

Abstract
The increase in cytosolic Ca2+ concentration ([Ca2+]cyt) and upregulation of calcium-sensing receptor (CaSR) and stromal interaction molecule 2 (STIM2) along with inhibition of voltage-gated K+ (KV) channels in pulmonary arterial smooth muscle cells (PASMC) have been implicated in the development of pulmonary arterial hypertension; however, the precise upstream mechanisms remain elusive. Activation of CaSR, a G protein-coupled receptor (GPCR), results in Ca2+ release from the endoplasmic/sarcoplasmic reticulum (ER/SR) and Ca2+ influx through receptor-operated and store-operated Ca2+ channels (SOC). Upon Ca2+ depletion from the SR, STIM forms clusters to mediate store-operated Ca2+ entry. Activity of KV channels, like KCNA5/KV1.5 and KCNA2/KV1.2, contributes to regulating membrane potential, and inhibition of KV channels results in membrane depolarization that increases [Ca2+]cyt by opening voltage-dependent Ca2+ channels. In this study, we show that activation of Notch by its ligand Jag-1 promotes the clustering of STIM2, and clustered STIM2 subsequently enhances the CaSR-induced Ca2+ influx through SOC channels. Extracellular Ca2+-mediated activation of CaSR increases [Ca2+]cyt in CASR-transfected HEK293 cells. Treatment of CASR-transfected cells with Jag-1 further enhances CaSR-mediated increase in [Ca2+]cyt. Moreover, CaSR-mediated increase in [Ca2+]cyt was significantly augmented in cells co-transfected with CASR and STIM2. CaSR activation results in STIM2 clustering in CASR/STIM2-cotransfected cells. Notch activation also induces significant clustering of STIM2. Furthermore, activation of Notch attenuates whole cell K+ currents in KCNA5- and KCNA2-transfected cells. Together, these results suggest that Notch activation enhances CaSR-mediated increases in [Ca2+]cyt by enhancing store-operated Ca2+ entry and inhibits KCNA5/KV1.5 and KCNA2/KV1.2, ultimately leading to voltage-activated Ca2+ entry.
Keywords: CaSR, GPCR, KCNA5 and KCNA2, Notch, STIM2
INTRODUCTION
Extracellular Ca2+-sensing receptor (CaSR) is a class III G protein-coupled receptor (GPCR) found to have inorganic ion, Ca2+, as its physiological agonist (7). The major physiological role of the CaSR was initially reported to adjust the extracellular Ca2+ concentration by regulating parathyroid hormone secretion and the rate of Ca2+ reabsorption by the kidney and bone (7). However, CaSR has also been implicated in the development and progression of lung airway (47) and vascular (40, 43, 44, 50) diseases. CaSR is a multifaceted receptor that can also bind to a plethora of ligands other than Ca2+, such as aromatic amino acids (e.g., l-phenylalanine) and antibiotics (e.g., aminoglycosides like neomycin) (48), which trigger conformational changes in the receptor responsible for the initiation of multiple signaling pathways (8). One of the demonstrated signaling pathways is intracellular Ca2+ signaling.
The ligand-mediated activation of CaSR signals through the Gq/11, Gi, and G12/13 phospholipase C (PLC) and phosphatidylinositol biphosphate (PIP2) pathways results in the production of two second messengers: diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). Together, DAG and IP3 contribute to an increase in [Ca2+]cyt by inducing Ca2+ influx through Ca2+ channels in the plasma membrane and Ca2+ release from the intracellular Ca2+ stores (4). DAG opens receptor-operated Ca2+ channels (ROC) and promotes Ca2+ influx to the cytosol. [Ca2+]cyt is increased due to receptor-operated Ca2+ entry (ROCE). IP3 binds to IP3 receptors (IP3R) on the membrane of the sarcoplasmic reticulum (SR) or endoplasmic reticulum (ER) and leads to Ca2+ release or mobilization from the SR/ER into the cytosol. Stromal-interacting molecular proteins, including STIM1 and STIM2, are Ca2+-sensitive proteins responsible for sensing the level of [Ca2+] in the SR/ER. Upon Ca2+ depletion or reduction from the intracellular stores (SR/ER), STIM1/2 undergoes conformational changes by multimerizing with each other and translocating to the SR/ER-plasma membrane (SR/ER-PM) junction (puncta), where the STIM1/2 multimers recruit Orai to form store-operated Ca2+ channels (SOC) allowing Ca2+ entry. Although both STIM1 and STIM2 are involved in recruiting Orai proteins to form SOC and induce store-operated Ca2+ entry (SOCE), STIM1 is a more potent activator of SOCE. [Ca2+]cyt is increased in this case due to SOCE (9, 16, 18). Expression or upregulated expression of CaSR has been implicated in augmenting extracellular Ca2+-induced increase in [Ca2+]cyt in pulmonary arterial smooth muscle cells (PASMC) isolated from patients with idiopathic pulmonary arterial hypertension (IPAH) and animals with experimental pulmonary hypertension (PH) (34, 40, 43–45, 50). Expression or upregulated expression of STIM2 has also been demonstrated to increase the resting [Ca2+]cyt due to SOCE in PASMC from IPAH patients. One of the reasons for this study is to study whether Jag-1-mediated Notch activation can induce STIM2 clustering, an important mechanism for SOCE.
We have recently demonstrated that enhanced downstream signaling cascades (including Ca2+ signaling and Akt/mTOR pathway), when CaSR is activated by extracellular ligands, contribute to the development and progression of pulmonary vascular remodeling in PH (40). Indeed, pharmacological blockade of CaSR or genetic deletion of the CaSR gene significantly attenuates the development and progression of experimental PH in rats and mice (40, 44). Upregulated CaSR and augmented CaSR-mediated [Ca2+]cyt increase in airway smooth muscle cells have also been demonstrated to be associated with the development and progression of airway remodeling and bronchospasm in asthma (47).
[Ca2+]cyt could be also increased due to Ca2+ influx through voltage-dependent Ca2+ channels (VDCC) in the plasma membrane. Voltage-gated K+ (KV) channels, highly expressed in normal human and animal PASMC, play an important role in the regulation of the membrane potential (Em) (49). When KV channels (e.g., KV1.5, KV1.2) are closed and/or KV channel protein expression is downregulated, the membrane depolarizes, which leads to a rise in [Ca2+]cyt by opening VDCC and inducing Ca2+ entry through VDCC. We (31, 49) and other investigators (5, 11, 25) have demonstrated that downregulated expression or decreased activity of KV channels in PASMC is associated with the development of sustained pulmonary vasoconstriction and vascular remodeling in patients with IPAH and animals with hypoxia-induced PH (5, 11, 25, 31, 49). However, it is unknown whether CaSR and Notch receptors functionally interact with KV channels to regulate membrane potential and Ca2+ influx through VDCC.
Our previous study has demonstrated that short-term activation of Notch signaling enhances SOCE in PASMC (46). Activation of Notch is upon Notch receptors binding with ligands, including Jagged1–3 (Jag-1–3) and delta-like ligand 1–4 (Dll1–4), and releasing the Notch intracellular domains (NICD) by γ-secretase. The released cytosol NICD then translocates to the nucleus, where it binds with the DNA-binding protein CSL [CBF1/RBPJκ/Su(H)/Lag-1] transcription factor complex, resulting in subsequent activation of the canonical Notch target genes such as the HES/Hay-family members as well as Myc and p21 (2). Activation of Notch signaling has been reported to contribute to the development of chronic PH and acute hypoxia-induced pulmonary vasoconstriction by augmenting Ca2+ signaling (15, 22, 32).
In this study, we aimed to investigate the cellular and molecular mechanisms involved in Notch-induced SOCE, focusing on CaSR-mediated Ca2+ influx and the potential involvement of STIM2. Furthermore, we also explored whether Notch activation has an effect on KV1.5/KV1.2 channel activity.
MATERIALS AND METHODS
Cell culture.
Human embryonic kidney 293 (HEK293) cells were cultured in high-glucose DMEM (Invitrogen, Grand Island, NY) containing 10% fetal bovine serum (FBS; Invitrogen), 100 IU/ml penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO) in a humidified atmosphere at 37°C and 5% CO2.
Constructs and transfection.
CASR construct was purchased from OriGene (Rockville, MD), yellow fluorescent protein (YFP)-STIM2 construct and green fluorescent protein (GFP) construct were purchased from Addgene (plasmid no. 18862, plasmid no. 19755), and KCNA5 construct was obtained from Harvard Medical School (plasmid HsCD00457881). HEK293 cells were transiently transfected with CASR expression construct (1 μg), YFP-STIM2 expression construct (1 μg), KCNA5 expression (1 μg), KCNA2 expression (1 μg), or GFP construct (1 μg) using X-tremeGENE 9 DNA Transfection Reagent (Roche, Germany). Electrophysiological and fluorescence microscopy experiments were performed 48 h after the transfection.
