatened the efficacy of even these “superdrugs”. The first β-lactamase inhibitor that was resistant to cleavage by carbapenemases, avibactam, was introduced to the market in combination with ceftazidime in 2015 (Fig. 3A).28 Since then, two additional β-lactam-β-lactamase inhibitor combinations with activity against CRE, imipenemrelebactam (Fig. 3B) and meropenem-vaborbactam (Fig. 3C), have been approved.29 Among these new β-lactamase inhibitors, avibactam inhibits most Ambler class A, C, and D serine carbapenemases, including KPC (class A) and OXA-48 (class D) enzymes. Vaborbactam and relebactam are also active against KPCs but not against OXA-48 carbapenemases, and none of these agents inhibit the class B metallo-β-lactamases (MBLs).30 Unfortunately, MBLs and OXA-48 carbapenemases are increasingly common throughout the world and no doubt will be seen with growing frequency in the United States, either in travelers or visitors returning from endemic areas or through establishment of endemicity. A dramatic recent example of an MBL isolate in the US was the so-called Nevada strain, which was recovered from a woman who had previously received health care in India and contained a New Delhi metallo-β-lactamase enzyme; the strain also possessed numerous other resistance elements and was determined to be non-susceptible to all antibiotics available for testing at the CDC when it was reported in a patient in Nevada in 2016.31 In the absence of available metallo-carbapenemase inhibitors, one strategy to treat such strains is the administration of the monobactam, aztreonam, in combination with ceftazidime-avibactam. This combination is effective because aztreonam is intrinsically immune to MBLs, while avibactam inhibits class A and D serine carbapenemase that would otherwise inactivate aztreonam, thereby restoring activity against the Nevada strain and similar isolates.32, 33
Figure 3. Combination Therapy, New β-lactamase Inhibitors.
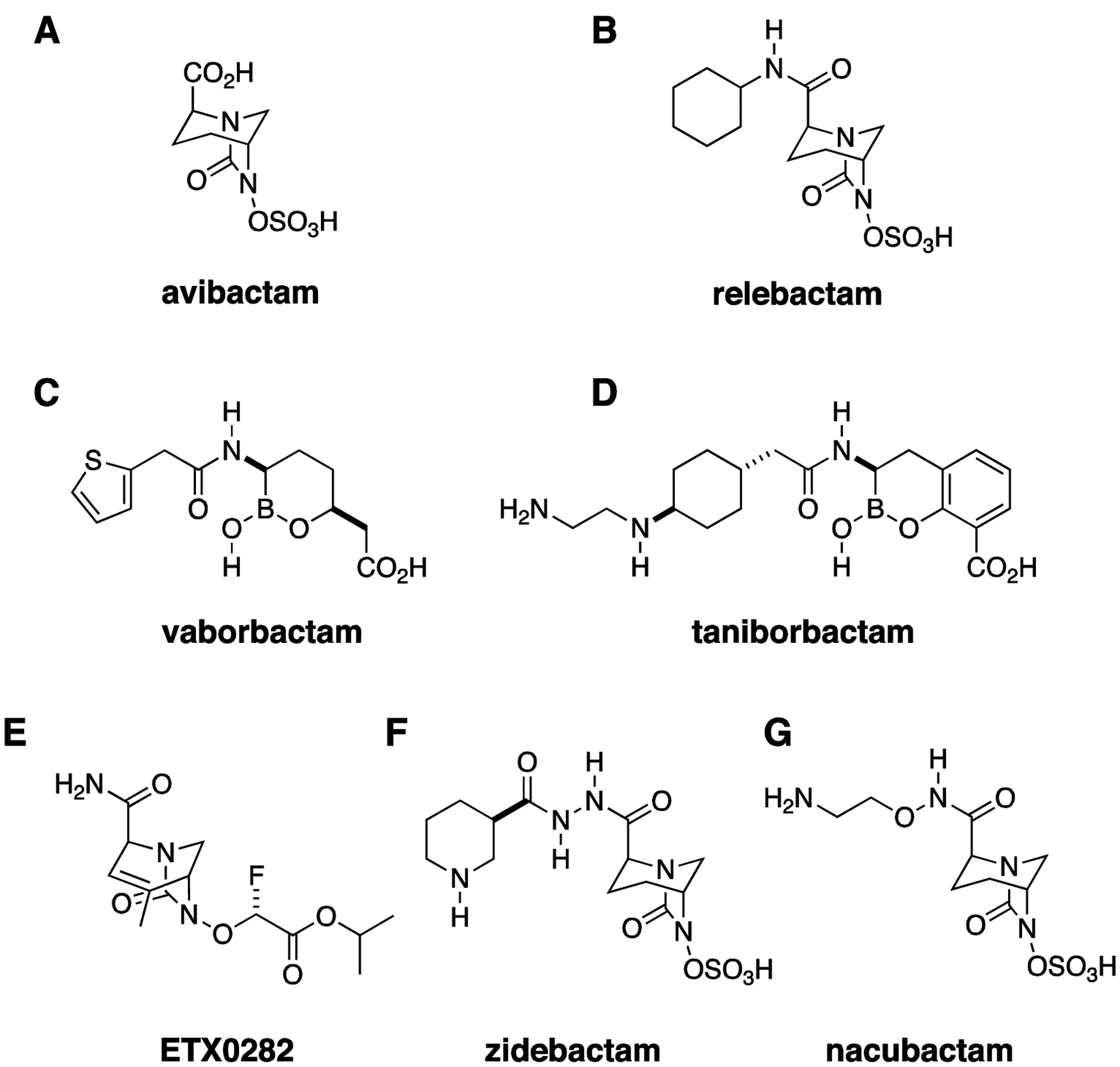
(A) Avibactam was the first approved diazabicyclooctane (DBO) β-lactamase inhibitor with activity against Ambler class A, C, and D β-lactamases. (B) Relebactam. (C) Vaborbactam, a boronic acid β-lactamase inhibitor with activity against Ambler class A and C β-lactamases. (D) Taniborbactam, a bicyclic boronic acid β-lactamase inhibitor with activity against metallo-carbapenemases. (E) ETX0282 is an orally bioavailable DBO. (F) Zidebactam and (G) nacubactam have β-lactamase inhibitory activity similar to avibactam. However, their direct penicillin binding protein inhibitory activity, which may synergize with their partner β-lactam, also confers activity against metallocarbapenemase-producing strains.
It is unsurprising, however, that resistance to even the latest and broadest-spectrum β-lactam-β-lactamase inhibitors has followed closely on the heels of their development. Emergence of resistance to ceftazidime-avibactam during treatment with this drug was reported within a year of its introduction,34 and cross-resistance between ceftazidime-avibactam and meropenem-vaborbactam has also been observed.35, 36 Meanwhile, a number of new β-lactam-β-lactamase inhibitor combinations are being actively developed. Taniborbactam (VNRX-5133, Fig. 3D), like vaborbactam, is a boronic-acid-based β-lactamase inhibitor. It is notable for being the first β-lactamase inhibitor active against MBLs.37, 38 and is currently being evaluated in combination with cefepime in a phase 3 trial for complicated urinary tract infections (https://clinicaltrials.gov/ct2/show/NCT03840148). One limitation to currently available β-lactam-β-lactamase inhibitor combinations is that there are no oral drugs in this class with activity against CRE. Oral agents facilitate decreased length of hospital admission and reduction in the risks associated with long-term IV antibiotic use, so it is encouraging that cefpodoxime-ETX0282 (Fig. 3E), an orally available β-lactam-β-lactamase inhibitor combination with in vitro activity against serine carbapenemases, including OXA-48 (https://www.entasistx.com/application/files/5615/6154/7872/Microbe_2019_McLeod_ETX0282_Final.pdf), is currently in phase 1 trials (https://www.entasistx.com/pipeline). Zidebactam and nacubactam (Figs. 3F, G) are notable new broad spectrum β-lactamase inhibitors, being investigated in combination with cefepime and meropenem, respectively, that are able to inhibit a broad range of serine carbapenemases and also demonstrate direct antimicrobial activity, through binding to penicillin binding protein (PBP) 2, and so-called enhancer effects (i.e., synergy with their β-lactam partners) that extend activity spectrum of the combinations to include metallo-β-lactamase and OXA-carbapenemase producing strains.39–42
Combination therapy beyond β-lactam-β-lactamase inhibitor combinations
The mechanism of action of β-lactam-β-lactamase inhibitor combinations is well understood, and these drugs, usually provided as pre-manufactured combinations, are routinely used to β-lactamase-producing bacteria causing various types of infections. There is also generally a straightforward correlation between in vitro AST and expected in vivo effects with these combinations: the addition of a β-lactamase inhibitor restores the activity of a β-lactam antibiotic to which a bacterial isolate is resistant by protecting the drug from the activity of bacterial β-lactamase enzymes.
