

Abstract
Reducing infarct size during a cardiac ischaemic‐reperfusion episode is still of paramount importance, because the extension of myocardial necrosis is an important risk factor for developing heart failure. Cardiac ischaemia‐reperfusion injury (IRI) is in principle a metabolic pathology as it is caused by abruptly halted metabolism during the ischaemic episode and exacerbated by sudden restart of specific metabolic pathways at reperfusion. It should therefore not come as a surprise that therapy directed at metabolic pathways can modulate IRI. Here, we summarize the current knowledge of important metabolic pathways as therapeutic targets to combat cardiac IRI. Activating metabolic pathways such as glycolysis (eg AMPK activators), glucose oxidation (activating pyruvate dehydrogenase complex), ketone oxidation (increasing ketone plasma levels), hexosamine biosynthesis pathway (O‐GlcNAcylation; administration of glucosamine/glutamine) and deacetylation (activating sirtuins 1 or 3; administration of NAD+‐boosting compounds) all seem to hold promise to reduce acute IRI. In contrast, some metabolic pathways may offer protection through diminished activity. These pathways comprise the malate‐aspartate shuttle (in need of novel specific reversible inhibitors), mitochondrial oxygen consumption, fatty acid oxidation (CD36 inhibitors, malonyl‐CoA decarboxylase inhibitors) and mitochondrial succinate metabolism (malonate). Additionally, protecting the cristae structure of the mitochondria during IR, by maintaining the association of hexokinase II or creatine kinase with mitochondria, or inhibiting destabilization of FOF1‐ATPase dimers, prevents mitochondrial damage and thereby reduces cardiac IRI. Currently, the most promising and druggable metabolic therapy against cardiac IRI seems to be the singular or combined targeting of glycolysis, O‐GlcNAcylation and metabolism of ketones, fatty acids and succinate.
Keywords: ischemia, metabolism, mitochondria
1. INTRODUCTION
Cardiac metabolism changes rapidly during a sudden ischaemic episode of the heart, with the oxygen shortage repressing oxidative metabolism of fatty acids (FA), carbohydrates, ketones and amino acids, and activating anaerobic glycolysis to spare the use of limited oxygen. During reperfusion with the wash‐in of oxygen and the wash‐out of ischaemic metabolites, there is an abrupt normalization of intracellular pH and a specific start‐up of oxidative metabolism for the various substrates, with ongoing changes in what is now aerobic glycolysis. These metabolic changes during ischaemia and early reperfusion are not merely passive bystanders, but determine to a large extent the actual injury developing in the heart following an ischaemic episode. Modulation of these metabolic changes, therefore, offers the opportunity for developing therapy against cardiac IRI, as was already shown by Sodi‐Pallares in 1962 using potassium‐insulin‐glucose administration during myocardial infarction. 1 Since these earlier studies, it has become clear that metabolic therapy for IRI has travelled a rather bumpy road, without the development of a proven effective clinical metabolic therapy as of yet. Here, we review the current literature concerning this topic, going from the well‐known metabolic pathways to novel metabolic targets that go beyond the general metabolism of glucose and FA, and focus on important processes such as acidosis, ketone oxidation, succinate accumulation, mitochondrial FOF1‐ATPase, energy transfer pathways, protein O‐GlcNAcylation and acetylation as novel metabolic targets for treating IRI.
2. GENERAL ASPECTS OF CARDIAC METABOLISM IN HEALTHY HEART
The healthy heart is a true omnivore in that it can degrade various energy‐containing substrates. The major cardiac fuels for respiration are fats (triglyceride and long‐chain fatty acids), carbohydrates (glucose, lactate and cardiac glycogen) and ketone bodies (acetoacetate and β‐hydroxybutyrate) (Figure 1). Regulation of substrate use by the heart is to a large extent determined by the amount of substrate delivered to the heart (ie plasma concentration), the number of specific substrate transporters present in the cell membrane (CD36/Fat for fatty acids, GLUT1/4 for glucose and MCT1/2 for lactate and ketone bodies) and the activities of the metabolic enzymes and substrate/products/cofactors present in the enzymatic pathways. 2 , 3 , 4 It should thereby be realized that a high plasma concentration of one substrate usually competes and inhibits the use of other substrates. For example, high plasma fatty acid levels will impair glycolysis and glucose oxidation, 4 or increasing plasma lactate levels will impair fatty acid oxidation and glycolysis. 5 Although in general fatty acids contribute more than carbohydrates to ATP generation in the heart, this depends critically on (patho) physiological, nutritional and hormonal state. For example, when insulin, lactate and fatty acids are present at normal physiological concentrations in the ex vivo‐perfused rodent heart, the contribution of carbohydrates can be higher than that of fatty acids. 6 , 7 , 8 Ketone bodies, when provided at physiological plasma concentrations (<0.3 mmol/L), contribute less than 5% to cardiac ATP generation. 9 Each substrate is finally broken down to acetyl coenzyme A (acetyl‐CoA) that feeds the tricarboxylic acid (TCA) cycle to produce reducing equivalents (NADH and FADH2) that then feed the electron transport chain to build a proton gradient to drive the FOF1‐ATP/synthase to make ATP. During normoxia, approximately 90% of the ATP produced is derived from mitochondrial oxidative breakdown of substrates, with cytosolic glycolysis only contributing ~5%‐10% of total ATP production. Only during total ischaemia does glycolysis become the major supplier of ATP, when glycogen stored in the heart starts feeding glycolysis. The malate/aspartate shuttle (MAS; Figure 1) is critical for maintaining glycolytic rate because it is the main pathway to recycle glycolysis‐produced NADH through the mitochondria into cytosolic NAD+ to maintain glycolytic activity.
Figure 1.
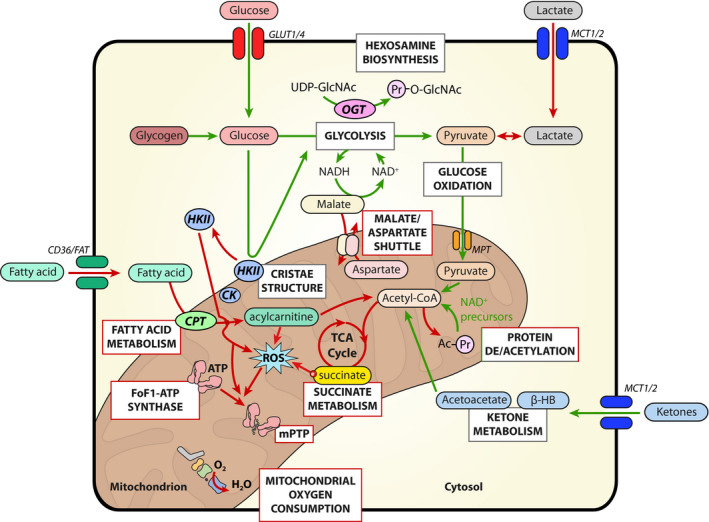
Summary of the proposed pathways of cardiac metabolism covered in this review. Many of the discussed pathways show protection against IRI (green arrows) or are protective if blocked (red arrows). CK, creatine kinase; CPT, carnitine palmitoyltransferase; HKII, hexokinase II; MCT, monocarboxylate transporter; MPT, mitochondrial pyruvate transporter; mPTP, mitochondrial permeability transition pore; OGT, O‐GlcNAc transferase
It should be noted that for cardiac metabolism, it is not only the flux through the metabolic pathway that regulates cardiac function and physiology, but also the level of its intermediary metabolites. Typical examples of regulating metabolic intermediates are as follows: a) acetyl‐CoA, partly regulating the acetylation status and thereby function of proteins, b) succinate, build‐up during ischaemia, partly determining reactive oxygen production upon reperfusion, and c) UDP‐GlcNAc, as end product of the hexosamine biosynthetic pathway. Although only a small part of glucose (<0.01% of glycolytic rate) 10 is shuttled into this pathway, the generated O‐linked attachment of N‐acetylglucosamine moiety onto proteins affects protein function significantly.
Finally, spatial and temporal distribution of metabolic enzymes can also be important effectors of cardiac metabolism. One classical example relates to hexokinase II (HKII) that shuttles between mitochondria and cytosol, depending on nutritional and pathophysiological status. When bound to mitochondria (mtHKII), HKII increases glycolysis, impairs in vivo cardiac oxygen consumption, impairs fatty acid oxidation, facilitates growth processes and protects mitochondria against injury. 7 , 11 , 12 Another example is creatine kinase (CK), the enzyme making ATP from phosphocreatine (PCr) and thereby functioning as an important local buffer for ATP breakdown. Cytosolic and mitochondrial isoforms of CK form a metabolic signalling network that is needed for rapid energy (ATP) transfer between ATP producers (mitochondria) and users (muscle contraction and ion pumps), preventing measurable decreases in ATP during changes in cardiac work. 13 , 14
3. TARGETING METABOLIC PATHWAYS TO COMBAT CARDIAC IRI
3.1. Glycolysis
3.1.1. Glycolysis during ischaemia
During ischaemia, glycolysis is increased due to (i) increased glucose extraction from the blood (going from 1% during normoxia to 30% extraction during severe low flow), (ii) increased translocation of GLUT transporters to the sarcolemma, (iii) increased glycogenolysis, due to stimulation of glycogen phosphorylase A by diminishing glucose, ATP, glucose 6‐phosphate and increases in AMP and Ca2+, (iv) removing citrate‐mediated inhibition of the glycolysis‐controlling enzyme 6‐phosphofructokinase‐1 (PFK‐1) and (v) AMP‐activated protein kinase (AMPK) activation, resulting in both increasing GLUT transport capacity at the membrane and activation of 6‐phosphofructokinase‐2 (PFK2), resulting in fructose 2,6‐bisphosphate production, the most potent allosteric stimulator of PFK1. 15 , 16 , 17 , 18 Activation of glycolysis during low‐flow conditions makes use of all these mechanisms, whereas, during no‐flow conditions, only mechanisms (iii), (iv) and (v) are active. Of note, although most pre‐clinical studies have employed the no‐flow ischaemia model, low‐flow ischaemia may better reflect the clinical condition of myocardial infarction, where residual flow is often present. 19 Following long‐lasting glycolytic activity, glycolysis can subsequently be inhibited by accumulation of its products: lactate, protons and NADH. 15 As long as glycolysis can proceed and generate ATP during the entire oxygen‐limited condition, it offers protection against IRI. Because during low‐flow conditions lactate and protons can still partly be removed and glucose uptake proceeds, inhibition of glycolysis is much delayed as compared to no‐flow conditions. This explains why during even extreme low‐flow ischaemia (5% of normal flow), an extended 40‐min period of low‐flow induces hardly any IRI (<3% cell death), 20 whereas 40‐minute no‐flow induces 20x more cell death. 21 Low‐flow ischaemia does induce damage when glucose is replaced by another substrate or taken out of the perfusate, supporting the concept that it is the glycolytically derived ATP during ischaemia that protects against IRI. 22 , 23 Increasing glycolysis during low‐flow ischaemia through, for example, increasing levels of glucose and insulin in the perfusate or boosting endogenous glycogen is associated with delayed contracture development and protection against IRI. 24 The start of contracture development during ischaemia is a read‐out of the time when glycolysis stops 22 and the free energy of ATP hydrolysis, the ∆G ATP, falls below a critical level to support the ATPase activity of ion pumps and cross‐bridge cycling. 26 Also during no‐flow ischaemia activating or prolonging glycolysis is most frequently associated with protection against IRI. In line, AMPK‐deficient mouse models are characterized by decreased glucose uptake and glycolysis, increased ATP depletion during ischaemia, more rapid and severe ischaemic contracture, increased cell death during reperfusion and poorer post‐ischaemic contractile recovery. 27 , 28 Previous studies also clearly showed that either increasing pre‐ischaemic glycogen 29 or activating glycolysis through redox control by niacin‐induced lowering of NADH/NAD 30 resulted in delayed ischaemic contracture and reduced IRI. Similarly, the pharmacologic overactivation of AMPK reduces cardiomyocyte death and ameliorates post‐ischaemic function recovery. 31 , 32
However, there are experimental conditions where increasing pre‐ischaemic glycogen and/or a delayed contracture development were actually associated with increased IRI. 23 , 33 , 34 Although it is not completely clear what sets these conditions apart, it could be related to excessive glycogen loading (and thus excessive accumulation of glycogen breakdown products and consequently low pH during ischaemia) 23 due to elevated insulin before ischaemia (thereby inhibiting non‐ischaemic glycogen breakdown) 16 as compared to no insulin before ischaemia. 33 The presence of more insulin before ischaemia can also result in impaired activation of the reperfusion injury salvage kinase (RISK) pathway during reperfusion, 35 possibly explaining the increased IRI in these experimental conditions. Mechanisms explaining increased infarct size with increased accumulation of glycolytic breakdown products during ischaemia are a) detachment of hexokinase II from mitochondria (mtHKII) due to high levels of G6P and low pH 11 , 33 , 36 and/or b) increased ischaemic Na+ loading due to increased proton production by excessive glycogen breakdown, resulting in increased Ca2+ overload upon reperfusion. 23
3.1.2. Glycolysis during reperfusion
The strongest cardioprotective intervention for protecting against cardiac IRI, observed across all species, concerns ischaemic pre‐conditioning (IPC). IPC relates to the application of short, non‐lethal periods of ischaemia before the long, lethal period of ischaemia, resulting in a 75% reduction in infarct size. 37 IPC activates glycolysis during normoxia and reperfusion, through activation of AMPK and Akt, translocation of GLUT4 transporters to the cell membrane and translocation of HKII from cytosol to mitochondria (Figure 1). 7 , 38 , 39 , 40 , 41 , 42 , 43 Interestingly, no protection by IPC is observed in the absence of glucose. 39 Taken together, the data strongly suggest that IPC protective mechanism is mediated through activated aerobic glycolysis during early reperfusion. An increased glycolysis may mediate protection through increased activity of mtHKII, knowing that glucose phosphorylation by mtHKII is needed for protection against mitochondrial damage and cell death. 44 , 45 Additionally, older literature has suggested that protection by glycolysis is due to the preferential use of glycolytically produced ATP by ion pumps and restoration of ionic homeostasis during reperfusion. 46 Finally, increasing glycolysis during early reperfusion contributes to maintain a low pH, which in turn exerts multiple protective effects, including the prevention of mitochondrial permeability transition pore (mPTP) opening, 47 one of the mechanisms proposed for the cardioprotective effect of post‐conditioning. 48 It is especially an increased ratio of glycolysis over glucose oxidation that induces this net proton production, 49 and may, for example, explain the decreased infarct size following IR in high‐fat diet‐induced obesity. 50 It is thereby noteworthy that this reduction in infarct size with increased glycolysis uncoupling from glucose oxidation is opposite to the negative effect this uncoupling exerts on cardiac mechanical function, resulting in a decreased cardiac performance and mechanical efficiency. 49 However, the decreased mechanical function upon reperfusion may actually contribute to the reduction in infarction, knowing that a slow recovery of energy requirement in early reperfusion is cardioprotective 51 and that there is often a dichotomy between recovery of mechanical function and cell death following IR. 52 , 53
Besides ‘conditioning interventions’ to activate aerobic glycolysis, several pharmacologic cardioprotective agents are also known to increase glucose uptake/glycolysis (metformin, insulin, volatile anaesthetics, adenosine, NO donors, fructose 1,6‐diphosphate, nicotinamide mononucleotide (NMN), HIF1α stabilizers and AMPK activators). Some of these agents should ideally be applied at the end of the ischaemic period or at the onset of reperfusion, in order to prevent excessive glycogen accumulation, resulting in excessive accumulation of glycogen breakdown products during the ischaemic episode, thereby offsetting the beneficial effects of an activated glycolysis during early reperfusion.
