

Abstract
Interactions between gut microbiota not only regulate physical health, but also form a vital bridge between the environment and the host, thus helping the host to better adapt to the environment. The improvement of modern molecular sequencing techniques enables in‐depth investigations of the gut microbiota of vertebrate herbivores without harming them. By sequencing the 16S rRNA V4‐V5 region of the gut microbiota of both the captive and wild kiang in winter and summer, the diversity and function of the microbiota could be compared. The reasons for observed differences were discussed. The results showed that the dominant phyla of the kiang were Bacteroidetes and Firmicutes, and the structure and abundance of the gut microbiota differed significantly between seasons and environments. However, the relatively stable function of the gut microbiota supplies the host with increased adaptability to the environment. The diversity of the intestinal flora of the kiang is relatively low in captivity, which increases their risk to catch diseases to some extent. Therefore, importance should be attached to the impact of captivity on wildlife.
Keywords: adaption, gut microbiota, Qinghai–Tibet Plateau, Tax4Fun, Tibetan wild ass (Equus kiang)
The alpha diversity of gut microbiomes in the wild group was significantly higher than that in the captive group. The content of pathogenic bacteria increased in the captive group. The health level of the captive kiang was lower than that of the wild kiang.

1. INTRODUCTION
Animal guts have a large number of bacterial communities, which play important roles in host immunity (Yun et al., 2017), health and disease (Pulikkan et al., 2018), nutrient metabolism (Kau, Ahern, Griffin, Goodman, & Gordon, 2011), and energy acquisition (Shepherd, DeLoache, Pruss, Whitaker, & Sonnenburg, 2018). The symbiotic relationship between microbes and host is critical for their mutual survival. Herbivores utilize gut symbionts to convert plant biomass into fermentable sugars, volatile fatty acids, and microbial proteins (Xiao et al., 2019). The composition of these intestinal microorganisms is not stable and is easily affected by host diet (David et al., 2014), age, sex, genetics (Ren et al., 2017), and environmental fluctuations (Amato et al., 2015).
Food is the main source of energy for the host (Leftwich, Hutchings, & Chapman, 2018), and the gut microbiome forms a key symbiotic relationship with the host. Prior studies identified diet as the main factor for gut microbial variations between mammalian species (David et al., 2014; Zhang, Ju, & Zuo, 2018). Temporal variations of the relative abundance of individual bacterial groups are closely related to changes in the host diet (Amato et al., 2015). In recent years, the relationship between host gut microbiota and their nutritional metabolism has been increasingly studied (Jeong, Jang, & Kim, 2019; Zhao et al., 2018). However, these studies still suffer from a number of shortcomings, such as lack of detailed dietary information for wild populations or only focusing on captive animals (Mariat et al., 2009).
The Tibetan wild ass (Equus kiang) is the only wild equid on the Tibetan Plateau (St‐Louis & Côté, 2009). Due to the harshness of the environment of the Qinghai–Tibet Plateau, wild herbivores need more efficient nutrient metabolism mechanism (Gibson et al., 2019). However, the diversity and function of the gut microbiota in the wild kiang have not been investigated exhaustively (Gao et al., 2019). This study examined the stability of the captive and wild kiang gut microbiomes during both summer and winter of one year. The unique living environment of the kiang provides a rare opportunity to study the gut microbiome and to assess how the kiang and its gut microbiota adapt to the Qinghai–Tibet Plateau. Analyzing the changes in their intestinal microbiota can help to understand the relationship between nutrients, health, and adaptive evolution. The results provide promising targeted methods for the conservation of the kiang.
2. MATERIALS AND METHODS
2.1. Collection of fecal samples
Feces samples from the wild kiang were collected from Maduo county, Qinghai province, and samples of animals in captivity were collected from the Qinghai–Tibet Plateau Wildlife Park. Winter samples were collected in January 2018 and summer samples were collected in July. Both in the wild and in the Qinghai–Tibet Plateau Wildlife Park, the kiang was observed. After defecating, fresh feces were quickly collected in a clean zipper bag (using PE gloves) and samples were stored in dry ice. Since this happened in their natural habitat, the kiang is very alert; therefore, they were observed for a long time from the distance, and after they had left an area, fresh feces were collected. After sampling, samples were stored at −80°C. All fecal samples were collected in their natural state.
Twenty‐one fecal samples were collected from the captive kiang in summer (SC), and 21 samples were collected in winter (WC). Moreover, 45 fecal samples were collected from the wild kiang in summer (SW) and 60 samples were collected in winter (WW).
2.2. DNA extraction, PCR amplification, and purification
Total genomic DNA from samples was extracted using the CTAB method (Miaomiao, Guangcheng, Xiaohong, Huailei, & Weizhen, 2008). 1% agarose gels were used to detect both DNA concentration and purity. The DNA was diluted to 1 ng/μl with sterile water, depending on the concentration. The V4‐V5 region of 16S rRNA gene was amplified using the following primers: 515F: 5′‐GTGCCAGCMGCCGCGGTAA‐3′ and 907R: 5′‐CCGTCAATTCCTTTGAGTTT‐3′ (Gao et al., 2019).
The PCR products were mixed with the same volume of 1× loading buffer (containing SYB green) and were detected by electrophoresis on a 2% agarose gel. The PCR products were mixed at equal density ratio. Then, mixed PCR products were purified using the GeneJET™ Gel Extraction Kit (Thermo Scientific). A sequencing library was generated using the Ion Plus Fragment Library Kit 48 rxns (Thermo Scientific) according to the manufacturer's recommendations. The library quality was assessed using a Qubit@ 2.0 Fluorometer (Thermo Scientific). The library was sequenced on an Ion S5TM XL platform (Thermo Scientific), and 400‐bp single‐end reads were generated.
2.3. Sequence analysis
The raw reads were filtered under specific conditions to obtain high‐quality clean reads according to the Cutadapt quality controlled process (Martin, 2011). Then, the clean reads of all samples were clustered using Uparse software (Uparse v7.0.1001; Edgar, 2013), and the sequences were clustered into operational taxonomic units (OTUs) with 97% identity (Identity). According to the results of OTU clustering, the Silva database (Version 132; Quast et al., 2012) was used based on the Mothur algorithm to annotate taxonomic information and to statistically calculate the community composition of each sample at each classification level.
