

Abstract
High-density lipoprotein (HDL) metabolism is facilitated in part by scavenger receptor class B, type 1 (SR-B1) that mediates HDL uptake into cells. Higher levels of HDL have been associated with protection in other diseases, however, its role in prostate cancer is not definitive. SR-B1 is up-regulated in prostate cancer tissue, suggesting a possible role of this receptor in tumor progression. Here, we report that knockout (KO) of SR-B1 in both human and mouse prostate cancer cell lines through CRISPR/Cas9-mediated genome editing reduces HDL uptake into the prostate cancer cells and reduces their proliferation in response to HDL. In vivo studies using syngeneic SR-B1 WT (SR-B1+/+) and SR-B1 KO (SR-B1−/−) prostate cancer cells in WT and apolipoprotein-AI KO (apoA1-KO) C57BL/6J mice revealed that WT hosts, containing higher levels of total and HDL-cholesterol, grew larger tumors than apoA1-KO hosts with lower levels of total and HDL-cholesterol. Furthermore, SR-B1−/− prostate cancer cells formed smaller tumors in WT hosts than SR-B1+/+ cells in the same host model. Increased tumor volume was overall associated with reduced survival. We conclude that knocking out SR-B1 in prostate cancer tumors reduces HDL-associated increases in prostate cancer cell proliferation and disease progression.
Keywords: cell proliferation; high-density lipoprotein (HDL); lipoprotein metabolism; lipoprotein receptor; prostate cancer; scavenger receptor; CRISPR/Cas; SCARB1; scavenger receptor class B, type 1
Introduction
Prostate cancer is the most common malignancy and second leading cause of cancer-related deaths among men in the United States (1). The association of high-density lipoprotein (HDL) levels with prostate cancer risk has been inconsistent, with some studies showing a positive association (2, 3), some showing an inverse association (4, 5), and others showing no association (6, 7). HDL biogenesis is mediated by ABC transporter A1 (ABCA1), which assembles cellular lipids with exogenous lipid-poor apolipoprotein A1 (apoA1) to generate nascent HDL (8). HDL uptake in tissues is facilitated by SR-B1 (9), which can also mediate bidirectional cholesterol transport between cells and HDL (10–13). SR-B1, encoded by the SCARB1 gene, is highly expressed in the liver, and even more so in steroidogenic tissues, such as the adrenals, testes, and ovaries where it mediates cholesterol uptake, to promote cholesterol ester (CE) storage used for steroid hormone synthesis (14, 15).
It has been reported that prostate cancer accumulates CE in lipid droplets that correlates with prostate cancer aggressiveness (16). This phenotype was attributed to phosphatase and tensin homolog (PTEN) deletion that ultimately resulted in up-regulation of the LDL receptor (LDLR) and subsequent uptake of LDL cholesterol (16). SR-B1 is inducible by androgens in human hepatoma cells and primary monocyte macrophage (17). Moreover, reports suggest that androgens during puberty are responsible for lower HDL levels in men versus women, most likely due to higher hepatic SR-B1 levels (18–21). A prior study found that SR-B1 is up-regulated in high grade versus low grade prostate cancer, and in metastatic versus primary prostate cancer, whereas the LDLR was not altered in high grade or metastatic prostate cancer (22). Furthermore, it was shown that high versus low SR-B1 expression in prostatectomy specimens was associated with decreased progression-free survival (22). In a small study, SR-B1 mRNA levels were significantly higher in prostate cancer tissue versus matched normal prostate tissue (23). In our study, we examined the effect of HDL on prostate cancer cell growth, proliferation, and tumor progression. We found that HDL, in an SR-B1–dependent manner, promoted increased prostate cancer cell growth in vitro. In a syngeneic mouse model, a high HDL environment promoted tumor progression in an SR-B1–dependent manner. These results suggest that SR-B1 and HDL uptake promote prostate cancer progression and that inhibiting HDL uptake may be a viable target for decreasing disease burden.
Results
SCARB1 is up-regulated in human prostate cancer
Previous studies have focused on lipoprotein receptors in prostate cancer, and how their changes may influence cholesterol transport in prostate cancer (16, 22–24). Therefore, we evaluated expression of SR-B1, LDLR, ABCA1, and ABCG1 in RNA-seq data from a total of 52 normal prostate and 498 prostate cancer tumor tissues from the TCGA PRAD dataset (25). Because the LDLR expression levels in normal prostates were not normally distributed, nonparametric statistics were used for all gene expression data. The median log2 expression levels of SCARB1 mRNA in normal prostate and prostate cancer were 9.82 and 10.48, respectively, representing a 58% increase in SR-B1 mRNA in prostate cancer (p < 0.0001, Fig. 1). In contrast, LDLR mRNA was 31.5% lower (p = 0.0006; Fig. 1) in prostate cancer tissue, congruent with a previous report (23). The mRNA for the cholesterol efflux protein ABCA1 was unchanged between normal and prostate cancer tissue, whereas ABCG1 expression was 77% higher in prostate cancer (p < 0.0001; Fig. 1).
Figure 1.
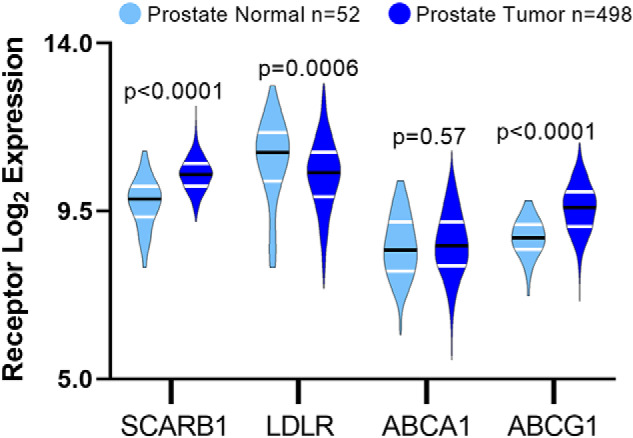
Expression of lipoprotein metabolism genes in normal prostate and prostate tumor tissue. mRNA expression of SCARB1, LDLR, ABCA1, and ABCG1 analyzed from TCGA PRAD RNA-Seq data (n = 52 normal samples and n = 498 tumor samples; median values shown (black line) and interquartiles (white lines); Mann-Whitney nonpaired, nonparametric test p values displayed).
HDL increased cell proliferation and cholesterol levels in prostate cancer cells
Since other studies (22, 23) and our analysis of the TCGA data set showed up-regulation of SR-B1 in prostate cancer (Fig. 1), we hypothesized that HDL may drive an increase of total cholesterol levels, similarly to what was previously illustrated for LDL (16). WT human DU145 and mouse TRAMP-C2, expressing SR-B1 (SR-B1+/+), prostate cancer cells were incubated with 200 μg/ml of HDL for 2 days leading to 48 (p < 0.01) and 15% (p < 0.05) increases in total cellular cholesterol levels (Fig. 2, A and B). We next determined if HDL could promote proliferation of prostate cancer cells by evaluating the impact on cell number. DU145 and TRAMP-C2 cells were treated with or without 300 μg/ml of HDL in LPDS for 4 days. HDL induced 29 (p = 0.0061) and 68% (p < 0.0001) increases in cell number in DU145 and TRAMP-C2 cells, respectively (Fig. 3, A and B).
Figure 2.