However, combination therapy has often been employed to treat CRE infections even in the absence of a known mechanism of action or of robust supportive clinical data. When treating a pan-drug-resistant isolate against which no single available antibiotic has activity, such salvage approaches offer the only possible hope of effective treatment.32 However, it is less clear what benefit, if any, is provided by the use of combination therapy for CRE strains against which one or more antibiotics retain activity, which, fortunately, is still the case for most CRE isolates. Frequently invoked regimens most often include some combination of a carbapenem, a polymyxin, an aminoglycoside, or tigecycline.43 Almost all data on the clinical efficacy of these combinations is retrospective, with conflicting results.44–46 A recent study, one of the very few randomized controlled trials in treatment for carbapenem-resistant Gram-negative bacteria, compared colistin to colistin plus meropenem for treatment of carbapenem-resistant Enterobacteriaceae, Acinetobacter baumannii and Pseudomonas aeruginosa.47 Overall, combination therapy was not superior to monotherapy in terms of clinical failure at 14 days or mortality at 14 or 28 days, but the study was not powered to address outcomes in CRE specifically. The authors note that they plan to combine their data with the results of a similar ongoing trial (https://clinicaltrials.gov/ct2/show/NCT01597973) to evaluate outcomes in patients with CRE infections specifically. Fosfomycin is an older cell wall synthesis inhibitor antibiotic to which many CRE retain susceptibility.48 When oral fosfomycin (the only formulation currently available in the US) is used for treatment or prevention of lower urinary tract infections, it is generally provided as monotherapy,49 but when administered intravenously for systemic multidrug-resistant Gram-negative infections, it is almost always used in combination with other antibiotics, including colistin, tigecycline, gentamicin, and meropenem. Although small observational studies have shown clinical efficacy with these combinations,50, 51 data comparing fosfomycin monotherapy to combination therapy or different fosfomycin combination regimens to each other is lacking.
Data on the most effective combinations in vitro have also been conflicting.52 One major barrier to our understanding of the possible utility of combination therapy for CRE is that very few studies53, 54 incorporate both in vitro synergy testing data and clinical outcomes, so that it is difficult to determine whether the improved outcomes are not seen consistently with combination therapy because there is truly no benefit from such an approach or because the combinations used to treat patients in these trials were not active against the specific isolates with which they were infected. The ideal approach to investigation of this question would involve a clinical trial in which patients with CRE infections were randomized to receive either a combination that demonstrates in vitro synergy against their infecting isolate or a single active drug, but the logistical challenges of carrying out a randomized trial among the medically complex, critically ill population that comprises the majority of patients infected with CRE along with the technical difficulty of performing synergy testing by conventional methods make such a study extremely difficult to carry out. However, the inevitable increase in rates of CRE infections combined with innovations in rapid synergy testing may ultimately facilitate such an approach.55
New Discovery of Old Drugs
Synthesis around existing scaffolds has clearly been successful at addressing liabilities of existing class members, for example, the addition of side chains to β-lactams to make them more stable to carbapenemases. Several other strategies provide potential opportunities. It is apparent that many antibiotics with compelling properties as future lead candidates or candidates in their own right, but were never developed as therapeutics, have the potential to address CRE and other Gram-negative resistance. An illustrative example would be the aminoglycoside apramycin (Fig. 4A), thus far relegated to use in agriculture, but more recently found, in contrast to approved aminoglycosides, to maintain activity in the presence of circulating RNA methylases, show a low propensity for ototoxicity in in vitro models, and demonstrate a compelling activity spectrum including CRE, carbapenem-resistant A. baumannii (CRAB) and Pseudomonas aeruginosa.32, 56–59 In one survey, approximately 30% of CRE encoded the AAC(3)-IV apramycin inactivating enzyme,59 a vulnerability which could potentially be addressed through further synthetic modifications.60 An upcoming phase I clinical trial of apramycin is listed in grants.gov (https://clinicaltrials.gov/ct2/show/NCT04105205) for first in human safety exploration.
Figure 4. New Discovery with Old Drugs.
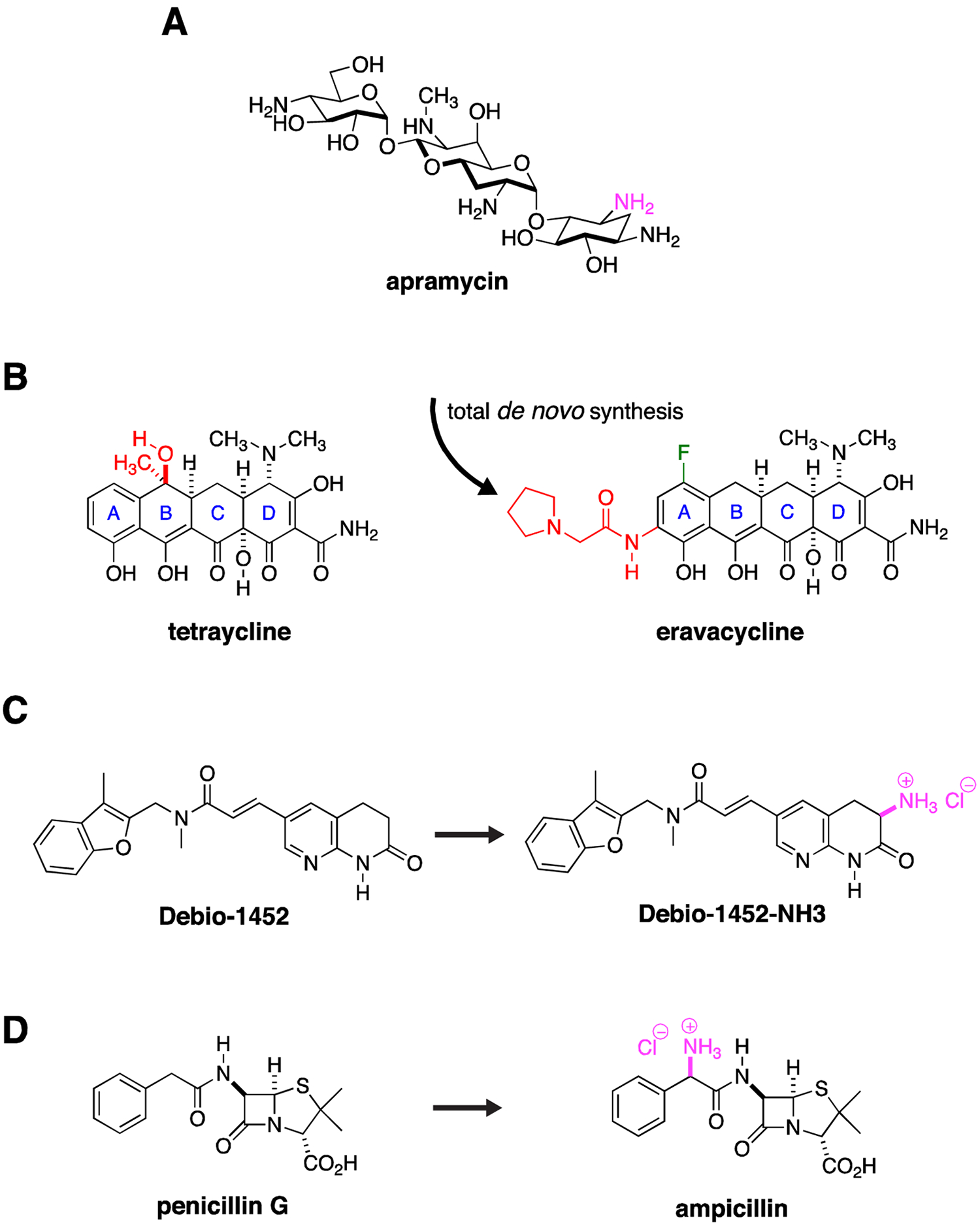
(A) Apramycin has activity against CRE. One amino group (purple) is modified by a circulating AAC(3)-IV acetylase. Derivatization around this site may significantly extend its activity spectrum by blocking acetylase activity. (B) Total de novo synthetic approaches allow exploration of a range of structural permutations not possible with semi-synthesis approaches that modify an existing natural product. The total synthesis of eravacycline allowed installation of a fluorine (green) and an pyrrolidinylacetylamino substituent (red) on the tetraycline A ring while removing methyl and hydroxy groups (red) on the tetraycline B ring, derivatization that would not have been possible by modification of the existing tetracycline scaffold. (C) The Gram-positive, FabI inhibitory activity of Debio-1452 was extended to Gram-negative pathogens through installation of a primary amine (purple), one of the so called eNTRyway rules that are associated with Gram-negative envelope penetration. (D) Similarly, the selective Gram-positive activity of penicillin G was extended to Gram-negative pathogens through installation of a primary amine (purple) to create ampicillin.