Food was collected from the captive kiang and the composition and nutritional content of the food were analyzed at the Center for Analysis and Testing of the Northwest Plateau Institute of Biology, Chinese Academy of Sciences (Qinghai, China).
2.4. Statistical analysis
QIIME (version 1.9.1) was used to calculate all diversity indexes in these samples, and R software (version 2.15.3) was used for visual analysis. LEfSe software (LEfSe 1.0) was used for linear discriminant analysis (LDA; Eckburg et al., 2005). Principal coordinate analysis (PCoA) was performed using Bray–Curtis. The functions of gut microbiota were predicted for the winter and summer groups and for both captive and wild groups using Tax4Fun in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Asshauer, Wemheuer, Daniel, & Meinicke, 2015).
3. RESULTS
3.1. 16S rDNA sequence diversity of the wild and captive kiang in summer
Via data quality control, 1,611,185 high‐quality reads were obtained in the summer captive group (SC), which were divided into 3,300 OTUs. In the summer wild group (SW), 3,402,962 high‐quality reads were obtained, which were divided into 3,995 OTUs. Rarefaction curves (Figure 1) can directly reflect the rationality of data and indirectly reflect the richness of samples. Each curve in the figure represents a different sample and is displayed using a different color. With increasing number of samples, the curve tended to flatten, which proved that the sequenced amount of these samples was reasonable.
Figure 1.
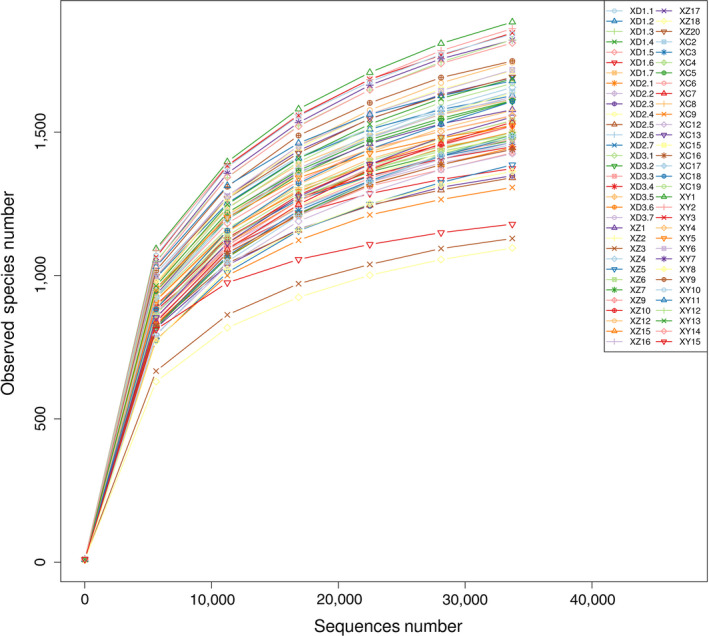
Rarefaction curves of all fecal samples of the kiang in summer
3.2. Dietary composition of the food provided for the captive kiang
In the Qinghai–Tibet Plateau Wildlife Park, the captive kiang mainly eats fresh alfalfa and fodder in summer, and mainly semidry oat grass, carrots, and fodder in winter. The nutritional composition of the food of the captive kiang is listed in Table 1. Their fodder contained more crude protein, carbohydrate, and energy, which provides rich nutrients for the captive kiang. Fresh alfalfa contains high crude fat in summer, and semidry oat grass has high levels of crude fiber in winter.
Table 1.
Composition of the food of the captive kiang
| Food composition | Fresh alfalfa | Fodder | Semidry oat grass | Carrots |
|---|---|---|---|---|
| Moisture (%) | 72.00 | 13.20 | 10.10 | 92.70 |
| Ash (%) | 1.70 | 6.60 | 4.50 | 42.42 |
| Crude fiber (%) | 13.83 | 8.49 | 35.31 | 0.95 |
| Crude protein (%) | 3.46 | 17.50 | 6.28 | 0.89 |
| Crude fat (%) | 0.28 | 0.20 | 0.06 | 0.06 |
| Total sugar (%) | 5.15 | 5.00 | 2.48 | 2.60 |
| Carbohydrate (%) | 8.75 | 54.00 | 43.80 | 4.55 |
| Energy (KJ/kg) | 2,170 | 12,230 | 8,530 | 950 |
| Na (g/kg) | 0.02 | 2.11 | 0.86 | 0.32 |
| P (g/kg) | 0.27 | 2.09 | 0.67 | 0.22 |
| Ca (g/kg) | 1.94 | 5.01 | 1.50 | 0.31 |
3.3. Bacterial composition and their relative abundances in summer
In the summer wild group, 25 phyla, 27 classes, 47 orders, 74 families, 127 genera, and 77 species were detected. In the summer captive group, 22 phyla, 25 classes, 40 orders, 72 families, 109 genera, and 57 species were detected. We have published the winter data in a previous article (Gao et al., 2019).
The gut bacterial community composition and structure were analyzed at different taxonomical levels and seasons. In the summer captive group (SC), Bacteroidetes (46.55%) and Firmicutes (41.51%) were the predominant phyla and Phascolarctobacterium (1.87%) was the predominant genus. In the summer wild group (SW), Bacteroidetes (49.43%) and Firmicutes (36.11%) were the predominant phyla and Phascolarctobacterium (1.23%) was the predominant genus. To visually assess the relative abundance of bacterial communities, the top 10 of each group were selected and the cumulative histogram percentage of their relative abundance at the phylum level is shown in Figure 2.
Figure 2.