Measurement of [Ca2+]cyt.
HEK293 cells were incubated with 4 μM fura-2 acetoxymethyl ester (fura-2 AM) (Invitrogen/Molecular Probes, Eugene, OR) for 60 min at room temperature (22–24°C). Fura-2 fluorescence was measured in single HEK293 cell with an EM-CC camera (Evolve; Photometrics, Tucson, AZ) and analyzed using NIS Elements 3.2 software (Nikon) in an inverted fluorescent microscope (Eclipse Ti-E; Nikon, Tokyo, Japan), as described previously (37). Cytosolic free Ca2+ concentration ([Ca2+]cyt) is expressed as 340/380 fluorescence ratio within a region of interest (ROI) in a cell recorded every 2 s. The HEPES-buffered bath solution contained 137 mM NaCl, 5.9 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 14 mM glucose, and 10 mM HEPES (pH was adjusted to 7.4 with 10 N NaOH). The Ca2+-free solution was prepared by replacing 1.8 mM CaCl2 with equimolar MgCl2 and adding 0.1 mM EGTA to chelate residual Ca2+. All experiments for measurement of [Ca2+]cyt were carried out at room temperature (22–24°C).
Western blotting.
HEK293 cells were lysed in 1× RIPA buffer (Bio-Rad, Hercules, CA) supplemented with protease inhibitor cocktail (Roche). Protein concentration was determined with the BCA protein assay kit (Thermo Scientific, Waltham, MA). Cell lysates were heated at 100°C for 7 min, electrophoresed on SDS-PAGE, and transferred to polyvinylidene difluoride membrane (Millipore). The blots were blocked with TBS-T containing 5% nonfat dry milk at room temperature for 1 h. CaSR was detected by a CaSR antibody (product no. PA1-37213, 1:1,000 dilution; Thermo Scientific), whereas KCNA5 (or KV1.5) was detected by a KCNA5 antibody (sc-377110, 1:1,000; Santa Cruz Biotechnology) using a Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific). The protein levels of target proteins were normalized to β-actin (sc-9104, 1:1,000; Santa Cruz Biotechnology) and expressed in arbitrary units.
Reverse transcription-PCR.
Total RNA was isolated from HEK293 cells by the TRIzol method. The extracted RNA was quantified by NanoDrop 1000 (Thermo Scientific). RNA was converted into first-strand cDNA using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Semiquantitative RT-PCR was carried out using PCR Nucleotide Mix (Roche) with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the internal control on a Thermal Cycler T100 (Bio-Rad, Hercules, CA). The PCR products were electrophoresed through 1.5% agarose gel, and amplified cDNA bands were visualized by Photo Imager (VWR, Radnor, PA).
STIM2 imaging in a single cell.
HEK293 cells were grown on 12-mm diameter coverslips and transfected with YFP-STIM2 construct for 48 h before imaging. The same cell was imaged before and after the application of experimental condition. Fluorescently transfected cells were imaged with a Nikon inverted fluorescence microscope (Eclipse Ti-E; Nikon, Tokyo, Japan) with ×100 objective. Fluorescence intensity analysis and line scans were carried out using ImageJ software. The same region of interest in a cell was analyzed before and after the application of agonists and chemicals or the change of experimental conditions.
Electrophysiological recording.
Whole cell K+ currents (IK) were measured at room temperature (22–24°C) using an Axopatch 200B amplifier and Digidata 1440A interface (Molecular Devices, Foster City, CA). Command-voltage protocols and data acquisition were performed using pCLAMP 10.5 software (Molecular Devices, Foster City, CA). Pipettes were fabricated from borosilicate glass using a P-97 Flaming/Brown Micropipette puller (Sutter Instruments, Novato, CA). Whole cell K+ currents through KV channels (KCNA5/KV1.5 and KCNA2/KV1.2) were recorded using a Ca2+-free bath (extracellular) solution containing 137 mM NaCl, 1.2 mM MgCl2, 5 mM HEPES, 1.5 mM EGTA, and 10 mM glucose (pH was adjusted to 7.4 with NaOH) and a pipette (intracellular) solution containing 140 mM KCl, 10 mM NaCl, 1 mM MgCl2, 5 mM HEPES, 1.5 mM EGTA, and 5 mM Na-ATP (pH was adjusted to pH 7.2 with NaOH). Ca2+-activated K+ currents were minimized by using Ca2+-free and EGTA-containing extracellular and intracellular solutions, whereas ATP-sensitive K+ currents were minimized by adding 5 mM ATP in the pipette (intracellular) solution. Data were collected at a filtering frequency of 2 kHz and sampling rate of 50 kHz.
Drugs and chemicals.
NPS-2143 (TOCRIS, Bristol, UK), cinacalcet (Santa Cruz Biotechnology, Dallas, TX), cyclopiazonic acid (CPA; Sigma-Aldrich, St. Louis, MO), 4-amiyopyridine (4-AP; TOCRIS, Minneapolis, MN), and U73122 (Sigma-Aldrich) were prepared as stock solutions in dimethyl sulfoxide (DMSO). Spermine (Sigma-Aldrich), streptomycin (Sigma-Aldrich), neomycin (Sigma-Aldrich), l-phenylalanine (Sigma-Aldrich), and Jagged-1 (AnaSpec, Fremont, CA) were dissolved in distilled water as stock solutions. All stock solutions (in water or DMSO) were aliquoted and kept frozen at −20°C until use.
Statistical analysis.
Data are expressed as means ± SE. Statistical significance between two or among multiple groups was examined using Student’s t test or Scheffé’s test after one-way ANOVA, respectively. Significant difference is expressed in the figures or figure captions when P < 0.05.
RESULTS
Physiological and pharmacological properties of CaSR-mediated increases in [Ca2+]cyt.
To characterize the CaSR-mediated increase in [Ca2+]cyt, we transiently transfected HEK293 cells with the human CASR gene and measured the changes of [Ca2+]cyt in response to extracellular Ca2+ (1.8 mM) 48 h after CASR transfection. As shown in Fig. 1, A and B, overexpression of CASR in HEK293 cells significantly enhanced extracellular Ca2+-induced increase in [Ca2+]cyt compared with that in nontransfected (control) and GFP-transfected cells. There was no difference between control and GFP groups. The extracellular Ca2+-induced increases in [Ca2+]cyt in CASR-transfected cells had three different patterns: 1) a transient increase followed by a sustained plateau phase that was observed in most (75%) of the cells tested (Fig. 1B, left), 2) a slowly-activating rise in [Ca2+]cyt upon application of extracellular Ca2+ that was detected in some (15%) cells (Fig. 1B, middle), and 3) a low-amplitude and high-frequency oscillatory increase in [Ca2+]cyt that was observed in some (10%) cells (Fig. 1B, right and inset). The different patterns of extracellular Ca2+-induced increases in [Ca2+]cyt indicate that Ca2+-mediated CaSR activation may functionally be coupled to different Ca2+ channels in the plasma membrane and the SR/ER membrane through different mechanisms and to different intracellular second messengers.
Fig. 1.

Calcium-sensing receptor (CaSR) overexpression enhances extracellular Ca2+-induced increase in cytosolic Ca2+ concentration ([Ca2+]cyt) in human embryonic kidney 293 (HEK293) cells. A: representative traces showing changes in [Ca2+]cyt in nontransfected (Control) and green fluorescent protein-transfected (GFP) and 3 different patterns in CASR-transfected (CASR) HEK293 cells before, during, and after extracellular application of 1.8 mM Ca2+. Summarized data (right, means ± SE) showing the amplitude of 1.8 mM Ca2+-induced increases in [Ca2+]cyt in control HEK293 cells and HEK293 cells transfected with GFP or CASR constructs, respectively; n = 5 cells for each group, *P < 0.05 vs. Control and GFP (ANOVA). B: normalized record of 1.8 mM Ca2+ (1.8Ca)-mediated sustained (left), transient (middle), and oscillatory (right) increases in [Ca2+]cyt in CASR-transfected cells. Inset: expanded record shown at right. C: representative traces showing changes in [Ca2+]cyt in CASR-transfected HEK293 cells before, during, and after extracellular sequential application of 1.8 mM Ca2+ and 1.8 mM Ca2+ plus vehicle (DMSO), NPS-2143 (CaSR inhibitor), or U73122 (PLC inhibitor). Summarized data (right, means ± SE) showing the amplitude of 1.8 mM Ca2+-induced increase in [Ca2+]cyt in the presence of vehicle, NPS-2143, or U73122 in HEK293 cells transfected with CASR construct; n = 6 cells for each group, *P < 0.05 vs. Vehicle (ANOVA). D: representative traces (left) showing changes in [Ca2+]cyt in nontransfected (control) and GFP-transfected (GFP) HEK293 cells as well as CASR-transfected (CaSR) cells with or without NPS-2143 (60 μM) before, during, and after extracellular application of 0.1, 0.5, 1.0, 1.8, and 10 mM of Ca2+. Summarized data (right, means ± SE) showing the dose-response curves of extracellular Ca2+-induced increases in [Ca2+]cyt in control and GFP-transfected HEK293 cells as well as in cells transfected with CASR constructs in the presence of NPS-2143; n = 5 cells for each group. E: representative RT-PCR images (left) showing the mRNA level of CaSR in control cells and cells transfected with CASR construct. Summarized data (right, means ± SE) showing mRNA level of CaSR in control cells and cells transfected with CASR construct; n = 5 cells for each group. F: representative Western blot images showing CaSR in control and CASR-transfected cells; summarized data (right, means ± SE) showing the protein levels of CaSR in control cells and cells transfected with CASR construct; n = 5 cells for each group. ***P < 0.001 vs. control (Student’s t test).