New Discovery with Existing Drugs
Cumulative advances in medicinal chemistry have also opened up further avenues for exploration. For example, increasingly more efficient total de novo synthetic techniques allow derivatization of several antibiotic natural product scaffolds to an extent not previously possible. These approaches allow exploration of permutations of already proven scaffolds against well-established antimicrobial targets such as the ribosome. Such approaches, for example, have led to the development of the fully synthetic, fluorinated tetracycline analogue, eravacycline (Fig. 4B),61 which maintains activity against the majority of CRE strains through its ability to avoid efflux and circumvent ribosome protection-based tetracycline-class resistance,62, 63 and in the future could provide a way to address inactivation by emerging high-level, tetracycline modification enzyme-based resistance.64 These total synthesis approaches distinctly allow for an increase in structural diversity of available pharmacophores through installation of unnatural functionality (e.g., fluorine) and stereochemistry (e.g., diastereoisomers). Thus through the total synthesis of novel unnatural natural product analogues and stereoisomers, unique structure-activity relationship (SAR) and stereochemical structure-activity relationship (S-SAR) studies are enabled. This latter S-SAR approach is particularly applicable for the introduction of unnatural sugars in carbohydrate natural products.65–72
Tapping Gram-positive agents for CRE
De novo synthetic strategies or more traditional semi-synthetic approaches can also be applied to extending the activity spectrum of traditional Gram-positive agents into the Gram-negative antimicrobial space with activity against CRE. The Gram-negative double cell membrane envelope presents a formidable barrier for antimicrobials with cytoplasmic targets. Studies with outer membrane permeabilizing agents or mutants with defects in the outer membrane barrier (in particular, tolC and lptD) highlight the potent Gram-negative activity of many classic Gram-positive agents when they are able to access their targets. For instance, we found that almost all CRE strains tested showed dramatic increased susceptibility to fusidic acid, linezolid, rifampin, macrolides, and lincosamides under these conditions.73 Respiratory Gram-negative pathogens with inherent lower permeability barriers also test intrinsically more susceptible to these agents.74, 75 The high rate of such permeability-enabled susceptibility in CRE suggests the absence or rarity of intrinsic or acquired resistance to Gram-positive agents, presumably as a consequence of lack of evolutionary selective pressure, and therefore provides a strong premise for exploration of modification of these Gram-positive agents to increase Gram-negative penetrance, or co-formulation with adjunctives that increase flux across the Gram negative cell membrane.76 It is possible that restoration of sufficient activity of existing β-lactams against CRE may prove achievable using the latter strategy where penetrance is limited by porin and efflux mechanisms.77
The concept of modification of Gram-positive agents has received much recent attention as rules of Gram-negative penetration are being formulated based on analysis of distinguishing Gram-negative antimicrobial properties. One such analysis defined three components of Gram-negative penetrant antimicrobials: low globularity or more planar structures, few rotatable bonds, and presence of primary amines. Together these properties define what have been called permeation or eNTRyway rules.78 Recent examples of their application have seen the conversion of a Gram-positive FabI inhibitor into a broad-spectrum inhibitor with broad Gram-negative activity, including potential, although not described, activity against CRE (Fig. 4C).79 An older and better-known example of Gram-negative conversion is the expanded spectrum conferred to the Gram-positive drug penicillin through addition of a primary amine to form ampicillin (Fig. 4D).80
New antibiotic targets
Most available antibiotics with activity against Gram-negative bacteria target a limited number of functional sites or processes: cell wall synthesis (β-lactams), protein synthesis (aminoglycosides, tetracyclines), DNA synthesis (quinolones), folate synthesis (trimethoprimsulfamethoxazole), or outer membrane integrity (polymyxins, including colistin). While a newly introduced anti-CRE agent may have a broader spectrum of activity or improved safety profile or pharmacokinetic properties relative to existing drugs in the same class, it is likely to be vulnerable to many of the same resistance mechanisms that affect other drugs in the class. Identifying and bringing to market a drug from a novel class is a lengthier and more challenging process than is optimization of a new β-lactam or tetracycline, for example, but antibiotics with novel bacterial targets may face less pre-existing resistance and thus may have a broader baseline spectrum of activity than drugs that are variants of available agents.
The outer membrane of Gram-negative bacteria is the target of the polymyxin antibiotics, including colistin, which were introduced in 194981 but had largely fallen out of favor by the early 1980s as a result of their nephrotoxicity, neurotoxicity, and low therapeutic index, as well as questions about their efficacy.25 Over the past two decades, however, they have experienced a resurgence in use as one of the few classes of drugs to which most CRE retain susceptibility,82 but emerging polymyxin resistance threatens their utility even in this role.83 Investigators and pharmaceutical companies are now starting to develop drugs that exploit vulnerabilities in outer membrane synthesis or stability in different ways. The compound SPR741, like SPR206 mentioned above, has structural similarities to polymyxins but does not demonstrate antibacterial activity on its own.76 Instead, it increases the permeability of Gram-negative bacteria, including CRE, to antibiotics such as azithromycin that otherwise have limited activity against Gram-negative cells as a result of their inability to cross the outer membrane in sufficient quantities.84 Colistin itself appears to have a similar permeabilizing effect even on colistin-resistant Gram-negative bacteria.73
Rather than directly targeting the outer membrane, compounds that inhibit the activity of the LolCDE transporter, preventing trafficking of lipoproteins to the outer membrane,85 have been shown to inhibit bacterial growth. Compounds of this type have shown activity in animal models of CRE infection during early pharmaceutical development (https://www.summitplc.com/app/uploads/2019/09/DDS-04-ASM-ESCMID-FINAL.pdf). Lipid A, part of the lipopolysaccharide component of the outer membrane, is the target of polymyxins; mutations in lipid A are responsible for polymyxin resistance. Inhibition of LpxC, an enzyme required for lipid A synthesis, presents a different method of attack against this key outer membrane component. LpxC-targeting compounds have shown antibacterial activity in in vitro studies and animal models86 and are being developed as possible therapeutic agents against CRE (https://www.abstractsonline.com/pp8/#!/7859/presentation/14971).
Most carbapenem resistance in Enterobacteriaceae is mediated by plasmid-borne carbapenemase enzymes. A compound that could inactivate or expel these plasmids would theoretically be able to reverse carbapenem resistance and allow co-treatment with narrow spectrum agents potentially avoiding extensive alterations of the microbiome associated with use of typical broad-spectrum antibiotics. Early work in this area has identified several agents with promising potential for plasmid eviction in CRE (https://www.abstractsonline.com/pp8/#!/7859/presentation/15044).
High throughput screening approaches to identify new CRE therapeutics
The existing Gram-negative antimicrobial space is dominated by natural products with complicated structures. Gram-negative antibiotics look remarkably different from human drugs. Compare the complicated structures of plazomicin, ceftazidime, eravacycline, and azithromycin (which has borderline Gram-negative activity), and to the orally bioavailable proton pump inhibitor, esomeprazole; the β-blocker, albuterol; the cholesterol lowering, hydroxymethylglutaryl coenzyme A reductase inhibitor, rosuvastatin; and pregabalin, used to treat neuropathy and epilepsy (Fig. 5). Commercial screening libraries are biased with two principles in mind - likeness to orally absorbed human drugs87 and, not surprisingly, synthetic simplicity which in general implies lipophilic and/or planar structures. Typically, commercial screening libraries comprised of compounds with a drug-like molecular molecular weight range occupy a physicochemical space different from known Gram-negative active antimicrobials.88 Therefore, it might be predicted a priori that yield of antimicrobials from screening these libraries might be low.
Figure 5. High Throughput Screening Libraries are Not Optimized to Identify CRE Therapeutics.