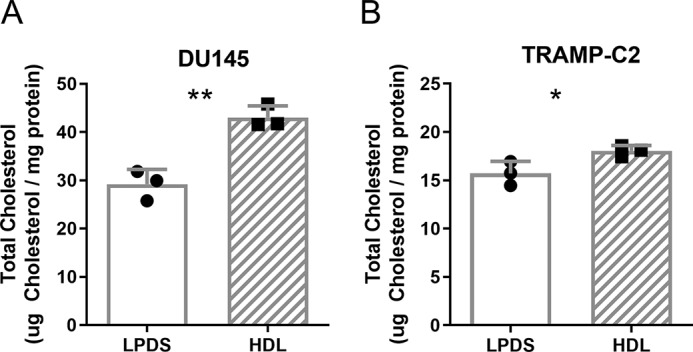
HDL effects on total cholesterol levels in prostate cancer cells. A, human DU145, and B, mouse TRAMP-C2 cells were incubated with ±200 μg/ml of HDL for 2 days in LPDS and total cholesterol levels normalized to the cell protein determined (n = 3; mean ± S.D.; *, p < 0.05; **, p < 0.01, by t test).
Figure 3.

HDL effects on cell accumulation and proliferation of prostate cancer cells. A, human DU145 (n = 5 over 2 independent experiments), and B, mouse TRAMP-C2 (n = 6 over 3 independent experiments) cells were treated ±300 μg/ml of HDL in LPDS media for 4 days and the final cell counts were normalized to the LPDS control. Values are expressed as the mean ± S.D.; **, p < 0.01; ****, p < 0.0001 by t test.
SR-B1 is required for HDL-mediated prostate cancer growth in vitro
Due to the response of prostate cancer cells to HDL, we next determined if these HDL effects were mediated by SR-B1. Therefore, we knocked out SR-B1 in both the human and mouse prostate cancer cell lines using CRISPR/Cas9 targeting exon 4, an early coding exon of the SCARB1 gene, to generate cell lines with complete knockout of SR-B1 expression. Western blotting demonstrated successful SR-B1 KO clones (SR-B1−/−) for both DU145 and TRAMP-C2 (Fig. 4A). Although HDL increased the relative cell number for both DU145 SR-B1+/+ (59.3% increase, p < 0.0003) and SR-B1−/− cells (23.4% increase, p = 0.011), the increase in cell accumulation in response to a 4-day 200 μg/ml of HDL treatment was significantly attenuated upon knockout of SR-B1 (Fig. 4B, p < 0.0003, % control cell number in HDL-treated SR-B1+/+ versus SR-B1−/−). We isolated three independent SR-B1−/− clonally-derived cell lines from edited TRAMP-C2 cells, and their cell accumulation in LPDS was evaluated, showing that the SR-B1−/− #5 line accumulated the least cells (p = 0.02 versus TRAMP-C2 SR-B1+/+), the SR-B1 #10 line accumulated the most cells (NS versus TRAMP-C2 SR-B1+/+), and the SR-B1−/− #17 line was most similar in cell number to SR-B1+/+ TRAMP-C2 cells (NS, Fig. 4C). The response of these cell lines to HDL was evaluated, normalized to the LPDS control for each of these lines. HDL significantly increased cell accumulation in WT cells (98% increase p = 0.011), but not in any of the three SR-B1−/− lines (Fig. 4D). Thus, we chose to further utilize and characterize HDL treatment on cellular processes in the SR-B1−/− #17 cell line, as its basal growth levels in LPDS were the most similar to that of the TRAMP-C2 SR-B1+/+ cells (Fig. 4C).
Figure 4.

HDL effects in WT and SR-B1 KO cells. A, Western blotting for SR-B1 in WT and SR-B1 KO DU145 and TRAMP-C2 cells. B, cell accumulation assay for DU145 WT and SR-B1 KO cells incubated ±300 μg/ml of HDL in LPDS for 3 days (n = 3–4; mean ± S.D.; t test with Bonferroni correction for 3 tests; p values displayed). C, cell number of SR-B1+/+ TRAMP-C2 cells and three independent SR-B1−/− clonally derived cell lines after incubation in 10% LPDS for 3 days (n = 3; mean ± S.D.; ANOVA with Dunnett's post test comparing to SR-B1+/+ cells; p value displayed). D, cell accumulation in TRAMP-C2 SR-B1+/+ and three SR-B1−/− clones incubated ±200 μg/ml of HDL for 3 days normalized to each cell lines LPDS control (n = 3; mean ± S.D.; t test with Bonferroni correction for 7 tests (4 tests ± HDL for each line and 3 tests of HDL treated SR-B1+/+ versus SR-B1−/− clones), significant p values displayed). E, cell cycle analysis in TRAMP-C2 SR-B1+/+ and SR-B1−/− cells treated ±300 μg/ml of HDL in LPDS media for 1 day (% of cells in S + G2 phases; n = 3; mean ± S.D.; t test with Bonferroni correction for 3 tests, p values displayed). NS, not significant.
We next investigated if the absence of SR-B1 impacts cell cycling upon HDL treatment by treating TRAMP-C2 SR-B1+/+ and SR-B1−/− cells with or without 300 μg/ml of HDL in LPDS for 1 day, then assessing the fraction of cycling cells in G2 + S phase via propidium iodide content. We demonstrated that HDL increased the proportion of SR-B1+/+ cells cycling by 38% versus the LPDS control (p = 0.002), whereas in SR-B1−/− cells there was only a 17.5% increase cycling cells in response to HDL (p = 0.012, Fig. 4E). The HDL effect on cell cycling was significantly greater in SR-B1+/+ versus SR-B1−/− cells (p = 0.001, Fig. 4E).
To confirm that HDL-uptake was impacted upon KO of SR-B1, we incubated TRAMP-C2 SR-B1+/+ and SR-B1−/− cells with Alexa 568-HDL. Fluorescent microscopy showed that SR-B1+/+ cells took up more Alexa 568-HDL as compared with SR-B1−/− cells (Fig. 5A). This was further confirmed by flow cytometry, where the cells were treated with 20 μg/ml of Alexa 568-HDL with or without 2 mg/ml of unlabeled-HDL competitor. The cellular uptake of Alexa 568-HDL was determined by median fluorescence intensity showing that the SR-B1−/− cells had reduced total and specific Alexa 568-HDL uptake compared with the SR-B1+/+ cells by 44.1 (p = 0.002) and 59.6% (p = 0.007), respectively (Fig. 5B). To investigate if cholesterol is involved in the HDL effect on cell accumulation, we treated SR-B1+/+ or SR-B1−/− cells with or without 1 μm lovastatin to reduce endogenous cholesterol biosynthesis, which significantly decreased cell accumulation in both cell lines (p < 0.05, Fig. 5C). HDL treatment for 4 days added to the lovastatin significantly rescued the cell accumulation only in the SR-B1+/+ cells (p < 0.05 versus lovastatin alone, Fig. 5C). This suggests that HDL provides cholesterol, in an SR-B1-dependent manner, to help cells grow when de novo cholesterol was reduced by statin treatment (Fig. 5C).
Figure 5.

HDL uptake and impact on statin-treated cells. TRAMP-C2 SR-B1+/+ or SR-B1−/− cells were incubated with 20 μg/ml of Alexa 568-HDL for 90 min, then counterstained with DAPI. A, epifluorescent microscopy. B, total and specific uptake analysis by flow cytometry with TRAMP-C2 SR-B1+/+ and SR-B1−/− cells treated with 20 μg/ml of Alexa 568-HDL and ±2 mg/ml of unlabeled HDL for 90 min in serum-free media (n = 3, mean ± S.D.; t test p values displayed). C, cell accumulation of TRAMP-C2 SR-B1+/+ and SR-B1−/− in LPDS treated with ±1 μm lovastatin and 100 μg/ml of HDL for 4 days normalized to untreated cells (n = 3, mean ± S.D.; different letters represent p < 0.05 by ANOVA with Tukey post-test within each cell type).
