

Abstract
During pregnancy, placental vascular growth, which is essential for supporting the rapidly growing fetus, is associated with marked elevations in blood flow. These vascular changes take place under chronic physiological low O2 (less than 2–8% O2 in human; chronic physiological normoxia, CPN) throughout pregnancy. O2 level below CPN pertinent to the placenta results in placental hypoxia. Such hypoxia can cause severe endothelial dysfunction, which is associated with adverse pregnancy outcomes (e.g., preeclampsia) and high risk of adult-onset cardiovascular diseases in children born to these pregnancy complications. However, our current knowledge about the mechanisms underlying fetoplacental endothelial function is derived primarily from cell models established under atmospheric O2 (~21% O2 at sea level, hyperoxia). Recent evidence has shown that fetoplacental endothelial cells cultured under CPN have distinct gene expression profiles and cellular responses compared with cells cultured under chronic hyperoxia. These data indicate the critical roles of CPN in programming fetal endothelial function and prompt us to re-examine the mechanisms governing fetoplacental endothelial function under CPN. Better understanding these mechanisms will facilitate us to develop preventive and therapeutic strategies for endothelial dysfunction-associated diseases (e.g., preeclampsia). This review will provide a brief summary on the impacts of CPN on endothelial function and its underlying mechanisms with a focus on fetoplacental endothelial cells.
Keywords: endothelial cells, hyperoxia, hypoxia, oxygen, physiological low oxygen
INTRODUCTION
Oxygen (O2) is essential to drive oxidative phosphorylation for generating energy in mitochondria in all eukaryotic organisms, including humans. Under physiological conditions, following partial O2 pressure (Po2) gradients, global O2 delivery to cells depends on cardiac output and arterial O2 content, the latter of which is tightly controlled by O2 uptake and O2 carrying capacity (9, 24, 47).
Under physiological conditions, vascular endothelial cells and most other types of cells in the human body reside in low-O2 environments in vivo, relative to atmospheric O2 (~21% O2 or 160 mmHg Po2 at sea level; Fig. 1; Refs. 9, 24, 47). These physiological low O2 levels (physiological normoxia), ranging from 13.5% O2 (or 100 mmHg Po2) in the pulmonary venous blood to 3–5% O2 (or 20–40 mmHg Po2) in many other tissues (4, 47), are considered normoxic to the cells living in these tissues (17). O2 deficiency (hypoxia) or excess (hyperoxia) in any given tissue can cause adverse cellular consequences, leading to the deleterious effects on human health (10, 75, 97). Indeed, disruption of the O2 homeostasis is associated with cardiovascular disease, cerebrovascular disease, chronic lung disease, atherosclerosis, and restenosis (23, 24, 33). Endothelial dysfunction (e.g., impaired endothelial growth and vasodilator production) is one of the hallmarks of cardiovascular diseases (5, 31) and has also been linked with other human diseases including preeclampsia, intrauterine growth retardation, diabetic retinopathy, rheumatoid arthritis, and tumor growth and metastasis (5, 6, 23, 24, 31). Under these pathological conditions, endothelial cells within the relevant tissues, such as placentas from women with preeclamptic pregnancies, could be exposed to severe hypoxia (<1% O2; Fig. 1; Refs. 5, 6, 24, 26, 31, 60). However, the vast majority of our current knowledge on controlling endothelial function has been derived from the endothelial cell models established under atmospheric O2 (47, 99). This atmospheric O2 level, for most mammalian cells in vivo, is extremely hyperoxic and toxic to cells possibly because of the increased generation of reactive oxygen species (ROS; Refs. 10, 20). In addition, prolonged culture of cells under atmospheric O2 programs surviving cells to adapt to this pathological hyperoxic condition. Thus it is critical to reveal endothelial responses and underlying mechanisms under appropriate chronic physiological normoxia (CPN), which will aid us in developing optimal therapies for preventing and treating endothelial dysfunction-associated diseases during pregnancy (e.g., preeclampsia, intrauterine growth retardation, and chorioangioma). Recently, a comprehensive review has addressed the importance of physiological normoxia in cellular function and biological processes (47). In the current review, we will primarily discuss the impact of CPN on fetoplacental endothelial cells including human umbilical vein endothelial cells (HUVECs) and human umbilical arterial endothelial cells (HUAECs), the former of which are widely used in human endothelial biology, especially for fetoplacental endothelial cells. We propose that functional and transcriptional phenotypes of fetoplacental endothelial cells cultured constantly under CPN immediately after isolation should much more closely reflect their in vivo state than cells derived from hyperoxic culture.