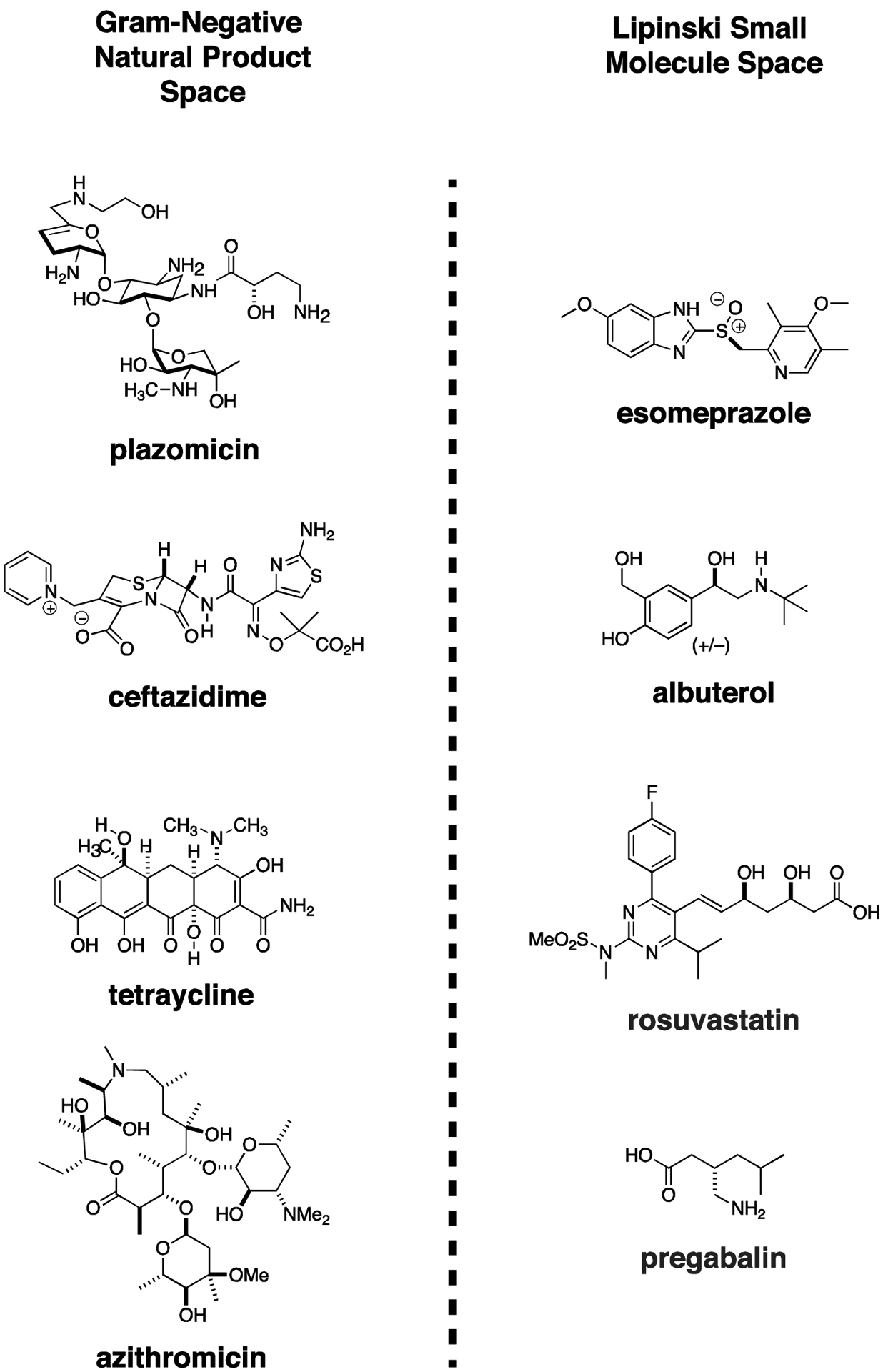
Gram-negative antibiotic natural products are complex molecules with numerous stereocenters, lack of rotatable bonds, and relatively planar structures. High throughput screening libraries used extensively in human drug discovery occupy a distinct chemical space and have not been especially productive in identifying new Gram-negative therapeutics. The difference in chemical structures between representative Gram-negative antibiotics and four of the most widely prescribed orally bioavailable human drugs can readily be appreciated. (Although azithromycin is not used as a drug for most Gram-negative infections, it is highly effective at sufficient concentrations against certain important Enterobacteriaceae species, such as Salmonella, and Shigella).
Nevertheless, in the hopes of identifying small molecules that either have direct activity against CRE or that would enhance the potency of carbapenems against CRE, we embarked on such a high throughput screening effort. A multidrug-resistant KPC-expressing Klebsiella pneumoniae was our target and the assay readout was growth inhibition.89 Our supposition was that minor antimicrobial activity could later be improved through medicinal chemistry efforts, for example, to improve antimicrobial penetrance. ~200,000 small molecules from several vendors were screened.90 Essentially no compounds had direct antimicrobial activity; only a few had carbapenem-potentiating activity. None provided dynamic and tractable lead candidates. This study illustrated that CRE are very difficult targets and highlighted the poor suitability of commercially available small molecule screening libraries for antimicrobial discovery. Undoubtedly, many prior unpublished efforts in the pharmaceutical industry and in academia seeking new Gram-negative agents through small molecule screening campaigns have been undertaken with hope for potential to discover new targets and with findings unfortunately relegated to the undisclosed graveyard of negative results.
Small molecule libraries enriched for molecules with Gram-negative antimicrobial properties would potentially provide improved yield. However, the nature of production of large numbers of such compounds is generally antithetical to the Gram-negative space; for example, small molecules with large numbers of rotatable bonds and/or with an increased planarity are much easier to prepare, while the converse is not. In the context of screening of specialized libraries of the future it may be prudent to screen with Gram-negative bacteria with decreased outer membrane barrier function to increase initial sensitivity for detection of activity. With the inherent low yield of such screening approaches, focused, hypothesis-driven derivatization, examples of which we have already described, and/or continued discovery of natural products may prove a more impactful strategy per effort expended. Although estimates of natural product discovery rate of novel antibiotics through screening soil actinomycetes has been low, this may provide an additional avenue of future CRE therapeutic discovery with appropriate automation and sampling of novel habitats.91
Non-antibiotic treatment approaches
Developing antibiotics that attack novel bacterial targets may succeed in increasing the time between introduction of the antibiotic and emergence of resistance. New derivatives of existing antibiotics may incrementally address each emerging resistance mechanism. However, in the end bacteria will find ways to develop resistance to any class of antibiotics, and treatment approaches that expand beyond the traditional small-molecule drug model offer the potential to meaningfully expand our antimicrobial armamentarium and to bypass typical forms of antimicrobial resistance.
The development of monoclonal antibody therapies has increased dramatically over the past twenty years, but most available monoclonal antibody drugs are designed for neoplastic and autoimmune conditions92, 93 while the use of antibody therapy for infectious diseases has been limited to date.94 The only antibacterial monoclonal antibodies available in the United States at present are used to treat anthrax95, 96 and to prevent recurrences of Clostridioides difficile colitis.97 In addition, hyperimmune globulin is used to treat babies with infantile botulism resulting from Clostridium botulinum neurotoxin production.98 These antibodies act by binding to the primary toxin produced by the pathogen. Most bacterial pathogens, however, including Enterobacteriaceae, cause disease through a variety of different virulence mechanisms, many of which are not toxin-mediated,99 and thus present a much more challenging target for antibody development. Approaches to developing antibodies against similarly complex bacteria have involved combinations of different antibodies and the use of bispecific antibodies that have two separate binding sites.100, 101 Antibodies can be selected to exert a variety effects upon binding to the bacterial cell, including initiation of phagocytosis, prevention of host cell binding, and toxin inhibition.100
Antibodies can also be conjugated to antibiotics. By inhibiting the activity of the antibiotic until it reaches its intended bacterial target, such conjugates may be able to enhance antibacterial activity and to slow the development of resistance by avoiding exposure of the entire microbiome to an antibiotic.102 While much recent anti-bacterial antibody investigation has focused on S. aureus,103 a monoclonal antibody has been identified that kills E. coli in vitro by targeting BamA, a required component of the β-barrel assembly machine in the Gram-negative outer membrane.104 In 2018, Diago-Navarro et al. described two monoclonal anticapsular antibodies that provided protection in in vitro and animal models against sequence type 258 carbapenem-resistant K. pneumoniae.105 In vitro investigations have explored the activity of other types of compounds complexed to antibiotics or antibiotic prodrugs in order to facilitate the drugs’ access to a specific anatomic region or bacterial population.102, 106 Nano-based delivery systems, which are currently in the early in vitro stage of investigation, offer another potential method of targeted drug distribution, chaperoning antibiotics or even bacteriophages to the interior of phagocytic cells or biofilms to attack bacteria that are normally hidden in these protected spaces.107, 108
A variety of other non-traditional approaches to treatment of bacterial infections are areas of active investigation. These approaches include the employment of bacteriophages that are highly selective against specific pathogens, in some cases targeted against the exact isolate infecting an individualized patient.109 Small investigations of the safety of phages targeted against Enterobacteriaceae have been performed in patients with diarrhea and healthy subjects in Bangladesh.110, 111 (Phage therapy is discussed in detail in the article by Chan in this edition.) Virulence inhibitors, which target the bacterial virulence factors without necessarily inhibiting or killing the organisms, can prevent bacteria from causing infection without selecting for resistance. Pinkner et al. showed, for example, that 2-pyridone compounds could inhibit biogenesis of the pili that are required by uropathogenic E. coli for adherence and establishment of urinary tract infections.112 Rasko et al. identified a small molecule compound that inhibited QseC, a membrane histidine sensor kinase that activates expression of bacterial virulence genes, in in vitro experiments.113 Should such therapies prove effective, it seems probable that their potential utility for CRE would be investigated. While Enterobacteriaceae have not typically been a target of antibacterial vaccines, recent studies are beginning to suggest a possible role for immunization against CRE, particularly K. pneumoniae. Glycoconjugate vaccines targeting capsular polysaccharides,114, 115 surface O polysaccharides,116 and outer-membrane vesicles117 of carbapenem-resistant and hypervirulent K. pneumoniae have shown immunogenicity in animal models.