We then aimed to determine whether pharmacological inhibition of CaSR is able to prevent or attenuate [Ca2+]cyt increases in response to extracellular Ca2+ in CaSR-transfected cells. Extracellular application of 60 μM NPS-2143 (21), an allosteric selective blocker of CaSR, almost abolished the extracellular Ca2+-induced increase in [Ca2+]cyt in CASR-transfected cells (Fig. 1C). To clarify the role of IP3 production in CaSR-mediated [Ca2+]cyt rise, we examined the effect of a key enzyme in IP3 production, phospholipase C (PLC), by using its specific inhibitor U73122 (24). Indeed, the application of U73122 (10 μM) significantly attenuated the extracellular Ca2+-induced increase in [Ca2+]cyt in CASR-transfected cells in comparison with cells treated with vehicle (Fig. 1C). In CASR-transfected cells, extracellular Ca2+ elicited increases in [Ca2+]cyt in a dose-dependent manner with an EC50 at ∼3.3 mM (Fig. 1D). Blockade of CaSR with NPS (Fig. 1D) and blockade of PLC with U73122 (Fig. 1C) significantly inhibited the extracellular Ca2+-mediated increases in [Ca2+]cyt in CASR-transfected cells. These data indicate that activation of CaSR by its cationic agonist Ca2+ increases intracellular second messengers IP3 and diacelglycerol (DAG) via PLC and subsequently induces 1) Ca2+ release from the SR/ER via Ca2+ release channels or IP3 receptors and 2) Ca2+ influx through various Ca2+ channels in the plasma membrane.
To further confirm our findings, we estimated transfection efficiency by quantifying CaSR expression in CASR-transfected cells. As expected, both protein and mRNA levels of CaSR were significantly increased in cells after CASR transfection in comparison with control cells (Fig. 1, E and F). These results suggest that CaSR-mediated increase in [Ca2+]cyt is indeed due, at least partially, to Ca2+ mobilization from intracellular stores like the SR/ER via the CaSR-PLC-IP3 pathway and Ca2+ influx through CaSR-coupled cation channels via the CaSR-PLC-DAG pathway, consistent with the fact that CaSR is a G protein-coupled receptor.
In addition to extracellular Ca2+, polyamines, antibiotics, and amino acids have been demonstrated to activate CaSR as well (7, 17). Overexpression of CaSR not only enhanced the extracellular Ca2+-induced increase in [Ca2+]cyt but also significantly augmented the extracellular spermine-, cinacalcet-, streptomycin-, neomycin-, and phenylalanine-mediated increases in [Ca2+]cyt (Fig. 2). Different activators of CaSR seemed to induce distinct patterns of changes (or increases) in [Ca2+]cyt; cinacalcet (Cina; 1 μM), streptomycin (Strep; 100 μM), neomycin (Neom; 100 μM), and l-phenylalanine (l-Phe, 3 mM) only caused Ca2+ oscillations or oscillatory increases in [Ca2+]cyt, whereas spermine (Sperm; 3 mM) caused a rapid transient increase followed by sustained increase in [Ca2+]cyt. In the absence of extracellular Ca2+, however, the amplitude and frequency of spermine-, cinacalcet-, and streptomycin-induced increases in [Ca2+]cyt were lower than those in the Ca2+-containing solution (Fig. 2, A–C). The frequency of neomycin-induced increases in [Ca2+]cyt was lower, but not amplitude in cells bathed in Ca2+-free condition (Fig. 2D). l-Phenylalanine was not able to induce an increase in [Ca2+]cyt in the absence of Ca2+ (Fig. 2E). These results indicate that CaSR-mediated increase in [Ca2+]cyt was due to both Ca2+ mobilization from the intracellular stores and Ca2+ influx through the plasmalemmal Ca2+ channels. The transient increase in [Ca2+]cyt was likely due to Ca2+ mobilization, whereas the sustained increase in [Ca2+]cyt was due to Ca2+ influx through various Ca2+ channels in the plasma membrane. The CaSR-mediated oscillatory increase in [Ca2+]cyt was likely caused by a coordinated mechanism involving Ca2+ release and uptake in the SR/ER as well as Ca2+ influx and extrusion through the plasma membrane (19, 36). Different patterns of CaSR-mediated increases in [Ca2+]cyt may result in different effects on cell functions (e.g., contraction, proliferation, migration, and protein synthesis) (10, 36).
Fig. 2.

Characterization of spermine-, cinacalcet-, streptomycin-, neomycin-, and l-phenylalanyl-induced cytosolic Ca2+ concentration ([Ca2+]cyt) increase in calcium-sensing receptor (CASR)-transfected human embryonic kidney 293 (HEK293) cells. Representative traces (a) showing changes in [Ca2+]cyt in control (left 3 curves), green fluorescent protein (GFP)-transfected (middle 3 curves), and CASR-transfected (right 3 curves) HEK293 cells before, during, and after extracellular sequentially application of 3 mM spermine (A), 1 μM cinacalcet (B), 100 μM streptomycin (C), 100 μM neomycin (D), and 3 mM l-phenylalanyl (E) in the presence or absence (0 Ca) of 1.8 mM extracellular Ca2+. Summarized data (means ± SE; b) showing the amplitude and frequency of spermine- (A), cinacalcet- (B), streptomycin- (C), neomycin- (D) and l-phenylalanyl-induced (E) increases in [Ca2+]cyt in presence or absence of 1.8 mM extracellular Ca2+ in HEK293 cells transfected with CASR constructs; n = 5 for each group. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. 1.8Ca2+ (Student’s t test).
Production and deposition of β-amyloid (Aβ) has been implicated in Alzheimer’s disease (42), and upregulated CaSR is found in animal models of Alzheimer’s disease (14). Upregulated mRNA and protein expression of amyloid protein precursor (APP) has also been recently identified in lung tissues from patients with Down syndrome as a risk factor for pulmonary hypertension (13). Overexpression of CaSR also enhanced the extracellular β-amyloid-induced oscillatory increase in [Ca2+]cyt, whereas removal of extracellular Ca2+ abolished β-amyloid-induced [Ca2+]cyt increases (Fig. 3). The pattern of β-amyloid-induced oscillatory increases in [Ca2+]cyt was similar to that induced by cinacalcet (Fig. 2B), streptomycin (Fig. 2C), and neomycin (Fig. 2D) in the presence of extracellular Ca2+. However, the β-amyloid-induced oscillatory increase in [Ca2+]cyt seemed to be more dependent on extracellular Ca2+ or Ca2+ influx than the cinacalcet-, streptomycin-, and neomycin-induced increases in [Ca2+]cyt. The dependence of extracellular Ca2+ of the β-amyloid-induced oscillatory increase in [Ca2+]cyt is similar to the l-phenylalanine-induced [Ca2+]cyt increase.
Fig. 3.

β-Amyloid-induced oscillatory increases in calcium-sensing receptor (CaSR)-transfected cells. A: representative traces showing changes in [Ca2+]cyt in control (left), green fluorescent protein (GFP)-transfected (middle), and CASR-transfected (right) human embryonic kidney 293 (HEK293) cells before, during, and after extracellular application of 10 μM β-amyloid in the presence or absence (0 Ca) of 1.8 mM extracellular Ca2+. B and C: summarized data (means ± SE) showing the amplitude (B) and frequency (C) of β-amyloid-induced oscillatory increases in [Ca2+]cyt in the presence (red) or absence (0 Ca; blue) of 1.8 mM extracellular Ca2+ in HEK293 cells transfected with CASR constructs; n = 5 cells for each group. ***P < 0.001 vs. 0 Ca (ANOVA and Student’s t test).
Notch activation enhances CaSR-mediated increases in [Ca2+]cyt.