HDL and SR-B1 effects on prostate cancer progression in vivo
We hypothesized that elevated HDL levels may promote tumor progression in an SR-B1-dependent manner. To test our hypothesis, we utilized C57BL/6J WT and apoA1-KO mice as high and low HDL models. WT mice had fasting total and HDL cholesterol levels of 97 ± 15 and 63 ± 17 mg/dl, respectively. ApoA1-KO mice had significantly lower levels of total and HDL-cholesterol (32 ± 8 and 18 ± 9 mg/dl, respectively) versus WT hosts (p < 0.0001 for both, Fig. S1A). WT mice weighed more than apoA1-KO mice (p < 0.0001, Fig. S1B); however, there were no differences in testes weights (Fig. S1C). We measured plasma testosterone, and the data were not normally distributed with several outliers. Nonparametric t tests found no effect on testosterone levels between WT and apA1-KO mice (Fig. S1D); however, removal of the two outliers in each group resulted in normally distributed data showing 42% reduced testosterone levels in apoA1-KO mice (p = 0.009, Fig. S1E). Dihydrotestosterone levels were undetectable in most mice (not shown).
To test our central hypothesis of the effects of host HDL and tumoral SR-B1 status on tumor progression, we performed a four-arm study using 2 × 106 syngeneic TRAMP-C2 SR-B1+/+ or SR-B1−/− cells that were subcutaneously injected into WT or apoA1-KO mice on the C57BL/6J background. Over an 8-week time course SR-B1+/+ and SR-B1−/− cells formed solid tumors in WT and apoA1-KO mice. Histology of H&E-stained tumors from all study arms showed unorganized sheets of cells with irregular shaped nuclei (Fig. S2). Tumors from all groups were characterized as aggressive by a clinical pathologist. Tumor volume (p < 0.0001, Fig. 6A) and survival (p = 0.0016, Fig. 6B) were significantly different in the treatment arms.
Figure 6.
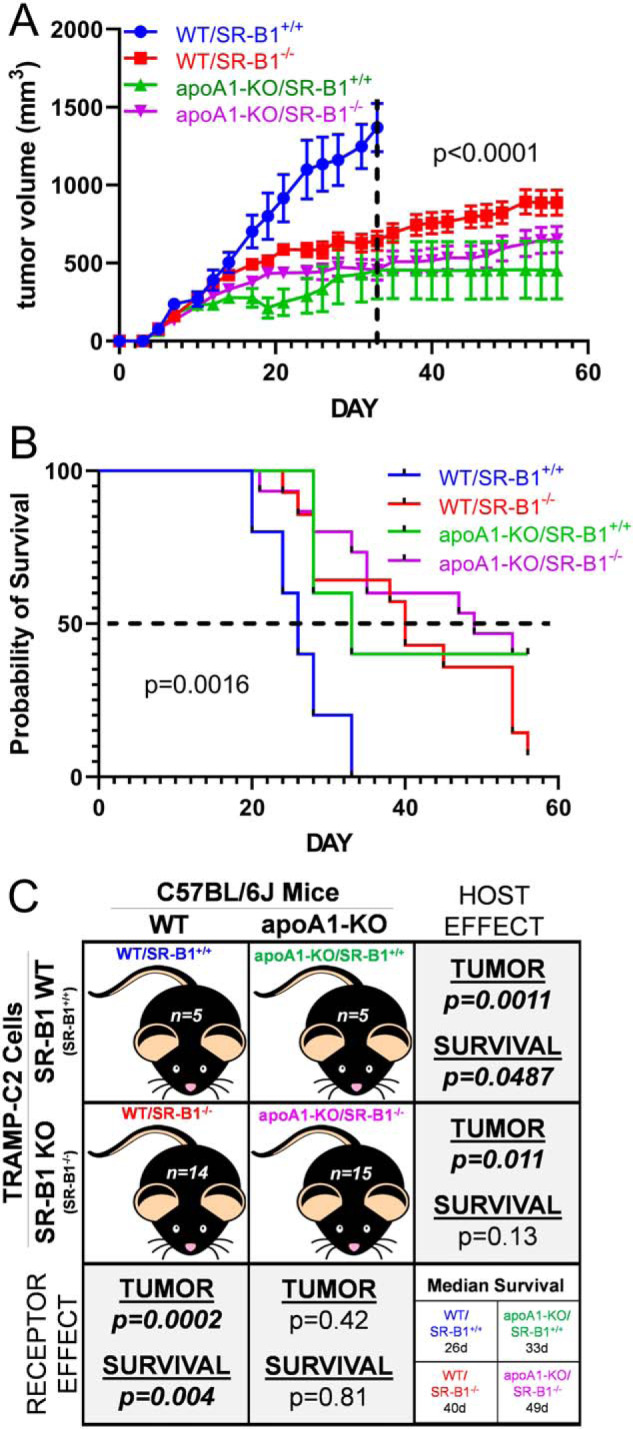
Tumor progression study in vivo. A, tumor volumes of mice injected with 2 × 106 cells in left and right flanks, then followed for 8 weeks. Tumor volume is expressed as mean ± S.E.; WT mice SR-B1+/+ cells (WT/SR-B1+/+, blue) n = 5; WT mice SR-B1−/− cells (WT/SR-B1−/−, red) n = 14; apoA1-KO mice SR-B1+/+ cells (apoA1-KO/SR-B1+/+, green) n = 5; apoA1-KO mice SR-B1−/− cells (apoA1-KO/SR-B1−/−, purple) n = 15. Two-way ANOVA up to day 33, when all groups survived, demonstrated group effects, time effects, and interaction effects all with p < 0.0001. B, Kaplan Meier survival plot (p = 0.0016 by Mantel Cox Log Rank Sum test for all four groups). C, summary of study design and pairwise statistical analysis of host mouse genotype and injected cell genotype effects tumor progression and survival. Tumor volume analyses involving the WT/SR-B1+/+ group, only used data up to day 33. Pairwise two-way ANOVA group effect on tumor volumes p values are displayed. Pairwise survival Mantel Cox Log Rank Sum group effect p values are displayed.
First, examining the host mouse effects (shown in the rows in Fig. 6C) using SR-B1+/+ cells, WT mice injected with SR-B1+/+ cells (WT/SR-B1+/+) had significantly larger tumors versus apoA1-KO mice injected with SR-B1+/+ cells (apoA1-KO/SR-B1+/+) (p < 0.0001, Fig. 6, A and C). Additionally, the WT/SR-B1+/+ group all reached their human end point by day 33 with median survival of 26 days, and their log-rank survival was significantly shorter versus the apoA1-KO/SR-B1+/+ group (median survival of 33 days, p = 0.049, Fig. 6, B and C). Examining the host effects using SR-B1−/− cells, WT mice injected with KO cells (WT/SR-B1−/−) had significantly larger tumors versus apoA1-KO mice injected with SR-B1−/− cells (apoA1-KO/SR-B1−/−) (p < 0.0001, Fig. 6, A and C). The WT/SR-B1−/− group (median survival of 40 days) trended toward shorter log-rank survival versus the apoA1-KO/SR-B1−/− group (median survival of 49 days, p = 0.13, Fig. 6, B and C). Thus the host effect was clear, mice with higher HDL levels promoted more rapid tumor progression, with shorter survival.
Next we examined the SR-B1 receptor effects in WT hosts (shown in the columns in Fig. 6C), the WT/SR-B1+/+ group had significantly larger tumors versus WT mice injected with SR-B1−/− cells (WT/SR-B1−/−) (p < 0.0001, Fig. 6, A and C). Log-rank survival for the WT/SR-B1+/+ group was significantly shorter versus the WT/SR-B1−/− group (p = 0.004, Fig. 6, B and C). Examining the SR-B1 receptor effects in apoA1-KO mice, apoA1-KO mice injected with SR-B1+/+ cells (apoA1-KO/SR-B1+/+) had similar rates of tumor progression and survival compared with the apoA1-KO mice injected with SR-B1−/− cells (apoA1-KO/SR-B1−/−) (NS, Fig. 6, A–C). Thus the SR-B1 receptor effect was only evident in WT mice, where receptor expression promoted more rapid tumor progression and shorter survival.
