

Abstract
Z-DNA-binding protein 1 (ZBP1) is an innate immune sensor of nucleic acids that regulates host defense responses and development. ZBP1 activation triggers inflammation and pyroptosis, necroptosis, and apoptosis (PANoptosis) by activating receptor-interacting Ser/Thr kinase 3 (RIPK3), caspase-8, and the NLRP3 inflammasome. ZBP1 is unique among innate immune sensors because of its N-terminal Zα1 and Zα2 domains, which bind to nucleic acids in the Z-conformation. However, the specific role of these Zα domains in orchestrating ZBP1 activation and subsequent inflammation and cell death is not clear. Here we generated Zbp1ΔZα2/ΔZα2 mice that express ZBP1 lacking the Zα2 domain and demonstrate that this domain is critical for influenza A virus–induced PANoptosis and underlies perinatal lethality in mice in which the RIP homotypic interaction motif domain of RIPK1 has been mutated (Ripk1mRHIM/mRHIM). Deletion of the Zα2 domain in ZBP1 abolished influenza A virus–induced PANoptosis and NLRP3 inflammasome activation. Furthermore, deletion of the Zα2 domain of ZBP1 was sufficient to rescue Ripk1mRHIM/mRHIM mice from perinatal lethality caused by ZBP1-driven cell death and inflammation. Our findings identify the essential role of the Zα2 domain of ZBP1 in several physiological functions and establish a link between Z-RNA sensing via the Zα2 domain and promotion of influenza-induced PANoptosis and perinatal lethality.
Keywords: influenza, influenza virus, RNA virus, negative-strand RNA virus, infection, microbiology
Introduction
Activation of inflammation and activation of cell death are interconnected cellular events that regulate host defense and immune responses. RIP homotypic interaction motif (RHIM)–family proteins play a major role in orchestrating inflammation and cell death to profoundly shape the immune response and host defense (1, 2). Z-DNA–binding protein 1 (ZBP1), also called DNA-dependent activator of IFN regulatory factor (DAI), is one such protein that interacts with the other RHIM proteins receptor–interacting protein kinase 3 (RIPK3) and receptor-interacting protein kinase 1 (RIPK1) by forming homotypic interactions (3). ZBP1 and RIPK3 have also been shown to participate in virus-induced necroptosis (4). We recently demonstrated that ZBP1 recognizes influenza A virus (IAV), an RNA virus, and triggers NLRP3 inflammasome activation and the pyroptosis, necroptosis, and apoptosis (PANoptosis) pathways (5). ZBP1 activation assembles RIPK3 and caspase-8 signaling complexes that mediate activation of the NLRP3 inflammasome and pyroptosis, RIPK3-mixed lineage kinase domain-like pseudokinase (MLKL)–driven necroptosis, and Fas-associated death domain protein (FADD)-caspase-8–driven apoptosis (5–8). These studies unraveled specific cellular functions that are regulated by ZBP1 in response to influenza infection. Further studies identified an endogenous physiological function of ZBP1 by demonstrating its role in perinatal lethality in mice (9, 10). These studies found that mice with mutations that abolish the RHIM activity of RIPK1 have perinatal lethality and skin inflammation and that activation of ZBP1-driven necroptosis is responsible for the pathological phenotypes seen in these mice (9, 10).
ZBP1 consists of two N-terminal Z-nucleic acid–binding domains (Zα1 and Zα2) followed by RHIM domains and a functionally undefined C-terminal region (11). The Zα1 and Zα2 domains of ZBP1 bind to DNA/RNA in the Z-conformation (12–15). Recognition of endogenous RNA, viral RNAs, and viral ribonucleoproteins (vRNPs) that might exist in the Z-nucleic acid conformation is thought to activate ZBP1-mediated inflammation and cell death (6, 8, 16). The Zα domains of ZBP1 show high affinity for Z-nucleic acids in vitro (12–14, 17); however, whether the recognition of Z-nucleic acids regulates cellular functions is not studied. In addition, the role of the Z-nucleic acid–binding Zα domains of ZBP1 in regulating cell death and inflammation remains poorly investigated. Here we identify the critical role of the Zα2 domain of ZBP1 in regulating IAV-induced NLRP3 inflammasome activation, cell death, and perinatal lethality during mouse development. Our observations define a pivotal role of the Zα2 domain of ZBP1 in vivo and provide evidence for the activation of ZBP1 by endogenous Z-RNAs.