Fig. 1.

Oxygen gradients at sea level in maternal and fetal tissues during pregnancy. NT, normotensive pregnancies; PE, preeclampsia; Umb., umbilical cord.
PHYSIOLOGICAL NORMOXIA IN PLACENTAS AND EMBRYOS
During pregnancy, there are marked elevations of placental blood flow in association with dramatic vascular growth, which is essential to support rapid growth of the fetus (29, 40, 65, 79). Human placental vasculogenesis and angiogenesis take place under physiological normoxia during normal pregnancy (29, 40, 65). Before 10 wk of gestation, extravillous trophoblast cells invade into the decidua, occluding uterine spiral arterioles and preventing blood flow into the intervillous space (37). The villous membrane during this period is approximately five times thicker than that in the terminal villi at term, which leads to a higher resistance for O2 diffusion. This restricted blood flow into the intervillous space, together with a higher O2 diffusion distance, results in a low-oxygen environment (approximately 1.5–3.3% O2 at up to 8–10 wk of gestation) in placentas. Embryos and placentas in early first trimester are especially vulnerable to ROS due to underdeveloped placental antioxidant defense systems (9). As such, physiological normoxia within the early placental tissue may not only promote embryonic survival and growth, but also enhance trophoblast cell invasion (29, 40, 78). Between 8 and 10 wk of gestation, as the trophoblast plugs loosen and maternal blood flow to the intervillous space surges, placental O2 levels elevate to ~8% O2; however, by the end of third trimester, placental O2 levels decrease to ~6% as the demand for O2 and O2 extraction from the placenta increase with rapid fetal growth (29, 40).
The physiological normoxia is critical to placental development and angiogenesis (9, 11, 78). Indeed, previous studies have demonstrated that physiological normoxia (2–3% O2) promotes outgrowth and proliferation of the extravillous trophoblast in placental explants obtained from the first trimester human placenta compared with 21% O2 (11, 30). In contrast, hypoxia (~1.5% O2) inhibits such outgrowth and proliferation (39). In addition, decreased perfusion and O2 delivery into the intervillous space induced by immature spiral artery remodeling could lead to severe placental hypoxia (10, 31). This severe placental hypoxia can induce excessive release of ROS, cytokines, and antiangiogenic factors, resulting in pregnancy complications, such as preeclampsia and miscarriage (10, 31). However, although preeclampsia may not change placental vascular densities compared with normotensive pregnancy (55, 62), it clearly impairs fetoplacental endothelial function such as increased cell permeability, defective calcium responses, and nitric oxide (NO) production (8). All of these data indicate that any disruption of O2 homeostasis could adversely affect placental growth and vascular function.
Evidence from high-altitude pregnancy studies further highlights the significant impacts of chronic hypoxia on the placental vasculature in vivo as placental villous vascularization is increased at high-altitude pregnancy in European women compared with those living at low altitudes or sea level in the United States (61, 92, 101). Specifically, increased vascularization in high-altitude pregnancies is associated with a lower Po2 in the uterine artery and intervillous space, decreased antioxidant activity, and lower oxidative stress in placentas (93, 100, 101). Studies of high-altitude pregnancies from different ancestries (i.e., Andean vs. European) indicate that genetic and epigenetic factors contribute to greater placental efficiency in oxygen transport in Andean pregnancies (3, 10, 24, 101).