We previously demonstrated that short-term (10 min) activation of Notch signaling with its ligand Jag-1 significantly enhanced store depletion-mediated Ca2+ influx in human PASMC (46), whereas Orai1/2 and STIM1/2 were all involved in eliciting store-operated Ca2+ entry (SOCE). We then sought to examine in this study whether Notch activation also affects the function of CaSR or the extracellular Ca2+-induced increase in [Ca2+]cyt.
As we showed earlier, in CASR-transfected HEK293 cells, extracellular Ca2+ resulted in different patterns of increases in [Ca2+]cyt (Fig. 1A). At first, we measured extracellular Ca2+-induced rise in [Ca2+]cyt in GFP-transfected HEK293 cells treated with or without Jag-1 (50 μM) for 10 min. Jag-1 failed to enhance the extracellular Ca2+-induced increase in [Ca2+]cyt, in HEK293 cells transfected with an empty GFP vector (Fig. 4A). To confirm the functional interaction between Notch and CaSR, we treated CaSR-transfected HEK cells with or without Jag-1 and repeated the experiments. Short-term (10 min) activation of Notch signaling significantly enhanced the amplitude of all the patterns of the extracellular Ca2+-induced increases in [Ca2+]cyt compared with control in CaSR-transfected HEK293 cells (Fig. 4, B and D). These results provide strong evidence that Notch signaling synergistically interacts with CaSR to enhance extracellular Ca2+-induced increases in [Ca2+]cyt.
Fig. 4.

Notch activation enhances extracellular Ca2+-induced increase in [Ca2+]cyt in calcium-sensing receptor (CASR)-transfected human embryonic kidney 293 (HEK293) cells. A: representative average traces showing changes in [Ca2+]cyt in green fluorescent protein (GFP)-transfected HEK293 cells before, during, and after extracellular application of 1.8 mM Ca2+ without (left) or with (middle) pretreatment (10 min) of 50 μM Notch receptor ligand Jagged1 (Jag-1). Summarized data (means ± SE; right) showing extracellular 1.8 mM Ca2+-induced increase in [Ca2+]cyt in GFP-transfected HEK293 cells before, during, and after extracellular application of 1.8 mM Ca2+ without or with pretreatment of 50 μM Notch receptor agonist Jag-1; n = 5 independent sets of transfected cells. B: representative individual (dark blue and dark red; top) and average (blue and red; bottom) traces showing changes in [Ca2+]cyt in CASR-transfected HEK293 cells before, during, and after extracellular application of 1.8 mM Ca2+ without (dark blue and blue; left) or with (dark red and red; right) pretreatment (10 min) of 50 μM Notch receptor ligand Jag-1. C: representative individual (dark green and pink; top) and average (green and pink, bottom) traces showing changes in [Ca2+]cyt in CASR/stromal interaction molecule 2 (STIM2)-cotransfected HEK293 cells before, during, and after extracellular application of 1.8 mM Ca2+ without (dark green and green; left) or with (dark pink and pink; right) pretreatment (10 min) of 50 μM Notch receptor agonist Jag-1. D: summarized data (means ± SE) showing the amplitude of extracellular 1.8 mM Ca2+-induced increases in [Ca2+]cyt in CASR-transfected (CASR) and CASR/STIM2-cotransfected (CASR + STIM2) HEK293 cells without or with pretreatment of 50 μM Jag-1; n = 6 independent sets of transfected cells, *P < 0.05 as indicated (ANOVA).
To investigate the role of STIM2, a critical Ca2+ sensor in the SR/ER and an important inducer of SOCE, in CaSR-mediated [Ca2+]cyt rise, we cotransfected HEK293 cells with CaSR and STIM2. In CASR-transfected HEK293 cells, the extracellular Ca2+-induced oscillatory increases in [Ca2+]cyt were enhanced and altered to be sustained increases by cotransfection of STIM2 (Fig. 4C). The amplitude of the extracellular Ca2+-induced increases in [Ca2+]cyt was significantly enhanced in CASR/STIM2-cotransfected HEK293 cells compared with the CASR-transfected cells (Fig. 4D). Short-term (10 min) Jag-1 treatment further enhanced the amplitude of extracellular Ca2+-induced increases in [Ca2+]cyt in CASR/STIM2-cotransfected HEK293 cells compared with control cells transfected with CASR alone (Fig. 4, C and D). Altogether, these data imply that STIM2 contributes to the enhanced CaSR-mediated Ca2+ influx and Notch activation functionally interacts with CaSR to facilitate the extracellular Ca2+-induced CaSR-mediated Ca2+ entry. These data suggest a novel mechanism by which Notch signaling or Notch ligands (e.g., Jag-1) functionally interact with CaSR and/or its downstream effectors to enhance Ca2+ release and Ca2+ influx via multiple pathways: Ca2+ mobilization (IP3 and IP3 receptor), SOCE (STIM1/2 or Orai1/2), and ROCE (TRPC6 and Orai1/2). Jag-1 may directly or indirectly activate CaSR and enhance Ca2+-mediated activation of CaSR; Jag-1-mediated NICD may also promote STIM2 clustering with Orai channels in the plasma membrane to enhance CaSR-associated SOCE. The next set of experiments was designed to examine how Notch activation exerts functional regulation of CaSR-associated SOCE.
Notch activation induces clustering of STIM2.
Given the important role of STIM2 in triggering SOCE (12, 27, 35), it is possible that STIM2 augments the component of SOCE in the CaSR-mediated Ca2+ entry (which is composed of, at least, ROCE and SOCE). Notch activation (by Jag-1) may exert a functional effect on STIM2 to amplify extracellular Ca2+-mediated CaSR activation and CaSR-associated SOCE. In response to Ca2+ depletion in the SR/ER, STIM2 undergoes conformational changes and translocates to specific SR/ER-PM junctions. At these sites, STIM2 accumulates as clusters, referred to as puncta, which in turn determine the recruitment of Orai1 into the SR/ER-PM junctions, resulting in STIM2-Orai1 interaction and activation of SOCE (27, 35).
To determine the effect of Notch on STIM2 clustering, we performed single-cell imaging experiments in HEK293 cells transfected with STIM2 or CaSR/STIM2 construct. Under control conditions, STIM2-transfected HEK293 cells showed evenly distributed STIM2 in Ca2+-free solution. Short-term (10 min) pretreatment with the SR/ER Ca2+ pump (SERCA) inhibitor cyclopiazonic acid (CPA), which passively depletes intracellularly stored Ca2+ in the SR/ER, resulted in significant clustering of STIM2 (Fig. 5, A and B). Next, we compared STIM2 fluorescent intensity before and after extracellular application of 1.8 mM Ca2+ in STIM2-transfected HEK293 cells. We found, however, that short-term pre-incubation (10 min) of the STIM2-transfected cells in 1.8 mM Ca2+-containing solution had no effect on STIM2 clustering compared with Ca2+-free solution (data not shown). Furthermore, short-term (10 min) Notch activation by Jag-1 induced apparent clustering of STIM2 in the SR/ER-PM junctions in both Ca2+-free and Ca2+-containing conditions (Fig. 5, C–F). These data indicate that Jag-1-mediated Notch activation causes similar STIM2 clustering, as does CPA-mediated store depletion.
Fig. 5.

Notch activation induces clustering of stromal interaction molecule 2 (STIM2) in STIM2-transfected human embryonic kidney 293 (HEK293) cells. A: representative fluorescent images (a) showing STIM2 clusters (green) in HEK293 cells transfected with yellow fluorescent protein (YFP)-STIM2 construct before (control) and during the treatment with cyclopiazonic acid (CPA). Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimensional images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. B: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of YFP-STIM2 before (control) and during application of cyclopiazonic acid (CPA) in STIM2-transfected HEK293 cells; n = 5 independent sets of transfected cells. C: representative fluorescent images (a) showing STIM2 clusters (green) in HEK293 cells transfected with YFP-STIM2 construct before (control) and during the treatment with 50 μM Jagged1 (Jag-1) for 10 min in Ca2+-free solution. Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimentsonal images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. D: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of YFP-STIM2 before (control) and during application of Jag-1 in Ca2+-free solution in STIM2-transfected HEK293 cells; n = 5 independent sets of transfected cells. E: representative fluorescent images (a) showing STIM2 (green) in HEK293 cells transfected with YFP-STIM2 construct before (control) and during the treatment with 50 μM Jag-1 for 10 min in Ca2+-containing solution. Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimentional images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. F: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of YFP-STIM2 before (control) and during application of Jag-1 in Ca2+-containing solution in STIM2-transfected HEK293 cells; n = 5 independent sets of transfected cells. We specifically selected ROIs (regions of interest) mostly close to the plasma membrane (i.e., the peripheral region of the cell) for analyzing the size, number, and intensity of STIM2 clusters. Summarized data (B, D, and F) show the overall intensity of the STIM2 clusters located in the peripheral regions of the cells. *P < 0.05 and ***P < 0.001 vs. control (Student’s t test).