In summary, most studies report that increasing glycolysis during ischaemia and early reperfusion confers protection against cardiac IRI, making glycolysis a potential important metabolic goal for IRI therapy.
3.2. Hexosamine biosynthesis pathway (HBP)
Beyond glycolysis, glucose can be used by accessory metabolic pathways such as the hexosamine biosynthesis pathway (HBP) (Figure 1). HBP concludes with O‐linked attachment of N‐acetylglucosamine moiety (O‐GlcNAc) onto serine and threonine residues of proteins. Similar to other post‐translational modifications, O‐GlcNAcylation is a dynamic and reversible molecular process involved in the regulation of metabolism, gene expression and protein‐protein interaction. 54 , 55 The rate of O‐GlcNAcylation is governed by (extra)cellular stresses or stimuli but also by nutrient type and availability. Next to glucose, glutamine, but also many other molecules, such as acetyl‐CoA and exogenous glucosamine, can enter into HBP to promote O‐GlcNAcylation. 56 , 57
Increases in O‐GlcNAcylation level occur and appear deleterious under chronic situations such as diabetes and cardiac hypertrophy. 54 , 55 On the other hand, it has been shown that O‐GlcNAcylation is central for the maintenance of cardiovascular functions. Cardiac‐specific deletion of the gene encoding for the O‐GlcNAc transferase (OGT), the enzyme responsible for the addition of O‐GlcNAc moiety on proteins, leads to progressive cardiomyopathy. 58 More importantly, increases in protein O‐GlcNAcylation are overall protective under acute settings. Using various in vitro, ex vivo and in vivo animal models, numerous studies demonstrate that O‐GlcNAcylation confers cardioprotection following acute IRI and other types of cardiac injuries. 54 , 59 Among the in vitro studies, protocols that mimic IR in isolated cardiomyocytes have been shown to promote protein O‐GlcNAcylation, and further increase in O‐GlcNAcylation using glucosamine treatment or OGT overexpression decreases cell death. 60 , 61 In line, IR performed in isolated perfused hearts increases O‐GlcNAc levels and, once again, treatment promoting O‐GlcNAcylation (glucosamine but also inhibitors of the β‐N‐acetylglucosaminidase OGA, the enzyme responsible for removing O‐GlcNAc moiety) reduces ischaemic contracture and improves post‐ischaemic contractile function recovery. 62 , 63 , 64 , 65 Notably, the treatment was also cardioprotective when applied at the onset of reperfusion, which increases its clinical relevance. 66 Finally, IPC performed in the ex vivo‐perfused heart or in vivo by left anterior descending artery ligation stimulates protein O‐GlcNAcylation and administration of an OGA inhibitor prior to surgery reduces infarct size. 66 , 67 This was further validated in humans submitted to remote IPC. 68 These data suggest that the cardioprotective effect of IPC may be mediated at least in part by an increase in OGT expression and activity. 67
The molecular mechanisms involved in the cardioprotective action of O‐GlcNAcylation are not fully understood. It has several effects, including (i) the modification of Ca2+ handling, 64 (ii) the increase in the mitochondrial translocation of the anti‐apoptotic Bcl‐2 protein, 69 (iii) the alteration of P38 MAPK signalling 63 and (iv) the attenuation of mitochondrial depolarization and mPTP opening, among others. Importantly, the cardioprotective action of O‐GlcNAcylation is lost under diabetic conditions, showing that the action of this post‐translational modification could differentially affect cardiac function when acutely or chronically induced. 54
In summary, most studies report that the acute increase in O‐GlcNAcylation elicits protection against cardiac IRI. The cardioprotective action of O‐GlcNAcylation could be promoted by acting pharmacologically on O‐GlcNAc enzymes such as OGA or OGT or by metabolically fuelling HBP via glucosamine and/or glutamine. It would be worthwhile to investigate these novel strategies in humans.
3.3. The malate‐aspartate shuttle
In the healthy adult myocardium, the malate‐aspartate shuttle (MAS) constitutes the main pathway for transportation of redox products from glycolysis in the cytosol into the mitochondrial matrix over the impermeable inner mitochondrial membrane (Figure 1). 70 Under normal conditions, the shuttle capacity is high. The shuttle also meets elevated glycolytic demand in a variety of physiological and pathophysiological processes including cardiac hypertrophy. 71 Hence, shuttle activity is modifiable. Because of its central role as a regulatory mechanism in the energy metabolism of the cardiomyocytes, it constitutes a potential target for induction of cardioprotection.
The MAS has gained conceptual interest as a means to modify mitochondrial function, because transient shut down of metabolism by blocking the cytosolic‐mitochondrial crosstalk via the MAS may induce cardioprotection (Figure 1). As a proof of concept, transient aminooxyacetate (AOA) administration can induce reversible MAS inhibition. AOA is a non‐specific competitive inhibitor of various amino acid transaminases 72 but in in situ heart models, AOA primarily leads to reversible inhibition of the MAS. 73 , 74 However, recent data also show that AOA directly reacts with alpha‐keto acids (pyruvate, alpha‐ketoglutarate, oxaloacetate, etc) to form stable oximes with unknown functional consequences as of yet. 75
Cardioprotection by MAS inhibition using AOA can be induced both prior to and during an ischaemic insult. 76 , 77 In the normal heart, MAS inhibition reduces mitochondrial complex I‐linked respiration and glycolysis. 76 Even though the effect of MAS inhibition predominantly involves the glycolytic pathway, MAS inhibition also yields cardioprotection in a setting with free FA as additional substrate next to glucose. 78 MAS inhibition reduced succinate‐induced ROS production (see 3.5.5. for mechanism) by limiting transport of substrate to the succinate build‐up. 79 The attenuated succinate oxidation during reperfusion reduces reverse electron flow at complex I and hence reduces oxidative stress and cellular damage. This key mechanism is further supported by the ability of MAS inhibition to preserve post‐ischaemic complex I respiration. 77
However, the mechanism does not explain that MAS inhibition also reduces infarct size when administrated in the late phase of an ischaemic event, when it is unlikely to reduce succinate substantially. 77 MAS inhibition may activate other mechanisms, depending on the timing of administration. The main effect of AOA is inhibition of aspartate aminotransferase, which is the pivotal enzyme in the MAS. The aspartate aminotransferase uses oxaloacetate as substrate, and inhibition by AOA may increase the intramitochondrial concentration of this tricarboxylic acid cycle intermediate. Because oxaloacetate serves as a strong inhibitor of succinate dehydrogenase thereby limiting post‐ischaemic oxidation of succinate, it is of interest to study whether the endogenously formed oxaloacetate conveys protection through reduced succinate dehydrogenase activity during reperfusion.
The MAS constitutes an important regulatory mechanism in the cardiomyocyte. The MAS may serve as a target for cardioprotection. However, drug toxicity remains a challenge and development of new reversible MAS inhibitors is necessary.
3.4. Lysine acetylation of cardiac proteins
The adduction of acyl groups to protein lysine residues (acylation) is a biologically significant post‐translational modification. Although many such acylations exist (eg succinylation, glutarylation and malonylation), here we focus on the most widely studied, acetylation. Early studies on the cardiac acetyl‐proteome revealed a preponderance of metabolic and mitochondrial targets, 80 , 81 leading to speculation that lysine acetylation is an important metabolic regulator. However, recent studies in hyperacetylation models have revealed a minimal impact on bioenergetics, 82 consistent with studies at the molecular level. 83 An important consideration in this regard is acetylation stoichiometry, with little understood about the precise relationship between site occupancy and enzyme activity.
While there are thousands of kinases and phosphatases in a typical cell, enzymes that regulate acetylation number far fewer. Most work on acetyltransferases has focused on those in the nucleus that regulate histones (see Ref. 84 for recent review). However, more recently other acetylation pathways have been uncovered. In particular, GCN5L1 was identified as a mitochondrial lysine acetyltransferase expressed in the heart, 85 and its deletion confers a number of cardiac pathologic phenotypes. 86 Furthermore, direct non‐enzymatic lysine acetylation from acetyl‐CoA (and corresponding acylation from other acyl‐CoAs) has been found to occur in the cell, 87 such that the concept of ‘acyl‐carbon stress’ is now well established to be partly mediated by excessive lysine acetylation. 88 , 89
On the deacetylation side, in addition to the histone deacetylases (HDACs), the sirtuin (SIRT) class of NAD+‐dependent lysine deacylases has emerged as key mediators of cardioprotection (Figure 1). Of the 7 mammalian SIRTs, SIRT1 and SIRT3 are robustly expressed in the heart and have been shown to play a direct role in several cardioprotective paradigms. 39 , 90 , 91 , 92 , 93 , 94 Unlike other tissues where SIRT1 is mostly nuclear, cardiomyocyte SIRT1 resides mostly in the cytosol 91 and is thus positioned to interact with the cardioprotective signalling machinery.