In the human fetus, O2 levels are even lower than those in the placenta. For instance, the mean O2 concentrations in the umbilical vein and umbilical artery are ~3.7% (28 mmHg Po2) and 2.5% O2 (19 mmHg Po2), respectively, at the end of the third trimester (65). The O2 level in embryos ranges from 2 to 9% in vivo (87). Before establishment of the vascular system and formation of placentas to transport O2 and nutrients from maternal to fetal circulation, mammalian embryos reside in an extremely low-O2 environment (2). This physiological normoxia stimulates the differentiation of early mesoderm into hemangioblasts and is necessary for maintaining the pluripotency of embryonic stem cells (26, 87). Thus the placenta and fetus grow and function under CPN throughout the entire pregnancy.
Detrimental effects of hyperoxia on embryos and oocytes are well documented in mammalian species including human, rabbit, sheep, and cow (6, 51, 91). Exposure of mouse embryos to 21% O2 consistently reduces the blastulation rate (defined as percentage of zygotes that develop into the blastocyst stage), possibly due to decreased cell growth (96). Superoxide dismutase, an enzyme that defends against the harmful effects of ROS in cells, improves the blastulation rate of mice under 21% O2, indicating that ROS damage occurs under 21% O2 (96). Such hyperoxic exposure may be permanently harmful to embryonic development as subsequent treatment of embryos with 5% O2 and/or superoxide dismutase cannot improve or reverse the reduced blastulation rate (96).
REGULATION OF HYPOXIA-INDUCIBLE FACTORS
Cells sense acute fluctuation of oxygen tension through a family of oxygen-sensitive transcriptional factors: hypoxia-inducible factors (HIFs; Refs. 46, 85). HIF-1α is ubiquitously expressed in many tissues including placentas (31, 56), whereas HIF-2α and HIF-3α are restrictedly expressed in certain cells such as endothelial cells and renal interstitial cells (7). It is believed that HIF-1α and HIF-2α differentially regulate a battery of genes in various cell types in response to hypoxia. For instance, HIF-1α, but not HIF-2α, mediates the transcription of the majority of genes in response to hypoxia in both endothelial and breast cancer cells, whereas HIF-2α regulates the gene transcription in renal carcinoma cells (89).
Similar to the fluctuation of O2 levels in human placentas during pregnancy (Fig. 1), expression of both HIF-1α and HIF-2α proteins is high in early pregnancy and decreased significantly afterward (77). Expression of HIF-1α and HIF-2α proteins is aberrantly elevated in term placentas from preeclampsia compared with normal pregnancy (11, 77), possibly due to further decreases in placental O2 levels in preeclampsia (Fig. 1). Besides O2 availability, HIF-1α protein expression can be regulated by other factors including estrogen, angiotensin II, interleukin-1β, transforming growth factor-β1/3, tumor necrosis factor-α, and insulin-like growth factor (11, 45, 71, 104). Thus abnormal expression of these factors could also contribute to elevated increased HIF-1α and HIF-2α protein expression in preeclampsia.
Both HIF-1α and HIF-2α are required for early vascular development during the embryonic stage (38, 48, 72, 81). This is evidenced by the fact that targeted disruption of either HIF-1α or HIF-2α gene in the mouse causes impaired vascular development and embryonic lethality (38, 48, 72, 81). However, HIF-1α and HIF-2α have unique physiological roles (75). For instance, HIF-1α is required for angiogenesis, whereas HIF-2α is critical for vascular remodeling in the embryonic stage (75). HIF-1α and HIF-2α are also key modulators for normal placental development as supported by reports that increased HIF-1α and/or HIF-2α expression in human placental tissues is associated with preeclampsia and fetal growth restriction (11, 29, 76, 78, 88).
Although global knockout of HIF-1α reveals its essential roles in vascular development, conditional deletion of HIF-1α in endothelial cells delays, but does not inhibit, angiogenesis in wound healing (90). Unlike global HIF-1α knockout mice, the mice with endothelial cell-specific deletion of HIF-1α are viable and developed normally (90), suggesting that endothelial HIF-1α is not essential in endothelial cell growth during embryonic vasculogenesis and angiogenesis.