Then, we performed the same experiments in HEK293 cells transfected with CaSR/STIM2 construct. Likewise, short-term (10 min) CPA treatment induced clustering of STIM2 in Ca2+-free solution in CaSR/STIM2-cotransfected HEK293 cells (Fig. 6, A and B). To examine whether Notch activation affects STIM2 clustering in CaSR/STIM2-overexpressed HEK293 cells, we measured STIM2 fluorescent intensity in cells pre-incubated with or without Jag-1 in Ca2+-free solution for 10 min. Single-cell imaging experiments showed that Jag-1 treatment resulted in significant STIM2 clustering in HEK293 cells co-transfected with CaSR/STIM2 construct (Fig. 6, C and D). Interestingly, short-term incubation of CaSR/STIM2-cotransfected cells in Ca2+-containing solution, in fact, induced a strong STIM2 clustering compared with that in the absence of extracellular Ca2+ (Fig. 6, E and F). We were, however, unable to observe Jag-1-enhanced STIM2 clustering in HEK293 cells cotransfected with CaSR/STIM2 in Ca2+-containing solution. Our findings indicate that Jag-1-mediated Notch activation and extracellular Ca2+-mediated CaSR induce both STIM2 oligomerization and clustering in the SR/ER membrane and subsequent STIM2 translocation to the SR/ER-PM junctions, which then recruit and open the store-operated Ca2+ channels (SOC) in the plasma membrane and elicit SOCE (27, 28, 38). The observations from these experiments imply that STIM2 is involved in mediating Ca2+ influx through SOC upon extracellular Ca2+-mediated activation of CaSR, and activation of Notch signaling functionally facilitates the clustering of STIM2 and thus enhances CaSR-mediated increase in [Ca2+]cyt by amplifying SOCE.
Fig. 6.

Notch activation or calcium-sensing receptor (CaSR) activation induces clustering of stromal interaction molecule 2 (STIM2) in CASR/STIM2-cotransfected human embryonic kidney 293 (HEK293) cells. A: representative fluorescent images (a) showing STIM2 clusters (green) in HEK293 cells transfected with CASR/YFP-STIM2 construct before (control) and during the treatment with cyclopiazonic acid (CPA). Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimensional images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. B: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of yellow fluorescent protein (YFP)-STIM2 before (control) and during application of CPA in CASR/STIM2-cotransfected HEK293 cells; n = 5 independent sets of transfected cells. C: representative fluorescent images (a) showing STIM2 (green) in cells transfected with CASR/YFP-STIM2 before (control) and during the treatment with 50 μM Jag-1 for 10 min in Ca2+-free solution. Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimentional images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. D: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of YFP-STIM2 before and during application of Jag-1 in Ca2+-free solution in CASR/STIM2-cotransfected HEK293 cells; n = 6 independent sets of transfected cells. E: representative fluorescent images (a) showing STIM2 clusters (green) in HEK293 cells transfected with CASR/YFP-STIM2 before (0 Ca) and after 1.8 mM extracellular Ca2+ treatment (Ca) for 10 min. Magnified images (b) showing STIM2 clusters (arrows) in the puncta area and 3-dimentional images (c) showing STIM2 clusters in the puncta area as indicated in the magnified images. F: line scan graph (left) showing distribution and fluorescent intensity of STIM2 along the white dashed lines of the region indicated in b. Summarized data (means ± SE; right) showing the changes in fluorescent density of YFP-STIM2 before and after 1.8 mM extracellular Ca2+ treatment for 10 min in CASR/STIM2-co-transfected HEK293 cells; n = 5 independent sets of transfected cells. We specifically selected regions of interest (ROIs) mostly close to the plasma membrane (i.e., the peripheral region of the cell) for analyzing the size, number, and intensity of STIM2 clusters. The summarized data (B, D, and F) show the overall intensity of the STIM2 clusters located in the peripheral regions of the cells. **P < 0.01 and ***P < 0.001 vs. control (Student’s t test).
Activation of Notch attenuates KCNA5/KV1.5 and KCNA2/KV1.2 channel activity.
To further determine the impact of Notch signaling in physiological or pathophysiological cell functions, we examined the effect of Notch activation on Kv1.5/KCNA5 channel activity using patch clamp techniques. HEK293 cells transfected with KCNA5 construct showed significant whole cell K+ currents through KCNA5 channels. Notch activation by Jag-1 significantly attenuated whole cell KCNA5 currents in a time-dependent manner (Fig. 7, A and B). The amplitudes of whole cell KCNA5 currents, elicited by a series of test potentials ranging from −80 mV to +80 mV (for 250 ms) from a holding potential of −70 mV, were reduced by >70% in Jag-1-treated cells (for 45 min) compared with control cells (Fig. 7, A and B). The Jag-1-mediated inhibition of KCNA5 currents or the Jag-1-sensitive KCNA5 currents (Fig. 7C) seemed to start from the test potential of −40 mV (see Fig. 7C, inset). However, the activating kinetics of KCNA5 channels was not significantly affected by Jag-1-mediated Notch activation (Fig. 7D). The time course of the Jag-1-mediated reduction of whole cell KCNA5 currents showed that it took 10–15 min for the currents to decrease during extracellular application of Jag-1 (Fig. 7E); the delayed inhibitory effect of Jag-1 or decrease in currents implied that 1) the Jag-1-mediated effect on KCNA5 channels was due to release of the Notch intracellular domain (NICD) and/or 2) there were too many KCNA5 channels in the cells due to overexpression or transfection. Although there was a delay in Jag-1-mediated decrease in KCNA5 currents (Fig. 7E), the results imply that activation of Notch signaling per se is able to functionally affect Kv1.5 channel activity without changing the expression of KCNA5 channels.
Fig. 7.

Notch activation or calcium-sensing receptor (CaSR) activation attenuates Kv1.5 channel activity in potassium voltage-gated channel 5 (KCNA5)-transfected and KCNA5/CASR-cotransfected human embryonic kidney 293 (HEK293) cells. A: representative records showing superimposed whole cell Kv1.5/KCNA5 currents, elicited by depolarizing the cells from a holding potential of −70 mV to a series of test potentials ranging from −80 to + 80 mV (in 20 mV increments) in KCNA5-transfected HEK293 cells before (control) and after 15, 30, and 45 min of extracellular application of 50 μM Jagged1 (Jag-1). B: summarized data (means ± SE) showing the current-voltage (I-V) relationship curves constructed from current recordings in HEK293 cells transfected with KCNA5 constructs before (control), and after 15, 30, and 45 min of extracellular application of 50 μM Jag-1; n = 3. P < 0.01 between control curve and Jag-1 (15 min) curve; P < 0.001 between control curve and Jag-1 (30 min) and Jag-1 (45 min) curves. C: representative record (left) and the I-V curve (inset) of Jag-1-sensitive KCNA5 currents, which were obtained by subtracting the currents recorded in Jag-1 (45 min) cells from the currents recorded in control cells. D: representative currents, elicited by depolarizing the cell from a holding potential to +80 mV in control (blue) and Jag-1-treated cells. E: data showing the mean time courses of changes in normalized whole cell KCNA5 currents measured at +80 mV in HEK293 cells transfected with KCNA5 construct before and during extracellular application of 50 μM Jag-1. F: Western blot analysis on KCNA5/Kv1.5 in control cells and cells transfected with an empty vector (Vector) or the KCNA5 gene (KCNA5). G: representative current records showing changes in whole cell K+ currents in KCNA5-transfected HEK293 cells before (0 Ca) and during extracellular application of 1.8 mM Ca2+ without (1.8 Ca) or with (1.8 Ca + 4-AP) the addition of a non-specific KV channel antagonist 4-aminopyridine (4-AP; 1 mM). H: summarized data (means ± SE) showing the current-voltage (I-V) relationship curves constructed from current recordings in HEK293 cells transfected with KCNA5 constructs before (0 Ca) and during extracellular application of 1.8 mM Ca2+ without (1.8Ca) or with (1.8Ca + 4-AP) the addition of 4-AP; n = 5 cells for each group. P < 0.01 between the 1.8Ca + 4-AP curve and the curves of 0 Ca and 1.8 Ca (ANOVA). I: representative current records showing changes in whole cell K+ currents in KCNA5/CaSR-cotransfected HEK293 cells before (0 Ca) and during extracellular application of 1.8 mM Ca2+ without (1.8 Ca) or with (1.8Ca + U73122) the addition of a specific phospholipase C inhibitor U73122 (10 μM). J: summarized data (means ± SE) showing the current-voltage (I-V) relationship curves constructed from current recordings in HEK293 cells co-transfected with KCNA5/CASR constructs before (0 Ca) and during extracellular application of 1.8 mM Ca2+ without (1.8 Ca) or with (1.8Ca + U73122) the addition of U73122; n = 5 cells for each group. P < 0.01 between the 1.8Ca curve and the curves of 0Ca and 1.8Ca + U73122 (ANOVA).