Results
Generation of C57BL/6 mice that lack the Zα2 domain of ZBP1
Experiments using ZBP1 knockout mice, which show complete loss of ZBP1 expression throughout the body (Zbp1−/−), have delineated the role of ZBP1 during in vivo infection and in mouse development (11). ZBP1 is a critical innate immune sensor for activating inflammatory signaling and PANoptosis in immune and nonimmune cells in response to IAV infection (5, 18). In addition, ZBP1 also activates necroptosis and inflammation during RIPK1-RHIM mutant mouse development, leading to perinatal lethality (9, 10). The N-terminal Zα domains of ZBP1 bind to nucleic acids in the Z-conformation. These Zα domains bind to Z-DNA or Z-RNA because of their conformational similarity when they attain the Z-conformation (12, 13, 15, 17, 19). Specific mutations in either of these Zα domains block their Z-nucleic acid–binding activity (12, 13, 19). Although several recent studies established the role of ZBP1 in innate immunity and development, specific contributions of the Z-nucleic acid–recognizing Zα domains of ZBP1 in vivo are not clear. To investigate this, we generated ZBP1 mutant mice with deletion of the Zα2 domain, which contains critical residues essential for Z-nucleic acid recognition, in the C57BL/6 background using CRISPR/Cas9 technology (hereafter referred to as Zbp1ΔZα2/ΔZα2 mice) (Fig. 1A and Table S1). At the amino acid sequence level, the CRISPR/Cas9 approach generated a Zα2 domain deletion in ZBP1 by joining residues at positions 76 and 151 and deleting residues at positions 77–150 (Fig. 1A). Zbp1ΔZα2/ΔZα2 mice were viable and did not show visible phenotypic defects.
Figure 1.
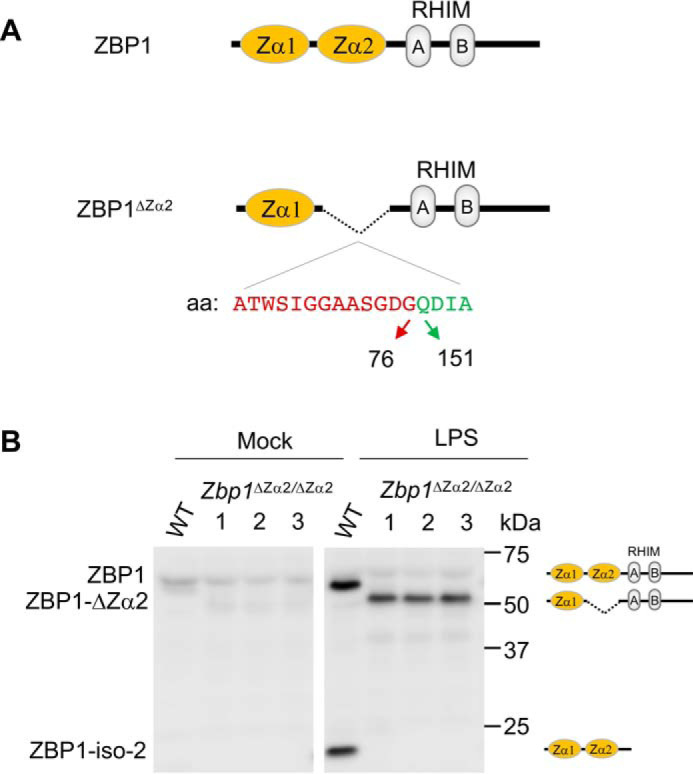
Generation of mice with Zα2 domain deletion in ZBP1 (Zbp1ΔZα2/ΔZα2). A, schematic of domain structure and deletion of the Zα2 domain in ZBP1. Amino acid residues (aa) present at the N terminus (red) and C terminus (green) of the Zα2 domain that are in sequence after deletion of this domain are represented. B, immunoblot analysis of ZBP1 after mock or LPS stimulation of WT and Zbp1ΔZα2/ΔZα2 BMDMs. ZBP1-iso-2, ZBP1 isoform 2.
To confirm deletion of the Zα2 domain at the protein level, we generated bone marrow–derived macrophages (BMDMs). Zbp1ΔZα2/ΔZα2 BMDMs showed similar expression of the myeloid markers CD11b and F4/80 compared with BMDMs generated from WT littermates, suggesting that the deletion caused no defects in the ability of bone marrow progenitors to differentiate into macrophages (Fig. S1A). Lipopolysaccharide (LPS) stimulation induced up-regulation of ZBP1 expression in WT and Zbp1ΔZα2/ΔZα2 BMDMs, indicating that Zα2 domain deletion did not interfere with the expression of ZBP1 protein (Fig. 1B). A shift in ZBP1 protein toward a lower molecular weight was observed in Zbp1ΔZα2/ΔZα2 BMDMs (ZBP1-ΔZα2), confirming deletion of the Zα2 domain in these mice (Fig. 1B). Isoform 2 of ZBP1, which consists of only the Zα1-Zα2 domains, was not seen in the Zbp1ΔZα2/ΔZα2 BMDMs (Fig. 1B). These results established deletion of the Zα2 domain of ZBP1 in mice with no detectable phenotypic differences in mice and macrophages.