PHYSIOLOGICAL NORMOXIA REGULATION OF ANGIOGENESIS IN VITRO
Proangiogenesis of Physiological Normoxia and Hypoxia
Angiogenesis, a process of de novo formation of blood vessels from pre-existing ones, is tightly regulated in several crucial steps including endothelial proliferation, migration, tube formation, and vessel maturation by numerous humoral factors including angiogenic factors and angiogenic inhibitors (e.g., angiostatin, endostatin, and thrombospondins; Ref. 12). Hypoxia is also a major stimulator of angiogenesis, which is generally thought to be mediated via increasing expression of angiogenic factors and their receptors (75, 85).
Fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor A (VEGFA) are two potent angiogenic factors that stimulate endothelial function on binding and activating their receptors containing tyrosine kinases (27, 59, 74, 99). Currently, there are four major fibroblast growth factor receptors (FGFRs) identified in humans: FGFR1, FGFR2, FGFR3, and FGFR4 (70). The expression of FGFRs seems to be tissue-specific as only FGFR1 and FGFR3 have been found in the chorionic villi of human placenta (35). FGFR1 mediates FGF2 function and is the major FGFR expressing in endothelial cells (95), including human umbilical vein endothelial cells (HUVECs) and human umbilical arterial endothelial cells (HUAECs; Refs. 42, 43). Vascular endothelial growth factor receptor-1 (VEGFR1) and receptor-2 (VEGFR2) are two key receptors responsible for VEGFA’s actions. VEGFR2 is the major signal transducer of VEGFA in endothelial cells and mediates most known VEGFA bioactivities (e.g., cell proliferation, migration, and permeability; Ref. 27). Both VEGFR1 and 2 are critical for regulating vasculogenesis and angiogenesis during embryonic development, since null mutation of either receptor in the mouse results in abnormal vascular formation and development, leading to embryonic death (28, 86).
FGF2- and VEGFA-induced cellular responses are mediated via activating a complex signaling network that includes mitogen-activated protein kinase kinase 1/2 (MEK1/2)/ERK1/2 and phosphatidylinositol-3 kinase (PI3K)/v-akt murine thymoma viral oncogene homolog 1 (Akt1) pathways (20, 74, 99), both of which are key signaling pathways mediating endothelial functions (66, 99).
Acute hypoxia (3–5% O2, 4–120 h) has been shown to promote major steps of angiogenesis, including cell proliferation in rat, porcine, and/or bovine aortic endothelial cells (36, 54, 64, 82, 94) and formation of capillary-like tube structures in bovine pulmonary microvascular endothelial cells (73) in vitro compared with hyperoxic (21%) O2. Similarly, stimulatory effects of physiological normoxia or hypoxia on angiogenesis are also observed in human endothelial cells from different types of blood vessels (Table 1; Refs. 41–43, 49, 57, 67, 83, 98, 102, 106, 107). Many of these human endothelial cells are derived from fetoplacental blood vessels such as placental arteries and umbilical cord veins (Table 1), the latter of which are one of most widely used cell models for fetoplacental cells. For instance, pre-exposing primary human placental artery endothelial cells (hPAECs) to 3% O2 for 48 h enhances FGF2- and VEGFA-stimulated cell proliferation compared with atmospheric O2 (Ref. 98; Table 1). Interestingly, these physiological normoxia-primed hPAECs are more sensitive to FGF2 and VEGFA stimulation as the minimum concentrations of FGF2 and VEGFA that stimulate cell proliferation under physiological normoxia are much lower (≤10-fold) than those under atmospheric O2.
Table 1.