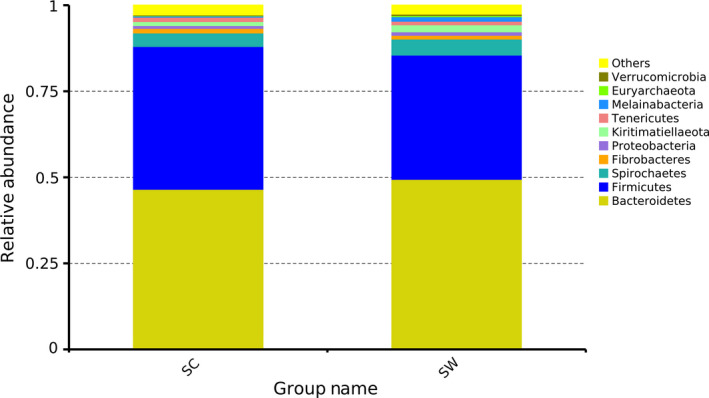
Stacked histograms of the relative abundance of gut microbiota phyla in the wild and captive kiang in summer. SC, summer captive group; SW, summer wild group
3.4. Analysis of between‑group discrepancies
The alpha diversity (observed species, Shannon, Simpson, Chao1, and ACE) of the captive and wild kiang was calculated in summer and winter (Table 2). The Shannon index and observed species index were higher in the wild winter group than in the summer group (P Shannon = 0.0094, P observed species = 0.0174), and the wild winter group had a higher Shannon index than the captive winter group (P Shannon = 0.0067). In addition, the ACE and Chao1 indexes of the summer group were significantly lower than in the winter group (P ACE = 0.0014, P Chao1 = 0.0094) and they were higher in the wild group than in the captive group (P ACE = 0.0001, P Chao1 = 0.0001) in all samples. Consequently, the diversity of gut bacteria in the winter group was higher than in the summer group, and their diversity in the wild group was higher than in the captive group.
Table 2.
Comparison of alpha diversity indices of gut microbiota from different groups
| alpha diversity | WC | WW | SC | SW |
|---|---|---|---|---|
| Observed_species | 1,745.1 ± 203.2 | 1,909.1 ± 123.9 | 1,490 ± 83.4 | 1,579.3 ± 186.1 |
| Shannon | 8.4 ± 0.64 | 8.8 ± 0.25 | 8.6 ± 0.21 | 8.4 ± 0.57 |
| Simpson | 0.984 ± 0.020 | 0.992 ± 0.002 | 0.993 ± 0.002 | 0.987 ± 0.022 |
| Chao1 | 2,154.5 ± 207.5 | 2,337.2 ± 169.3 | 1,684.3 ± 130.4 | 1,837.2 ± 248.5 |
| ACE | 2,145.1 ± 206.44 | 2,329.7 ± 163.2 | 1,682.1 ± 119.9 | 1,833.0 ± 237.9 |
Abbreviations: SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group.
Multi‐response permutation procedure (MRPP) testing between the summer and winter groups and the captive and wild groups yielded A > 0. The intergroup differences were larger than the intragroup differences (O'Reilly & Mielke, 1980; Sullivan, Skeffington, Gormally, & Finn, 2010), which further indicate that the grouping of this study was reasonable. Combined with the nonmetric multi‐dimensional scaling (NMDS) plot (Figure 3a) and PCoA plots (Figure 3b), it was shown that the summer captive group, summer wild group, winter captive group, and winter wild group were significantly different.
Figure 3.
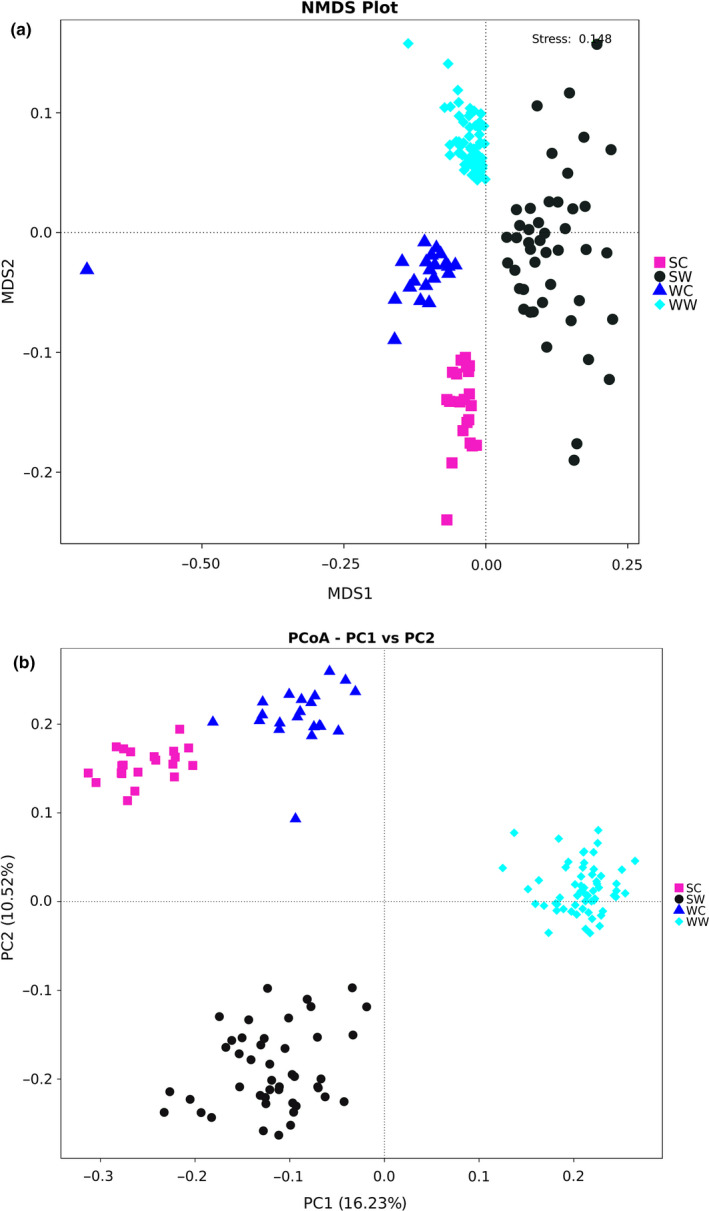
Cluster analysis result for both winter group and summer group according to NMDS (a) and Principal coordinate analysis (PCoA) (b). PCoA was performed using Bray‐Curtis. SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group
3.5. Analysis of different species among groups
Significant differences were found between the diversity and abundance of intestinal microbiota for the different seasons. To investigate the intestinal bacteria in different seasons and environments, a difference analysis of two aspects was conducted, using Metastat analysis. As shown in Table 3, major differences were found for intestinal microbiota at the phylum level between different seasons and environments. The results listed in Table 4 describe the major differences of intestinal microbiota at the genus level for different seasons and environments.