The Zα2 domain of ZBP1 triggers inflammasome activation and PANoptosis during IAV infection
ZBP1 plays a crucial role in the activation of the NLRP3 inflammasome and PANoptosis upon recognizing IAV infection (5, 6). To understand the role of the Zα2 domain in ZBP1-induced NLRP3 inflammasome activation and PANoptosis, we infected WT and Zbp1ΔZα2/ΔZα2 BMDMs with mouse-adapted IAV (influenza A/Puerto Rico/8/34 (PR8; H1N1)). Similar to LPS stimulation, IAV infection up-regulated the expression of ZBP1-ΔZα2 to a similar extent that ZBP1 was up-regulated (Fig. 2A). In addition, there was no defect in IAV replication in Zbp1ΔZα2/ΔZα2 BMDMs, as indicated by NS1 protein expression (Fig. 2A). IAV infection triggers specific activation of the NLRP3 inflammasome (5, 6, 20, 21). Zbp1ΔZα2/ΔZα2 BMDMs showed a complete lack of caspase-1 (CASP1) activation compared with WT BMDMs, suggesting a reduction in IAV-induced NLRP3 inflammasome activation in these BMDMs (Fig. 2A). Cleavage of gasdermin D (GSDMD) and release of the GSDMD N-terminal domain (p30), which is a measure of GSDMD activation and pyroptosis, was also lost in Zbp1ΔZα2/ΔZα2 BMDMs in response to IAV infection (Fig. 2A). These observations suggest that the Zα2 domain is critical for ZBP1-mediated activation of the NLRP3 inflammasome and GSDMD-induced pyroptosis.
Figure 2.
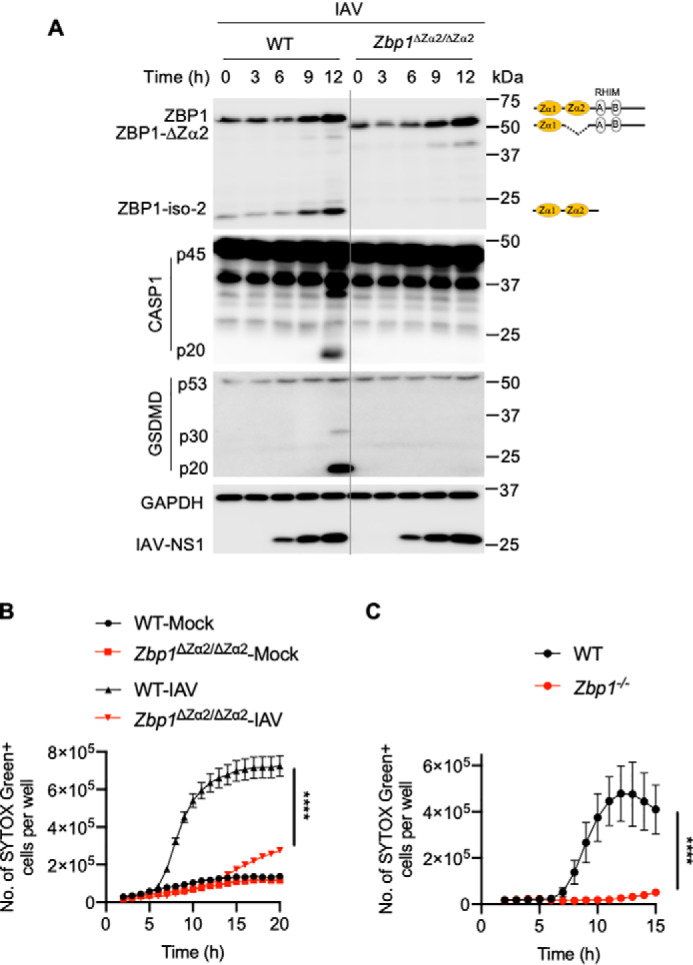
The Zα2 domain of ZBP1 is critical for triggering IAV-induced NLRP3 inflammasome activation and PANoptosis. A, immunoblot analysis of ZBP1, CASP1, GSDMD, IAV-NS1, and GAPDH in WT and Zbp1ΔZα2/ΔZα2 BMDMs infected with IAV (mouse-adapted, influenza A/Puerto Rico/8/34 (PR8; H1N1)). B and C, Cell death as measured by the number of SYTOX Green+ cells. BMDMs were infected with IAV, and cell death was monitored at regular intervals. ****, p < 0.0001 (one-way analysis of variance) for B; ****, p < 0.0001 (unpaired two-sided t test) for C. Data are represented as mean ± S.E. (B and C) and are representative of three independent experiments.