Discussion
The effects of HDL are mixed on the prevalence of many cancers including, prostate, breast, endometrial, gynecologic, colorectal, biliary tract, lung, and hematological cancers (reviewed in Ref. 26). Prostate cancer is a disease typically driven by androgens in which surgical and chemical castration have proven to slow progression of the disease (27, 28). However, prostate cancer often becomes resistant to such treatments (29). The effects of HDL on prostate cancer development and progression are controversial, with both positive and inverse associations found in various studies (2–5). A meta-analysis of six studies found no significant effect of HDL-cholesterol on the risk of developing prostate cancer (30). However, the meta-analysis of three studies of high-grade prostate cancer found a nonsignificant trend with a 1.21-fold increased relative risk per 1 mm increase in HDL-cholesterol (30). Additionally, a Mendelian randomization study showed that the 35 SNPs associated with HDL-cholesterol did not generate a genetic risk score for prostate cancer (31). Thus, HDL levels may not affect the development of prostate cancer, but may still influence its progression.
HDL-cholesterol is sexually dimorphic, being ∼10 mg/dl higher in adult women versus men (32). However, before puberty, boys and girls have similar (high) HDL, which drop as boys go through puberty and stay unchanged as girls go through puberty (18, 19). Androgens lower HDL in humans, and this is thought to be mediated by androgen induction of hepatic SR-B1 (21). Thus, the effect of androgens on lowering HDL-cholesterol may partially obscure a potential positive association between HDL and prostate cancer, as androgens drive the early stages of prostate cancer. In a prospective study, prostate cancer patients treated with androgen deprivation therapy had increased HDL-cholesterol 1, 3, and 6 months later versus baseline levels (33). Thus, the HDL-raising effects of this therapy may be contra-indicated based on the current findings that HDL promotes prostate cancer progression via SR-B1.
HDL is synthesized from lipid-poor apoA1 by ABCA1, which can accept more cell cholesterol through ABCG1, and HDL cellular uptake is mediated by SR-B1 (8, 9). Lee et al. (24) found that ABCA1 expression was lower and ABCA1 promoter had higher DNA methylation in more versus less advanced prostate cancer, and that ABCA1 gene expression was epigenetically silenced by DNA methylation in the LNCaP cell line. Schörghofer et al. (22) demonstrated that SR-B1 mRNA expression was higher in high grade versus low grade prostate cancer biopsies, and also higher in metastatic versus primary prostate cancer. In contrast, this study reported no difference in LDLR expression in these tissues (22). Gordon et al. (23) also reported increased SR-B1 mRNA expression, but lower LDLR mRNA expression, in prostate cancer versus normal prostatic tissue. Our TCGA data analysis showed that SR-B1 and ABCG1 mRNAs were up-regulated, LDLR mRNA was decreased, and ABCA1 mRNA was unchanged in prostate cancer versus normal tissue.
We found that HDL treatment of cells cultured in LPDS led to an increase in cell number and proliferation in both human and mouse prostate cancer cell lines. A similar growth promoting effect of HDL was observed by Sekine et al. (34) using prostate cancer cell lines cultured in 1% FBS. This HDL effect was associated with increased phospho-ERK1 and phospho-AKT after 30 min of HDL incubation. In both TRAMP-C2 and DU145 cells, we found that SR-B1 knockout abolished the growth promoting effects of HDL. However, Sekine et al. (34) showed that siRNA-mediated knockdown of SR-B1 in PC3 cells did not abolish the growth promoting effects of HDL, which they attributed to ABCA1 expression. Gordon et al. (23) reported that the growth of C4-2 human prostate cancer cell line in 10% FBS is inhibited by SR-B1 siRNA or by the anti-SR-B1 drug BLT-1, agreeing with our finding that SR-B1 is growth promoting in the presence of HDL.
Sekine et al. (34) demonstrated that HDL promotes cholesterol uptake by PC3 prostate cancer cells, yet 100 μg/ml of HDL in 1% FBS for 24 h did not promote an increase in total cholesterol levels in PC3, DU145, or LNCaP cell lines. However, we demonstrated that 200 μg/ml of HDL in LPDS for 48 h increased total cholesterol in DU145 and TRAMP-C2 cells (Fig. 2, A and B). Furthermore, we showed that Alexa 568-HDL uptake by TRAMP-C2 cells was in part mediated by SR-B1 (Fig. 5, A and B), a finding that was demonstrated by Gordon et al. (23) in C4-2 prostate cancer cells in which DiI-HDL uptake was partially reduced upon SR-B1 knockdown or chemical inhibition by BLT-1. We found that inhibiting de novo cholesterol synthesis in TRAMP-C2 cells by lovastatin led to reduced cell accumulation, which was rescued by HDL treatment in an SR-B1–dependent fashion, suggesting that the cholesterol content of HDL may play a role in the recovery of cell accumulation (Fig. 5C). Additionally, various statins were found to promote cell cycle arrest of PC3 cells, also indicating the need for de novo cholesterol biosynthesis to drive prostate cancer cell proliferation (35). Four meta-analyses of human observational studies on statin use and prostate cancer incidence, progression, and mortality have been published in 2016 or later, with three finding beneficial effects of statins (36–39). For example, prostate cancer-specific mortality was significantly reduced in statin users pre- and post-diagnosis with prostate cancer (HR = 0.53 and 0.64, respectively) (36). Thus, cholesterol metabolism may play an important role in prostate cancer therapeutics.
A large meta-analysis demonstrated that increased HDL-cholesterol was associated with lower incidence of cancer; although this study did not examine different types of cancer (40). This finding was corroborated in C57BL/6J mice using syngeneic B16F10 melanoma and Lewis lung carcinoma cells injected into (going from low to high HDL-cholesterol levels) apoA1-KO, WT, and human apoA1 transgenic mice; and, for both of these cancer types, higher HDL levels were associated with smaller tumors (41). The protective effect of HDL was associated with increased tumor-associated macrophages, cytotoxic CD8 T-cells, and decreased recruitment of myeloid-derived suppressor cells (41, 42). In addition, B16F10 melanoma tumor progression was slower in Scarb1 KO mice, another model of high plasma HDL (42). In C57BL/6J WT and apoA1-KO mice, we found an opposite effect of host HDL on syngeneic TRAMP-C2 prostate cancer cells, where higher HDL led to larger tumors, and decreased survival.
Why do the TRAMP-C2 cells respond differently than the B16F10 and Lewis lung cells? Perhaps it is due to the relative expression levels of SR-B1 and the role of HDL in providing lipids required for cell cycling. Llaverias et al. (43) reported that feeding a Western-type versus chow diet to C57BL/6J TRAMP transgenic mice, which are prone to spontaneously develop prostate tumors, results in increased total and HDL-cholesterol, and a higher prostatic tumor incidence with larger tumors. The tumors from Western-type versus chow diet-fed TRAMP mice also have higher levels of SR-B1 expression (43). The SNP rs4765623, intronic in the SCARB1 gene, is associated with clear cell renal cell carcinoma (ccRCC), a cancer characterized by excessive lipid loading (44). This same SNP is associated with SCARB1 expression levels in human left ventricle, with the T allele associated with increased expression (25). rs4765623 is in linkage disequilibrium with rs12582221 (d′ = 1, r2 = 0.39) (45), and this SNP is highly associated with SR-B1 expression in testes (25). SR-B1 expression is increased in ccRCC tissue versus normal kidney tissue (46, 47), similar to what we and others observed in prostate cancer versus normal prostate. Also in alignment with our SR-B1 KO findings in prostate cancer cells, Velagapudi et al. (48) showed that antibodies against SR-B1 reduce cellular uptake of 125I-HDL into, and HDL-induced proliferation of, a ccRCC cells line (48). Thus, SR-B1 and HDL may drive proliferation of specific lipid accumulating cancers. Whether HDL promotes or inhibits tumor progression may depend on tumor lipid delivery versus host immune effects.