Table 3.
Major differences of gut microbiota at the phylum level for different groups
| Phylum/group | WC‐vs.‐SC | WW‐vs.‐SW | SW‐vs.‐SC | WC‐vs.‐WW |
|---|---|---|---|---|
| Acidobacteria | — | ** | — | — |
| Spirochaetes | — | ** | — | ** |
| Planctomycetes | — | ** | — | ** |
| Verrucomicrobia | ** | — | * | — |
| Deferribacteres | ** | ** | ** | — |
| Thermus | — | ** | ** | — |
| Actinobacteria | ** | ** | — | * |
| Armatimonadetes | * | ** | — | — |
| Thaumarchaeota | — | * | — | — |
| Chloroflexi | ** | — | ** | — |
| Firmicutes | ** | — | * | ** |
| Melainabacteria | — | — | ** | ** |
| Fusobacteria | ** | — | — | — |
| Cyanobacteria | — | ** | ** | — |
| Synergistetes | — | ** | ** | ** |
| Bacteroidetes | ** | ** | — | ** |
| Tenericutes | ** | ** | — | ** |
| Kiritimatiellaeota | ** | ** | ** | — |
| Gemmatimonadetes | ** | — | — | ** |
| Fibrobacteres | ** | ** | — | ** |
| Elusimicrobia | ** | * | — | ** |
| Lentisphaerae | ** | ** | * | ** |
| Chlamydiae | ** | — | — | — |
| Gracilibacteria | ** | ** | — | — |
| Proteobacteria | — | — | — | ** |
Abbreviations: SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group.
p < .01.
p < .001.
Table 4.
Major differences of gut microbiota at the genus level for different groups
| Name | WC‐vs.‐SC | WW‐vs.‐SW | SW‐vs.‐SC | WC‐vs.‐WW |
|---|---|---|---|---|
| Phascolarctobacterium | ** | ** | ** | — |
| Fibrobacter | ** | ** | — | ** |
| Alloprevotella | ** | ** | * | ** |
| Anaerovibrio | — | ** | — | ** |
| Saccharofermentans | — | * | ** | ** |
| Papillibacter | — | * | — | — |
| Sphaerochaeta | ** | ** | — | * |
| Akkermansia | ** | — | * | — |
| Methanocorpusculum | ** | ** | ** | ** |
| Agathobacter | — | ** | * | ** |
| Flexilinea | * | — | ** | — |
| Oribacterium | * | — | ** | ** |
| Pseudobutyrivibrio | — | ** | ** | ** |
| Mycoplasma | ** | — | — | — |
| Candidatus_Soleaferrea | — | ** | — | — |
| Bacteroides | ** | ** | * | — |
| Marvinbryantia | ** | — | — | ** |
| Erysipelatoclostridium | — | ** | * | — |
| Parabacteroides | ** | — | * | — |
| Streptococcus | ** | ** | ** | ** |
| Gillisia | ** | ** | ** | ** |
| Fusobacterium | * | — | — | — |
| Lachnoclostridium | ** | * | — | — |
Abbreviations: SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group.
p < .01.
p < .001.
The results of the LDA Effect Size analysis show species with significantly different abundance among groups (Eckburg et al., 2005). The results have two parts: LDA value distribution histogram (Figure 4a) and evolutionary branch diagram (phylogenetic distribution; Figure 4b). Lachnospiraceae is the most important family in the summer captive group (SC). Bacteroidetes and Spirochaetes are more important phyla and Bacteroidia and Spirochaetia are more important classes in the summer wild group (SW). In the winter wild group (WW), Kiritimatiellaeota, Tenericutes, and Kiritimatiellaeota are more important. In the winter captive group (WC), Firmicutes is the most important phylum.
Figure 4.
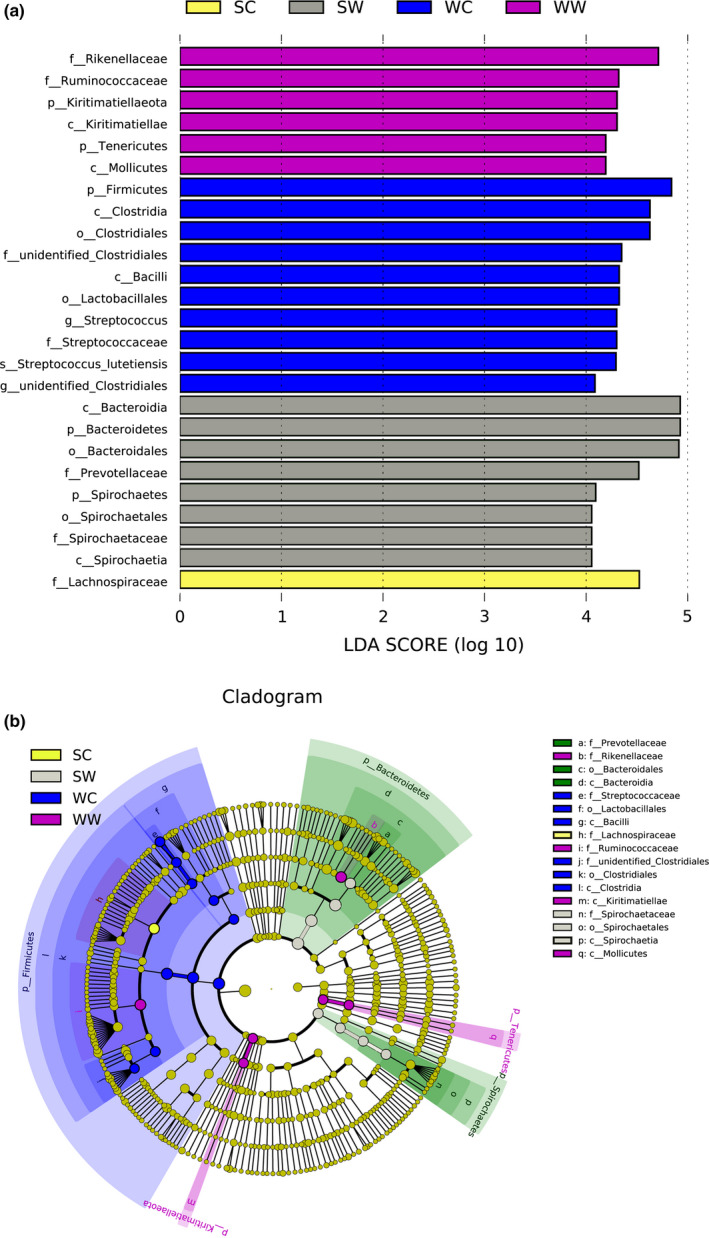
Results of LEfSe analysis. The histogram shows species whose LAD Score exceeded the default value of 4. The length of the histogram represents the impact of the different species. The species without significant difference are marked in yellow, and different species have the same color as the group in the evolutionary branch diagram. SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group
3.6. Analysis of gut microbiome function