Antimicrobial susceptibility testing and new antibiotics
Even when new anti-CRE antibiotics are successfully brought to market, the lack of readily available antimicrobial susceptibility testing (AST) constitutes a major barrier to their use in patients. There are numerous factors contributing to this delay. First, interpretive criteria (“breakpoints” that classify a minimal inhibitory concentration (MIC) as susceptible, intermediate, or resistant) must be established and accepted by the relevant advisory bodies in order for AST to be able to provide a clinically meaningful result. Recent harmonization of interpretive criteria promulgation between the Clinical and Laboratory Standards Institute (CLSI) and the Food and Drug Administration (FDA), have eliminated much of the delay and ambiguity surrounding adoption of breakpoints for new antibiotics and the FDA clearance of commercial AST products.118
However, clinical microbiology laboratories cannot simply commence susceptibility testing as soon as interpretive criteria and AST materials are available. The generally recognized requirements for verification studies that individual laboratories must perform prior to addition of any new drug to the lab’s AST platform119 are prohibitively time-consuming for many labs. Furthermore, such pre-implementation studies require access to isolates with a range of MICs to the new agent, as determined by a reference method; such a collection can be difficult to obtain.120 Because antimicrobial stewardship principles and the high cost of new drugs limit the number of patients for whom the newest antibiotics are used, many labs simply decide that the time and cost involved in a verification study is not warranted by the volume of use of the drug, yet for those patients infected by a multidrug-resistant organism with few or no other treatment options, the lack of readily available AST can be highly detrimental. It would be helpful if the burden of such verification requirements were lowered. We have argued elsewhere that the requirements for additional statistically unpowered pre-implementation accuracy and precision verification studies in clinical laboratories above and beyond the extensive, statistically powered studies performed by antimicrobial susceptibility test manufacturers as a pre-requisite for FDA-clearance serves no purpose, and should be eliminated to foster introduction of AST for new drugs into clinical labs. We believe that such new testing should be implemented immediately in concert with ongoing quality control testing that is a standard part of clinical microbiology laboratory practice.120
As novel types of antibiotics and antimicrobial therapeutics are introduced, they bring additional complications to AST. For example, AST for cefiderocol, the first approved siderophore antibiotic, which has broad-spectrum activity against CRE2 and was approved by the FDA in November 2019 [https://www.fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug-treat-complicated-urinary-tract-infections-part-ongoing-efforts] requires the preparation and use of iron-depleted media by chelation of standard cation-adjusted Mueller-Hinton broth followed by supplementation with specific concentrations of magnesium, calcium, and zinc ions,14, 121 a procedure that seems likely to be beyond the practical capacity of most clinical laboratories. Furthermore, the drive to develop ever more rapid AST methods, while clearly of significant clinical value, has resulted in complex test platforms to which new individual drugs cannot be readily added. Similarly, laboratories that rely heavily on molecular susceptibility testing to identify resistance genes may not be able to predict susceptibility to new drugs based on these results, because the phenotypic susceptibility patterns to a new drug of bacteria possessing various combinations of resistance mechanisms require time to define and may never be fully predictable.122 Rapid, flexible AST methods that are based on simplified or semi-automated adaptations to traditional dilution MIC or diffusion-based testing may ultimately provide the best balance between the need for fast, accurate AST results and the necessity to promptly incorporate new antibiotics into AST panels. The automated inkjet printer-based dilution testing method adopted by the Centers for Disease Control’s Antibiotic Resistance Lab Network (ARLN) (https://www.cdc.gov/drugresistance/laboratories.html) is an example of a platform that is based on traditional microdilution testing but uses automation to allow addition of new antibiotics, including initial validation and quality control procedures, in a timely manner.123, 124 Through its Expanded Antimicrobial Susceptibility Testing for Hard-to-Treat Infections (ExAST) program (https://www.cdc.gov/drugresistance/laboratories/ar-lab-network-testing-details/expanded-ast.html), the ARLN even offers susceptibility testing for the combination aztreonam-avibactam, which, as described above, has demonstrated activity against MBL-expressing organisms.
New antimicrobial modalities such as phage and antibody testing, which may only function against a subset of isolates, further complicate the testing landscape, emphasizing that the burden of AST product verification should be assumed by test manufacturers during FDA-clearance and ongoing test performance ensured by existing quality systems manufacturing regulation and practices.125, 126 These measures are already in place and should suffice. Proverbially, if new agents are brought to market, but are practically unavailable, because we cannot direct these agents to patients who will benefit through lack of available AST in local laboratories, do they really exist? AST of bacterial isolates in specialized reference laboratories is associated with up to a week delay in obtaining results and is far from an ideal solution.127 In order to save lives during the emergence of antimicrobial resistance, the regulation and thought process behind pre-implementation verification requirements for AST in local clinical laboratories must be reconsidered and minimized to remove roadblocks for making such testing (and therefore new antimicrobials that are being developed to address resistance threats such as CRE) widely availability at sites of patient care.120, 128
Acknowledgements
All authors have read the journal’s authorship agreement, have reviewed and approved the manuscript, and have read the journal’s policy on disclosure of potential conflicts of interest. All authors have disclosed any financial or personal relationships with organizations that could potentially be perceived as influencing subject matter reviewed in this article. J.E.K. is on the clinical advisory board of First Light Biosciences (Chelmsford, MA) and is the scientific co-founder of AstraDx, LLC. An HP D300 digital dispenser and supplies for synergy studies described in the manuscript were provided for use of the Kirby Laboratory by TECAN (Morrisville, NC). T.B.-K. provided consulting services to ClearPath Vaccines, LLC in 2017. None of these companies had a role in manuscript preparation or decision to publish.
Abbreviations:
- AST
antimicrobial susceptibility testing
- CDC
United States Centers for Disease Control and Prevention
- CLSI
Clinical and Laboratory Standards Institute
- CRAB
carbapenem-resistant A. baumannii
- CRE
carbapenem-resistant Enterobacteriaceae
- cryo-EM
cryogenic electron microscopy
- ESBLs
extended-spectrum β-lactamases
- FDA
United States Food and Drug Administration
- KPC
Klebsiella pneumoniae carbapenemase
- MBLs
metallo-β-lactamases
- MIC
minimal inhibitory concentration
- PBPs
penicillin binding proteins
- S-SAR
stereospecific structure-activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.CDC. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services, CDC; 2019. [Google Scholar]
- 2.Bonomo RA. Cefiderocol: A Novel Siderophore Cephalosporin Defeating Carbapenem-resistant Pathogens. Clin Infect Dis 2019;69(Supplement_7):S519–s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silver LL. Challenges of Antibacterial Discovery. Clinical Microbiology Reviews 2011;24(1):71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoddard SF, Smith BJ, Hein R, Roller BR, Schmidt TM. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res 2015;43:D593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connor M, Dahlberg AE. Isolation of spectinomycin resistance mutations in the 16S rRNA of Salmonella enterica serovar Typhimurium and expression in Escherichia coli and Salmonella. Curr Microbiol 2002;45(6):429–33. [DOI] [PubMed] [Google Scholar]
- 6.Sparling PF, Davis BD. Bactericidal action of streptomycin and comparison with spectinomycin in heterozygotes of Escherichia coli. Antimicrob Agents Chemother 1972;1(3):252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salian S, Matt T, Akbergenov R, et al. Structure-activity relationships among the kanamycin aminoglycosides: role of ring I hydroxyl and amino groups. Antimicrob Agents Chemother 2012;56(12):6104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aggen JB, Armstrong ES, Goldblum AA, et al. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother 2010;54(11):4636–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox G, Ejim L, Stogios PJ, et al. Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes. ACS Infect Dis 2018;4(6):980–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vickers A, Mushtaq S, Woodford N, Doumith M, Livermore DM. Activity of RX-04 Pyrrolocytosine Protein Synthesis Inhibitors against Multidrug-Resistant Gram-Negative Bacteria. Antimicrob Agents Chemother 2018;62(8):e00689–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer N, Neumann P, Konevega AL, et al. Structure of the E. coli ribosome-EF-Tu complex at <3 A resolution by Cs-corrected cryo-EM. Nature 2015;520(7548):567–70. [DOI] [PubMed] [Google Scholar]
- 12.Khusainov I, Vicens Q, Bochler A, et al. Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res 2016;44(21):10491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan CE, Huang W, Rudin SD, et al. Cryo-EM structure of the Acinetobacter baumannii 70S ribosome and implications for new antibiotic development. mBio 2020;11(1):e03117–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamano Y In Vitro Activity of Cefiderocol Against a Broad Range of Clinically Important Gram-negative Bacteria. Clin Infect Dis 2019;69(Supplement_7):S544–s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Page MGP. The Role of Iron and Siderophores in Infection, and the Development of Siderophore Antibiotics. Clin Infect Dis 2019;69(Supplement_7):S529–s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato T, Yamawaki K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin Infect Dis 2019;69(Supplement_7):S538–s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhanel GG, Golden AR, Zelenitsky S, et al. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs 2019;79(3):271–89. [DOI] [PubMed] [Google Scholar]
- 18.Shionogi Inc. Antimicrobial Drugs Advisory Committee: Cefiderocol Briefing Document, NDA # 209445. October 16, 2019. Accessed January 5, 2020 https://www.fda.gov/media/131705/download.