We further investigated the role of the Zα2 domain of ZBP1 in regulating IAV-induced PANoptosis in BMDMs. We found that IAV infection induced robust cell death in WT BMDMs compared with mock treatment (Fig. 2B). This IAV-induced cell death was significantly reduced in Zbp1ΔZα2/ΔZα2 BMDMs, suggesting a lack of ZBP1-mediated signaling (Fig. 2B). Consistent with previous reports (5, 6, 8), Zbp1−/− BMDMs showed a complete loss of IAV-induced cell death (Fig. 2C). In addition, immunoblot analysis indicated a lack of caspase-8 (CASP8) and caspase-3 (CASP3) activation in Zbp1ΔZα2/ΔZα2 BMDMs after IAV infection (Fig. S1B). Lack of activation of RIPK3, as measured by its phosphorylation when caspase activity was inhibited, was also observed in IAV-infected Zbp1ΔZα2/ΔZα2 BMDMs compared with WT BMDMs (Fig. S1C). These results suggest that the Zα2 domain of ZBP1 is necessary and sufficient for ZBP1 sensing of IAV infection to engage PANoptosis. Deletion of the Zα2 domain of ZBP1 caused defects in cellular function similar to those seen with loss of complete ZBP1 expression.
The Zα2 domain is crucial for ZBP1-induced necroptosis and perinatal lethality in RIPK1-RHIM mutant mice
Previous studies unraveled an important role of ZBP1 by studying RIPK1-RHIM mutant mice (9, 10). Mutations in the RHIM domain of RIPK1 (Ripk1mRHIM/mRHIM) inhibit its ability to form homotypic interactions with other RHIM-containing proteins, and this disruption causes perinatal lethality in mice. This RIPK1-RHIM mutation triggers necroptosis, inflammation, and skin lesions related to the activation of RIPK3. The perinatal lethality in RIPK1-RHIM mutant mice is driven by ZBP1-induced activation of RIPK3-MLKL–mediated necroptosis and inflammation. Deletion of ZBP1 or RIPK3 rescues Ripk1mRHIM/mRHIM mice from excessive cell death and perinatal lethality. This rescue denotes a critical role of RIPK1 in controlling activation of ZBP1 during development. These findings also demonstrate an important role of ZBP1 in regulating cell death and inflammation under physiological conditions and during mouse development. However, how ZBP1 is activated in Ripk1mRHIM/mRHIM mice and whether sensing of endogenous Z-RNAs via Zα domains triggers perinatal lethality is not clear. To understand this, we tested whether deletion of the Zα2 domain of ZBP1 is sufficient to rescue Ripk1mRHIM/mRHIM mice from perinatal lethality. Consistent with recent studies (9, 10), deletion of ZBP1 rescued Ripk1mRHIM/mRHIM mice from lethality, and viable mice were produced only when both the alleles of Zbp1 were absent (Fig. S2A). We found that deletion of the Zα2 domain of ZBP1 also rescued perinatal lethality in Ripk1mRHIM/mRHIM mice, and the mice became viable (Fig. 3, A and B). Similar to WT and Ripk1mRHIM/mRHIMZbp1−/− mice, Ripk1mRHIM/mRHIMZbp1ΔZα2/ΔZα2 mice also survived and developed normally into adulthood (Fig. 3C). Deletion of the Zα2 domain in both the alleles of Zbp1 was necessary to make Ripk1mRHIM/mRHIM mice viable (Fig. 3, A–C). This suggests that complete inhibition of the Z-RNA–sensing activity of ZBP1 through deletion of the Zα2 domain is necessary to inhibit ZBP1-driven necroptosis and perinatal lethality in Ripk1mRHIM/mRHIM mice. These observations identify that the Zα2 domain of ZBP1 is critical for driving cell death during development in Ripk1mRHIM/mRHIM mice, which, in turn, indicates potential sensing of endogenous RNA ligands by ZBP1.
Figure 3.