Inhibition of SIRT1 either pharmacologically 91 or genetically 39 , 90 prevents cardioprotection via ischaemic pre‐conditioning (IPC). Conversely, activation of SIRT1 either pharmacologically 95 , 96 , 97 or genetically 39 is sufficient to confer cardioprotection against acute IRI. Furthermore, a decline in SIRT1 activity is proposed to play a role in the loss of IPC protection with ageing. 98 In the context of metabolism, it was shown that the metabolic remodelling that occurs in acute IPC is critically dependent on SIRT1. 99 SIRT1 is also proposed to be an intermediate signal in cardioprotection via other stimuli such as phosphodiesterase (PDE) inhibitors. 10 Given the original discovery of the sirtuins as putative mediators of the longevity effects of caloric restriction (CR) 11 it has also been posited that the cardioprotective benefits of CR may be mediated via SIRT1. 12 Similar cardioprotective benefits have also been described for mitochondrial SIRT3, 92 , 93 with its pharmacologic activation also shown to confer acute cardioprotection. 13 Among the more prominent SIRT3 deacetylation targets is the mPTP regulator cyclophilin D (CypD), with deacetylation at lysine 166 (mouse) required for the cardioprotective effects of SIRT3. 14
Although activation of SIRT1 or SIRT3 may appear attractive as a pharmacologic protective strategy, it should be cautioned that the specificity of many SIRT‐activating drugs is uncertain, with stilbenes such as resveratrol plus several commercial SIRT1‐activating drugs called into question. 15 In addition, inactivation of the NAD+‐recycling enzyme NAMPT phenocopies SIRT1 inhibition in preventing cardioprotection, 93 , 106 suggesting that NAD+ availability is a critical determinant for the cardioprotective efficacy of SIRT1. However, while the potential of boosting NAD+ bioavailability via delivery of precursors such as nicotinamide mononucleotide (NMN) or nicotinamide riboside (NR) has shown considerable promise in pre‐clinical studies, 95 , 107 the wide variety of non‐SIRT metabolic pathways that rely on the NAD+/NADH redox couple 18 suggests that side effects of such compounds may outweigh their cardioprotective benefits. 19 Interestingly, it may well be that part of the cardioprotective effects of NAD+‐boosting strategies mainly involves redox‐controlled activation of glycolysis. 19 In line, we recently showed that increased protein acetylation by metabolic over‐fuelling dramatically reduced both insulin‐ and AMPK‐mediated glucose uptake in cardiomyocytes, presuming the importance of reducing protein acetylation for promoting glucose metabolism during IRI. 110 , 111 Overall, while promoting deacetylation, for example by targeting SIRT1 activation, is an attractive avenue for cardioprotection, considerable effort is required for the development of specific drugs to achieve this end.
3.5. Mitochondrial metabolism
3.5.1. Oxygen consumption
A general characteristic of irreversible ischaemia, that is long (≥25 minutes) periods of ischaemia with cell death occurring, is the fast and complete recovery of oxygen consumption and therefore metabolic recovery during early reperfusion. At the same time, the recovery of mechanical function is lagging behind and still severely depressed. Thus, there is metabolic recovery that is uncoupled from mechanical recovery. In contrast, with reversible ischaemia, thus short (≤20 minutes) periods of ischaemia without cell death, recovery of oxygen consumption is much slower and still coupled to mechanical function following reversible ischaemia. 112 , 113 , 114 This fast recovery of post‐ischaemic metabolism was associated with increased mitochondrial Ca2+ in early reperfusion with irreversible ischaemia. 112 , 113 , 114 , 116 The fast recovery of oxygen consumption can possibly be explained by Ca2+ activation of pyruvate dehydrogenase, resulting in increased carbohydrate (glucose, lactate, pyruvate) oxidation during the first hour of reperfusion. 116 At high FA levels (>1.0 mmol/L), increased glucose oxidation is not observed, 117 likely because high FA impair pyruvate dehydrogenase (PDH) activation. Thus, fast metabolic recovery uncoupled from mechanical function is a signature of damaging irreversible IR, although it is unknown whether this is causal to, or just an epiphenomenon of, IRI. When causal, it offers the possibility that imposing slower metabolic recovery in early reperfusion will protect from IRI. Indeed, interventions directed at an attenuated recovery of blood flow, 118 or impaired mitochondrial activity by rotenone or mitochondrial complex I by nitrosating/NO agents are protective, 120 , 121 , 122 supporting the notion of slow metabolic wake‐up 51 as a primary mechanism to combat IRI.
3.5.2. Glucose oxidation
Glucose oxidation, which actually is oxidation of the end product of glycolysis, that is pyruvate, occurs in the mitochondria. The flux from glycolysis to glucose oxidation is mostly controlled by the activity of the PDH complex located in the mitochondrial matrix. Glucose oxidation is completely shut down during ischaemia; its degree of reactivation during reperfusion depends, for example, on the availability of FA for oxidation because FA compete with glucose for oxidation. During early reperfusion, FA oxidation is increased due to activation of AMPK by the ischaemia. 123 , 124 FA oxidation reduces glucose oxidation because of the Randle cycle, in which acetyl‐CoA derived from FA oxidation inhibits PDH. 124 However, activation of PDH by the mitochondrial Ca2+ overload occurring at reperfusion may overcome this inhibition and result in higher glucose oxidation than in normoxic conditions. 125 Thus, glucose oxidation is reduced during early reperfusion following moderate ischaemia, but may increase following more severe or longer ischaemia. 114 , 126
Several ex vivo (ie in isolated perfused hearts) and in vivo studies have shown that stimulation of glucose oxidation during reperfusion is associated with a better recovery of myocardial function and a lesser reperfusion injury. In the ex vivo setting, interventions at reperfusion that increase glucose oxidation and improve the recovery of myocardial function may directly target the PDH flux, by activating PDH activity with dichloroacetate, 127 , 128 , 129 , 130 or by the law of mass action, provisioning extra pyruvate 129 or increasing glucose uptake by administration of high glucose/high insulin at reperfusion 131 or through overexpression of the GLUT1 transporter. 132 Indirectly, inhibition of FA oxidation 133 , 134 , 135 or uptake 136 , 137 may prevent inhibition of glucose oxidation by reversing the Randle mechanism. Importantly, IPC, the most robust cardioprotective intervention, was also shown to increase glucose oxidation during reperfusion, 138 , 139 although other studies found no changes. 140
A major limitation of ex vivo experiments is that they do not allow assessment of long‐term post‐ischaemic myocardial salvage. Indeed, the improved recovery of function observed during the short reperfusion period (usually one hour) might reflect only speeding up of the mechanical recovery, associated with the improved re‐energization observed. 132 , 133 , 136 Nevertheless, several ex vivo studies observed a reduction of myocardial necrosis biomarkers associated with increased glucose oxidation in response to glucose addition 141 or FA uptake blockade. 136
Fewer in vivo studies have addressed this question; yet, they have consistently reported a reduction in infarct size associated with stimulation of glucose oxidation by various mechanisms. Thus, one to several days after reperfusion following 30 minutes of coronary occlusion, the infarct size was reduced in response to acute administration prior to reperfusion of dichloroacetate, 142 phosphonate compounds 143 or reconstituted high‐density lipoproteins (HDL), 144 which all stimulated glucose oxidation. Although administration of GLP‐1 analogue albiglutide 145 or rosiglitazone days before the ischaemic event in diabetic rats 146 also increased post‐ischaemic myocardial glucose oxidation and reduced infarct size, such approach is less relevant for the emergency room situation where patients cannot be treated beforehand.
Thus, most studies concur that stimulation of glucose oxidation during reperfusion improves the recovery of function and reduces infarct size. Conversely, in situations with reduced glucose oxidation, such as in pathologically hypertrophied hearts, 147 a lesser recovery of function and/or larger infarct size is observed. There are, however, a few discordant observations, with studies indicating that stimulating PDH activity or reversing the Randle cycle may not be sufficient to limit post‐ischaemic injury. 131 , 148 Also, administration of Ruthenium Red, an inhibitor of mitochondrial Ca2+ uptake, after severe IR reduced PDH activation but improved the recovery of function and reduced creatine kinase release. 125 Possibly, following severe ischaemia inducing massive post‐ischaemic Ca2+, the nefarious effects of mitochondrial Ca2+ overload overcome the positive effects of stimulating glucose oxidation.
Mainly two hypotheses are invoked to explain the positive impact of stimulating glucose oxidation on post‐ischaemic recovery of function and myocardial salvage: 1. efficiency of oxygen use and 2. reduction of proton overload. 1. Depending on the activity of the MAS, the complete oxidation of one glucose yields 31‐33 ATP, with a P/O ratio (ie moles of ATP produced divided by the moles of oxygen atoms used) of 2.6‐2.8. 149 For complete oxidation of palmitate or oleate, the P/O ratio is only 2.45‐2.47. This 5% to 15% better oxygen efficiency of glucose oxidation over FA oxidation may not seem impressive, but may become critical when oxygen supply is limited or when mitochondrial function is deteriorated. 2. At least in isolated perfused hearts, the rate of glycolytic flux exceeds the rate of glucose oxidation, by up to one order of magnitude in the presence of FA. 128 , 130 Although complete glucose metabolism, that is glycolysis + oxidation, is proton neutral, glycolysis not followed by pyruvate oxidation generates two protons. 150 During reperfusion, the glycolysis/oxidation uncoupling worsens, aggravating the proton overload, which drives the Na+ and Ca2+ overload leading to cardiomyocyte necrosis and/or opening of the mPTP. 151 Stimulation of glucose oxidation without a concomitant stimulation of glycolysis, such as achieved with dichloroacetate, for example, reduces uncoupling and the generation of protons, thereby possibly limiting reperfusion injury. This interpretation puts uncoupled glycolysis as a driver of myocardial reperfusion injury, seemingly in contradiction with what was said in the previous section. Thus, stimulation of glycolysis could perhaps be a double‐edged sword, but the self‐harming edge can be blunted by concomitant stimulation of glucose oxidation.
In conclusion of this section, glucose oxidation appears to be a promising cardioprotection target that seems to have been overlooked by clinical studies. A search of the ClinicalTrials.gov database with the keywords ‘myocardial infarction’ or ‘reperfusion injury’ and ‘glucose oxidation’ failed to retrieve a single study. Acute stimulation of glucose oxidation at reperfusion with well‐tolerated agents such as dichloroacetate or reconstituted HDL would be worth trying in a clinical context.
3.5.3. Fatty acid metabolism
The energy metabolism in heart heavily relies on fat oxidation. Transmembrane proteins such as CD36 and fatty acid transport proteins (FATPs) are involved in transport of nonesterified (free) long‐chain FA from circulation to cardiac tissues (Figure 1). Following cellular uptake, FA can either be stored in the form of triglycerides or undergo metabolism in mitochondria. Long‐chain (LC) FA metabolism proceeds through multiple steps ensuring transfer of LC acyl groups into mitochondria. The first step in this process is synthesis of acetyl‐CoA in the outer mitochondrial membrane. 152 In the next step, carnitine palmitoyltransferase 1 (CPT‐1) catalyses synthesis of LC acylcarnitine which is necessary for transportation of fatty acid intermediates through the inner mitochondrial membrane (Figure 1). 153 LC acylcarnitine synthesis rate is the FA metabolism rate‐limiting step which is regulated by concentrations of malonyl‐CoA, an endogenous inhibitor of CPT‐1. 154 Malonyl‐CoA is synthesized by acetyl‐CoA carboxylase (ACC) from acetyl‐CoA using biotin and ATP as cofactors. ACC activity is regulated by its phosphorylation (inactivation) and dephosphorylation (activation). Accordingly, stimulation of fatty acid metabolism by AMPK is achieved by phosphorylation of ACC, yielding inhibition of malonyl‐CoA synthesis to ensure rapid LC acylcarnitine synthesis by CPT‐1. 155 In contrast, activation of insulin signalling prevents ACC phosphorylation, stimulates malonyl‐CoA synthesis and results in CPT‐1 inhibition. 156 Another enzyme, malonyl‐CoA decarboxylase (MCD), catalyses the reverse reaction and converts malonyl‐CoA into acetyl‐CoA. MCD activity is inhibited by SIRT4‐mediated deacetylation of the enzyme ensuring high malonyl‐CoA concentrations and facilitated LC acylcarnitine synthesis. 157 LC acylcarnitines are further transferred from the intermembrane space into mitochondrial matrix and are converted to acetyl‐CoA by CPT‐2 to enter β‐oxidation in mitochondria.
The shift towards LC acylcarnitine accumulation in the mitochondria is a result of unbalanced AC synthesis and mitochondrial oxidation rates. As a consequence, CPT‐1 generates LC acylcarnitines at amounts which mitochondria cannot fully metabolize. During cardiac ischaemia, mitochondrial malfunction combined with energy deficiency‐driven activation of CPT‐1 results in even higher content of LC acylcarnitines. At high levels, LC acylcarnitines inhibit oxidative phosphorylation (OXPHOS), which in turn induces mitochondrial membrane hyperpolarization and stimulates the production of reactive oxygen species (ROS) in cardiac mitochondria (Figure 1). 158 , 159 Therefore, FA metabolism regulation approaches aiming at cardioprotection in IR settings should be carefully evaluated for effects on overall energy homeostasis and possible cardiotoxicity because of harmful fatty acid intermediate accumulation (ie ‘acyl‐carbon stress’). Some knockout mouse models altering transporters or enzymes involved in FA metabolism have resulted in disturbances in cardiac function. 160 This highlights the importance of FA as essential substrates for the heart energy production to maintain normal cardiac function under chronic conditions. Nevertheless, the use of small molecular inhibitors of enzymes in fatty acid transport and metabolism pathways has demonstrated that inhibition of FA oxidation in general reduces damage induced by IR 160 , 161 , 162 , 163 , 164 , 165 , 166 , 167 (Table 1). It should be noted, however, that effects of FA on cardiac IRI are critically dependent on FA concentrations, with detrimental effects commonly only observed at rather high levels (>0.8 mmol/L) of FA. 168 Although older literature has reported much higher FA plasma levels in cardiac patients, these high levels were likely the result of ongoing lipolysis in the test tube due to the use of heparin in these patients; preventing test tube lipolysis shows that these high FA levels are usually not observed during human in vivo IR episodes. 169 Finally, FA may also contribute to cardiac IRI due to its dislodging effects on hexokinase II‐mitochondrial binding in the heart. 170
Table 1.