- 19.Reck F, Bermingham A, Blais J, et al. Optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae - Identification of LYS228. Bioorg Med Chem Lett 2018;28(4):748–55. [DOI] [PubMed] [Google Scholar]
- 20.Blais J, Lopez S, Li C, et al. In Vitro Activity of LYS228, a Novel Monobactam Antibiotic, against Multidrug-Resistant Enterobacteriaceae. Antimicrob Agents Chemother 2018;62(10): e00552–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dean CR, Barkan DT, Bermingham A, et al. Mode of Action of the Monobactam LYS228 and Mechanisms Decreasing In Vitro Susceptibility in Escherichia coli and Klebsiella pneumoniae. Antimicrob Agents Chemother 2018;62(10):e01200–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones AK, Ranjitkar S, Lopez S, et al. Impact of Inducible blaDHA-1 on Susceptibility of Klebsiella pneumoniae Clinical Isolates to LYS228 and Identification of Chromosomal mpl and ampD Mutations Mediating Upregulation of Plasmid-Borne blaDHA-1 Expression. Antimicrob Agents Chemother 2018;62(10):e01202–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akhoundsadegh N, Belanger CR, Hancock REW. Outer Membrane Interaction Kinetics of New Polymyxin B Analogs in Gram-Negative Bacilli. Antimicrob Agents Chemother 2019;63(10):e00935–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown P, Abbott E, Abdulle O, et al. Design of Next Generation Polymyxins with Lower Toxicity: The Discovery of SPR206. ACS Infect Dis 2019;5(10):1645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falagas ME, Kasiakou SK. Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin Infect Dis 2005;40(9):1333–41. [DOI] [PubMed] [Google Scholar]
- 26.Bush K, Bradford PA. Beta-Lactams and Beta-Lactamase Inhibitors: An Overview. Cold Spring Harb Perspect Med 2016;6(8):a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev 2010;23(1):160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bush K A resurgence of beta-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents 2015;46(5):483–93. [DOI] [PubMed] [Google Scholar]
- 29.Tamma PD, Hsu AJ. Defining the Role of Novel beta-Lactam Agents That Target Carbapenem-Resistant Gram-Negative Organisms. J Pediatric Infect Dis Soc 2019;8(3):251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Bano J, Gutierrez-Gutierrez B, Machuca I, Pascual A. Treatment of Infections Caused by Extended-Spectrum-Beta-Lactamase-, AmpC-, and Carbapenemase-Producing Enterobacteriaceae. Clin Microbiol Rev 2018;31(2):e00079–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen LF, Todd R, Kiehlbauch J, Walters M, Kallen A. Notes from the Field: Pan-Resistant New Delhi Metallo-Beta-Lactamase-Producing Klebsiella pneumoniae - Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep 2017;68(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brennan-Krohn T, Kirby JE. Synergistic Combinations and Repurposed Antibiotics Active against the Pandrug-Resistant Klebsiella pneumoniae Nevada Strain. Antimicrob Agents Chemother 2019;63(9):e01374–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall S, Hujer AM, Rojas LJ, et al. Can Ceftazidime-Avibactam and Aztreonam Overcome beta-Lactam Resistance Conferred by Metallo-beta-Lactamases in Enterobacteriaceae? Antimicrob Agents Chemother 2017;61(4):e02243–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shields RK, Potoski BA, Haidar G, et al. Clinical Outcomes, Drug Toxicity, and Emergence of Ceftazidime-Avibactam Resistance Among Patients Treated for Carbapenem-Resistant Enterobacteriaceae Infections. Clin Infect Dis 2016;63(12):1615–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hackel MA, Lomovskaya O, Dudley MN, Karlowsky JA, Sahm DF. In Vitro Activity of Meropenem-Vaborbactam against Clinical Isolates of KPC-Positive Enterobacteriaceae. Antimicrob Agents Chemother 2018;62(1):e01904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaibani P, Re MC, Campoli C, Viale PL, Ambretti S. Bloodstream infection caused by KPC-producing Klebsiella pneumoniae resistant to ceftazidime/avibactam: epidemiology and genomic characterization. Clin Microbiol Infect 2019:pii: S1198–743X(19)30610-X. [DOI] [PubMed] [Google Scholar]
- 37.Liu B, Trout REL, Chu GH, et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-beta-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J Med Chem 2019:[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamrick JC, Docquier JD, Uehara T, et al. VNRX-5133 (Taniborbactam), a broad-spectrum inhibitor of serine- and metallo-beta-lactamases, restores activity of cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob Agents Chemother 2019:AAC.01963–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monogue ML, Tabor-Rennie J, Abdelraouf K, Nicolau DP. In Vivo Efficacy of WCK 5222 (Cefepime-Zidebactam) against Multidrug-Resistant Pseudomonas aeruginosa in the Neutropenic Murine Thigh Infection Model. Antimicrob Agents Chemother 2019;63(7):e00233–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnes MD, Taracila MA, Good CE, et al. Nacubactam Enhances Meropenem Activity against Carbapenem-Resistant Klebsiella pneumoniae Producing KPC. Antimicrob Agents Chemother 2019;63(8):e00432–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mushtaq S, Vickers A, Woodford N, Haldimann A, Livermore DM. Activity of nacubactam (RG6080/OP0595) combinations against MBL-producing Enterobacteriaceae. J Antimicrob Chemother 2019;74(4):953–60. [DOI] [PubMed] [Google Scholar]
- 42.Sader HS, Rhomberg PR, Flamm RK, Jones RN, Castanheira M. WCK 5222 (cefepime/zidebactam) antimicrobial activity tested against Gram-negative organisms producing clinically relevant beta-lactamases. J Antimicrob Chemother 2017;72(6):1696–703. [DOI] [PubMed] [Google Scholar]
- 43.Tamma PD, Cosgrove SE, Maragakis LL. Combination therapy for treatment of infections with Gram-negative bacteria. Clin Microbiol Rev 2012;25(3):450–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tumbarello M, Trecarichi EM, De Rosa FG, et al. Infections caused by KPC-producing Klebsiella pneumoniae: differences in therapy and mortality in a multicentre study. J Antimicrob Chemother 2015;70(7):2133–43. [DOI] [PubMed] [Google Scholar]
- 45.Gutierrez-Gutierrez B, Salamanca E, de Cueto M, et al. Effect of appropriate combination therapy on mortality of patients with bloodstream infections due to carbapenemase-producing Enterobacteriaceae (INCREMENT): a retrospective cohort study. Lancet Infect Dis 2017;17(7):726–34. [DOI] [PubMed] [Google Scholar]
- 46.Durante-Mangoni E, Andini R, Zampino R. Management of carbapenem-resistant Enterobacteriaceae infections. Clin Microbiol Infect 2019;25(8):943–50. [DOI] [PubMed] [Google Scholar]
- 47.Paul M, Daikos GL, Durante-Mangoni E, et al. Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: an open-label, randomised controlled trial. Lancet Infect Dis 2018;18(4):391–400. [DOI] [PubMed] [Google Scholar]
- 48.Falagas ME, Vouloumanou EK, Samonis G, Vardakas KZ. Fosfomycin. Clin Microbiol Rev 2016;29(2):321–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falagas ME, Vouloumanou EK, Togias AG, et al. Fosfomycin versus other antibiotics for the treatment of cystitis: a meta-analysis of randomized controlled trials. J Antimicrob Chemother 2010;65(9):1862–77. [DOI] [PubMed] [Google Scholar]
- 50.Pontikis K, Karaiskos I, Bastani S, et al. Outcomes of critically ill intensive care unit patients treated with fosfomycin for infections due to pandrug-resistant and extensively drug-resistant carbapenemase-producing Gram-negative bacteria. Int J Antimicrob Agents 2014;43(1):52–9. [DOI] [PubMed] [Google Scholar]
- 51.Michalopoulos A, Virtzili S, Rafailidis P, Chalevelakis G, Damala M, Falagas ME. Intravenous fosfomycin for the treatment of nosocomial infections caused by carbapenem-resistant Klebsiella pneumoniae in critically ill patients: a prospective evaluation. Clin Microbiol Infect 2010;16(2):184–6. [DOI] [PubMed] [Google Scholar]
- 52.Brennan-Krohn T, Kirby JE. When One Drug Is Not Enough: Context, Methodology, and Future Prospects in Antibacterial Synergy Testing. Clin Lab Med 2019;39(3):345–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Maio Carrillho CM, Gaudereto JJ, Martins RC, et al. Colistin-resistant Enterobacteriaceae infections: clinical and molecular characterization and analysis of in vitro synergy. Diagn Microbiol Infect Dis 2017;87(3):253–57. [DOI] [PubMed] [Google Scholar]