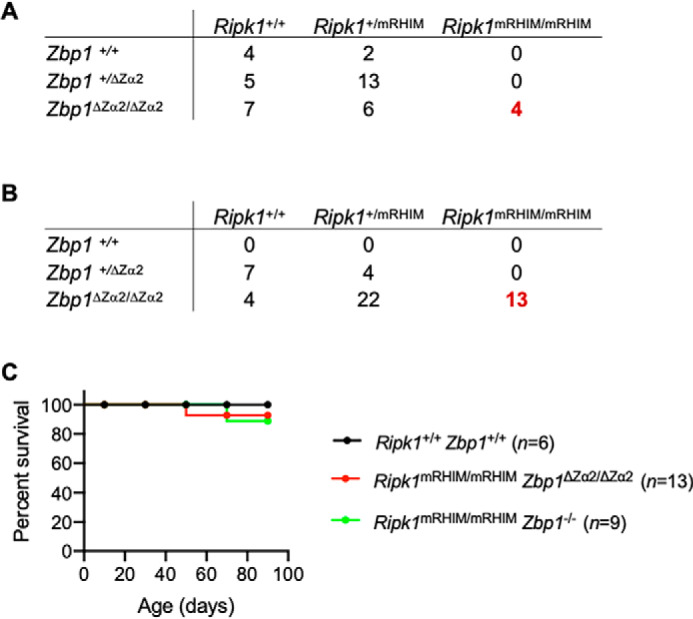
The Z-nucleic acid–binding Zα2 domain of ZBP1 is essential for embryonic development and acts as an activator of RIPK1-RHIM mutation-induced perinatal lethality in mice. A, the number of offspring of each genotype generated from intercrossing Ripk1+/mRHIM Zbp1+/ΔZα2 (heterozygote) parents. B, the number of offspring of each genotype generated from intercrossing Ripk1+/mRHIM Zbp1ΔZα2/ΔZα2 parents. In A and B, the numbers of Ripk1mRHIM/mRHIM mice rescued from perinatal lethality are shown in red. C, Kaplan–Meier plot showing survival of Ripk1mRHIM/mRHIM Zbp1ΔZα2/ΔZα2 mice and Ripk1mRHIM/mRHIM Zbp1−/− mice compared with WT Ripk1+/+ Zbp1+/+ mice.
To further investigate the impact of deleting the Zα2 domain of ZBP1, we studied the immune cell population in circulation. The numbers and percentages of circulating lymphocytes, monocytes, and red blood cells were unaltered in viable Ripk1mRHIM/mRHIMZbp1ΔZα2/ΔZα2 mice compared with WT mice (Fig. S2B). We observed a higher percentage of circulating neutrophils in Ripk1mRHIM/mRHIMZbp1ΔZα2/ΔZα2 mice and Ripk1mRHIM/mRHIMZbp1−/− mice (Fig. S2B). Consistent with these observations, a previous study reported neutrophilic inflammation in Ripk1mRHIM/mRHIMZbp1−/− mice (10). Ex vivo differentiated BMDMs from Ripk1mRHIM/mRHIM Zbp1ΔZα2/ΔZα2 mice and Ripk1mRHIM/mRHIMZbp1−/− mice showed similar CD11b and F4/80 myeloid expression markers compared with WT BMDMs (Fig. S2C). In addition, IAV infection did not induce cell death in Ripk1mRHIM/mRHIMZbp1ΔZα2/ΔZα2 or Ripk1mRHIM/mRHIMZbp1−/− BMDMs (Fig. S2D). These results suggest that deletion of the Zα2 domain of ZBP1 prevents cell death and perinatal lethality in Ripk1mRHIM/mRHIM mice and facilitates normal immune cell development in these mice, with a mild increase in circulating neutrophils.
Discussion
Several recent studies unraveled an important role of ZBP1 in regulating cell death and inflammation and uncovered its role as an innate immune sensor. ZBP1 was first described as a tumor-associated protein that is up-regulated in macrophages (22). Identification of a RHIM domain in ZBP1 facilitated its characterization in cell death and inflammation (3, 4, 11). RHIM-containing proteins regulate inflammatory signaling, apoptosis, and inflammatory cell death (1). Initial reports on ZBP1 suggested that it plays a role in virus-induced necroptosis via its RHIM-mediated interaction with RIPK3 (3, 4). We identified that IAV infection and its RNA genome products activate the NLRP3 inflammasome to mount inflammation and lung repair responses in vivo (20, 21, 23). Identification of ZBP1's role in sensing IAV and activating the inflammasome and multiple programmed cell death pathways expanded its functions in diverse cellular activities and illustrated potential regulation of inflammation in vivo (5, 11). Although Zα domains interact with Z-nucleic acids in vitro, activation and cellular functions of these Zα domains of ZBP1 and in vivo relevance were not studied.
Our findings here suggest that the Zα2 domain is critical for activation of ZBP1's functions. The Zα2 domain is required for activation of ZBP1 to drive NLRP3 inflammasome activation, PANoptosis, and perinatal lethality in mice. It was interesting to find that deletion of the Zα2 domain was sufficient to block ZBP1's functions. Our observations also suggest that the Zα1 domain might be dispensable for ZBP1 regulation in vivo. The Zα2 domain of ZBP1 shows a distinct way of binding to Z-nucleic acids, unlike other Zα domains from various other proteins (12). This suggests that the Zα2 domain of ZBP1 might have evolved to control cellular functions by executing distinct mechanisms, and our findings showing its critical role in regulating host innate responses support this hypothesis. Earlier in vitro studies also demonstrated that deletion of the Zα1 domain is not sufficient to inhibit ZBP1-induced cell death (8). Our results also suggest that the Zα2 domain of ZBP1 may recognize viral or endogenous RNAs that attain Z-conformation to activate ZBP1-driven cell death and perinatal lethality. The Asn-122 and Tyr-126 residues in the Zα2 domain of ZBP1 are essential for the Z-nucleic acid interaction, and mutations in these positions have been shown to abolish the Z-nucleic acid–binding potential of ZBP1 (12). Thus, the Zα2 domain of ZBP1 is a crucial determinant of ZBP1 for sensing Z-nucleic acids and subsequent engagement of inflammation and cell death. Observations from IAV infection experiments suggest that deletion of the Zα2 domain phenocopies complete ZBP1 deletion in regulating PANoptosis and activation of the NLRP3 inflammasome.