According to the results of the predicted function, the main functional information of maximum abundance was selected for each sample of the KEGG classification level (level 1 and level 2, where level 1 showed all six categories of functional information), and a relative functional abundance column diagram was generated (Table 5). At level 1 (Figure 5a), the main functions were metabolism, genetic information processing, and environmental information processing. The function of human diseases in the captive summer group was significantly 1ower than that in the captive winter group (P human diseases = 0.0460), while genetic information processing was significantly higher (P genetic information processing = 0.0479) in the wild summer group compared with the wild winter group. In winter, metabolism was higher (P metabolism = 0.0005) in the wild group; however, genetic information processing, environmental information processing, and cellar processes were higher (P genetic information processing = 0.0336, P environmental information processing = 0.0149, and P cellar processes = 0.0324) in the captive group.
Table 5.
Relative abundance of predicted function of the gut microbiota of the kiang
| KO hierarchy | WC (%) | WW (%) | SC (%) | SW (%) |
|---|---|---|---|---|
| Level 1 | ||||
| Metabolism | 44.66 | 45.47 | 44.90 | 44.39 |
| Genetic information processing | 23.17 | 22.74 | 23.13 | 23.52 |
| Environmental information processing | 13.48 | 13.18 | 13.36 | 13.32 |
| Cellular processes | 8.43 | 8.20 | 8.34 | 8.55 |
| Human diseases | 2.77 | 2.77 | 2.73 | 2.74 |
| Organismal systems | 2.00 | 2.01 | 1.98 | 2.02 |
| Level 2 | ||||
| Carbohydrate metabolism | 11.28 | 11.53 | 11.35 | 11.12 |
| Membrane transport | 10.10 | 9.80 | 9.97 | 9.97 |
| Replication and repair | 9.61 | 9.38 | 9.64 | 9.79 |
| Translation | 9.26 | 9.11 | 9.22 | 9.40 |
| Amino acid metabolism | 9.09 | 9.43 | 9.18 | 9.02 |
| Energy metabolism | 4.38 | 4.54 | 4.40 | 4.42 |
| Nucleotide metabolism | 4.24 | 4.16 | 4.23 | 4.30 |
| Signal transduction | 3.26 | 3.26 | 3.28 | 3.24 |
| Metabolism of cofactors and vitamins | 3.13 | 3.21 | 3.17 | 3.17 |
| Glycan biosynthesis and metabolism | 3.10 | 3.08 | 3.09 | 3.06 |
Abbreviations: SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group.
Figure 5.
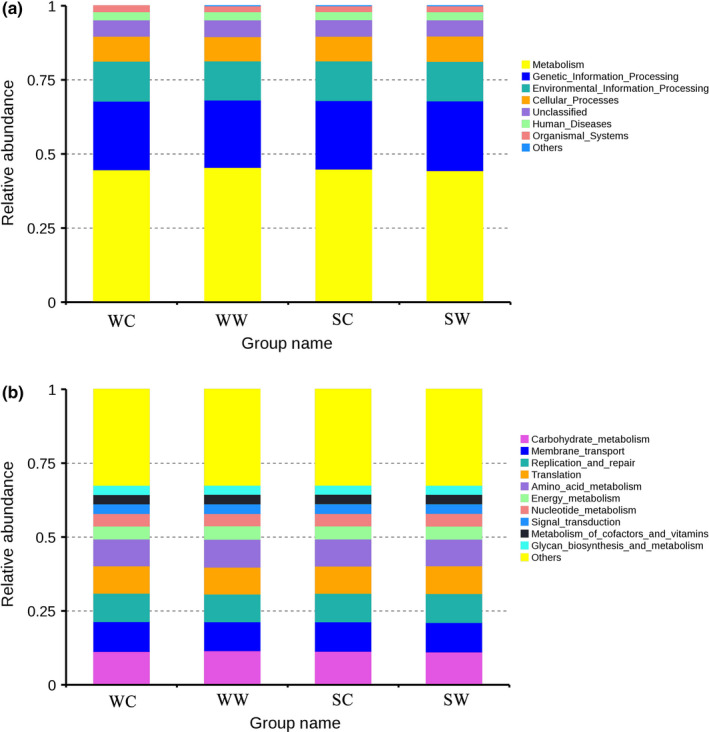
Relative abundance column diagram of microbiota functions based on the KEGG database. (a) Microbiota functions are shown on the first level; (b) top‐ten microbiota functions are shown on the second level. SC, summer captive group; SW, summer wild group; WC, winter captive group; WW, winter wild group
At level 2 (Figure 5b), carbohydrate metabolism, membrane transport, and replication and repair were the main functions of the gut microbiome of the captive kiang. The function of metabolism of cofactors and vitamins in the captive summer group was higher than in the captive winter group (P metabolism of cofactors and vitamins = 0.0068). Carbohydrate metabolism was higher (P Carbohydrate metabolism = 0.0460) in the wild winter group than in the wild summer group. In winter, carbohydrate metabolism, amino acid metabolism, energy metabolism and cofactors, and vitamins were higher (P carbohydrate metabolism = 0.0224, P amino acid metabolism = 0.0104, P Energy metabolism = 0.0064, P cofactors and vitamins = 0.0151) in the wild group. Membrane transport and replication and repair were higher (P Membrane transport = 0.0071, P replication and repair = 0.0140) in the captive group. In summer, carbohydrate metabolism was higher (P carbohydrate metabolism = 0.0466) in the captive group than that in the wild group.