In general, SR-B1 expression is highest in the major HDL metabolizing organ, the liver, and in steroidogenic tissues. In humans SR-B1 expression is highest in adrenals, liver, and ovary (25). In 9 tested tissues of C57BL/6J mice, the liver and testes had the highest expression of SR-B1 (49). The role of HDL uptake to support CE storage for steroidogenic tissue was demonstrated in apoA1-KO mice, where adrenal and ovarian CE stores, as wells as stress-induced plasma corticosteroid levels, are significantly reduced (50). Although total testes CE storage was not different between WT and apoA1-KO mice, histological analysis showed markedly reduced neutral lipid loading in the Leydig cells, the site of de novo androgen biosynthesis (50). We measured plasma testosterone levels in 2 cages each of WT and apoA1-KO mice, and in each cage, there was one high outlier, which may be due to the well-known effect of higher testosterone levels in the socially dominant male of group-housed mice (51, 52). Excluding these outliers, we found 42% higher plasma testosterone in the WT versus apoA1-KO mice, which is a confounder for the pro-tumor effect of HDL in our study.
We found that SR-B1−/− versus SR-B1+/+ TRAMP-C2 cells injected into WT hosts with high HDL-cholesterol led to reduced tumor progression and increased survival, without the caveat of decreased androgen levels observed in apoA1-KO mice. Thus, SR-B1 promotes prostate cancer tumor growth in vivo, similar to SR-B1's growth-promoting effects we observed in cell culture. However, SR-B1 is a multiligand receptor that can also mediate uptake of non-HDL ligands such as LDL and fat-soluble vitamins (53–55). Thus, it is possible that the loss of SR-B1 in TRAMP-C2 cells reduced tumor progression due to decreased uptake of these other ligands. It appears that the growth promoting effects of SR-B1 in TRAMP-C2 cells may be primarily mediated by its ligand HDL, as SR-B1 status (KO versus WT) has no effect on tumor progression in apoA1-KO mice; but, we cannot exclude the role of other SR-B1 ligands that may play a role in tumor progression in WT mice. Our findings on the role of SR-B1 in prostate cancer are similar to the findings of Gordon et al. (23) who showed that treatment of mice with the SR-B1 inhibitor BLT-1 led to reduced human PC3 cell xenograft progression. These findings also align well with our TGCA analysis and prior analyses showing that SR-B1 expression is higher in prostate cancer versus controls, higher in high grade versus low-grade prostate cancer, and higher in metastatic versus localized prostate cancer (22, 23).
In conclusion, our findings are in alignment in with Llaverias et al. (43) where a high fat diet increased autochthonous tumor prevalence and burden in the TRAMP transgenic mouse model, associated with increased HDL levels. Additionally, our findings corroborated the in vivo findings of Gordon et al. (23) who demonstrated that treatment with an SR-B1 inhibitor decreased prostate cancer progression. Our study was more specific and could differentiate cancer cell versus host effects due to genetic ablation of SR-B1 in the cancer cells, whereas Gordon et al. (23) used a chemical inhibitor of SR-B1, having specific and nonspecific effects on the host as well as the xenograft cancer cells. Our study employed immunocompetent mice, which may be a better model to study HDL effects on prostate cancer because HDL has been shown to modulate tumor infiltrating leukocytes (41).
Experimental procedures
Cells and chemicals
The human prostate cancer cell line DU145 and mouse prostate cancer cell line TRAMP-C2 were purchased from American Type Culture Collection (Manassas, VA, USA). The DU145 cell line was authenticated by Genetica cell line testing. DU145 was cultured in RPMI1640 (supplemented with 10% FBS (Sigma) and TRAMP-C2 was cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS (Sigma) and 10 nm dihydrotestosterone (Sigma) at 37 °C in 5% CO2. Both cell lines grown were treated with a low-dose mycoplasma removal agent (MP Biomedicals) as a preventative measure. Mycoplasma testing was performed regularly to confirm the absence of mycoplasma contamination on cells with SR-B1 rabbit polyclonal antibody (NB400-104), purchased from Novus Biologicals. GAPDH rabbit polyclonal antibody (sc-25778) was purchased from Santa Cruz. Lipoprotein-deficient serum (LPDS) was prepared as previously described (56). In brief, FBS was adjusted to 1.21 g/ml, and centrifuged to separate lipoprotein from sera. LPDS was collected and dialyzed against PBS (pH 7.4), and then sterilized using a 0.22-μm filter. LPDS was adjusted to 30 mg/ml of protein using PBS.
Isolation and labeling of lipoproteins
Expired de-identified human plasma was collected from normal healthy volunteers acquired from the Cleveland Clinic Blood Bank under an exempt Institutional Review Board protocol. LDL (1.019–1.063 g/ml) and HDL (1.063–1.21 g/ml) were isolated by potassium bromide density gradient centrifugation as described (56). Lipoproteins were dialyzed against PBS (pH 7.4), sterilized using a 0.22-μm filter, and then the protein concentration was determined by the alkaline-Lowry method (57). HDL was labeled with Alexa 568 by incubating 4.5 mg of human HDL in 90 μl of 1 m sodium bicarbonate with Alexa Fluor 568 succinimidyl ester (A20003, Thermo Fisher) for 1 h at room temperature (8:1 dye:estimated apoA1 mole ratio). The reaction was stopped by incubating the conjugate with 0.1 ml of 1.5 m hydroxylamine (pH 8.5) for 1 h at room temperature. The conjugate was purified by extensive dialysis with PBS. For murine lipoprotein isolation and quantification, blood from the retro-orbital venous sinus was collected and centrifuged at 12,000 × g for 30 min.
Cell accumulation and proliferation by cell cycle analysis of prostate cancer cells
For cell accumulation, proliferation, and cell cycle analysis, cells were seeded into a 24-well–plate at densities of 10,000 and 20,000 cells/well for TRAMP-C2 and DU145 cells, respectively, in 10% FBS containing medium overnight, then incubated with serum-free media for 30 min. Cells were then incubated in 10% LPDS with or without 300 μg/ml of HDL in the absence or presence of 1 μm lovastatin for 4 days. Thereafter, cells were lifted by trypsin and counted using using a Beckman Coulter automated cell counter (Z series). For cell cycle analysis, cells were treated with or without 300 μg/ml of HDL in 2% LPDS media for 1 day, ethanol fixed, stained with 50 μg/ml of propidium iodide, and then subjected to flow cytometry.
Generation of SR-B1 KO cells
TRAMP-C2 cell were transfected with the Cas9 expression plasmid, pSpCas9(BB)-2A-Puro (Addgene No. 48139) using Lipofectamine LTX and Plus Reagent (Invitrogen) according to the manufacturer's instructions, then treated with 5 μg/ml of puromycin for 2 days to clonally select for Cas9 stably transfected TRAMP-C2 cells. Nucleofection (Amaxa), according to the manufacturer, was used to transfect 1 × 106 Cas9 stable TRAMP-C2 cells with 0.6 nm mouse SR-B1 sgRNA targeting exon 4. Human DU145 cells (1 × 106) were co-transfected with 0.6 nm human SR-B1 sgRNA targeting exon 4 complexed with 0.07 nm Cas9 protein (Synthego) by nucleofection. Both sgRNAs were designed using CRISPOR online software (58) and synthesized by Synthego. sgRNAs sequences were: mouse sgScarb1 reverse strand, 5′-ugcgguucauaaaagcacgc-3′; human sgSCARB1 reverse strand, 5′-caugaaggcacguucgccga-3′. Transfected cells were plated in 96-well–dishes to ∼1 cell/well, then clonally expanded and screened via PCR-Sanger sequencing to detect targeted sequence disruptions, and then by Western blotting for SR-B1. PCR screening primers were: human fwd, 5′-ccagtgggttctgagtttccca-3′, rev, 5′-gatccccagccagctacaaagc-3′; mouse fwd, 5′-ggttccatttaggcctcaggt-3′, rev, 5′-ctctctgaagggacagaagacac-3′.