While this manuscript was in preparation, new studies appeared, demonstrating that sensing of influenza Z-RNAs or endogenous retroviral Z-nucleic acids via the Zα domains of ZBP1 is essential for activating cell death and inflammation (24–26). These findings from independent groups corroborate our results presented here and also our previous reports on ZBP1 sensing of an RNA virus (IAV) to activate cell death and inflammation (5, 6, 24–26). Overall, our findings, in conjunction with these new studies, establish a critical and specific role of the Zα2 domain of ZBP1 in regulating PANoptosis, inflammation, and perinatal lethality in mice and provide genetic evidence of the functional relevance of Z-nucleic acid–binding domains in vivo.
Experimental procedures
Mice
The Zα2 domain of ZBP1 was deleted in C57BL/6 mice (Zbp1ΔZα2/ΔZα2) using CRISPR/Cas9 gene editing. Briefly, C57BL/6J (The Jackson Laboratory, Bar Harbor, ME, USA) fertilized zygotes were injected into the pronucleus with 15 ng/ml of each chemically modified single guide RNA (sgRNA) (Synthego), 60 ng/ml SpCas9 protein (Berkeley Microlab), and 10 ng/ml single-stranded oligodeoxynucleotide (ssODN) donor (IDT) diluted in 0.1 mm EDTA. Founder mice were genotyped by targeted next-generation sequencing on a MiSeq Illumina sequencer and analyzed with CRIS.py (27). Editing construct sequences and relevant primers are listed in Table S1. After confirming the deletion, mice were backcrossed to C57BL/6 genetic background mice at least three times. Ripk1+/mRHIM mice were a gift from Dr. Vishva M. Dixit (Genentech) (10). These mice were crossed to Zbp1ΔZα2/ΔZα2 mice and Zbp1−/− mice to generate Ripk1mRHIM/mRHIM Zbp1ΔZα2/ΔZα2 mice and Ripk1mRHIM/mRHIMZbp1−/− mice. Offspring were monitored for survival and rescue of perinatal lethality in Ripk1mRHIM/mRHIM mice. Mutations or deletions were confirmed by PCR. All mice were bred and housed in the Animal Resource Center at St. Jude Children's Research Hospital. Animal study protocols were approved by the St. Jude Children's Research Hospital Committee on the Care and Use of Animals.
BMDM cultures and IAV infection
Primary BMDMs from mouse bone marrow were grown for 6 days in Iscove's modified Dulbecco's medium (Gibco) supplemented with 30% L929-conditioned medium, 10% FBS (Atlanta Biologicals), 1% penicillin and streptomycin, and 1% nonessential amino acids (Gibco). BMDMs were seeded and cultured overnight before using them for stimulations or infections. For LPS stimulation experiments, BMDMs were mock-treated (medium only) or stimulated with 100 ng/ml LPS (InvivoGen) for 4 h. The IAV-PR8 strain was generated as described previously (5, 6). BMDMs were infected with IAV (multiplicity of infection, 10) for 2 h in serum-free DMEM (Sigma). After 2 h of infection, 10% FBS was added. For IAV + benzyloxycarbonyl-VAD (zVAD) treatment, 30 μm zVAD was added to BMDMs after 2 h of infection. Whole-cell lysates were collected at the indicated time points for immunoblot analysis, or cell death was monitored.
Immunoblot analysis
Immunoblotting and detection were performed as described previously (6). BMDMs were washed with PBS once, cells were lysed in radioimmune precipitation assay buffer, and sample loading buffer containing SDS and 2-mercaptoethanol was added. Proteins were separated on 8%–12% polyacrylamide gels and transferred onto PVDF membranes (Millipore). Membranes were blocked in 5% skim milk, followed by incubation with primary antibodies and secondary HRP-conjugated antibodies. Primary antibodies used were anti-ZBP1 (AG-20B-0010-C100, AdipoGen), anti-caspase-1 (AG-20B-0042, AdipoGen), anti-caspase-3 (9662, CST), anti-cleaved caspase-3 (9661, CST), anti-caspase-8 (AG-20T-0138-C100, AdipoGen), anti-cleaved caspase-8 (8592, CST), anti-GSDMD (Ab209845, Abcam), anti-GAPDH (5174, CST), anti-RIPK3 (2283, ProSci), anti-IAV M1 (ab20910, Abcam), and anti-IAV NS1 (sc-130568, Santa Cruz Biotechnology Inc.). HRP-conjugated secondary antibodies (anti-rabbit (111-035-047) and anti-mouse (315-035-047), Jackson ImmunoResearch Laboratories) were used. Protein detection was done by using Luminata Forte Western HRP substrate (Millipore, WBLUF0500).