FAO inhibitors for IRI
| Compound | Mechanism/target | Activity in MI models | Model | Ref |
|---|---|---|---|---|
| Sulfo‐N‐succinimidyl oleate (SSO) | Inhibition of sarcolemmal FAT/CD36 | Prevented cardiac dysfunction after ischaemia | Isolated diabetic and control male Wistar rat hearts | [21] |
|
CBM‐301940 CBM‐300864 |
Inhibition of malonyl‐CoA decarboxylase | Improved cardiac function during and after ischaemia |
Isolated rat hearts Pigs in vivo |
[22, 23] |
| Methyl‐GBB | Decreased accumulation of long‐chain acylcarnitines | Decreased MI size, improved survival |
Ligation of LAD, rats Isolated perfused rat hearts |
[24] |
| Trimetazidine | Long‐chain 3‐ketoacyl‐CoA thiolase inhibitor | |||
| AMPK and ERK signalling pathways | Reduced MI size and oxidative stress | In vivo regional ischaemia and reperfusion, mice | [25] | |
| Carvedilol | Adrenergic receptor blocker; modulator of cardiac AMPK signalling pathway | MI size reduction, improved cardiac functions | Ligation of LAD, mice | [26] |
Abbreviations: LAD, left anterior descending coronary artery; MI, myocardial infarction.
3.5.4. Ketones
Beta‐hydroxybutyrate (BHB) and acetoacetate (AcAc) represent the two main ketone bodies. 171 BHB, which is produced by degradation of FA in liver and then transported to extrahepatic tissues (including the heart), is one of the important metabolic substrates for energy production during fasting. 172 BHB is not only a metabolic intermediate, but also possesses a variety of signalling functions. 173 It has been shown to inhibit Class I histone deacetylases (HDACs), increasing therefore histone acetylation, and thereby induces the expression of genes that restrain oxidative stress. 174 BHB also suppresses sympathetic nervous system activity and reduces total energy expenditure and heart rate by inhibiting short‐chain fatty acid signalling through G protein‐coupled receptor 41 (GPR41). 175 The myocardium is the highest ketone body consumer per unit mass and under physiological conditions oxidizes ketone bodies in proportion to their delivery, at the expense of fatty acid and glucose oxidation. 176 , 177 Ketone bodies have an intermediate energetic efficiency, yielding more ATP per molecule of oxygen used (P/O ratio) in comparison with fatty acid oxidation, but less when compared to glucose. 178 , 179 Additionally, the oxidation of ketone bodies also yields potentially higher energy than fatty acid oxidation, keeping ubiquinone oxidized, which raises redox span in the electron transport chain (ETC) and makes more energy available to synthesize ATP. A recent ex vivo cardiac study employing direct measurements of cardiac efficiency showed no effects of ketones on cardiac efficiency. 9 However, increasing the supply of ketones to the heart did increase total ketone body oxidation without a decrease in any other metabolic pathway. 9 Therefore, ketone bodies are considered an alternative and efficient energy source in myocardium, especially in failing hearts. 180 Increased circulating ketone bodies have been previously reported in patients with congestive heart failure. 181
In addition to the beneficial effects of ketone bodies in heart failure, they have been shown to have beneficial effects in IRI by reducing myocardial infarct size, either by increased levels during fasting or when they were administered exogenously. Because of concentration‐dependent dynamics, increases in ketone bodies during fasting can elevate the rate of BHB utilization. 182 It has been reported that fasting increased the myocardial BHB/AcAc ratio reflecting altered mitochondrial redox state and fasting of rats for only 24 hours improved the post‐ischaemic recovery of contractile function and reduced the lactate dehydrogenase release in isolated hearts subjected to global IR. 183 In another study, short‐term fasting increased the concentration of BHB and BHB/AcAc ratio compared to controls, limited the infarct size and reduced the total number of premature ventricular complexes and the duration of ventricular tachycardia occurring at early reperfusion. 184 However, low concentration of endogenous ketone bodies failed to preserve the myocardial ATP levels whereas exogenous supplementation (to 40 times the original concentration) prevented the loss of ATP by ischaemic injury. 185
Administration of exogenous BHB 60 minutes before the start of ischaemia reduced in vivo myocardial infarct size and apoptosis in rats subjected to IR. 186 It was recently demonstrated that starting in vivo BHB treatment at reperfusion and continuing administration for the next 24 hours of reperfusion using minipumps reduced infarct size, attenuated apoptosis in myocardium and preserved cardiac function of IR in mice. The above‐mentioned beneficial effects were attributed to reduced mitochondrial formation of ROS, enhanced ATP production, attenuated mitochondrial swelling and partly restored mitochondrial membrane potential in myocardium. 187
In summary, in the experimental IRI context, ketone bodies may confer cardioprotective effects possibly due to altered mitochondrial redox state resulting from increased ketogenesis, up‐regulation of crucial OXPHOS mediators and reduction of oxidative stress.
3.5.5. Succinate and ROS
A new mechanism is emerging whereby the production of mitochondrial ROS is considered a highly orchestrated, metabolite‐driven process early in IRI. The citric acid cycle metabolite, succinate, is extensively accumulated during ischaemia and is rapidly oxidized upon reperfusion. 79 , 188 , 189 In vivo, succinate likely accumulates via the reduction of fumarate by succinate dehydrogenase (SDH or complex II) reversal. A lack of the terminal electron acceptor, oxygen, maintains a reduced CoQ pool, and additionally, the pH of ischaemic tissue is lowered. 51 , 79 , 190 Upon reperfusion, succinate is rapidly oxidized to fumarate, and together with ETC activity restarting, the reduced CoQ pool provides electrons for the ETC complex to proton pump, establishing a large proton motive force (Δp). The large Δp and highly reduced CoQ pool, together with depleted adenine nucleotides, drive reverse electron transport (RET) through mitochondrial complex I, resulting in the production of superoxide at the flavin mononucleotide (FMN) site (Figure 1). 191 , 192 The mitochondrial ROS produced, together with impaired calcium handling, activate downstream pathways, resulting in mitochondrial permeability pore formation and, ultimately, cell death.
Not all of the succinate that has accumulated at ischaemia is oxidized by SDH, as it has been suggested that a proportion is released from the cell. 75 Succinate was significantly elevated in the blood of patients with an acute ST‐elevation myocardial infarct 193 suggesting its release into the bloodstream upon reperfusion. This opens the intriguing possibility that changes in mitochondrial metabolites during IRI could be involved in paracrine signalling, complementary to the signalling role succinate plays in the immune system. 194
Accumulation of succinate, with its derivative succinyl‐CoA, also leads to protein succinylation. 195 The Sirt5, which has limited deacylase activity, also catalyses the removal of succinyl groups from proteins. In line, it has been shown that the increase in IRI in Sirt5‐deficient mouse heart can be reversed by preventing succinate accumulation. 196
Blocking succinate metabolism during ischaemia or reperfusion with malonate, a competitive SDH inhibitor, has been found to be protective in multiple pre‐clinical models of cardiac IRI in mouse, rat or pig. 51 , 188 Therefore, targeting succinate metabolism through SDH inhibition is twofold, either by preventing the succinate rise during ischaemia or by reducing the oxidation upon reperfusion, highlighting a potential therapeutic target. 197
At present, the metabolic source for succinate in vivo appears to be fumarate. However, a key question remains as to the mechanism of fumarate production during ischaemia. Firstly, adenosine monophosphate (AMP) build‐up during ischaemia can be broken down to fumarate via the purine nucleotide cycle (PNC). Secondly, the conversion of aspartate to oxaloacetate and then reduction to malate in the MAS could provide the fumarate required. 51 , 112 Alternatively, a recent mechanism has suggested that canonical TCA cycle activity may result in succinate accumulation, by aminotransferase anaplerosis, as opposed to SDH reversal. 75 Differences in the models used to investigate metabolic changes may be resulting in discrepancy as to which direction of the TCA cycle contributes most to ischaemic succinate accumulation. Further work is required to elucidate accumulation pathways. However, the mechanistic insights produced by further investigating succinate in IRI will provide key targets for the design of cardioprotective drugs, thus providing many lucrative avenues for future therapies in many pathologies.
Importantly, the role of succinate as a proximal source of electrons for ROS generation during reperfusion is undisputed. As such, the acute delivery of complex II inhibitors at the onset of reperfusion, regardless of succinate accumulation, appears to be a potentially promising clinical approach for treatment of MI in the acute setting (eg during PCI, percutaneous coronary intervention).
3.5.6. FOF1‐ATPase during ischaemia
Mitochondrial ATP synthase or FoF1‐ATP/synthase transforms the electrical power generated during respiration (ΔΨm) into ATP‐containing chemical energy, following the chemiosmotic principle that governs the life of all organisms. It contains an H+ channel domain (Fo) embedded within the inner mitochondrial membrane and a catalytic domain (F1) protruding towards the mitochondrial matrix, interconnected by central and peripheral stalks. 198 High‐resolution cryoelectron microscopy of native mitochondrial membranes has revealed that FoF1‐ATP/synthase self‐associates into long rows of dimers that shape the cristae of mitochondria of all eukaryotic cells into elongated tubular cristae. 199 Adequate shaping of mitochondrial cristae determines the respiratory fitness. 20 The FoF1‐ATP/synthase is extremely efficient in generating ATP (30 kg/day in a healthy heart), but when cardiomyocytes are challenged by an anoxic episode, the catalytic subunit may paradoxically reverse into an energy‐dissipating machine, favouring H+ extrusion at the expense of ATP hydrolysis. 21
Under physiological conditions, a fraction of FoF1‐ATP/synthase remains blocked by the inhibitory factor 1 (IF1), a 12 kDa protein that translocates exclusively to the mitochondria. Remarkably, these tissues with high‐energy demand, like the heart, have the highest content of IF1, probably because in these tissues a relevant fraction of the FoF1‐ATP/synthase only becomes activated upon demand, acting as a reservoir for ATP synthesis. Expression of IF1 varies between species, and as a general rule, it is higher in animals with a high rate of heart contraction, like mice, and lower in species with low rate of heart contraction, like humans. 22 Under ischaemic conditions, reversion of FoF1‐ATP/synthase into a hydrolase precipitates energy exhaustion and rigour contracture in cells already jeopardized by the lack of oxygen. 23 Therefore, species‐dependent expression levels of IF1 can affect the susceptibility of cardiomyocytes to ischaemic damage. 24 Moreover, IF1 has been involved in metabolic reprogramming: By impeding ATP synthesis, it can inhibit OXPHOS and drive the cell towards a more glycolytic metabolism.
Cardiomyocytes from aged mice exhibit a partial failure of FoF1‐ATP/synthase to revert its catalytic mode of operation 34 ; this alteration delays the development of ischaemic‐rigour contracture secondary to ATP exhaustion but accelerates (ΔΨm) decline during ischaemia and impairs (ΔΨm) recovery upon reperfusion in the aged cardiomyocytes, a response that is paralleled by more pronounced mPTP opening, hypercontracture and cardiomyocyte death. 34 Indeed, recent evidence suggests that FoF1‐ATP/synthase could be the true molecular entity of the mPTP (Figure 1), the opening of which has been consistently associated with the extension of myocardial necrosis during ischaemia‐reperfusion injury (IRI) in different experimental models. 25 A structural alteration in the C‐ring within FoF1‐ATP/synthase 26 or in the molecule dimerization 27 has been proposed to act as ‘death channel’, with the latter receiving a greater consensus and more experimental support. 28 An intermediate model includes the dissociation of FoF1‐ATP/synthase dimers followed by the rearrangement of the C‐ring. 29 The concept that FoF1‐ATP/synthase is the anatomical support for H+ dissipation and cell death integrates other well‐accepted mPTP regulators, 210 including CypD, which has been recently proposed to reduce mito‐ATP synthase supramolecular assembly, thereby increasing mPTP opening probability. 211 Mass spectrometry methods have detected several types of post‐translational modifications in different subunits of FoF1‐ATP/synthase, 212 some of them, that is, phosphorylation of β‐subunit, have been detected in response to adenosine‐induced conditioning strategy. 213 Nevertheless, to date, the impact of FoF1‐ATP/synthase post‐translation modifications on the susceptibility of cardiomyocytes to undergo mPTP and death upon IRI has not been established. Because mPTP is increasingly recognized as a prominent therapeutic target in the context of myocardial IRI, the elucidation of the role of FoF1‐ATP/synthase as a modulator of cardiomyocyte death and survival may help to identify new pharmacological strategies for cardioprotection.