HDL uptake assay
Approximately 100,000 cells/well were seeded onto a 24-well–plate and incubated overnight in 10% FBS containing media. Cells were then incubated with serum-free media for 30 min and then treated with 20 μg/ml of Alexa 568-HDL, as described above, for 90 min. Cells were fixed, then counterstained with DAPI for microscopy, or stained first, then formalin fixed for flow cytometry.
Cholesterol mass assay
Cells were plated on a 6-well–plate and incubated overnight in the medium, 10% FBS containing media. Cells were incubated in serum-free media for 30 min and treated with or 200 μg/ml of HDL for 2 days. Cholesterol concentrations were measured by an enzymatic fluorescent assay and normalized to protein as described in Ref. 59.
Western blotting assays
For protein expression via Western blotting, cell lysates were prepared in RIPA buffer (Pierce), containing 1 mm phenylmethylsulfonyl fluoride and 10% protease inhibitor mixture (Sigma). Equal amounts of proteins were electrophoresed on 4–20% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Each membrane was incubated with a 1:2000 dilution of primary antibodies described above. Blots were developed with a 1:5000 dilution of the horseradish peroxidase-conjugated secondary antibody (Bio-Rad). Proteins were visualized, using Amersham Biosciences Hyperfilm ECL (GE Healthcare).
Syngeneic in vivo studies
Our in vivo studies used 20–22-week–old, age-matched, apoA1-KO and WT C57BL/6J male mice that were subcutaneously injected in both flanks with 2 × 106 SR-B1 KO or WT TRAMP-C2 (syngeneic) prostate cancer tumor cells per site. Tumor progression and body weights were assessed three times per week for 8 weeks or until reaching an experimental end point of tumor reaching 15 mm in diameter, tumor ulcerations, impaired mobility, or 20% loss in body weight. Tumor volume, based on caliper measurements, was calculated according to the ellipsoid volume formula, tumor volume = (the shortest diameter)2 × the largest diameter × 0.525. After a mouse reached its end point, its final tumor volumes were included in the analysis up to the day that all mice from that group reached its end point, after which data were no longer plotted. Other analyses from in vivo studies are described in supporting Experimental procedures. All experiments and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic, Cleveland, OH, USA (protocol No. 2016-1722).
Bioinformatics
Bioinformatic information was acquired and viewed by The UCSC Xena browser (60) to visualize and extract expression data for SCARB1, LDLR, ABCA1, and ABCG1 in normal and prostate cancer tissue from the TCGA PRAD dataset. The Genotype-Tissue Expression (GTEx) Project (25) was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the following independent searches: SCARB1, rs4765623, and rs12582221 on the GTEx Portal on 01/08/2020; dbGaP accession number phs000424.v8.p2. LDlink (45) was used to evaluate the linkage disequilibrium of SCARB1 SNPs (rs12582221 and rs4765623) in the CEU population of the 1000 Genomes Project, with reference genome GRCh37/gh19 on 01/08/2020; LDlink phase 3, version 5.
Statistical analysis
Statistical analysis of TGCA PRAD gene expression data from prostate cancer and normal tissue was analyzed by the Mann-Whitney non-non-parametric test, and % changes were calculated from the anti-log2 of the median values. All in vitro data are expressed as the mean ± S.D. of at least triplicates. Differences between the values were evaluated by either Student's t test, with Bonferroni correction for multiple tests when appropriate, or one-way analysis of variance (one-way ANOVA) with Tukey's or Dunnett's post hoc analysis, with p < 0.05 considered statistically significant. ANOVA annotation uses lettering (a, b, c, etc.), where groups not sharing the same letter indicates p < 0.05. All in vivo tumor volume data are expressed as mean ± S.E. Differences between the tumor volumes during the time course were evaluated by two-way analysis of variance (two-way ANOVA). Survival curve data were evaluated by Mantel-Cox Log Rank analysis. Statistics were performed using GraphPad Prism version 8.1.1 software.
Data availability
All data are contained within the article and supporting information.
Supplementary Material
Acknowledgments
We thank the following cores at the Cleveland Clinic Lerner Research Institute: The Biological Research Unit (BRU) for assistance with mouse handling, the Flow Cytometry Core advice and help with flow cytometry instrumentation, and the Histology Core for tissue staining. We also thank Dr. Erinn Downs Kelly for comments on tumor pathology.
This article contains supporting information.
Author contributions—C. A. T. and J. D. S. conceptualization; C. A. T., E. O., G. B., J. M., H. L., and Y.-M. C. data curation; C. A. T. and J. D. S. formal analysis; C. A. T., G. B., J. M., K. G., N. S., J. M. B., and J. D. S. supervision; C. A. T., K. G., N. S., J. M. B., and J. D. S. funding acquisition; C. A. T. validation; C. A. T., E. O., G. B., H. L., and C. N. investigation; C. A. T. and C. N. visualization; C. A. T., E. O., G. B., J. M., H. L., S. W. L., C. N., A. H., Y.-M. C., K. G., and J. D. S. methodology; C. A. T. and J. D. S. writing-original draft; C. A. T., G. B., J. M., and J. D. S. project administration; C. A. T., E. O., G. B., J. M., H. L., S. W. L., C. N., A. H., Y.-M. C., K. G., N. S., J. M. B., and J. D. S. writing-review and editing; C. N. software; J. D. S. resources.
Funding and additional information—This work was supported by National Institutes of Health Grants R01HL128628 (to J. D. S.), R01DK120679 (to J. M. B.), P50AA024333 (to J. M. B.), P01HL147823 (to J. M. B.), R01CA172382 (to N. S.), R01CA190289 (to N. S.), R01CA236780 (to N. S.), and F31HL134231 (to C. A. T.), American Heart Association Grant SDG25710128 (to K. G.), the Case Comprehensive Cancer Center pilot Grant RES511106 (to J. D. S.), the VeloSano Foundation (to J. D. S.), National Institutes of Health Grant R01HL148158 (to K. G.), and Case Comprehensive Cancer Center pilot grant program supported by NCI National Institutes of Health Grant P30CA043703. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—N. S. has been a paid advisor to Celgene.
- HDL
- high-density lipoprotein
- apoA1
- apolipoprotein-AI
- BLT-1
- block lipid transport-1
- ccRCC
- clear cell renal cell carcinoma
- CE
- cholesterol ester
- KO
- knockout
- LDLR
- low-density lipoprotein receptor
- LPDS
- lipoprotein-deficient serum
- PRAD
- prostate adenocarcinoma
- SR-B1
- scavenger receptor class B, type 1
- sgRNA
- single guide RNA
- TCGA
- The Cancer Genome Atlas
- TRAMP
- transgenic adenocarcinoma mouse prostate
- FBS
- fetal bovine serum
- SNP
- single-nucleotide polymorphism
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole
- ANOVA
- analysis of variance
- ABCA1
- ABC transporter A1
- SCARB1
- scavenger receptor class B type 1.