4. DISCUSSION
Bacteroidetes and Firmicutes were identified as the most dominant phyla in all samples. In herbivores, these organisms are vital for the digestion and fermentation of fibrous forage (Flint & Bayer, 2008). They can derive energy and carbon from the degradation of complex carbohydrates (Williams et al., 2013). The phyla Bacteroidetes and Firmicutes are major components of all fecal microbial communities, and their presence is vitally important for host animals (Brice et al., 2019; Gao et al., 2019).
The relative abundance of the phylum Firmicutes was significantly higher (P SC‐SW = 0.002, P WC‐WW = 0.001) in the captive group (WC group was 49.74%) than in the wild group (WW group was 37.6%; Gao et al., 2019). The abundance of Bacteroidetes in the wild winter group (42.59%) was higher (P WC‐WW = 0.001) than that of the captive winter group (32.88%). The main food of the wild kiang is Poaceae and Cyperaceae (Yin, Huai, Zhang, Le, & Wei, 2007). Research has shown that in summer, the average content of Poaceae crude protein accounts for 9.16%, crude fat accounts for 2.17%, crude fiber accounts for 38.41% (Li, Wang, Kong, Jiang, & Wang, 2018), the average crude protein of Cyperaceae accounts for 12.58%, crude fat accounts for 2.56%, and crude fiber accounts for 35.72% (Song, 2002). Numerous bacteria of Bacteroidetes have a degrading function of organics with high molecular weight (Abdallah Ismail et al., 2011), such as fat, plant cell walls, and complex carbohydrates (Coelho et al., 2018). Scientific evidence suggests that the proportion of Firmicutes to Bacteroidetes is related to fat accumulation; however, the effect of Firmicutes is more apparent (Abdallah Ismail et al., 2011).
In any environment, a higher (P SC‐WC = 0.001, PS W‐WW = 0.001) abundance of Bacteroidetes was observed in the summer group in comparison with the winter group (the relative abundance of Bacteroidetes of the WC group was 32.88% and that of the WW group was 42.59%; Gao et al., 2019). These results showed that Firmicutes play an important role in the degradation of fiber and cellulose (Ben David et al., 2015), which is consistent with the result that the food of the winter group contains more fiber than that in summer group, and an increased proportion of Firmicutes enhances the kiang to obtain the nutrition and energy from their food in winter (Boutton, Tieszen, & Imbamba, 1988). Dietary fiber fermentation typically happens through butyrogenic bacteria, most of which belong to Firmicutes (Klement & Pazienza, 2019). The increase in the relative abundance of this phylum during summer is consistent with the observation that fresh alfalfa has a lower carbohydrate and energy but higher fat contents. In addition, the other bacterial flora showed significant differences between both groups. The nutritional interaction between the gut microbiome and the host is complex and requires further research (Abenavoli et al., 2019).
The alpha diversity of gut microbiomes in the wild group was significantly higher than that in the captive group, and in the winter group, this was significantly higher than in the summer group. Interestingly, a decrease in diversity has been reported to be associated with a number of diseases (Kau et al., 2011). However, the content of pathogenic bacteria increased in the captive group, such as Fusobacteria (Roggenbuck et al., 2014) and Tenericutes (Ludwig, Euzéby, & Whitman, 2010), which increases their risk to catch diseases to some extent. Industrialized food affects the diversity and function of the gut microbiome (Sonnenburg & Sonnenburg, 2019). This suggests that the kiang in the wild winter group had the best health, followed by the wild summer group, while the captive summer group had the worst health. This result is consistent with recent research (Chi et al., 2019). Therefore, in the captive environment, breeders should focus on disease prevention and control of wild animals.
In the captive environment, significant differences in composition and abundance of intestinal flora were found; however, their functions are basically identical, indicating that the taxonomic composition of microbial communities may differ, while being similar in function (Phillips et al., 2017). Different combinations of microbial lineages may achieve comparable community functions (Xiao et al., 2019). These results suggest that diet only alters the intestinal flora. To adapt to a new environment, its function remains unchanged. However, between the wild winter group and the captive winter group, significant differences in function were found. This suggests that the wild environment is more complex and offers less food. Consequently, the wild kiang requires more energy to survive; therefore, the function of metabolism, energy metabolism, and carbohydrate was enhanced. In contrast, the captive kiang has a higher fat and protein diet as well as contact with many humans; consequently, the function of environment information and amino acid metabolism is stronger.
In summary, the composition and abundance of the gut microbiome of the host can rapidly change their metabolic activity in response to changes in the environment. This has enhanced the flexibility of the gut microbiome, helped to maintain normal physiological needs, and improved the adaptability of the host. This study provides a preliminary examination of the seasonal and environmental changes of intestinal flora in the kiang. It not only emphasizes the commonalities of mammals, but also provides a basis for the interaction between intestinal flora and host.
5. CONCLUSION
In summer and winter, the composition and abundance of the intestinal flora of both the captive and wild kiang differed significantly. Environment was identified as the main reason for the observed differences. However, under the same habitat conditions, the function of the gut flora did not change significantly, which plays an important role in the adaptability of the kiang. During the same season, the diversity of the intestinal flora in the captive environment decreased and the content of pathogenic bacteria increased. This suggests that in an environment affected by industrial food, the health level of the captive kiang was lower than that of the wild population and great importance should be attached to the impact of captivity on wildlife.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTION
Hongmei Gao: conceptualization; data curation; formal analysis; methodology; visualization; writing‐original draft; writing‐review & editing. Xiangwen Chi: data curation; formal analysis; writing‐original draft. Guanying Li: conceptualization; visualization; writing‐original draft. Wen Qin: conceptualization; data curation; writing‐original draft. Pengfei Song: data curation; formal analysis; methodology. Feng Jiang: data curation; formal analysis; visualization. Daoxin Liu: data curation; methodology; visualization. Jingjie Zhang: data curation; methodology; visualization. Xiaowen Zhou: conceptualization; methodology; writing‐original draft; Writing‐review and editing. Shengqing Li: conceptualization; formal analysis; methodology. Tongzuo Zhang: conceptualization; writing‐original draft; writing‐review and editing.