3.6. Energy transfer pathways
Mitochondria are central players of cellular energy metabolism, especially in striated oxidative muscles and heart. This is much more complicated than only production of ATP via OXPHOS, located on mitochondrial inner membrane (MIM). Mitochondria are also source of ROS, proapoptotic factors; they synthesize different metabolites, regulate cellular redox potential and play an important role in ion homeostasis regulation and thermogenesis. Severe myocardial infarction leads to heart failure due to a marked loss of functional activities of cardiomyocytes, where reorganization of energy transport pathways is an important component.
Studies in the last decades have led to an understanding that in cardiomyocytes, the cellular energy metabolism is a precisely organized system where mitochondria and ATPases are linked to each other by specialized energy transfer pathways formed by isoenzymes of creatine kinase (CK) and adenylate kinase (AK) and glycolytic enzymes like hexokinase (HK). In addition to the regulation of cellular respiration by calcium homeostasis, CK and AK energy transfer pathways ensure precise feedback signalling between contraction workload and oxygen consumption in mitochondria. Each sarcomere has its own corresponding mitochondrion, which together with phosphotransfer system and feedback metabolic signalling creates the intracellular energy unit (ICEU). 214 , 215 , 216
An important characteristic of the heart is its metabolic stability, as reflected by the apparent invariability of intracellular concentration of ATP and phosphocreatine (PCr) in spite of the variable workloads, corresponding rates of ATP oxidative synthesis and myofibrillar hydrolysis. 217 Under conditions of total ischaemia, the PCr concentration falls rapidly and heart contraction ceases, but ATP concentration stays almost stable decreasing only by 10% at the end of the first minute of ischaemia. 217 Colocalization of BetaII‐tubulin and VDAC in heart muscle is functionally related to the ability of creatine to stimulate OXPHOS due to functional coupling between mitochondrial creatine kinase (MtCK) and adenine nucleotide translocase (ANT). 218 Key events observed after acute ischaemia‐reperfusion (IR) and chronic ischaemia are the decrease (or loss) in the stimulatory effect of creatine and decrease in diffusion restrictions for ATP and ADP at the level of the mitochondrial outer membrane (MOM), which is mediated by BetaII‐tubulin 219 , 220 The disruption of mitochondrial interactions with cytoskeleton will result in decreased intracellular compartmentalized energy transfer and the loss of probability of interaction between mitochondria and BetaII‐tubulin. In adult cardiomyocytes, octameric mitochondrial creatine kinase (MtCK) binds electrostatically to the negatively charged cardiolipins of the mitochondrial inner membrane sharing the same cardiolipin patches with ANT. 221 It is possible that the unaltered octameric isoenzyme of MtCK ensures the stability of its molecular interaction with ANT after IR. 220 Alternatively, the IR‐induced alteration of mitochondrial increased MOM permeability might influence kinetic properties of the MtCK. 220
The predominant HK isoform in adult heart, HK2, dynamically shuttles between the mitochondria and cytoplasm, resulting in increased glycolysis when bound to mitochondria. 222 Besides that, mitochondrially bound HK2 is an important player in the field of voltage‐dependent anion channel (VDAC) interactions with regulatory proteins and its functional coupling with OXPHOS. It was recently shown that IR disrupts interactions between VDAC, ANT and HK2 through nitration of tyrosine residues in VDAC and ANT, contributing to mitochondrial and cellular dysfunction following IR. 223 Contact sites between MIM and MOM have a substantial role in transporting ATP, generated within the mitochondria, to the cytosol as PCr. Binding between CK and HK2 may stabilize these contact sites, and loss of HK2 during ischaemia leads to contact site breakage and decreased rates of extramitochondrial PCr synthesis. 36 , 224 , 225 The interplay between energy transfer pathways and different binding sites for tubulin and hexokinase to VDAC may be one of the targets of IPC.
The connections of mechanisms of energy transfer pathways and cardioprotection are not clear yet; one component participating in this system is the AK system together with KATP channel. The AK‐catalysed phosphotransfer system would promote KATP channel opening primarily by accelerating conversion of ATP to ADP, whereas CK systems would predominantly facilitate conversion of ADP to ATP and KATP channel closure. 226
The alterations in MOM permeability for adenine nucleotides seem to be an important feature of cardiac ischaemic and IR injuries, as it regulates the energy transfer pathways. IPC induced redistribution of high‐energy phosphoryl transfer and increased phosphotransfer reactions (creatine kinase, glycolysis), leading to improved intracellular metabolic communication and preservation of cellular ATP synthesis and ATP consumption following IR. 227 These phosphoryl fluxes correlated tightly with post‐ischaemic functional recovery 227 and pre‐conditioning‐induced energetic remodelling, improving contractile performance following IR. The study of intracellular phosphotransfer reactions is still not fully unravelled and will need more sophisticated studies, before it can be used in the development of an injury‐tolerant state in cardiomyocytes.
4. CONCLUSION
We have outlined some of the important metabolic changes occurring during ischaemia and subsequent reperfusion. While many aspects happen in the entire cell, the mitochondria are a clear focus within many of these metabolic changes. Such changes include alterations in fatty acid and succinate metabolism, along with FOF1‐ATP/synthase activity, resulting in increased mitochondrial ROS production and subsequent opening of mPTP. Additionally, glycolysis, hexosamine biosynthesis, glucose oxidation ketone metabolism and the malate/aspartate shuttle are all processes which directly affect metabolite levels and mitochondrial pathways. All of which have shown to elicit cardioprotective effects when altered.
While many of the outlines metabolic processes are ideal drug targets for ischaemia/reperfusion injury, further studies are necessary to fully understand underlying mechanisms and establish potential therapies. In this context, it is vital to test possible pharmacologic interventions at a clinically relevant time‐point at the end of ischaemia as well as in pre‐diseased and aged models. 228 Some of the outlined mechanisms have already been shown to be relevant in man 193 and targeted successfully in many species, including large animals. 189 , 197 Others are still awaiting a relevant drug target suitable for translation in patient with an acute MI. Recent methodological advances in detecting metabolic changes within the heart will make these efforts easier to achieve. Furthermore, the observed metabolic changes are not limited to cardiac I/R injury, but could play important roles in many physiological and pathophysiological situations, such as exercise, inflammation and cancer.
DISCLOSURES
None.
AUTHOR CONTRIBUTIONS
Each author wrote part of the review and edited the entire manuscript.
Zuurbier CJ, Bertrand L, Beauloye CR, et al. Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J Cell Mol Med. 2020;24:5937–5954. 10.1111/jcmm.15180
Contributor Information
Coert J. Zuurbier, Email: c.j.zuurbier@amc.uva.nl.
Thomas Krieg, Email: c.j.zuurbier@amc.uva.nl, Email: tk382@medschl.cam.ac.uk.
REFERENCES
- 1. Sodi‐Pallares D, Testelli M, Fishleder F. Effects of intravenous infusion of a potassium‐insulin‐glucose solution on the electrographic signs of myocardial infarction. Am J Cardiol. 1962;9:166‐181. [DOI] [PubMed] [Google Scholar]
- 2. Geraets IME, Glatz JFC, Luiken JJFP, Nabben M. Pivotal role of membrane substrate transporters on the metabolic alteration in the pressure‐overloaded heart. Cardiovasc Res. 2019;115:1000‐1012. [DOI] [PubMed] [Google Scholar]
- 3. Taegtmeyer H, Young ME, Lopaschuk GD, et al. Assessing cardiac metabolism: a scientific statement from the American Heart Association. Circ Res. 2016;118:1659‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093‐1129. [DOI] [PubMed] [Google Scholar]
- 5. van der Vusse GJ, de Groot MJ. Interrelationship between lactate and cardiac fatty acid metabolism. Mol Cell Biochem. 1992;116:11‐17. [DOI] [PubMed] [Google Scholar]
- 6. Vincent G, Bouchard B, Khairallah M, Des Rosiers C. Differential modulation of citrate synthesis and release by fatty acids in perfused working rat hearts. Am J Physiol Heart Circ Physiol. 2004;286:H257‐H266. [DOI] [PubMed] [Google Scholar]
- 7. Nederlof R, Denis S, Lauzier B, et al. Acute detachment of hexokinase II from mitochondria modestly increases oxygen consumption of the intact mouse heart. Metabolism. 2017;72:66‐74. [DOI] [PubMed] [Google Scholar]
- 8. Van Weeghel M, Abdurrachim D, Nederlof R, et al. Increased fatty acid oxidation in a mouse model with decreased malonyl‐CoA sensitivity of CPT1B. Cardiovasc Res. 2018;114:1324‐1334. [DOI] [PubMed] [Google Scholar]
- 9. Ho KL, Zhang L, Wagg C, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 2019;115:1606‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olson AK, Bouchard B, Zhu WZ, et al. First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart. J Biol Chem 2020;295:2018‐2033.. 10.1074/jbc.RA119.010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nederlof R, Eerbeek O, Hollmann MW, et al. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischemia‐reperfusion injury in heart. Brit J Pharmacol. 2014;171:2067‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nederlof R, Gurel‐Gurevin E, Eerbeek O, et al. Reducing mitochondrial bound hexokinase II mediates transition from non‐injurious into injurious ischemia/reperfusion of the intact heart. J Physiol Biochem. 2016;73:323‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saks V, Dzeja P, Schlattner U, et al. Cardiac system bioenergetics: metabolic basis of the Frank‐Starling law. J Physiol. 2006;571:253‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Beek JH, Tian X, Zuurbier CJ, et al. The dynamic regulation of myocardial oxidative phosphorylation: analysis of the response time of oxygen consumption. Mol Cell Biochem. 1998;184:321‐344. [PubMed] [Google Scholar]
- 15. King LM, Opie LH. Glucose delivery is a major determinant of glucose metabolism in the ischemic myocardium with a residual coronary flow. Cardiovasc Res. 1998;39:381‐392. [DOI] [PubMed] [Google Scholar]
- 16. King LM, Opie LH. Glucose and glycogen utilization in myocardial ischemia‐ Changes in metabolism and consequences for the myocyte. Mol Cell Biochem. 1998;180:3‐26. [PubMed] [Google Scholar]
- 17. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horman S, Beauloye C, Vanoverschelde JL, Bertrand L. AMP‐activated protein kinase in the control of cardiac metabolism and remodeling. Curr Heart Fail Rep. 2012;9:164‐173. [DOI] [PubMed] [Google Scholar]
- 19. Sabia PJ, Powers ER, Ragosta M, et al. An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N Engl J Med. 1992;327:1825‐1832. [DOI] [PubMed] [Google Scholar]
- 20. Smeele KM, ter Horst LH, Koeman A, et al. The effect of standard chow and reduced hexokinase II on growth, cardiac and skeletal muscle hexokinase and low‐flow cardiac ischemia‐reperfusion injury. Lab Animal. 2011;45:160‐166. [DOI] [PubMed] [Google Scholar]
- 21. Wu R, Smeele KM, Wyatt E, et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodelling after ischemia‐reperfusion injury. Circ Res. 2011;108:60‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lochner A, Pentz A, Williams K, et al. Substrate effects on sarcolemmal permeability in the normoxic and hypoxic perfused rat heart. Bas Res Cardiol. 1996;91:64‐78. [DOI] [PubMed] [Google Scholar]
- 23. Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res. 1996;78:482‐491. [DOI] [PubMed] [Google Scholar]
- 24. Vanoverschelde JJ, Janier MF, Bakke JE, et al. Rate of glycolysis during ischemia determines extent of ischemic injury and functional recovery after reperfusion. Am J Physiol Heart Circ Physiol. 1994;267:H1785‐H1794. [DOI] [PubMed] [Google Scholar]
- 25. Kingsley PB, Sako EY, Yang MQ, et al. Ischemic contracture begins when anaerobic glycolysis stops: a 31P‐NMR study of isolated rat hearts. Am J Physiol. 1991;261:H469‐H478. [DOI] [PubMed] [Google Scholar]
- 26. Fiolet JWT, Baartscheer A. Cellular calcium homeostasis during ischemia: a thermodynamic approach. Cardiovasc Res. 2000;45:100‐106. [DOI] [PubMed] [Google Scholar]