References
- 1. Siegel R. L., Miller K. D., and Jemal A. (2019) Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 2. Jamnagerwalla J., Howard L. E., Allott E. H., Vidal A. C., Moreira D. M., Castro-Santamaria R., Andriole G. L., Freeman M. R., and Freedland S. J. (2018) Serum cholesterol and risk of high-grade prostate cancer: results from the REDUCE study. Prostate Cancer Prostatic Dis. 21, 252–259 10.1038/s41391-017-0030-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kok D. E., van Roermund J. G., Aben K. K., den Heijer M., Swinkels D. W., Kampman E., and Kiemeney L. A. (2011) Blood lipid levels and prostate cancer risk: a cohort study. Prostate Cancer Prostatic Dis. 14, 340–345 10.1038/pcan.2011.30 [DOI] [PubMed] [Google Scholar]
- 4. Hammarsten J., and Högstedt B. (2004) Clinical, haemodynamic, anthropometric, metabolic and insulin profile of men with high-stage and high-grade clinical prostate cancer. Blood Press 13, 47–55 10.1080/08037050310025735 [DOI] [PubMed] [Google Scholar]
- 5. Van Hemelrijck M., Walldius G., Jungner I., Hammar N., Garmo H., Binda E., Hayday A., Lambe M., and Holmberg L. (2011) Low levels of apolipoprotein A-I and HDL are associated with risk of prostate cancer in the Swedish AMORIS study. Cancer Causes Control 22, 1011–1019 10.1007/s10552-011-9774-z [DOI] [PubMed] [Google Scholar]
- 6. Jacobs E. J., Stevens V. L., Newton C. C., and Gapstur S. M. (2012) Plasma total, LDL, and HDL cholesterol and risk of aggressive prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Causes Control 23, 1289–1296 10.1007/s10552-012-0006-y [DOI] [PubMed] [Google Scholar]
- 7. Martin R. M., Vatten L., Gunnell D., Romundstad P., and Nilsen T. I. (2009) Components of the metabolic syndrome and risk of prostate cancer: the HUNT 2 cohort, Norway. Cancer Causes Control 20, 1181–1192 10.1007/s10552-009-9319-x [DOI] [PubMed] [Google Scholar]
- 8. Wang S., and Smith J. D. (2014) ABCA1 and nascent HDL biogenesis. Biofactors 40, 547–554 10.1002/biof.1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rigotti A., Miettinen H. E., and Krieger M. (2003) The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 24, 357–387 10.1210/er.2001-0037 [DOI] [PubMed] [Google Scholar]
- 10. Acton S. L., Scherer P. E., Lodish H. F., and Krieger M. (1994) Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J. Biol. Chem. 269, 21003–21009 [PubMed] [Google Scholar]
- 11. Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., and Krieger M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520 10.1126/science.271.5248.518 [DOI] [PubMed] [Google Scholar]
- 12. Calvo D., and Vega M. A. (1993) Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem. 268, 18929–18935 [PubMed] [Google Scholar]
- 13. de la Llera-Moya M., Rothblat G. H., Connelly M. A., Kellner-Weibel G., Sakr S. W., Phillips M. C., and Williams D. L. (1999) Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J. Lipid Res. 40, 575–580 [PubMed] [Google Scholar]
- 14. Rigotti A., Edelman E. R., Seifert P., Iqbal S. N., DeMattos R. B., Temel R. E., Krieger M., and Williams D. L. (1996) Regulation by adrenocorticotropic hormone of the in vivo expression of scavenger receptor class B type I (SR-BI), a high density lipoprotein receptor, in steroidogenic cells of the murine adrenal gland. J. Biol. Chem. 271, 33545–33549 10.1074/jbc.271.52.33545 [DOI] [PubMed] [Google Scholar]
- 15. Shen W. J., Azhar S., and Kraemer F. B. (2018) SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu. Rev. Physiol. 80, 95–116 10.1146/annurev-physiol-021317-121550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yue S., Li J., Lee S. Y., Lee H. J., Shao T., Song B., Cheng L., Masterson T. A., Liu X., Ratliff T. L., and Cheng J. X. (2014) Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 10.1016/j.cmet.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langer C., Gansz B., Goepfert C., Engel T., Uehara Y., von Dehn G., Jansen H., Assmann G., and von Eckardstein A. (2002) Testosterone up-regulates scavenger receptor BI and stimulates cholesterol efflux from macrophages. Biochem. Biophys. Res. Commun. 296, 1051–1057 10.1016/S0006-291X(02)02038-7 [DOI] [PubMed] [Google Scholar]
- 18. Kirkland R. T., Keenan B. S., Probstfield J. L., Patsch W., Lin T. L., Clayton G. W., and Insull W. Jr. (1987) Decrease in plasma high-density lipoprotein cholesterol levels at puberty in boys with delayed adolescence: correlation with plasma testosterone levels. JAMA 257, 502–507 10.1001/jama.257.4.502, 10.1001/jama.1987.03390040118029 [DOI] [PubMed] [Google Scholar]
- 19. Eissa M. A., Mihalopoulos N. L., Holubkov R., Dai S., and Labarthe D. R. (2016) Changes in fasting lipids during puberty. J. Pediatr. 170, 199–205 10.1016/j.jpeds.2015.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tilly-Kiesi M., Lichtenstein A. H., Joven J., Vilella E., Cheung M. C., Carrasco W. V., Ordovas J. M., Dolnikowski G., and Schaefer E. J. (1997) Impact of gender on the metabolism of apolipoprotein A-I in HDL subclasses LpAI and LpAI:AII in older subjects. Arterioscler. Thromb. Vasc. Biol. 17, 3513–3518 10.1161/01.ATV.17.12.3513 [DOI] [PubMed] [Google Scholar]
- 21. Wu F. C., and von Eckardstein A. (2003) Androgens and coronary artery disease. Endocr. Rev. 24, 183–217 10.1210/er.2001-0025 [DOI] [PubMed] [Google Scholar]
- 22. Schörghofer D., Kinslechner K., Preitschopf A., Schütz B., Röhrl C., Hengstschläger M., Stangl H., and Mikula M. (2015) The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. 13, 88 10.1186/s12958-015-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gordon J. A., Noble J. W., Midha A., Derakhshan F., Wang G., Adomat H. H., Tomlinson Guns E. S., Lin Y. Y., Ren S., Collins C. C., Nelson P. S., Morrissey C., Wasan K. M., and Cox M. E. (2019) Upregulation of scavenger receptor B1 is required for steroidogenic and nonsteroidogenic cholesterol metabolism in prostate cancer. Cancer Res. 79, 3320–3331 10.1158/0008-5472.CAN-18-2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee B. H., Taylor M. G., Robinet P., Smith J. D., Schweitzer J., Sehayek E., Falzarano S. M., Magi-Galluzzi C., Klein E. A., and Ting A. H. (2013) Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Cancer Res. 73, 1211–1218 10.1158/0008-5472.CAN-12-3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pirro M., Ricciuti B., Rader D. J., Catapano A. L., Sahebkar A., and Banach M. (2018) High density lipoprotein cholesterol and cancer: marker or causative? Prog. Lipid Res. 71, 54–69 10.1016/j.plipres.2018.06.001 [DOI] [PubMed] [Google Scholar]
- 27. Huggins C., and Hodges C. V. (1941) Studies on prostatic cancer: I. the effect of castation of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1, 293–297 [DOI] [PubMed] [Google Scholar]
- 28. Huggins C., Stevens R. E., and Hodges C. V. (1941) Studies on prostatic cancer: II. the effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 43, 209–223 10.1001/archsurg.1941.01210140043004 [DOI] [Google Scholar]
- 29. Debes J. D., and Tindall D. J. (2004) Mechanisms of androgen-refractory prostate cancer. N. Engl. J. Med. 351, 1488–1490 10.1056/NEJMp048178 [DOI] [PubMed] [Google Scholar]
- 30. YuPeng L., YuXue Z., PengFei L., Cheng C., YaShuang Z., DaPeng L., and Chen D. (2015) Cholesterol levels in blood and the risk of prostate cancer: a meta-analysis of 14 prospective studies. Cancer Epidemiol. Biomarkers Prev. 24, 1086–1093 10.1158/1055-9965.EPI-14-1329 [DOI] [PubMed] [Google Scholar]