SYTOX Green staining and cell death analysis
Real-time cell death assays of IAV-infected BMDMs were performed using an IncuCyte Zoom incubator imaging system (Essen Biosciences). BMDMs were seeded in 12-well tissue culture plates in the presence of 100 nm SYTOX Green (Thermo Fisher Scientific, S7020), a cell-impermeable DNA-binding fluorescent dye that enters dying cells upon membrane permeabilization. Analysis was done using the software package supplied with the IncuCyte imager. Using this software, a precise analysis of the number of SYTOX Green+ cells present in each image can be performed. The number of dead cells was acquired by counting SYTOX Green+ cells and plotted using GraphPad Prism v8 software.
Flow cytometry
Flow cytometry analysis was performed to monitor differentiation of bone marrow cultures into macrophages. For analysis, the following monoclonal antibodies were used: anti-mouse CD45.2 (clone 104, BioLegend, 109806), anti-CD11b (M1/70, Affymetrix eBioscience, 48-0112-82), and anti-F4/80 (BM8, BioLegend, 123116). These antibodies were diluted 1:300 to stain BMDMs for 15 min at 4 °C in FACS buffer (2% (v/v) of FBS in PBS containing 0.04% (w/v) sodium azide). Flow cytometry data were acquired on an LSR II flow cytometer (BD Biosciences) and analyzed with FlowJo software (FlowJo, LLC, and Illumina, Inc.).
Data availability
All data generated for this study are included within this manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Vishva M Dixit from Genentech for sharing Ripk1+/mRHIM mice. We also thank Rebecca Tweedell, Ph.D., for scientific editing.
This article contains supporting information.
Author contributions—S. K. and T.-D. K. conceptualization; S. K. and R. K. S. M. data curation; S. K., R. K. S. M., and T.-D. K. formal analysis; S. K., A. R. B., and P. V. investigation; S. K., R. K. S. M., and T.-D. K. methodology; S. K. and T.-D. K. writing-original draft; S. K., R. K. S. M., A. R. B., and T.-D. K. writing-review and editing; T.-D. K. funding acquisition; S. N. P. and S. M. P.-M. generated mice critical for the study.
Funding and additional information—This study was supported by National Institutes of Health Grants AR056296, CA163507, AI101935, and AI124346 and the American Lebanese Syrian Associated Charities (to T.-D. K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- RHIM
- RIP homotypic interaction motif
- DAI
- DNA-dependent activator of IFN regulatory factor
- IAV
- influenza A virus
- BMDM
- bone marrow–derived macrophage
- LPS
- lipopolysaccharide
- GSDMD
- gasdermin D.
References
- 1. Kesavardhana S., Malireddi R. K. S., and Kanneganti T. D. (2020) Caspases in cell death, inflammation, and gasdermin-induced pyroptosis. Annu. Rev. Immunol. 38, 567–595 10.1146/annurev-immunol-073119-095439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pasparakis M., and Vandenabeele P. (2015) Necroptosis and its role in inflammation. Nature 517, 311–320 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- 3. Rebsamen M., Heinz L. X., Meylan E., Michallet M. C., Schroder K., Hofmann K., Vazquez J., Benedict C. A., and Tschopp J. (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 10, 916–922 10.1038/embor.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Upton J. W., Kaiser W. J., and Mocarski E. S. (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 10.1016/j.chom.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuriakose T., Man S. M., Malireddi R. K., Karki R., Kesavardhana S., Place D. E., Neale G., Vogel P., and Kanneganti T. D. (2016) ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1, aag2045 10.1126/sciimmunol.aag2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kesavardhana S., Kuriakose T., Guy C. S., Samir P., Malireddi R. K. S., Mishra A., and Kanneganti T. D. (2017) ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 2217–2229 10.1084/jem.20170550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nogusa S., Thapa R. J., Dillon C. P., Liedmann S., Oguin T. H. 3rd, Ingram J. P., Rodriguez D. A., Kosoff R., Sharma S., Sturm O., Verbist K., Gough P. J., Bertin J., Hartmann B. M., Sealfon S. C., et al. (2016) RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24 10.1016/j.chom.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thapa R. J., Ingram J. P., Ragan K. B., Nogusa S., Boyd D. F., Benitez A. A., Sridharan H., Kosoff R., Shubina M., Landsteiner V. J., Andrake M., Vogel P., Sigal L. J., tenOever B. R., Thomas P. G., et al. (2016) DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681 10.1016/j.chom.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin J., Kumari S., Kim C., Van T. M., Wachsmuth L., Polykratis A., and Pasparakis M. (2016) RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128 10.1038/nature20558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Newton K., Wickliffe K. E., Maltzman A., Dugger D. L., Strasser A., Pham V. C., Lill J. R., Roose-Girma M., Warming S., Solon M., Ngu H., Webster J. D., and Dixit V. M. (2016) RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133 10.1038/nature20559 [DOI] [PubMed] [Google Scholar]