- 27. Carvajal K, Zarrinpashneh E, Szarszoi O, et al. Dual cardiac contractile effects of the alpha2‐AMPK deletion in low‐flow ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2007;292:H3136‐H3147. [DOI] [PubMed] [Google Scholar]
- 28. Russell RR, Coven DL, Pypaert M, et al. AMP‐activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Askenasy N. Glycolysis protects sarcolemmal membrane integrity during total ischemia in the rat heart. Bas Res Cardiol. 2001;96:612‐622. [DOI] [PubMed] [Google Scholar]
- 30. Trueblood NA, Ramasamy R, Wang LW, Schaeffer S. Niacin protects the isolated heart from ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol. 2000;279:H764‐H771. [DOI] [PubMed] [Google Scholar]
- 31. Kim AS, Miller EJ, Wright TM, et al. A small molecule AMPK activator protects the heart against ischemia‐reperfusion injury. J Mol Cell Cardiol. 2011;51:24‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Timmermans AD, Balteau M, Gelinas R, et al. A‐769662 potentiates the effect of other AMPK‐activated protein kinase activators on cardiac glucose uptake. Am J Physiol Heart Circ Physiol. 2014;306:H1619‐H1630. [DOI] [PubMed] [Google Scholar]
- 33. Uthman L, Nederlof R, Eerbeek O, et al. Delayed ischemic contracture onset by empagliflozin associated with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res. 2019;115:1533‐1545. [DOI] [PubMed] [Google Scholar]
- 34. Fernandez‐Sanz C, Ruiz‐Meana M, Castellano J, et al. Altered FoF1 ATPase synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost. 2015;113:441‐451. [DOI] [PubMed] [Google Scholar]
- 35. Nagoshi T, Matsui T, Aoyama T, et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlated with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2012;2:e005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal call injury in ischemic myocardium. Circulation. 1986;74:1124‐1136. [DOI] [PubMed] [Google Scholar]
- 38. Tong H, Chen W, London RE, et al. Preconditioning enhanced glucose uptake is mediated by p38 MAP kinase not by phosphatidylinositol 3‐kinase. J Biol Chem. 2000;275:11981‐11986. [DOI] [PubMed] [Google Scholar]
- 39. Nadtochiy SM, Yao H, McBurney MW, et al. SIRT1‐mediated acute cardioprotection. Am J Physiol Circ Physiol. 2015;301:H1506‐H1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ji L, Zhang X, Liu W, et al. AMPK‐regulated and Akt‐dependent enhancement of glucose uptake is essential in ischemic preconditioning‐alleviated reperfusion injury. PLoS One. 2013;8:e69910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496‐499. [DOI] [PubMed] [Google Scholar]
- 42. Smeele KMA, Soutworth R, Wu R, et al. Disruption of hexokinase II‐mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ Res. 2011;108:1165‐1169. [DOI] [PubMed] [Google Scholar]
- 43. Gurel E, Smeele KM, Eerbeek O, et al. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia‐reperfusion. J Appl Physiol. 2009;106:1909‐1916. [DOI] [PubMed] [Google Scholar]
- 44. Sun L, Shukair S, Naik TJ, et al. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinase I and II. Mol Cell Biol. 2008;28:1007‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gottlob K, Majewski N, Kennedy S, et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeremy RW, Ambrosio G, Pike MM, et al. The functional recovery of post‐ischemic myocardium requires glycolysis during early reperfusion. J Mol Cell Cardiol. 1993;25:261‐276. [DOI] [PubMed] [Google Scholar]
- 47. Halestrap AP. Calcium‐dependent opening of a non‐specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of a low pH against chemical and hypoxic cell damage. Biochem J. 1991;278:715‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohen MV, Yang XM, Downey JM. The pH hypothesis of Postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895‐1903. [DOI] [PubMed] [Google Scholar]
- 49. Liu Q, Docherty JC, Rendell JC, et al. High levels of FA delay the recovery of intracellular pH and cardiac efficiency in post‐ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol. 2002;39:718‐725. [DOI] [PubMed] [Google Scholar]
- 50. Inserte J, Aluja D, Barba I, et al. High‐fat diet improves tolerance to myocardial ischemia by delaying normalization of intracellular pH at reperfusion. J Mol Cell Cardiol. 2019;133:164‐173. [DOI] [PubMed] [Google Scholar]
- 51. Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut‐down and gradual wake‐up. J Mol Cell Cardiol. 2009;46:804‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peart L, Headrick JP. Adenosine‐mediated early preconditioning in mouse: protective signaling and concentration dependent effects. Cardiovasc Res. 2003;58:589‐601. [DOI] [PubMed] [Google Scholar]
- 53. Toyoda Y, Friehs I, Parker RA, et al. Differential role of sarcolemmal and mitochondrial K(ATP) channels in adenosine‐enhanced ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000;279:H2694‐H2703. [DOI] [PubMed] [Google Scholar]
- 54. Mailleux F, Gélinas R, Beauloye C, et al. O‐GlcNAcylation, enemy or ally during cardiac hypertrophy development? Biochim Biophys Acta. 2016;1862:2232‐2243. [DOI] [PubMed] [Google Scholar]
- 55. Wright JN, Collins HE, Wende AR, Chatham JC. O‐GlcNAcylation and cardiovascular disease. Biochem Soc Trans. 2017;45:545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bond MR, Hanover JA. O‐GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dassanayaka S, Jones SP. O‐GlcNAc and the cardiovascular system. Pharmacol Ther. 2014;142:62‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watson LJ, Long BW, DeMartino AM, et al. Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol. 2014;306:H142‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jensen RV, Andreadou I, Hausenloy DJ, Bøtker HE. The Role of O‐GlcNAcylation for Protection against Ischemia‐Reperfusion Injury. Int J Mol Sci. 2019;20:E404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia‐reperfusion injury via increased protein‐associated O‐GlcNAc. Am J Physiol Cell Physiol. 2007;292:C178‐C187. [DOI] [PubMed] [Google Scholar]
- 61. Ngoh GA, Watson LJ, Facundo HT, et al. Non‐canonical glycosyltransferase modulates post‐hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu J, Pang Y, Chang T, et al. Increased hexosamine biosynthesis and protein O‐GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303‐312. [DOI] [PubMed] [Google Scholar]
- 63. Fulop N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat hearts associated with increased O‐linked N‐acetylglucosamine protein modification and altered p38 activation. Am J Physiol Heart Circ Physiol. 2007;292:H2227‐2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu J, Marchase RB, Chatham JC. Increased O‐GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol. 2007;293:H1391‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu J, Marchase RB, Chatham JC. Glutamine‐induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O‐GlcNAc levels. J Mol Cell Cardiol. 2007;42:177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jones SP, Zachara NE, Ngoh GA, et al. Cardioprotection by N‐acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172‐1182. [DOI] [PubMed] [Google Scholar]
- 67. Vibjerg Jensen R, Johnsen J, Buus Kristiansen S, et al. Ischemic preconditioning increases myocardial O‐GlcNAc glycosylation. Scand Cardiovasc J. 2013;47:168‐174. [DOI] [PubMed] [Google Scholar]
- 68. Jensen RV, Zachara NE, Nielsen PH, et al. Impact of O‐GlcNAc on cardioprotection by remote ischaemic preconditioning in non‐diabetic and diabetic patients. Cardiovasc Res. 2013;97:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia‐reperfusion injury via increased protein O‐GlcNAc and increased mitochondrial Bcl‐2. Am J Physiol Cell Physiol. 2008;294:C1509‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nielsen TT, Stottrup NB, Lofgren B, Botker HE. Metabolic fingerprint of ischaemic cardioprotection: importance of the malate‐aspartate shuttle. Cardiovasc Res. 2011;91:382‐391. [DOI] [PubMed] [Google Scholar]
- 71. Rupert BE, Segar JL, Schutte BC, Scholz TD. Metabolic adaptation of the hypertrophied heart: role of the malate/aspartate and alpha‐glycerophosphate shuttles. J Mol Cell Cardiol. 2000;32:2287‐2297. [DOI] [PubMed] [Google Scholar]
- 72. Rej R. Aminooxyacetate is not an adequate differential inhibitor of aspartate aminotransferase isoenzymes. Clin Chem. 1977;23:1508‐1509. [PubMed] [Google Scholar]
- 73. Barron JT, Gu L, Parrillo JE. Malate‐aspartate shuttle, cytoplasmic NADH redox potential, and energetics in vascular smooth muscle. J Mol Cell Cardiol. 1998;30:1571‐1579. [DOI] [PubMed] [Google Scholar]
- 74. Bunger R, Glanert S, Sommer O, Gerlach E. Inhibition by (aminooxy)acetate of the malate‐aspartate cycle in the isolated working guinea pig heart. Hoppe Seylers Z Physiol Chem. 1980;361:907‐914. [DOI] [PubMed] [Google Scholar]
- 75. Zhang J, Wang YT, Miller JH, et al. Accumulation of succinate in cardiac ischemia primarily occurs via canonical Krebs Cycle activity. Cell Rep. 2018;23:2617‐2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stottrup NB, Lofgren B, Birkler RD, et al. Inhibition of the malate‐aspartate shuttle by pre‐ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Res. 2010;88:257‐266. [DOI] [PubMed] [Google Scholar]
- 77. Jespersen NR, Yokota T, Stottrup NB, et al. Pre‐ischaemic mitochondrial substrate constraint by inhibition of malate‐aspartate shuttle preserves mitochondrial function after ischaemia‐reperfusion. J Physiol. 2017;595:3765‐3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dalgas C, Povlsen JA, Lofgren B, et al. Effects of FA on cardioprotection by pre‐ischaemic inhibition of the malate‐aspartate shuttle. Clin Exp Pharmacol Physiol. 2012;39:878‐885. [DOI] [PubMed] [Google Scholar]
- 79. Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Foster DB, Liu T, Rucker J, et al. The cardiac acetyl‐lysine proteome. PLoS ONE. 2013;8:e67513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nguyen TTM, Wong R, Menazza S, et al. Cyclophilin D modulates mitochondrial acetylome. Circ Res. 2013;113:1308‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fisher‐Wellman KH, Draper JA, Davidson MT, et al. Respiratory phenomics across multiple models of protein hyperacylation in cardiac mitochondria reveals marginal impact on bioenergetics. Cell Rep. 2019;26: 1557‐1572.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nadtochiy SM, Wang YT, Zhang J, et al. Potential mechanisms linking SIRT activity and hypoxic 2‐hydroxyglutarate generation: no role for direct enzyme (De)acetylation. Biochem J. 2017;474:2829‐2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. 2020;17:96‐115. [DOI] [PubMed] [Google Scholar]
- 85. Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443:655‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Scott I, Wang L, Wu K, et al. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol. 2018;28:346‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wagner GR, Bhatt DP, O’Connell TM, et al. A class of reactive acyl‐CoA species reveals the non‐enzymatic origins of protein acetylation. Cell Metabol. 2017;25:823‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell. 2014;54:5‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fisher‐Wellmann KH, Lin CT, Ryan TE, et al. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy consuming redox circuit. Biochem J. 2015;467:271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170‐2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischemic preconditioning: the role of SIRT1. Cardiovasc Res. 2011;89:643‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exarcerbates ischemia‐reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. 2014;306:H1602‐H1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Parodi‐Rullan RM, Chapa‐Dubocq X, Rullan PJ, et al. High sensitivity of SIRT3 deficient hearts to ischemia‐reperfusion is associated with mitochondrial abnormalities. Front Pharmacol. 2017;8:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sundaresan NR, Pillai VP, Gupta MP. Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J Mol Cell Cardiol. 2011;51:614‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yamamoto T, Byun J, Zhai P, et al. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE. 2014;9:e98972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen CJ, Yu W, Fu YC, et al. Resveratrol protects cardiomyocytes from hypoxia‐induced apoptosis through the SIRT1‐FoxO1 pathway. Biochem Biophys Res Commun. 2009;378:389‐393. [DOI] [PubMed] [Google Scholar]
- 97. Guo Y, Zhang L, Li F, et al. Restoration of sirt1 function by pterostilbene attenuates hypoxia‐reoxygenation injury in cardiomyocytes. Eur J Pharmacol. 2016;776:26‐33. [DOI] [PubMed] [Google Scholar]