- 31. Bull C. J., Bonilla C., Holly J. M., Perks C. M., Davies N., Haycock P., Yu O. H., Richards J. B., Eeles R., Easton D., Kote-Jarai Z., Amin Al Olama A., Benlloch S., Muir K., Giles G. G., MacInnis R. J., et al. (2016) Blood lipids and prostate cancer: a Mendelian randomization analysis. Cancer Med. 5, 1125–1136 10.1002/cam4.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davis C. E., Williams D. H., Oganov R. G., Tao S. C., Rywik S. L., Stein Y., and Little J. A. (1996) Sex difference in high density lipoprotein cholesterol in six countries. Am. J. Epidemiol. 143, 1100–1106 10.1093/oxfordjournals.aje.a008686 [DOI] [PubMed] [Google Scholar]
- 33. Oka R., Utsumi T., Endo T., Yano M., Kamijima S., Kamiya N., Shirai K., and Suzuki H. (2016) Effect of androgen deprivation therapy on arterial stiffness and serum lipid profile changes in patients with prostate cancer: a prospective study of initial 6-month follow-up. Int. J. Clin. Oncol. 21, 389–396 10.1007/s10147-015-0891-7 [DOI] [PubMed] [Google Scholar]
- 34. Sekine Y., Demosky S. J., Stonik J. A., Furuya Y., Koike H., Suzuki K., and Remaley A. T. (2010) High-density lipoprotein induces proliferation and migration of human prostate androgen-independent cancer cells by an ABCA1-dependent mechanism. Mol. Cancer Res. 8, 1284–1294 10.1158/1541-7786.MCR-10-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sivaprasad U., Abbas T., and Dutta A. (2006) Differential efficacy of 3-hydroxy-3-methylglutaryl CoA reductase inhibitors on the cell cycle of prostate cancer cells. Mol. Cancer Ther. 5, 2310–2316 10.1158/1535-7163.MCT-06-0175 [DOI] [PubMed] [Google Scholar]
- 36. Meng Y., Liao Y. B., Xu P., Wei W. R., and Wang J. (2016) Statin use and mortality of patients with prostate cancer: a meta-analysis. OncoTargets Ther. 9, 1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murtola T. J., Peltomaa A. I., Talala K., Määttänen L., Taari K., Tammela T. L. J., and Auvinen A. (2017) Statin use and prostate cancer survival in the Finnish randomized study of screening for prostate cancer. Eur. Urol. Focus 3, 212–220 10.1016/j.euf.2016.05.004 [DOI] [PubMed] [Google Scholar]
- 38. Raval A. D., Thakker D., Negi H., Vyas A., Kaur H., and Salkini M. W. (2016) Association between statins and clinical outcomes among men with prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 19, 151–162 10.1038/pcan.2015.58 [DOI] [PubMed] [Google Scholar]
- 39. Tan P., Zhang C., Wei S. Y., Tang Z., Gao L., Yang L., and Wei Q. (2017) Effect of statins type on incident prostate cancer risk: a meta-analysis and systematic review. Asian J. Androl 19, 666–671 10.4103/1008-682X.190327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jafri H., Alsheikh-Ali A. A., and Karas R. H. (2010) Baseline and on-treatment high-density lipoprotein cholesterol and the risk of cancer in randomized controlled trials of lipid-altering therapy. J. Am. Coll. Cardiol. 55, 2846–2854 10.1016/j.jacc.2009.12.069 [DOI] [PubMed] [Google Scholar]
- 41. Zamanian-Daryoush M., Lindner D., Tallant T. C., Wang Z., Buffa J., Klipfell E., Parker Y., Hatala D., Parsons-Wingerter P., Rayman P., Yusufishaq M. S., Fisher E. A., Smith J. D., Finke J., DiDonato J. A., and Hazen S. L. (2013) The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J. Biol. Chem. 288, 21237–21252 10.1074/jbc.M113.468967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zamanian-Daryoush M., Lindner D. J., DiDonato J. A., Wagner M., Buffa J., Rayman P., Parks J. S., Westerterp M., Tall A. R., and Hazen S. L. (2017) Myeloid-specific genetic ablation of ATP-binding cassette transporter ABCA1 is protective against cancer. Oncotarget 8, 71965–71980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Llaverias G., Danilo C., Wang Y., Witkiewicz A. K., Daumer K., Lisanti M. P., and Frank P. G. (2010) A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am. J. Pathol. 177, 3180–3191 10.2353/ajpath.2010.100568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Purdue M. P., Johansson M., Zelenika D., Toro J. R., Scelo G., Moore L. E., Prokhortchouk E., Wu X., Kiemeney L. A., Gaborieau V., Jacobs K. B., Chow W. H., Zaridze D., Matveev V., Lubinski J., Trubicka J., et al. (2011) Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat. Genet. 43, 60–65 10.1038/ng.723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Machiela M. J., and Chanock S. J. (2018) LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 34, 887–889 10.1093/bioinformatics/btx561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu G. H., Lou N., Shi H. C., Xu Y. C., Ruan H. L., Xiao W., Liu L., Li X., Xiao H. B., Qiu B., Bao L., Yuan C. F., Zhou Y. L., Hu W. J., Chen K., Yang H. M., and Zhang X. P. (2018) Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer 18, 88 10.1186/s12885-017-3761-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Thompson B., Han S., Lotan Y., McDonald J. G., and Ye J. (2019) Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1864, 158525 10.1016/j.bbalip.2019.158525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Velagapudi S., Schraml P., Yalcinkaya M., Bolck H. A., Rohrer L., Moch H., and von Eckardstein A. (2018) Scavenger receptor BI promotes cytoplasmic accumulation of lipoproteins in clear-cell renal cell carcinoma. J. Lipid Res. 59, 2188–2201 10.1194/jlr.M083311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petryszak R., Keays M., Tang Y. A., Fonseca N. A., Barrera E., Burdett T., Füllgrabe A., Fuentes A. M., Jupp S., Koskinen S., Mannion O., Huerta L., Megy K., Snow C., Williams E., et al. (2016) Expression Atlas update: an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44, D746–D752 10.1093/nar/gkv1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Plump A. S., Erickson S. K., Weng W., Partin J. S., Breslow J. L., and Williams D. L. (1996) Apolipoprotein A-I is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. J. Clin. Invest. 97, 2660–2671 10.1172/JCI118716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Machida T., Yonezawa Y., and Noumura T. (1981) Age-associated changes in plasma testosterone levels in male mice and their relation to social dominance or subordinance. Horm. Behav. 15, 238–245 10.1016/0018-506X(81)90013-1 [DOI] [PubMed] [Google Scholar]
- 52. Williamson C. M., Lee W., Romeo R. D., and Curley J. P. (2017) Social context-dependent relationships between mouse dominance rank and plasma hormone levels. Physiol. Behav. 171, 110–119 10.1016/j.physbeh.2016.12.038 [DOI] [PubMed] [Google Scholar]
- 53. Calvo D., Gómez-Coronado D., Lasunción M. A., and Vega M. A. (1997) CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler. Thromb. Vasc. Biol. 17, 2341–2349 10.1161/01.ATV.17.11.2341 [DOI] [PubMed] [Google Scholar]
- 54. Gillotte-Taylor K., Boullier A., Witztum J. L., Steinberg D., and Quehenberger O. (2001) Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J. Lipid Res. 42, 1474–1482 [PubMed] [Google Scholar]
- 55. Witt W., Kolleck I., Fechner H., Sinha P., and Rüstow B. (2000) Regulation by vitamin E of the scavenger receptor BI in rat liver and HepG2 cells. J. Lipid Res. 41, 2009–2016 [PubMed] [Google Scholar]
- 56. Li K., Wong D. K., Luk F. S., Kim R. Y., and Raffai R. L. (2018) Isolation of plasma lipoproteins as a source of extracellular RNA. Methods Mol. Biol. 1740, 139–153 10.1007/978-1-4939-7652-2_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 58. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J. S., and Concordet J. P. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robinet P., Wang Z., Hazen S. L., and Smith J. D. (2010) A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J. Lipid Res. 51, 3364–3369 10.1194/jlr.D007336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goldman M., Craft B., Hastie M., Repečka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A. N., Zhu J., and Haussler D. (2019) The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 10.1101/326470 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and supporting information.