ETHICS STATEMENT
We got permission to enter and collect samples in the Qinghai–Tibet Plateau Wildlife Park. In this study, the process of collecting feces of wild animals did not involve issues of animal ethics and animal welfare.
ACKNOWLEDGMENTS
We would like to express our heartfelt thanks to the keepers of the Qinghai–Tibet Plateau Wildlife Park for their help in collecting fecal samples from the kiang in captivity. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (grant No. XDA23060602 and XDA2002030302), the National Key R&D Program of China (grant No. 2017YFC0506405), and the Qinghai Key R&D and Transformation Program (grant No. 2019‐SF‐150).
Gao H, Chi X, Li G, et al. Gut microbial diversity and stabilizing functions enhance the plateau adaptability of Tibetan wild ass (Equus kiang). MicrobiologyOpen. 2020;9:e1025 10.1002/mbo3.1025
DATA AVAILABILITY STATEMENT
The sequence data have been submitted to the GenBank database under accession number PRJNA558941 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA558941).
REFERENCES
- Abdallah Ismail, N. , Ragab, S. H. , Abd Elbaky, A. , Shoeib, A. R. , Alhosary, Y. , & Fekry, D. (2011). Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Archives of Medical Science, 7(3), 501–507. 10.5114/aoms.2011.23418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abenavoli, L. , Scarpellini, E. , Colica, C. , Boccuto, L. , Salehi, B. , Sharifi‐Rad, J. , … Capasso, R. (2019). Gut microbiota and obesity: A role for probiotics. Nutrients, 11(11), 2690 10.3390/nu11112690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato, K. R. , Leigh, S. R. , Kent, A. , Mackie, R. I. , Yeoman, C. J. , Stumpf, R. M. , … Garber, P. A. (2015). The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microbial Ecology, 69(2), 434–443. 10.1007/s00248-014-0554-7 [DOI] [PubMed] [Google Scholar]
- Asshauer, K. P. , Wemheuer, B. , Daniel, R. , & Meinicke, P. (2015). Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics, 31(17), 2882–2884. 10.1093/bioinformatics/btv287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben David, Y. , Dassa, B. , Borovok, I. , Lamed, R. , Koropatkin, N. M. , Martens, E. C. , … Moraïs, S. (2015). Ruminococcal cellulosome systems from rumen to human. Environmental Microbiology, 17(9), 3407–3426. 10.1111/1462-2920.12868 [DOI] [PubMed] [Google Scholar]
- Boutton, T. W. , Tieszen, L. L. , & Imbamba, S. K. (1988). Seasonal changes in the nutrient content of East African grassland vegetation. African Journal of Ecology, 26(2), 103–115. 10.1111/j.1365-2028.1988.tb00961.x [DOI] [Google Scholar]
- Brice, K. L. , Trivedi, P. , Jeffries, T. C. , Blyton, M. D. J. , Mitchell, C. , Singh, B. K. , & Moore, B. D. (2019). The Koala (Phascolarctos cinereus) faecal microbiome differs with diet in a wild population. PeerJ, 7, e6534 10.7717/peerj.6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi, X. , Gao, H. , Wu, G. , Qin, W. , Song, P. , Wang, L. , … Zhang, T. (2019). Comparison of gut microbiota diversity between wild and captive bharals (Pseudois nayaur). BMC Veterinary Research, 15(1), 243 10.1186/s12917-019-1993-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, L. P. , Kultima, J. R. , Costea, P. I. , Fournier, C. , Pan, Y. , Czarnecki‐Maulden, G. , … Bork, P. (2018). Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome, 6(1), 72 10.1186/s40168-018-0450-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, L. A. , Maurice, C. F. , Carmody, R. N. , Gootenberg, D. B. , Button, J. E. , Wolfe, B. E. , … Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505(7484), 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg, P. B. , Bik, E. M. , Bernstein, C. N. , Purdom, E. , Dethlefsen, L. , Sargent, M. , … Relman, D. A. (2005). Diversity of the human intestinal microbial flora. Science, 308(5728), 1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10(10), 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Flint, H. J. , & Bayer, E. A. (2008). Plant cell wall breakdown by anaerobic microorganisms from the Mammalian digestive tract. Annals of the New York Academy of Sciences, 1125, 280–288. 10.1196/annals.1419.022 [DOI] [PubMed] [Google Scholar]
- Gao, H. , Chi, X. , Qin, W. , Wang, L. , Song, P. , Cai, Z. , … Zhang, T. (2019). Comparison of the gut microbiota composition between the wild and captive Tibetan wild ass (Equus kiang). Journal of Applied Microbiology, 126, 1869–1878. 10.1111/jam.14240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, K. M. , Nguyen, B. N. , Neumann, L. M. , Miller, M. , Buss, P. , Daniels, S. , … Pukazhenthi, B. (2019). Gut microbiome differences between wild and captive black rhinoceros ‐ implications for rhino health. Scientific Reports, 9(1), 7570 10.1038/s41598-019-43875-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, M. Y. , Jang, H. M. , & Kim, D. H. (2019). High‐fat diet causes psychiatric disorders in mice by increasing Proteobacteria population. Neuroscience Letters, 698, 51–57. 10.1016/j.neulet.2019.01.006 [DOI] [PubMed] [Google Scholar]
- Kau, A. L. , Ahern, P. P. , Griffin, N. W. , Goodman, A. L. , & Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature, 474(7351), 327–336. 10.1038/nature10213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement, R. J. , & Pazienza, V. (2019). Impact of different types of diet on gut microbiota profiles and cancer prevention and treatment. Medicina, 55(4), 84 10.3390/medicina55040084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leftwich, P. T. , Hutchings, M. I. , & Chapman, T. (2018). Diet, gut microbes and host mate choice: Understanding the significance of microbiome effects on host mate choice requires a case by case evaluation. BioEssays, 40(12), 1800053 10.1002/bies.201800053 [DOI] [PubMed] [Google Scholar]