- 54.Oliva A, Scorzolini L, Castaldi D, et al. Double-carbapenem regimen, alone or in combination with colistin, in the treatment of infections caused by carbapenem-resistant Klebsiella pneumoniae (CR-Kp). J Infect 2017;74(1):103–06. [DOI] [PubMed] [Google Scholar]
- 55.Brennan-Krohn T, Truelson KA, Smith KP, Kirby JE. Screening for synergistic activity of antimicrobial combinations against carbapenem-resistant Enterobacteriaceae using inkjet printer-based technology. J Antimicrob Chemother 2017;72(10):2775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juhas M, Widlake E, Teo J, et al. In vitro activity of apramycin against multidrug-, carbapenem- and aminoglycoside-resistant Enterobacteriaceae and Acinetobacter baumannii. J Antimicrob Chemother 2019;74(4):944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang AD, Smith KP, Berg AH, et al. Efficacy of Apramycin against Multidrug-Resistant Acinetobacter baumannii in the Murine Neutropenic Thigh Model. Antimicrob Agents Chemother 2018:e02585–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang AD, Smith KP, Eliopoulos GM, Berg AH, McCoy C, Kirby JE. In vitro Apramycin Activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Diagn Microbiol Infect Dis 2017;88(2):188–91. [DOI] [PubMed] [Google Scholar]
- 59.Smith KP, Kirby JE. Evaluation of apramycin activity against carbapenem-resistant and -susceptible strains of Enterobacteriaceae. Diagn Microbiol Infect Dis 2016;86(4):439–41. [DOI] [PubMed] [Google Scholar]
- 60.Quirke JCK, Rajasekaran P, Sarpe VA, et al. Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J Am Chem Soc 2019;142(1):530–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu F, Myers AG. Development of a platform for the discovery and practical synthesis of new tetracycline antibiotics. Curr Opin Chem Biol 2016;32:48–57. [DOI] [PubMed] [Google Scholar]
- 62.Livermore DM, Mushtaq S, Warner M, Woodford N. In Vitro Activity of Eravacycline against Carbapenem-Resistant Enterobacteriaceae and Acinetobacter baumannii. Antimicrob Agents Chemother 2016;60(6):3840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arenz S, Nguyen F, Beckmann R, Wilson DN. Cryo-EM structure of the tetracycline resistance protein TetM in complex with a translating ribosome at 3.9-A resolution. Proc Natl Acad Sci USA 2015;112(17):5401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He T, Wang R, Liu D, et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat Microbiol 2019;4(9):1450–56. [DOI] [PubMed] [Google Scholar]
- 65.Zhou M, O’Doherty G. De Novo Asymmetric Synthesis of Digitoxin via a Palladium Catalyzed Glycosylation Reaction. Org Lett 2006;8:4339–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Borisova SA, Guppi SR, Kim HJ, et al. A de novo approach to the synthesis of glycosylated methymycin analogues with structural and stereochemical diversity. Org Lett 2010;12(22):5150–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou M, O’Doherty G. The de novo synthesis of oligosaccharides: application to the medicinal chemistry SAR-study of digitoxin. Curr Top Med Chem 2008;8(2):114–25. [DOI] [PubMed] [Google Scholar]
- 68.Azalim P, do Monte FM, Rendeiro MM, et al. Conformational states of the pig kidney Na(+)/K(+)-ATPase differently affect bufadienolides and cardenolides: A directed structure-activity and structure-kinetics study. Biochem Pharmacol 2020;171:113679. [DOI] [PubMed] [Google Scholar]
- 69.Mulzer M, Tiegs BJ, Wang Y, Coates GW, O’Doherty GA. Total synthesis of tetrahydrolipstatin and stereoisomers via a highly regio- and diastereoselective carbonylation of epoxyhomoallylic alcohols. J Am Chem Soc 2014;136(30):10814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goins CM, Sudasinghe TD, Liu X, Wang Y, O’Doherty GA, Ronning DR. Characterization of Tetrahydrolipstatin and Stereoderivatives on the Inhibition of Essential Mycobacterium tuberculosis Lipid Esterases. Biochemistry 2018;57(16):2383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Y, O’Doherty GA. Cryptocaryols A and B: total syntheses, stereochemical revision, structure elucidation, and structure-activity relationship. J Am Chem Soc 2013;135(25):9334–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aljahdali AZ, Foster KA, O’Doherty GA. The asymmetric syntheses of cryptocaryols A and B. Chem Commun (Camb) 2018;54:3428–35. [DOI] [PubMed] [Google Scholar]
- 73.Brennan-Krohn T, Pironti A, Kirby JE. Synergistic Activity of Colistin-Containing Combinations against Colistin-Resistant Enterobacteriaceae. Antimicrob Agents Chemother 2018;62(10):e00873–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chiaraviglio L, Kirby JE. High-Throughput Intracellular Antimicrobial Susceptibility Testing of Legionella pneumophila. Antimicrob Agents Chemother 2015;59(12):7517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kang YS, Kirby JE. A Chemical Genetics Screen Reveals Influence of p38 Mitogen-Activated Protein Kinase and Autophagy on Phagosome Development and Intracellular Replication of Brucella neotomae in Macrophages. Infect Immun 2019;87(8):e00044–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corbett D, Wise A, Langley T, et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob Agents Chemother 2017;61(8):e00200–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mendes RE, Rhomberg PR, Lister T, Cotroneo N, Rubio A, Flamm RK. Evaluation of Antimicrobial Effects of a New Polymyxin Molecule (SPR741) When Tested in Combination with a Series of beta-Lactam Agents Against a Challenge Set of Gram-Negative Pathogens. Microb Drug Resist 2019: [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 78.Richter MF, Drown BS, Riley AP, et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017;545:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parker EN, Drown BS, Geddes EJ, et al. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat Microbiol 2019;5(1):67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Antonoplis A, Zang X, Wegner T, Wender PA, Cegelski L. Vancomycin-Arginine Conjugate Inhibits Growth of Carbapenem-Resistant E. coli and Targets Cell-Wall Synthesis. ACS Chem Biol 2019;14(9):2065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ordooei Javan A, Shokouhi S, Sahraei Z. A review on colistin nephrotoxicity. Eur J Clin Pharmacol 2015;71(7):801–10. [DOI] [PubMed] [Google Scholar]
- 82.Nation RL, Li J. Colistin in the 21st century. Curr Opin Infect Dis 2009;22(6):535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ah YM, Kim AJ, Lee JY. Colistin resistance in Klebsiella pneumoniae. Int J Antimicrob Agents 2014;44(1):8–15. [DOI] [PubMed] [Google Scholar]
- 84.Stainton SM, Abdelraouf K, Utley L, Pucci MJ, Lister T, Nicolau DP. Assessment of the In Vivo Activity of SPR741 in Combination with Azithromycin against Multidrug-Resistant Enterobacteriaceae Isolates in the Neutropenic Murine Thigh Infection Model. Antimicrob Agents Chemother 2018;62(7):e00239–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nickerson NN, Jao CC, Xu Y, et al. A Novel Inhibitor of the LolCDE ABC Transporter Essential for Lipoprotein Trafficking in Gram-Negative Bacteria. Antimicrob Agents Chemother 2018;62(4):e02151–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tomaras AP, McPherson CJ, Kuhn M, et al. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in Gram-negative pathogens. mBio 2014;5(5):e01551–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental And Computational Approaches To Estimate Solubility And Permeability In Drug Discovery And Development Settings. Adv Drug Deliv Rev 2001;46(1–3):3–26. [DOI] [PubMed] [Google Scholar]
- 88.O’Shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 2008;51(10):2871–8. [DOI] [PubMed] [Google Scholar]
- 89.Smith KP, Kirby JE. Validation of a High-Throughput Screening Assay for Identification of Adjunctive and Directly Acting Antimicrobials Targeting Carbapenem-Resistant Enterobacteriaceae. Assay Drug Dev Technol 2016;14(3):194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith KP, Dowgiallo MG, Chiaraviglio L, et al. A Whole-Cell Screen for Adjunctive and Direct Antimicrobials Active against Carbapenem-Resistant Enterobacteriaceae. SLAS Discov 2019;24(8):842–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baltz RH. Natural product drug discovery in the genomic era: realities, conjectures, misconceptions, and opportunities. J Ind Microbiol Biotechnol 2019;46(3–4):281–99. [DOI] [PubMed] [Google Scholar]