Increased protein level of Kv1.5/KCNA5 channels in KCNA5-transfected cells (Fig. 7F) indicate that KCNA5 constructs were successfully transfected into cells, and the observed whole cell K+ currents are KV1.5/KCNA5 currents. To determine whether extracellular Ca2+ affects KV1.5 channel function, we measured channel activity in the absence (0Ca) of extracellular Ca2+ or the presence of 1.8 mM Ca2+. As shown in Fig. 7, G and H, the extracellular Ca2+ produced little to no effect on KV1.5 channel activity in KCNA5-transfected HEK293 cells compared with Ca2+-free (0Ca) conditions. 4-Aminopyridine (4-AP; 1mM), a specific KV channel blocker, in the presence of 1.8 mM Ca2+ significantly reduced the currents (Fig. 7, G and H). 4-AP-sensitive current is considered as currents through KV channels.
It is reported that GPCR stimulation suppresses KV1.5 channel activity via the tyrosine-protein kinase Src pathway (3, 39). To study whether extracellular Ca2+-mediated CaSR activation and/or CaSR-mediated Ca2+ influx are able to inhibit Kv1.5 channel activity, we measured whole cell KCNA5 current in HEK293 cells cotransfected with both KCNA5 and CASR construct (KCNA5 + CASR). Here, in cotransfected cells, we observed the similar whole cell KCNA5 currents in the absence of Ca2+. Activation of CaSR by adding extracellular Ca2+ significantly reduced the amplitude of whole cell KCNA5 currents (Fig. 7I and J). U73122, a PLC specific inhibitor, efficiently restored the whole cell KCNA5 currents in the presence of extracellular Ca2+ to the level detected in the Ca2+-free condition (Fig. 7, I and J). These data suggest that extracellular Ca2+-mediated activation of CaSR functionally inhibits Kv1.5 channels via a CaSR/G protein/PLC pathway. However, it is unclear how Notch activation exerts the functional inhibitory effect on KV1.5 channel activity. Our observations from this study indicate a potential possibility: Notch activation may exert an inhibiting effect on KV1.5 channel activity by way of regulating G protein and/or its downstream effectors via NICD.
To examine whether the inhibitory effect of Jag-1 on KV channels was specific to KCNA5/KV1.5 channels, we also examined the effect of Jag-1 on whole cell K+ currents in HEK293 cells transfected with the KCNA2 gene (Fig. 8). Similar to its effect on KCNA5 currents (Fig. 7, A–E), short-term Jag-1 treatment (50 μM for 20 min) significantly and reversibly decreased whole cell K+ currents through KCNA2/KV1.2 channels (Fig. 8, A–C). Jag-1 caused a 34% reduction of KCNA2 currents at +80 mV (Fig. 8B), and the Jag-1-mediated reduction of KCNA2/KV1.2 was more prominent at positive potentials (Fig. 8C). The current-voltage relationship of the Jag-1-sensitive KCNA2 currents (Fig. 8D) was similar to that of Jag-1-sensitive KCNA5 currents (Fig. 7C). However, the KCNA2 channels’ activation, inactivation, and deactivation kinetics seemed not to be affected by Jag-1 treatment by comparing the normalized currents at +80 mV before (control), during (Jag-1), and after (wash) application of Jag-1 (Fig. 8E). These data indicate that short-term treatment with Jag-1 significantly decreases the amplitude of whole cell K+ currents through either KCNA5/KV1.5 or KCNA2/KV1.2 channels but appears to have little effect on the current kinetics.
Fig. 8.
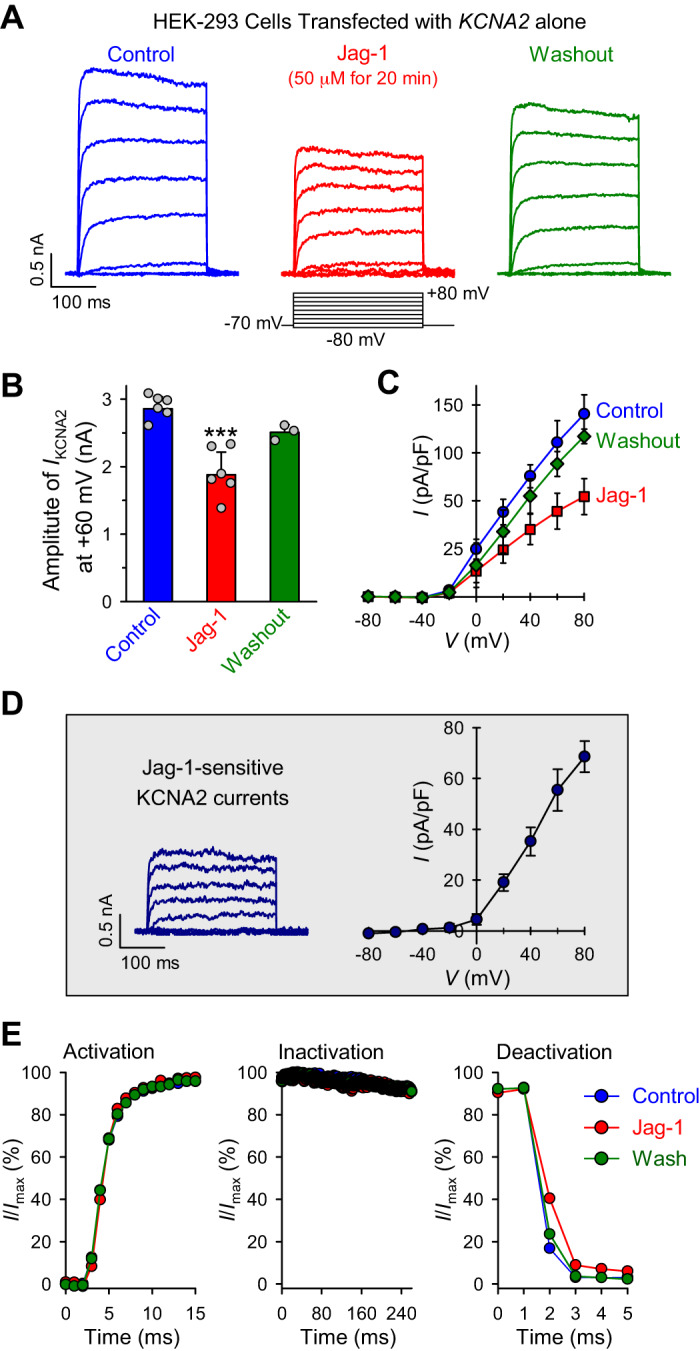
Jagged1 (Jag-1) reduces whole cell K+ currents in potassium voltage-gated channel 2 (KCNA2)-transfected human embryonic kidney 293 (HEK293) cells. A: representative records showing superimposed whole cell KV1.2/KCNA2 currents, elicited by depolarizing the cells from a holding potential of −70 mV to a series of test potentials ranging from −80 to + 80 mV (in 20-mV increments), in KCNA2-transfected HEK293 cells before (control), during (Jag-1), and after (washout) extracellular application of 50 μM Jag-1 for 20 min. B: summarized data (means ± SE; n = 8 cells) showing the amplitudes of KV1.2/KCNA2 currents at +60 mV in KCNA2-transfected cells before (control), during (Jag-1), and after (washout) Jag-1 treatment (50 μM for 20 min). ***P < 0.001 vs. control and washout (ANOVA). C: summarized data (means ± SE) showing the current-voltage (I-V) relationship curves constructed from current density in KCNA2-transfected cells before (control), during (Jag-1), and after (washout) Jag-1 treatment; P < 0.01 between control curve and Jag-1 curve (ANOVA). D: representative record (left) and the I-V curve (right) of Jag-1-sensitive KV1.2/KCNA2 currents, which were obtained by subtracting the currents recorded in KCNA2-transfected cells during Jag-1 (20 min) treatment from the currents recorded in the cells before Jag-1 treatment. E: normalized currents (I/Imax) at +80 mV (constructed from the current recordings in A) showing the activation (left), inactivation (middle), and deactivation (right) kinetics of the currents recorded before (control; blue), during (Jag-1; red), and after (wash; green) Jag-1 treatment in KCNA2-transfected cells.