- 98. Adam T, Sharp S, Opie LH, Lecour S. Loss of cardioprotection with ischemic preconditioning in aging hearts: role of sirtuin 1? J Cardiovasc Pharmacol Ther. 2013;18:46‐53. [DOI] [PubMed] [Google Scholar]
- 99. Nadtochiy SM, Urciuoli W, Zhang W, et al. Metabolic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J Mol Cell Cardiol. 2015;88:64‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Shalwala M, Zhu SG, Das A, et al. Sirtuin1 (SIRT1) activation mediates sildenafil induced delayed cardioprotection against ischemia‐reperfusion injury in mice. PLoS ONE. 2014;9:e86977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lin SJ, Kaeberlein M, Andalis AA, et al. Calorie restriction extends Saccharomyces Cerevisiae lifespan by increasing respiration. Nature. 2002;418:344‐348. [DOI] [PubMed] [Google Scholar]
- 102. Yamamoto T, Tamaki K, Shirakawa K, et al. Cardiac Sirt1 mediates the cardioprotective effects of caloric restriction by suppressing local complement system activation after ischemia‐reperfusion. Am J Physiol Heart Circ Physiol. 2016;310:H1003‐H1014. [DOI] [PubMed] [Google Scholar]
- 103. Pillai VB, Samant S, Sundaresan NR, et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Comm. 2015;6:6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bochaton T, Crola‐Da‐Silva C, Pillot B, et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3‐mediated deacetylation of cyclophilin D. J Mol Cell Cardiol. 2015;84:61‐69. [DOI] [PubMed] [Google Scholar]
- 105. Kumar A, Chauhan S. How much successful are the medicinal chemists in modulation of SIRT1: A critical review. Eur J Med Chem. 2016;119:45‐69. [DOI] [PubMed] [Google Scholar]
- 106. Hsu CP, Oka S, Shao D, et al. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gomes AP, Price NL, Ling AJY, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell. 2013;155:1624‐1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kulkarni CA, Brookes PS. Cellular compartmentation and the redox/nonredox functions of NAD. Antioxid Redox Signal. 2019;31:623‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nadtochiy SM, Wang YT, Nehrke K, et al. Cardioprotection by nicotinamide mononucleotide (NMN): involvement of glycolysis and acidic pH. J Mol Cell Cardiol. 2018;121:155‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Renguet E, Bultot L, Beauloye C, et al. The regulation of insulin‐stimulated cardiac glucose transport via protein acetylation. Front Cardiovasc Med. 2018;5:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Renguet E, Ginion A, Gellinas R, et al. Metabolism and acetylation contribute to leucine‐mediated inhibition of cardiac glucose uptake. Am J Physiol Heart Circ Physiol. 2017;313:H432‐H445. [DOI] [PubMed] [Google Scholar]
- 112. Gorge G, Chatelain P, Schaper J, Lerch R. Effect of increasing degrees of ischemic injury on myocardial oxidative metabolism early after reperfusion in isolated rat hearts. Circ Res. 1991;68:1681‐1692. [DOI] [PubMed] [Google Scholar]
- 113. Zuurbier CJ, Ince C. Post‐ischaemic changes in the response time of oxygen consumption to demand in the isolated rat heart are mediated partly by calcium and glycolysis. Pflug Arch. 2002;443:908‐916. [DOI] [PubMed] [Google Scholar]
- 114. Lerch R, Tamm C, Papageorgiou I, Benzi RH. Myocardial fatty acid oxidation during ischemia and reperfusion. Mol Cell Biochem. 1992;116:103‐109. [DOI] [PubMed] [Google Scholar]
- 115. Görge G, Chatelain P, Schaper J, Lerch R. Effect of increasing degrees of ischemic injury on myocardial oxidative metabolism early after reperfusion in isolated rat hearts. Circ Res. 1991;68:1681‐1692. [DOI] [PubMed] [Google Scholar]
- 116. Benzi RH, Lerch R. Dissociation between contractile function and oxidative metabolism in postischemic myocardium. Circ Res. 1992;71:567‐576. [DOI] [PubMed] [Google Scholar]
- 117. Terrand J, Papageorgiou I, Rosenblatt‐Velin N, Lerch R. Calcium‐activation of pyruvate dehydrogenase in severely injured postischemic myocardium. Am J Physiol Heart Circ Physiol. 2001;281:H722‐H730. [DOI] [PubMed] [Google Scholar]
- 118. Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243‐257. [DOI] [PubMed] [Google Scholar]
- 119. Yamazaki S, Fujibayashi Y, Rajagopalan RE, et al. Effects of staged versus sudden reperfusion after acute coronary occlusion in the dog. J Am Coll Cardiol. 1986;7:564‐572. [DOI] [PubMed] [Google Scholar]
- 120. Lefsnesky EJ, Chen Q, Moghaddas S, et al. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961‐47967. [DOI] [PubMed] [Google Scholar]
- 121. Chen Q, Moghaddas S, Hoppel CL, Lefnesky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;316:200‐207. [DOI] [PubMed] [Google Scholar]
- 122. Chouchani ET, Methner C, Nadtochiy SM, et al. Cardioprotection by S‐nitrosation of a cysteine switch on mitochondrial complex 1. Nat Med. 2013;19:753‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl‐CoA levels due to an increase in 5'‐AMP‐activated protein kinase inhibition of acetyl‐CoA carboxylase. J Biol Chem. 1995;270:17513‐17520. [DOI] [PubMed] [Google Scholar]
- 124. Kudo N, Gillespie JG, Kung L, et al. Characterization of 5'AMP‐activated protein kinase activity in the heart and its role in inhibiting acetyl‐CoA carboxylase during reperfusion following ischemia. Biochim Biophys Acta. 1996;1301:67‐75. [DOI] [PubMed] [Google Scholar]
- 125. Lopaschuk GD, Spafford MA, Davies NJ, Wall SR. Glucose and palmitate oxidation in isolated working rat hearts reperfused after a period of transient global ischemia. Circ Res. 1990;66:546‐553. [DOI] [PubMed] [Google Scholar]
- 126. Terrand J, Papageorgiou I, Rosenblatt‐Velin N, Lerch R. Calcium‐mediated activation of pyruvate dehydrogenase in severely injured postischemic myocardium. Am J Physiol Heart Circ Physiol. 2001;281:H722‐H730. [DOI] [PubMed] [Google Scholar]
- 127. McVeigh JJ, Lopaschuk GD. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am J Physiol Heart Circ Physiol. 1990;259:H1079‐H1085. [DOI] [PubMed] [Google Scholar]
- 128. Lopaschuk GD, Wambolt RB, Barr RL. An imbalance between glycolysis and glucose oxidation is a possible explanation for the detrimental effects of high levels of FA during aerobic reperfusion of ischemic hearts. J Pharmacol Exp Ther. 1993;264:135. [PubMed] [Google Scholar]
- 129. Lewandowski ED, White LT. Pyruvate dehydrogenase influences postischemic heart function. Circulation. 1995;91:2071‐2079. [DOI] [PubMed] [Google Scholar]
- 130. Liu B, El Alaoui‐Talibi Z, Clanachan AS, et al. Uncoupling of contractile function from mitochondrial TCA cycle activity and MVO2 during reperfusion of ischemic hearts. Am J Physiol Heart Circ Physiol. 1996;39:H72‐H80. [DOI] [PubMed] [Google Scholar]
- 131. Wang P, Lloyd Steven G, Chatham JC. Impact of high glucose/high insulin and dichloroacetate treatment on carbohydrate oxidation and functional recovery after low‐flow ischemia and reperfusion in the isolated perfused rat heart. Circulation. 2005;111:2066‐2072. [DOI] [PubMed] [Google Scholar]
- 132. Luptak I, Yan J, Cui L, et al. Long‐term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116:901‐909. [DOI] [PubMed] [Google Scholar]
- 133. Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid‐induced injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036‐1043. [DOI] [PubMed] [Google Scholar]
- 134. Lopaschuk GD, McNeil GF, McVeigh JJ. Glucose oxidation is stimulated in reperfused ischemic hearts with the carnitine palmitoyltransferase 1 inhibitor, Etomoxir. Mol Cell Biochem. 1989;88:175‐179. [DOI] [PubMed] [Google Scholar]
- 135. McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135‐142. [DOI] [PubMed] [Google Scholar]
- 136. Ramasamy R, Hwang Y, Bakr S, Bergmann SR. Protection of ischemic hearts perfused with an anion exchange inhibitor, DIDS, is associated with beneficial changes in substrate metabolism. Cardiovasc Res. 2001;51:275‐282. [DOI] [PubMed] [Google Scholar]
- 137. Mansor LS, Sousa Fialho MDL, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo‐N‐succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017;113:737‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Støttrup NB, Løfgren B, Birkler RD, et al. Inhibition of the malate–aspartate shuttle by pre‐ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Res. 2010;88:257‐266. [DOI] [PubMed] [Google Scholar]
- 139. Dalgas C, Povlsen JA, Løfgren B, et al. Effects of FA on cardioprotection by pre‐ischaemic inhibition of the malate–aspartate shuttle. Clin Exp Pharmacol Physiol. 2012;39:878‐885. [DOI] [PubMed] [Google Scholar]
- 140. Finegan BA, Lopaschuk GD, Gandhi M, Clanachan AS. Ischemic preconditioning inhibits glycolysis and proton production in isolated working rat hearts. Am J Physiol Heart Circ Physiol. 1995;269:H1767‐H1775. [DOI] [PubMed] [Google Scholar]
- 141. Tamm C, Benzi R, Papageorgiou I, et al. Substrate competition in postischemic myocardium. Effect of substrate availability during reperfusion on metabolic and contractile recovery in isolated rat hearts. Circ Res. 1994;75:1103‐1112. [DOI] [PubMed] [Google Scholar]
- 142. Ussher JR, Wang W, Gandhi M, et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. 2012;94:359‐369. [DOI] [PubMed] [Google Scholar]
- 143. Pham V, Zhang W, Chen V, et al. Design and synthesis of novel pyridoxine 5‘‐phosphonates as potential antiischemic agents. J Med Chem. 2003;46:3680‐3687. [DOI] [PubMed] [Google Scholar]
- 144. Heywood SE, Richart AL, Henstridge DC, et al. High‐density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci Transl Med. 2017;9:eaam6084. [DOI] [PubMed] [Google Scholar]
- 145. Bao W, Aravindhan K, Alsaid H, et al. Albiglutide, a long lasting glucagon‐like peptide‐1 analog, protects the rat heart against ischemia/reperfusion injury: evidence for improving cardiac metabolic efficiency. PLoS ONE. 2011;6:e23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yue T‐L, Bao W, Gu J‐L, et al. Rosiglitazone treatment in Zucker diabetic fatty rats Is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion‐induced myocardial injury. Diabetes. 2005;54:554‐562. [DOI] [PubMed] [Google Scholar]
- 147. Zhabyeyev P, Gandhi M, Mori J, et al. Pressure‐overload‐induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res. 2013;97:676‐685. [DOI] [PubMed] [Google Scholar]
- 148. Wang P, Fraser H, Lloyd SG, et al. A Comparison between Ranolazine and CVT‐4325, a novel inhibitor of fatty acid oxidation, on cardiac metabolism and left ventricular function in rat isolated perfused heart during ischemia and reperfusion. J Pharmacol Exp Ther. 2007;321:213. [DOI] [PubMed] [Google Scholar]
- 149. Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2017;292:7189‐7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Opie LH. Myocardial ischemia—Metabolic pathways and implications of increased glycolysis. Cardiovasc Drugs Ther. 1990;4:777‐790. [DOI] [PubMed] [Google Scholar]
- 151. Clanachan AS. Contribution of protons to post‐ischemic Na+ and Ca2+ overload and left ventricular mechanical dysfunction. J Cardiovasc Electrophysiol. 2006;17:S141‐S148. [DOI] [PubMed] [Google Scholar]
- 152. Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl‐coenzyme A synthetases in metabolic control. Curr Opin Lipidol. 2010;21:212‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yates DW, Garland PB. Carnitine palmitoyltransferase activities (EC 2.3.1.‐) of rat liver mitochondria. Biochem J. 1970;119:547‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl‐CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. 1977;60:265‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Witters LA, Kemp BE. Insulin activation of acetyl‐CoA carboxylase accompanied by inhibition of the 5'‐AMP‐activated protein kinase. J Biol Chem. 1992;267:2864‐2867. [PubMed] [Google Scholar]
- 157. Laurent G, German NJ, Saha AK, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. 2013;50:686‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tominaga H, Katoh H, Odagiri K, et al. Different effects of palmitoyl‐L‐carnitine and palmitoyl‐CoA on mitochondrial function in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;295:H105‐H112. [DOI] [PubMed] [Google Scholar]
- 159. Liepinsh E, Makrecka‐Kuka M, Volska K, et al. Long‐chain acylcarnitines determine ischaemia/reperfusion‐induced damage in heart mitochondria. Biochem J. 2016;473:1191‐1202. [DOI] [PubMed] [Google Scholar]
- 160. Abdurrachim D, Luiken JJ, Nicolay K, et al. Good and bad consequences of altered fatty acid metabolism in heart failure: evidence from mouse models. Cardiovasc Res. 2015;106:194‐205. [DOI] [PubMed] [Google Scholar]
- 161. Mansor LS, Sousa Fialho MDL, Yea G, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo‐N‐succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017;113:737‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Dyck JR, Cheng JF, Stanley WC, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94:e78‐84. [DOI] [PubMed] [Google Scholar]