- 11. Kuriakose T., and Kanneganti T. D. (2018) ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134 10.1016/j.it.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ha S. C., Kim D., Hwang H. Y., Rich A., Kim Y. G., and Kim K. K. (2008) The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc. Natl. Acad. Sci. U.S.A. 105, 20671–20676 10.1073/pnas.0810463106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim K., Khayrutdinov B. I., Lee C. K., Cheong H. K., Kang S. W., Park H., Lee S., Kim Y. G., Jee J., Rich A., Kim K. K., and Jeon Y. H. (2011) Solution structure of the Zβ domain of human DNA-dependent activator of IFN-regulatory factors and its binding modes to B- and Z-DNAs. Proc. Natl. Acad. Sci. U.S.A. 108, 6921–6926 10.1073/pnas.1014898107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Placido D., Brown B. A. 2nd, Lowenhaupt K., Rich A., and Athanasiadis A. (2007) A left-handed RNA double helix bound by the Zα domain of the RNA-editing enzyme ADAR1. Structure 15, 395–404 10.1016/j.str.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz T., Behlke J., Lowenhaupt K., Heinemann U., and Rich A. (2001) Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol. 8, 761–765 10.1038/nsb0901-761 [DOI] [PubMed] [Google Scholar]
- 16. Maelfait J., Liverpool L., Bridgeman A., Ragan K. B., Upton J. W., and Rehwinkel J. (2017) Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 10.15252/embj.201796476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rich A., and Zhang S. (2003) Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet. 4, 566–572 10.1038/nrg1115 [DOI] [PubMed] [Google Scholar]
- 18. Malireddi R. K. S., Kesavardhana S., and Kanneganti T. D. (2019) ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9, 406 10.3389/fcimb.2019.00406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deigendesch N., Koch-Nolte F., and Rothenburg S. (2006) ZBP1 subcellular localization and association with stress granules is controlled by its Z-DNA binding domains. Nucleic Acids Res. 34, 5007–5020 10.1093/nar/gkl575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanneganti T. D., Body-Malapel M., Amer A., Park J. H., Whitfield J., Franchi L., Taraporewala Z. F., Miller D., Patton J. T., Inohara N., and Núñez G. (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 10.1074/jbc.M607594200 [DOI] [PubMed] [Google Scholar]
- 21. Thomas P. G., Dash P., Aldridge J. R. Jr., Ellebedy A. H., Reynolds C., Funk A. J., Martin W. J., Lamkanfi M., Webby R. J., Boyd K. L., Doherty P. C., and Kanneganti T. D. (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575 10.1016/j.immuni.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fu Y., Comella N., Tognazzi K., Brown L. F., Dvorak H. F., and Kocher O. (1999) Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 240, 157–163 10.1016/S0378-1119(99)00419-9 [DOI] [PubMed] [Google Scholar]
- 23. Kanneganti T. D., Ozören N., Body-Malapel M., Amer A., Park J. H., Franchi L., Whitfield J., Barchet W., Colonna M., Vandenabeele P., Bertin J., Coyle A., Grant E. P., Akira S., and Núñez G. (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 10.1038/nature04517 [DOI] [PubMed] [Google Scholar]
- 24. Jiao H., Wachsmuth L., Kumari S., Schwarzer R., Lin J., Eren R. O., Fisher A., Lane R., Young G. R., Kassiotis G., Kaiser W. J., and Pasparakis M. (2020) Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395 10.1038/s41586-020-2129-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang T., Yin C., Boyd D. F., Quarato G., Ingram J. P., Shubina M., Ragan K. B., Ishizuka T., Crawford J. C., Tummers B., Rodriguez D. A., Xue J., Peri S., Kaiser W. J., López C. B., et al. (2020) Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129 e1113 10.1016/j.cell.2020.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang R., Li H., Wu J., Cai Z. Y., Li B., Ni H., Qiu X., Chen H., Liu W., Yang Z. H., Liu M., Hu J., Liang Y., Lan P., Han J., and Mo W. (2020) Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580, 386–390 10.1038/s41586-020-2127-x [DOI] [PubMed] [Google Scholar]
- 27. Connelly J. P., and Pruett-Miller S. M. (2019) CRIS.py: a versatile and high-throughput analysis program for CRISPR-based genome editing. Sci. Rep. 9, 4194 10.1038/s41598-019-40896-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated for this study are included within this manuscript.