DISCUSSION
Using CaSR- and CaSR/STIM2-overexpressed HEK293 cells, we found in this study that 1) Notch activation by Jag-1 or STIM2 overexpression enhances extracellular Ca2+-mediated increase in [Ca2+]cyt by activation of CaSR; 2) CaSR stimulation by extracellular Ca2+ or Notch stimulation by Jag-1 induces clustering of STIM2; and 3) Notch activation or CaSR activation inhibits KCNA5/Kv1.5 and KCNA2/KV1.2 channel activity. These observations suggest that activation of Notch functionally enhances CaSR-mediated increases in [Ca2+]cyt by promoting STIM2 clustering and, subsequently, augmenting SOCE. The inhibitory effect of Notch activation on KV1.5/KV1.2 channels indicates that Notch or Notch signaling can exert multiple effects on different receptors and/or ion channels. The study models have also allowed us to understand the precise cellular and molecular mechanisms of CaSR-associated Ca2+ influx and provide an insight into the noncanonical impact of Notch signaling in the regulation of ion channel functions involved in cell physiological and pathophysiological processes.
Oligomerization and clustering of STIM proteins (STIM1/STIM2) in the SR/ER membrane and translocation to the SR/ER-PM junctions when Ca2+ in SR/ER are depleted or decreased have been demonstrated to be the trigger to recruit Orai channels in the plasma membrane, form SOC, and induce SOCE (12, 29). It has been reported that STIM proteins also functionally interact with transient receptor potential canonical (TRPC) channels, resulting in SOCE (1, 12, 26), whereas TRPC is also reported to physically and functionally interact with Orai1 channels (23). Furthermore, TRPC channels have been established to form both ROC and SOC in various types of cells (4, 26). In addition, downregulation of TRPC6 significantly attenuates SOCE induced by active or passive depletion of Ca2+ from the SR/ER, whereas overexpression of TRPC6 enhances SOCE induced by SR/ER depletion (12). These data imply that there is a potential functional interaction between TRPC channels and the STIM- and Orai-mediated store-coupling process. The STIM-mediated SR/ER-PM junctions may coordinate activation of multiple proteins in organized loci (41).
Recently, Guo et al. (15) showed that Notch signaling positively regulates CaSR function and expression in hypoxia-induced PH. Although it is still unknown, we speculate that the cytosol NICD may functionally interact with STIM2 to induce or facilitate their clustering in response to CaSR-mediated store depletion. We previously reported that the Notch activation enhances SOCE, and Notch activation-released cytosolic NICD forms protein-protein interaction with TRPC6 in human PASMC (32). These data suggest that, in addition to the canonical long-term regulation of Notch-dependent gene expression, Notch activation or NICD may also functionally exert short-term effect on ion channel functions. Functional interaction of NICD with TRPC6 (and Orai channels) to facilitate its opening may be another possible mechanism by which Notch activation enhances CaSR-induced Ca2+ influx.
STIM2 is an SR/ER-resident Ca2+ sensor with low Ca2+ affinity that can sense small changes or decreases in the SR/ER [Ca2+]. Small fluctuations of the SR/ER [Ca2+] would cause STIM2 oligomerization, clustering, and translocation to the SR/ER-PM junction to trigger SOCE (6, 35). It is thus possible that NICD may lead to the decrease of [Ca2+] in the SR/ER, which subsequently activates STIM2 to induce SOCE. Therefore, there are two plausible mechanisms by which Notch activation induces STIM2 clustering and enhances SOCE: 1) NICD directly interacts with STIM2 to promote its clustering, and 2) NICD decreases the SR/ER [Ca2+] to indirectly induce STIM2 clustering. Each of these pathways would facilitate Ca2+ influx through SOC and, therefore, enhance CaSR-mediated increase in [Ca2+]cyt.
Furthermore, the extracellular domain of Notch receptor consists largely of up to 36 tandemly repeated epidermal growth factor (EGF) modules. The structural integrity of these modules in the extracellular domain and the heterodimeric stability of the Notch receptor are dependent on the presence of extracellular Ca2+ (in millimolar range). The extracellular Ca2+ is required, for example, for maintaining the stability of the noncovalent association of the Notch extracellular domain (NECD) with the transmembrane domain (TM) (30). Removal or chelation of extracellular Ca2+ disrupts the NECD/TM noncovalent association. The S2 and S3 cleavage during acute activation of Notch, or binding of Jag-1 to the Notch receptors, would release the bound Ca2+ from the NECD EGF modules to activate colocalized CaSR in the plasma membrane and facilitate CaSR-associated Ca2+ influx and release. It is also possible that the cleaved NICD containing Ca2+-bound EFG modules can function as a facilitator (or activator) of CaSR to sensitize CaSR to its extracellular ligands.
In this study, we also observed that Notch activation by Jag-1 or CaSR activation by extracellular Ca2+ attenuates KV1.5/KV1.2 channel activity. Previous studies have reported that activation of GPCR suppressed Kv1.5/KV1.2 channel activity via the tyrosine-protein kinase Src pathway (3, 39). Moreover, new reports suggest that activation of canonical Notch signaling decreases function and expression of KV channels (including KV1.5/KV1.2) in mouse cardiac myocytes through transcriptional and epigenetic changes (20). Here, we propose that Notch activation or NICD, either indirectly exerting an effect on GPCR (i.e., CaSR) and/or its downstream effectors (e.g., PIP2) or directly interacting with KV1.5/KV1.2 channel proteins, can functionally regulate Kv channel activity without changing its expression.
In this study, we used HEK293 cells transiently transfected with CaSR, STIM2, and KCNA5 (or KCNA2) to study functional interactions among these proteins. One of the limitations in the study is that receptors and ion channels may function differently in different cell models. The mechanism revealed in the overexpression cell models may not be exactly the same as in fully differentiated cells like pulmonary arterial smooth muscle and endothelial cells. The HEK293 cell transfection system is an efficient investigation cell model for characterizing the function of an individual protein and the interaction of two or multiple proteins. However, the overexpression cell model cannot mimic physiological conditions due to its high protein expression level and relatively simple cytosol microenvironment. In the future, we will try to optimize the transfection process in primary cells and characterize the functional effects of Notch activation on CaSR-mediated Ca2+ signaling and KV1.5/KV1.2 channel activity in pulmonary vascular smooth muscle cells.
In conclusion, extracellular Ca2+-mediated activation of CaSR increases [Ca2+]cyt by inducing Ca2+ release from IP3-sensitive stores and Ca2+ influx through SOC and ROC. Enhancement (function or expression) of the components of each Ca2+ signaling processes, such as STIM2 of SOC and TRPC6 of ROC, would augment CaSR-mediated increase in [Ca2+]cyt. Here, we demonstrate a conceptual coupling model between CaSR and Notch, in which Notch activation enhances CaSR-induced SOCE by promoting clustering of STIM2. The results from this study also provide strong evidence that Notch signaling can functionally regulate its effectors, in addition to the canonical regulation of transcription of Notch-dependent genes. The “noncanonical” functional effect of Jag-1-mediated Notch activation on Ca2+ channels and K+ channels gives Notch signaling a new definition on its regulating role in cell functions.
GRANTS
This work was supported in part by the grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL-135807 and HL-146764).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S., A.B., A.M., and J.X.-J.Y. conceived and designed research; S.S., A.B., T.Z., R.J.A., M.R., A.M., and J.X.-J.Y. performed experiments; S.S., A.B., T.Z., R.J.A., M.R., S.R., F.B., A.H., J.Y.-J.S., P.A.T., A.M., and J.X.-J.Y. analyzed data; S.S., A.B., T.Z., R.J.A., M.R., S.R., F.B., A.H., J.Y.-J.S., P.A.T., A.M., and J.X.-J.Y. interpreted results of experiments; S.S., A.B., T.Z., R.J.A., M.R., S.R., F.B., A.H., A.M., and J.X.-J.Y. prepared figures; S.S., A.B., and J.X.-J.Y. drafted manuscript; S.S., A.B., T.Z., R.J.A., F.B., A.H., J.Y.-J.S., P.A.T., A.M., and J.X.-J.Y. edited and revised manuscript; S.S., A.B., T.Z., R.J.A., M.R., S.R., F.B., A.H., J.Y.-J.S., P.A.T., A.M., and J.X.-J.Y. approved final version of manuscript.