- 163. Fillmore N, Lopaschuk GD. Malonyl CoA: a promising target for the treatment of cardiac disease. IUBMB Life. 2014;66:139‐146. [DOI] [PubMed] [Google Scholar]
- 164. Liepinsh E, Makrecka‐Kuka M, Kuka J, et al. Inhibition of L‐carnitine biosynthesis and transport by methyl‐gamma‐butyrobetaine decreases fatty acid oxidation and protects against myocardial infarction. Br J Pharmacol. 2015;172:1319‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Liu Z, Chen JM, Huang H, et al. The protective effect of trimetazidine on myocardial ischemia/reperfusion injury through activating AMPK and ERK signaling pathway. Metabolism. 2016;65:122‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hu H, Li X, Ren D, et al. The cardioprotective effects of carvedilol on ischemia and reperfusion injury by AMPK signaling pathway. Biomed Pharmacother. 2019;117:109106. [DOI] [PubMed] [Google Scholar]
- 167. Jaswal JS, Keung W, Wang W, et al. Targeting fatty acid and carbohydrate oxidation ‐ A novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. 2011;1813:1333‐1350. [DOI] [PubMed] [Google Scholar]
- 168. Dalgas C, Povlsen JA, Lofgern B, et al. Effects of FA on cardioprotection by pre‐ischaemic inhibition of the malate‐aspartate shuttle. Clin EXp Pharmacol Physiol. 2012;39:878‐885. [DOI] [PubMed] [Google Scholar]
- 169. Zuurbier CJ, Hoek FJ, van Dijk J, et al. Perioperative hyperinsulinaemic normoglycemic clamp causes hypolipidemia after coronary artery surgery. Brit J Anaesth. 2008;100:442‐450. [DOI] [PubMed] [Google Scholar]
- 170. Southworth R, Davey KAB, Warley A, Garlick PB. A reevaluation of the roles of hexokinase I and II in the heart. Am J Physiol Heart Circ Physiol. 2007;292:H378‐H386. [DOI] [PubMed] [Google Scholar]
- 171. Mitchell GA, Kassovka‐Bratinova S, Boukaftane Y, et al. Medical aspects of ketone body metabolism. Clin Invest Med. 1995;18:193‐216. [PubMed] [Google Scholar]
- 172. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060‐H1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Rojas‐Morales P, Tapia E, Pedraza‐Chaverri J. β‐Hydroxybutyrate: a signalling metabolite in starvation response? Cell Signal. 2016;28:917‐923. [DOI] [PubMed] [Google Scholar]
- 174. Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kimura I, Inoue D, Maeda T, et al. Short‐chain FA and ketones directly regulate sympathetic nervous system via G protein‐coupled receptor 41 (GPR41). Proc Nat Acad Sci. 2011;108:8030‐8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Pelletier A, Tardif A, Gingras MH, et al. Chronic exposure to ketone bodies impairs glucose uptake in adult cardiomyocytes in response to insulin but not vanadate: the role of PI3‐K. Mol Cell Biochem. 2007;296:97‐108. [DOI] [PubMed] [Google Scholar]
- 177. Yan J, Young ME, Cui L, et al. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet‐induced obesity. Circulation. 2009;119:2818‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Kashiwaya Y, Pawlosky R, Markis W, et al. A ketone ester diet increases brain malonyl‐CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem. 2010;285:25950‐25956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent FA. 2004;70:309‐319. [DOI] [PubMed] [Google Scholar]
- 180. Nielsen R, Møller N, Gormsen LC, et al. Cardiovascular effects of treatment with the ketone body 3‐hydroxybutyrate in chronic heart failure. Circulation. 2019;139:2129‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Lommi J, Kupari M, Koskinen P, et al. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665‐672. [DOI] [PubMed] [Google Scholar]
- 182. Sultan AMN. The effect of fasting on D‐3‐hydroxybutyrate metabolism in the perfused rat heat. Mol Cell Biochem. 1990;93:107‐118. [DOI] [PubMed] [Google Scholar]
- 183. Marina Prendes MG, Garcia JV, Fernandez MA, et al. Effects of 5‐hydroxydecanoate and ischemic preconditioning on the ischemic‐reperfused heart of fed and fasted rats. J Physiol Biochem. 2005;61:447‐456. [DOI] [PubMed] [Google Scholar]
- 184. Snorek M, Hodyc D, Sedivý V, et al. Short‐term fasting reduces the extent of myocardial infarction and incidence of reperfusion arrhythmias in rats. Physiol Res. 2012;61:567‐574. [DOI] [PubMed] [Google Scholar]
- 185. Hron WT, Menahan LA, Lech JJ. Inhibition of hormonal stimulation of lipolysis in perfused rat heart by ketone bodies. J Mol Cell Cardiol. 1978;10:161‐174. [DOI] [PubMed] [Google Scholar]
- 186. Zou Z, Sasaguri S, Rajesh KG, Suzuki R. dl‐3‐Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1968‐H1974. [DOI] [PubMed] [Google Scholar]
- 187. Yu Y, Yu Y, Zhang Y, et al. Treatment with D‐β‐hydroxybutyrate protects heart from ischemia/reperfusion injury in mice. Eur J Pharmacol. 2018;829:121‐128. [DOI] [PubMed] [Google Scholar]
- 188. Valls‐Lacalle L, Barba I, Miro‐Casas E, et al. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res. 2016;109:374‐384. [DOI] [PubMed] [Google Scholar]
- 189. Valls‐Lacalle L, Barba I, Miro‐Casas E, et al. Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep. 2018;8:2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chouchani ET, Pell VR, James AM, et al. A unifying mechanism for mitochondrial superoxide production during ischemia‐reperfusion injury. Cell Metab. 2016;23:254‐263. [DOI] [PubMed] [Google Scholar]
- 191. Stepanova A, Kahl A, Konrad C, et al. Reverse electron transfer results in a loss of flavin from mitochondrial complex I: Potential mechanism for brain ischemia reperfusion injury. J Cereb Blood Flow Metab. 2017;37:3649‐3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Kahl A, Stepanova A, Konrad C, et al. Critical role of flavin and glutathione in complex I‐mediated bioenergetic failure in brain ischemia/reperfusion injury. Stroke. 2018;49:1223‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kohlhauer M, Dawkins S, Costa ASH, et al. Metabolomic profiling in acute ST‐segment‐elevation myocardial infarction identifies succinate as an early marker of human ischemia‐reperfusion injury. J Am Heart Assoc. 2018;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Murphy MP, O'Neill LAJ. Krebs cycle reimagined: The emerging roles of succinate and itaconate as signal transducers. Cell. 2018;174:780‐784. [DOI] [PubMed] [Google Scholar]
- 195. Gao J, Shao K, Chen X, et al. The involvement of post‐translational modifications in cardiovascular pathologies: focus on SUMOylation, Neddylation, Succinylation, and Prenylation. J Mol Cell Cardiol. 2019;138:49‐58. [DOI] [PubMed] [Google Scholar]
- 196. Boylston JA, Sun J, Chen Y, et al. Characterization of the cardiac succinylome and its role in ischemia‐reperfusion injury. J Mol Cell Cardiol. 2015;88:73‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kula‐Alwar D, Prag HA, Krieg T. Targeting succinate metabolism in ischemia/reperfusion injury. Circulation. 2019;140:1968‐1970. [DOI] [PubMed] [Google Scholar]
- 198. Watt IN, Montgomery MG, Runswick MJ, et al. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci. 2010;39:16823‐16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Strauss M, Hofhaus G, Schroder RR, Kuhlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Jennings RB, Reimer KA, Steenbergen C. Effect of inhibition of the mitochondrial ATPase on net myocardial ATP in total ischemia. J Mol Cell Cardiol. 1991;23:1383‐1395. [DOI] [PubMed] [Google Scholar]
- 202. Esparza‐Molto PB, Nuevo‐Tapioles C, Chamorro M, et al. Tissue‐specific expression and post‐transcriptional regulation of the ATPase inhibitory factor 1 (IF1) in human and mouse tissue. Faseb J. 2019;33:1836‐1851. [DOI] [PubMed] [Google Scholar]
- 203. Rouslin W, Erickson JL, Solaro RJ. Effects of oligomycin and acidosis on rates of ATP depletion in ischemic heart muscle. Am J Physiol. 1986;250:H503‐H508. [DOI] [PubMed] [Google Scholar]
- 204. Garcia‐Bermudez J, Cuezva JM. The ATPase inhibitory factor 1 (IF1): a master regulator of energy metabolism and of cell survival. Biochim Biophys Acta. 2016;1857:1167‐1182. [DOI] [PubMed] [Google Scholar]
- 205. Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia‐reperfusion injury. J Mol Cell Cardiol. 2015;78:23‐34. [DOI] [PubMed] [Google Scholar]
- 206. Jonas EA, Porter GA, Beutner G, et al. Cell death disguised: The mitochondrial permeability transition pore as the C‐subunit of the F(1)F(0) ATP synthase. Pharmacol Res. 2015;99:382‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Giorgio V, von Stockum S, Antoniet M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci. 2013;110:5887‐5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Chinopoulos C. ATP synthase complex and the mitochondrial permeability transition pore: poles of attraction. EMBO Rep. 2017;18:1041‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Bonora M, Morganti C, Morciano G, et al. Mitochondrial permeability transition involves dissociation of F1Fo ATP synthase dimers and C‐ring conformation. EMBO Rep. 2017;18:1077‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Long Q, Yang K, Yang Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am J Cardiovasc Dis. 2015;5:19‐32. [PMC free article] [PubMed] [Google Scholar]
- 211. Beutner G, Alanzalon RE, Porter GA. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci Rep. 2017;7:14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Kane LA, van Eyk JE. Post‐translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr. 2009;41:145‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Arrell DK, Elliott ST, Kane LA, et al. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706‐714. [DOI] [PubMed] [Google Scholar]
- 214. Guzun R, Gonzalez‐Granillo M, Karu‐Varikmaa M, et al. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within mitochondrial interactosome. Biochim Biophys Acta. 2012;1818:1545‐1554. [DOI] [PubMed] [Google Scholar]
- 215. Saks VA, Kaambre T, Sikk P, et al. Intracellular energetic units in red muscle cells. Biochem J. 2001;356:643‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Anmann T, Varikmaa M, Timohhina N, et al. Formation of highly organized intracellular structure and energy metabolism in cardiac muscle cells during postnatal development of rat heart. Biochim Biophys Acta. 2014;1837:1350‐1361. [DOI] [PubMed] [Google Scholar]
- 217. Guzun R, Kaambre T, Bagur R, et al. Modular organization of cardiac energy metabolism: energy conversion, transfer and feedback regulation. Acta Physiol (Oxf). 2015;213:84‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Varikmaa M, Bagur R, Kaambre T, et al. Role of mitochondria‐cytoskeleton interactions in respiration regulation and mitochondrial organization in striated muscles. Biochim Biophys Acta. 2014;1837:232‐245. [DOI] [PubMed] [Google Scholar]
- 219. Dos Santos P, Laclau MN, Boudina S, et al. Alterations of the bioenergetics systems of the cell in acute and chronic myocardial ischemia. Mol Cell Biochem. 2004;256–257:157‐166. [DOI] [PubMed] [Google Scholar]
- 220. Bagur R, Tanguy S, Foriel S, et al. The impact of cardiac ischemia/reperfusion on the mitochondria‐cytoskeleton interactions. Biochim Biophys Acta. 2016;1862:1159‐1171. [DOI] [PubMed] [Google Scholar]
- 221. Schlattner U, Tokarska‐Schlattner M, Wallimann T. Mitochondrial creatine kinase in human health and disease. Biochim Biophys Acta. 2006;1762:164‐180. [DOI] [PubMed] [Google Scholar]
- 222. John S, Weiss JN, Ribalet B. Subcellular localisation of hexokinase I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yang M, Xu Y, Heisner JS, et al. Peroxynitrite nitrates adenine nucleotide translocase and voltage‐dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria. Mitochondrion. 2019;46:380‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Halestrap AP, Pereira GC, Pasdois P. The role of hexokinase in cardioprotection ‐ mechanism and potential for translation. Br J Pharmacol. 2015;172:2085‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Zuurbier CJ, Smeele KM, Eerbeek O. Mitochondrial hexokinase and cardioprotection of the intact heart. J Bioenerg Biomembr. 2009;41:181‐185. [DOI] [PubMed] [Google Scholar]
- 226. Dzeja PP, Terzic A. Phosphotransfer reactions in the regulation of ATP‐sensitive K+ channels. FASEB J. 1998;12:523‐529. [DOI] [PubMed] [Google Scholar]
- 227. Pucar D, Dzeja PP, Bast P, et al. Cellular energetics in the preconditioned state: protective role for phosphotransfer reactions captured by 18O‐assisted 31P NMR. J Biol Chem. 2001;276:44812‐44819. [DOI] [PubMed] [Google Scholar]
- 228. Boetker HE, Hausenloy D, Andreadou I, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018;113:39. [DOI] [PMC free article] [PubMed] [Google Scholar]