- Li, L. I. , Wang, Y. , Kong, L. , Jiang, W. U. , & Wang, S. (2018). Study on nutritional components of 35 common gramineous forage plants in Guizhou Province. Journal of Grassland and Forage Science, 3, 13–18. 10.3969/j.issn.2096-3971.2018.03.004 [DOI] [Google Scholar]
- Ludwig, W. , Euzéby, J. , & Whitman, W. B. (2010). Taxonomic outlines of the phyla Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes In Whitman W. B. (Ed.), Bergey's manual® of systematic bacteriology (pp. 21–24). New York, NY: Springer. [Google Scholar]
- Mariat, D. , Firmesse, O. , Levenez, F. , Guimarăes, V. D. , Sokol, H. , Doré, J. , … Furet, J.‐P. (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiology, 9, 123 10.1186/1471-2180-9-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17(1), 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Miaomiao, Y. , Guangcheng, W. , Xiaohong, P. , Huailei, M. , & Weizhen, L. (2008). A method suitable for extracting genomic DNA from animal and plant‐modified CTAB method. Agricultural Science and Technology, 9(2), 39–41. 10.1007/s11442-008-0073-x [DOI] [Google Scholar]
- O'Reilly, F. J. , & Mielke, P. W., Jr. (1980). Asymptotic normality of MRPP statistics from invariance principles of U‐statistics. Communications in Statistics‐Theory and Methods, 9(6), 629–637. 10.1080/03610928008827907 [DOI] [Google Scholar]
- Phillips, C. D. , Hanson, J. , Wilkinson, J. E. , Koenig, L. , Rees, E. , Webala, P. , & Kingston, T. (2017). Microbiome structural and functional interactions across host dietary niche space. Integrative and Comparative Biology, 57(4), 743–755. 10.1093/icb/icx011 [DOI] [PubMed] [Google Scholar]
- Pulikkan, J. , Maji, A. , Dhakan, D. B. , Saxena, R. , Mohan, B. , Anto, M. M. , … Sharma, V. K. (2018). Gut microbial dysbiosis in indian children with autism spectrum disorders. Microbial Ecology, 76(4), 1102–1114. 10.1007/s00248-018-1176-2 [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , … Glöckner, F. O. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web‐based tools. Nucleic Acids Research, 41(D1), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, T. , Boutin, S. , Humphries, M. M. , Dantzer, B. , Gorrell, J. C. , Coltman, D. W. , … Wu, M. (2017). Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome, 5(1), 163 10.1186/s40168-017-0382-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggenbuck, M. , Bærholm Schnell, I. , Blom, N. , Bælum, J. , Bertelsen, M. F. , Sicheritz‐Pontén, T. , … Hansen, L. H. (2014). The microbiome of New World vultures. Nature Communications, 5, 5498 10.1038/ncomms6498 [DOI] [PubMed] [Google Scholar]
- Shepherd, E. S. , DeLoache, W. C. , Pruss, K. M. , Whitaker, W. R. , & Sonnenburg, J. L. (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature, 557(7705), 434–438. 10.1038/s41586-018-0092-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Q. (2002). Quality analysis of several forage grasses in Cyperaceae in Qinghai. Journal of Sichuan Grassland, 4, 37–39. 10.3969/j.issn.1673-8403.2002.04.007 [DOI] [Google Scholar]
- Sonnenburg, J. L. , & Sonnenburg, E. D. (2019). Vulnerability of the industrialized microbiota. Science, 366(6464), eaaw9255 10.1126/science.aaw9255 [DOI] [PubMed] [Google Scholar]
- St‐Louis, A. , & Côté, S. D. (2009). Equus kiang (Perissodactyla: Equidae). Mammalian Species, 835, 1–11. 10.1644/835.1 [DOI] [Google Scholar]
- Sullivan, C. A. , Skeffington, M. S. , Gormally, M. J. , & Finn, J. A. (2010). The ecological status of grasslands on lowland farmlands in western Ireland and implications for grassland classification and nature value assessment. Biological Conservation, 143(6), 1529–1539. 10.1016/j.biocon.2010.03.035 [DOI] [Google Scholar]
- Williams, C. L. , Willard, S. , Kouba, A. , Sparks, D. , Holmes, W. , Falcone, J. , … Brown, A. (2013). Dietary shifts affect the gastrointestinal microflora of the giant panda (Ailuropoda melanoleuca). Journal of Animal Physiology and Animal Nutrition, 97(3), 577–585. 10.1111/j.1439-0396.2012.01299.x [DOI] [PubMed] [Google Scholar]
- Xiao, Y. , Xiao, G. , Liu, H. , Zhao, X. , Sun, C. , Tan, X. , … Feng, J. (2019). Captivity causes taxonomic and functional convergence of gut microbial communities in bats. PeerJ, 7, e6844 10.7717/peerj.6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, B. , Huai, H. , Zhang, Y. , Le, Z. , & Wei, W. (2007). Trophic niches of Pantholops hodgsoni, Procapra picticaudata and Equus kiang in Kekexili region. Ying yong sheng tai xue bao=. The Journal of Applied Ecology, 18(4), 766–770. [PubMed] [Google Scholar]
- Yun, D. , Qi, W. , Yibo, H. , Xiao, W. , Yonggang, N. , Xiaoping, W. , & Fuwen, W. (2017). Advance and prospects of gut microbiome in wild mammals. Acta Theriologica Sinica, 37(4), 399–406. 10.16829/j.slxb.201704010 [DOI] [Google Scholar]
- Zhang, N. , Ju, Z. , & Zuo, T. (2018). Time for food: The impact of diet on gut microbiota and human health. Nutrition, 51–52, 80–85. 10.1016/j.nut.2017.12.005 [DOI] [PubMed] [Google Scholar]
- Zhao, L. , Zhang, F. , Ding, X. , Wu, G. , Lam, Y. Y. , Wang, X. , … Zhang, C. (2018). Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science, 359(6380), 1151–1156. 10.1126/science.aao5774 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequence data have been submitted to the GenBank database under accession number PRJNA558941 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA558941).