REFERENCES
- 1.Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol 583: 25–36, 2007. doi: 10.1113/jphysiol.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development 138: 3593–3612, 2011. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- 3.Benians A, Leaney JL, Tinker A. Agonist unbinding from receptor dictates the nature of deactivation kinetics of G protein-gated K+ channels. Proc Natl Acad Sci USA 100: 6239–6244, 2003. doi: 10.1073/pnas.1037595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berna-Erro A, Galan C, Dionisio N, Gomez LJ, Salido GM, Rosado JA. Capacitative and non-capacitative signaling complexes in human platelets. Biochim Biophys Acta 1823: 1242–1251, 2012. doi: 10.1016/j.bbamcr.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 6.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131: 1327–1339, 2007. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366: 575–580, 1993. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 8.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81: 239–297, 2001. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 9.Chow JY, Estrema C, Orneles T, Dong X, Barrett KE, Dong H. Calcium-sensing receptor modulates extracellular Ca2+ entry via TRPC-encoded receptor-operated channels in human aortic smooth muscle cells. Am J Physiol Cell Physiol 301: C461–C468, 2011. doi: 10.1152/ajpcell.00389.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933–936, 1998. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 11.Fan Z, Liu B, Zhang S, Liu H, Li Y, Wang D, Liu Y, Li J, Wang N, Liu Y, Zhang B. YM155, a selective survivin inhibitor, reverses chronic hypoxic pulmonary hypertension in rats via upregulating voltage-gated potassium channels. Clin Exp Hypertens 37: 381–387, 2015. doi: 10.3109/10641963.2014.987390. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol 308: C581–C593, 2015. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galambos C, Minic AD, Bush D, Nguyen D, Dodson B, Seedorf G, Abman SH. Increased lung expression of anti-angiogenic factors in down syndrome: potential role in abnormal lung vascular growth and the risk for pulmonary hypertension. PLoS One 11: e0159005, 2016. doi: 10.1371/journal.pone.0159005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardenal E, Chiarini A, Armato U, Dal Prà I, Verkhratsky A, Rodríguez JJ. Increased Calcium-sensing receptor immunoreactivity in the hippocampus of a triple transgenic mouse model of alzheimer’s disease. Front Neurosci 11: 81, 2017. doi: 10.3389/fnins.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Q, Xu H, Yang X, Zhao D, Liu S, Sun X, Huang JA. Notch activation of Ca2+-sensing receptor mediates hypoxia-induced pulmonary hypertension. Hypertens Res 40: 117–129, 2017. doi: 10.1038/hr.2016.118. [DOI] [PubMed] [Google Scholar]
- 16.Handlogten ME, Huang C, Shiraishi N, Awata H, Miller RT. The Ca2+-sensing receptor activates cytosolic phospholipase A2 via a Gqα-dependent ERK-independent pathway. J Biol Chem 276: 13941–13948, 2001. doi: 10.1074/jbc.M007306200. [DOI] [PubMed] [Google Scholar]
- 17.Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4: 530–538, 2003. doi: 10.1038/nrm1154. [DOI] [PubMed] [Google Scholar]
- 18.Hu F, Pan L, Zhang K, Xing F, Wang X, Lee I, Zhang X, Xu J. Elevation of extracellular Ca2+ induces store-operated calcium entry via calcium-sensing receptors: a pathway contributes to the proliferation of osteoblasts. PLoS One 9: e107217, 2014. doi: 10.1371/journal.pone.0107217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob R, Merritt JE, Hallam TJ, Rink TJ. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature 335: 40–45, 1988. doi: 10.1038/335040a0. [DOI] [PubMed] [Google Scholar]
- 20.Khandekar A, Springer S, Wang W, Hicks S, Weinheimer C, Diaz-Trelles R, Nerbonne JM, Rentschler S. Notch-mediated epigenetic regulation of voltage-gated potassium currents. Circ Res 119: 1324–1338, 2016. doi: 10.1161/CIRCRESAHA.116.309877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JW, Park HA, Kwon OK, Park JW, Lee G, Lee HJ, Lee SJ, Oh SR, Ahn KS. NPS 2143, a selective calcium-sensing receptor antagonist inhibits lipopolysaccharide-induced pulmonary inflammation. Mol Immunol 90: 150–157, 2017. doi: 10.1016/j.molimm.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289–1297, 2009. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA 105: 2895–2900, 2008. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacMillan D, McCarron JG. The phospholipase C inhibitor U-73122 inhibits Ca2+ release from the intracellular sarcoplasmic reticulum Ca2+ store by inhibiting Ca2+ pumps in smooth muscle. Br J Pharmacol 160: 1295–1301, 2010. doi: 10.1111/j.1476-5381.2010.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moudgil R, Michelakis ED, Archer SL. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13: 615–632, 2006. doi: 10.1080/10739680600930222. [DOI] [PubMed] [Google Scholar]
- 26.Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR. TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J Physiol 587: 2429–2442, 2009. doi: 10.1113/jphysiol.2009.172254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S, Ambudkar IS. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci Signal 8: ra3, 2015. doi: 10.1126/scisignal.2005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136: 876–890, 2009. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature 443: 230–233, 2006. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 30.Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster JC. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol 20: 1825–1835, 2000. doi: 10.1128/MCB.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O’connor DT, Yuan JX. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C1837–C1853, 2007. doi: 10.1152/ajpcell.00405.2006. [DOI] [PubMed] [Google Scholar]
- 32.Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated Ca entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song S, Carr SG, McDermott KM, Rodriguez M, Babicheva A, Balistrieri A, Ayon RJ, Wang J, Makino A, Yuan JX. STIM2 (stromal interaction molecule 2)-mediated increase in resting cytosolic free Ca2+ concentration stimulates PASMC proliferation in pulmonary arterial hypertension. Hypertension 71: 518–529, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song S, Li J, Zhu L, Cai L, Xu Q, Ling C, Su Y, Hu Q. Irregular Ca2+ oscillations regulate transcription via cumulative spike duration and spike amplitude. J Biol Chem 287: 40246–40255, 2012. doi: 10.1074/jbc.M112.417154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song S, Yamamura A, Yamamura H, Ayon RJ, Smith KA, Tang H, Makino A, Yuan JX. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am J Physiol Cell Physiol 307: C373–C383, 2014. doi: 10.1152/ajpcell.00115.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135: 110–122, 2008. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Takesono A, Nowak MW, Cismowski M, Duzic E, Lanier SM. Activator of G-protein signaling 1 blocks GIRK channel activation by a G-protein-coupled receptor: apparent disruption of receptor signaling complexes. J Biol Chem 277: 13827–13830, 2002. doi: 10.1074/jbc.M201064200. [DOI] [PubMed] [Google Scholar]
- 40.Tang H, Yamamura A, Yamamura H, Song S, Fraidenburg DR, Chen J, Gu Y, Pohl NM, Zhou T, Jiménez-Pérez L, Ayon RJ, Desai AA, Goltzman D, Rischard F, Khalpey Z, Black SM, Garcia JG, Makino A, Yuan JX. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 310: L846–L859, 2016. doi: 10.1152/ajplung.00050.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Deng X, Hewavitharana T, Soboloff J, Gill DL. Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin Exp Pharmacol Physiol 35: 1127–1133, 2008. doi: 10.1111/j.1440-1681.2008.05018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med 7: 612–618, 2001. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 43.Xiao R, Su Y, Feng T, Sun M, Liu B, Zhang J, Lu Y, Li J, Wang T, Zhu L, Hu Q. Monocrotaline induces endothelial injury and pulmonary hypertension by targeting the extracellular calcium-sensing receptor. J Am Heart Assoc 6: e004854, 2017. doi: 10.1161/JAHA.116.004865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca2+-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012. doi: 10.1161/CIRCRESAHA.112.266361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca -activated Cl channels contributes to regulating receptor- and store-operated Ca entry in human pulmonary artery smooth muscle cells. Pulm Circ 1: 269–279, 2011. doi: 10.4103/2045-8932.83447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A, Powell FL, Thistlethwaite PA, Yuan JX. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca2+ entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 306: C871–C878, 2014. doi: 10.1152/ajpcell.00221.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yarova PL, Stewart AL, Sathish V, Britt RD Jr, Thompson MA, P Lowe AP, Freeman M, Aravamudan B, Kita H, Brennan SC, Schepelmann M, Davies T, Yung S, Cholisoh Z, Kidd EJ, Ford WR, Broadley KJ, Rietdorf K, Chang W, Bin Khayat ME, Ward DT, Corrigan CJ, T Ward JP, Kemp PJ, Pabelick CM, Prakash YS, Riccardi D. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci Transl Med 7: 284ra60, 2015. doi: 10.1126/scitranslmed.aaa0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye C, Rogers K, Bai M, Quinn SJ, Brown EM, Vassilev PM. Agonists of the Ca2+-sensing receptor (CaR) activate nonselective cation channels in HEK293 cells stably transfected with the human CaR. Biochem Biophys Res Commun 226: 572–579, 1996. doi: 10.1006/bbrc.1996.1396. [DOI] [PubMed] [Google Scholar]
- 49.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98: 1400–1406, 1998. doi: 10.1161/01.CIR.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 50.Zeng X, Zhu L, Xiao R, Liu B, Sun M, Liu F, Hao Q, Lu Y, Zhang J, Li J, Wang T, Wei X, Hu Q. Hypoxia-induced mitogenic factor acts as a nonclassical ligand of calcium-sensing receptor, therapeutically exploitable for intermittent hypoxia-induced pulmonary hypertension. Hypertension 69: 844–854, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08743. [DOI] [PubMed] [Google Scholar]