- 92.Liu JK. The history of monoclonal antibody development - Progress, remaining challenges and future innovations. Ann Med Surg (Lond) 2014;3(4):113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010;9(10):767–74. [DOI] [PubMed] [Google Scholar]
- 94.Pelfrene E, Mura M, Cavaleiro Sanches A, Cavaleri M. Monoclonal antibodies as anti-infective products: a promising future? Clin Microbiol Infect 2019;25(1):60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Migone TS, Subramanian GM, Zhong J, et al. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med 2009;361(2):135–44. [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto BJ, Shadiack AM, Carpenter S, et al. Obiltoxaximab Prevents Disseminated Bacillus anthracis Infection and Improves Survival during Pre- and Postexposure Prophylaxis in Animal Models of Inhalational Anthrax. Antimicrob Agents Chemother 2016;60(10):5796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for Prevention of Recurrent Clostridium difficile Infection. N Engl J Med 2017;376(4):305–17. [DOI] [PubMed] [Google Scholar]
- 98.Payne JR, Khouri JM, Jewell NP, Arnon SS. Efficacy of Human Botulism Immune Globulin for the Treatment of Infant Botulism: The First 12 Years Post Licensure. J Pediatr 2018;193:172–77. [DOI] [PubMed] [Google Scholar]
- 99.Croxen MA, Finlay BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 2010;8(1):26–38. [DOI] [PubMed] [Google Scholar]
- 100.Wagner EK, Maynard JA. Engineering therapeutic antibodies to combat infectious diseases. Curr Opin Chem Eng 2018;19:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DiGiandomenico A, Keller AE, Gao C, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med 2014;6(262):262ra155. [DOI] [PubMed] [Google Scholar]
- 102.Cheng AV, Wuest WM. Signed, Sealed, Delivered: Conjugate and Prodrug Strategies as Targeted Delivery Vectors for Antibiotics. ACS Infect Dis 2019;5(6):816–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lehar SM, Pillow T, Xu M, et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015;527(7578):323–8. [DOI] [PubMed] [Google Scholar]
- 104.Storek KM, Auerbach MR, Shi H, et al. Monoclonal antibody targeting the beta-barrel assembly machine of Escherichia coli is bactericidal. Proc Natl Acad Sci U S A 2018;115(14):3692–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diago-Navarro E, Motley MP, Ruiz-Perez G, et al. Novel, Broadly Reactive Anticapsular Antibodies against Carbapenem-Resistant Klebsiella pneumoniae Protect from Infection. mBio 2018;9(2):e00091–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Albayati ZA, Sunkara M, Schmidt-Malan SM, et al. Novel Bone-Targeting Agent for Enhanced Delivery of Vancomycin to Bone. Antimicrob Agents Chemother 2015;60(3):1865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang L, Lin J, Taggart CC, Bengoechea JA, Scott CJ. Nanodelivery strategies for the treatment of multidrug-resistant bacterial infections. J Interdiscip Nanomed 2018;3(3):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singla S, Harjai K, Katare OP, Chhibber S. Encapsulation of Bacteriophage in Liposome Accentuates Its Entry in to Macrophage and Shields It from Neutralizing Antibodies. PLoS One 2016;11(4):e0153777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schooley RT, Biswas B, Gill JJ, et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob Agents Chemother 2017;61(10):e00954–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sarker SA, Sultana S, Reuteler G, et al. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine 2016;4:124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCallin S, Alam Sarker S, Barretto C, et al. Safety analysis of a Russian phage cocktail: from metagenomic analysis to oral application in healthy human subjects. Virology 2013;443(2):187–96. [DOI] [PubMed] [Google Scholar]
- 112.Pinkner JS, Remaut H, Buelens F, et al. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci USA 2006;103(47):17897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rasko DA, Moreira CG, Li de R, et al. Targeting QseC signaling and virulence for antibiotic development. Science 2008;321(5892):1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seeberger PH, Pereira CL, Khan N, et al. A Semi-Synthetic Glycoconjugate Vaccine Candidate for Carbapenem-Resistant Klebsiella pneumoniae. Angew Chem Int Ed Engl 2017;56(45):13973–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feldman MF, Mayer Bridwell AE, Scott NE, et al. A promising bioconjugate vaccine against hypervirulent Klebsiella pneumoniae. Proc Natl Acad Sci USA 2019;116(37):18655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hegerle N, Choi M, Sinclair J, et al. Development of a broad spectrum glycoconjugate vaccine to prevent wound and disseminated infections with Klebsiella pneumoniae and Pseudomonas aeruginosa. PLoS One 2018;13(9):e0203143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu G, Ji H, Guo X, et al. Nanoparticle reinforced bacterial outer-membrane vesicles effectively prevent fatal infection of carbapenem-resistant Klebsiella pneumoniae. Nanomedicine 2019;24:102148. [DOI] [PubMed] [Google Scholar]
- 118.Humphries RM, Ferraro MJ, Hindler JA. Impact of 21st Century Cures Act on Breakpoints and Commercial Antimicrobial Susceptibility Test Systems: Progress and Pitfalls. J Clin Microbiol 2018;56(5):e00139–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clark RB, Lewisnski ML, Loeffelholtz MJ, Tibbets RJ. Verification and Validation of Procedures in the Clinical Microbiology Laboratory In: Sharp SE, editor. Cumitech. Washington, D.C.: American Society of Microbiology; 2009. [Google Scholar]
- 120.Kirby JE, Brennan-Krohn T, Smith KP. Bringing Antimicrobial Susceptibility Testing for New Drugs into the Clinical Laboratory: Removing Obstacles in Our Fight against Multidrug-Resistant Pathogens. J Clin Microbiol 2019;57(12):e01270–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing; twenty-ninth informational supplement. CLSI document M100-S29 Wayne, PA: Clinical and Laboratory Standards Institute; 2019. [Google Scholar]
- 122.Nordmann P, Poirel L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin Infect Dis 2019;69(Supplement_7):S521–S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smith KP, Kirby JE. Verification of an Automated, Digital Dispensing Platform for At-Will Broth Microdilution-Based Antimicrobial Susceptibility Testing. J Clin Microbiol 2016;54(9):2288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brennan-Krohn T, Kirby JE. Antimicrobial Synergy Testing by the Inkjet Printer-assisted Automated Checkerboard Array and the Manual Time-kill Method. J Vis Exp 2019(146). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.U.S. Food and Drug Administration. Guidance for Industry and FDA: Class II Special Controls Guidance Document: Antimicrobial Susceptibility Test (AST) Systems, <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/class-ii-special-controls-guidance-document-antimicrobial-susceptibility-test-ast-systems>; Published 2009. Accessed July 27, 2019.
- 126.Code of Federal Regulations. Quality System Regulation, Part 820, 21CFR820; Accessed December 26, 2019. [Google Scholar]
- 127.Smith KP, Kirby JE. Rapid Susceptibility Testing Methods. Clin Lab Med 2019;39(3):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee RA, Kirby JE. We Cannot Do It Alone: The Intersection of Public Health, Public Policy, and Clinical Microbiology. Clin Lab Med 2019;39(3):499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shields RK, Chen L, Cheng S, et al. Emergence of Ceftazidime-Avibactam Resistance Due to Plasmid-Borne blaKPC-3 Mutations during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob Agents Chemother 2017;61(3):e02097–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Niu S, Wei J, Zou C, et al. In vitro selection of aztreonam/avibactam resistance in dual-carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother 2020;75(3):559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Past Bush K. and Present Perspectives on beta-Lactamases. Antimicrob Agents Chemother 2018;62(10):e01076–18. [DOI] [PMC free article] [PubMed] [Google Scholar]