Effects of oxygen on angiogenic activities of human endothelial cells in vitro*
| Vessel Origins | O2 Used to Establish Cell Cultures | O2 Treatment | Cellular Responses to O2 Changes | Ref. |
|---|---|---|---|---|
| Umbilical vein and adult dermal microvasculature | ~21% | 4.6%, ≤2 days | Cell division ↑; VEGFA mRNA/protein and VEGFR2 phosphorylation ↑ | 67 |
| Umbilical vein | ~21% | 2%, ≤2 days | Gene and protein expression (microarray and proteomics): 3,996 differentially expressed genes | 83 |
| Placental artery | ~21% | 3%, 2 days | FGF2- and VEGFA-stimulated cell proliferation ↑; FGF2- and VEGFA-induced activation of Akt1 and ERK1/2 ↔ | 98 |
| Umbilical vein | ~21% | 5%, ≤3 days | Cell proliferation ↑; extracellular matrix secretion, intercellular adhesion, NO/PGI2 production ↑ | 102 |
| Umbilical vein and artery | 3 and 21%, ≤25 days | 3 and 21%, ≤5 days | Gene expression (microarray): 41 genes ↑ and 21 genes ↓ in vein endothelial cells; 74 genes ↑ and 86 genes ↓ in artery endothelial cells; cell surface area ↓; HIF-1α protein ↑; VEGFA-induced VEGFR2 activation ↑; expression of FGF2, VEGFA, and their major receptors ↔; FGF2-induced FGFR1 activation ↔; FGF2- and VEGFA-stimulated cell proliferation and migration ↑; FGF2- and VEGFA-induced Akt1 and ERK1/2 ↑; switching O2 levels rapidly (3 to 21% O2 or 21 to 3% O2 for 1 day) attenuates O2-programmed cell responses but does not completely diminish such programming for ≤5 days | 42, 43 |
| Umbilical vein | 3 and 21%, ≤25 days | 3 and 21%, ≤4 days | FGF2- and VEGFA-stimulated cell proliferation and migration ↑; HIF-1α does not mediate FGF2- and VEGFA-stimulated CPN cell proliferation and migration | 41 |
| Umbilical vein | ~21% | 5%, 1–4 days | Gene expression (microarray): proliferation ↓; induction of antioxidant defense proteins by electrophilic agents ↓; cell ultrastructure, viability, adhesion, basal redox status, or HIF-1α protein levels ↔ | 15 |
| Umbilical vein | 3%, ≤25 days | 3%, ≤4 days | FGF2- and VEGFA-stimulated cell migration and proliferation ↑; G protein α-subunits 11 and 14 mediate FGF2- and/or VEGFA-stimulated cell migration, proliferation, and/or permeability in part via PLC-3β | 106, 107 |
| Saphenous vein | 20% | 5%, ≤14 days | Cell growth ↓; antioxidant activities ↓ | 53 |
| Foreskin microvasculature | 20% | 1%, 3–4 days | FGF2/TNF-α- and VEGFA/TNF-α- induced tube formation ↑; VEGFA expression ↔ | 49 |
| Dermal microvasculature | 5 and 21% | 5 vs. 21%, ≤3 days | Cell proliferation ↑; FGF2-stimulated cell proliferation ↔; PMA-stimulated cell proliferation ↑; IL-1β-inhibited cell proliferation | 105 |
| Type IV collagen and CD31 ↓; tube formation ↓ | ||||
| Pulmonary artery | 21% | 1%, 1 day | Gene expression (microarray): 245 genes ↑ and 325 ↓ genes; cell migration and capillary tube formation ↑ | 57 |
| Coronary artery | ~21% | 1%, 1 day | VEGFA-stimulated endothelial cell proliferation, migration, and tube formation ↓; VEGFA-stimulated activation of eNOS and NO production ↓; VEGFA-induced activation of VEGFR2, Akt, ERK1/2, p38, p70S6 kinases, and S6 ribosomal protein ↓ | 69 |
Inhibitory effects of physiological normoxia or hypoxia on human endothelial angiogenesis are in boldface.
↑, Increase; ↓, decrease; ↔, no change; Akt1, v-akt murine thymoma viral oncogene homolog 1; CPN, chronic physiological normoxia; eNOS, endothelial nitric oxide synthase; FGFR1, fibroblast growth factor receptor-1; HIF-1α, hypoxia-inducible factor-1α; PGI2, prostacyclin; redox, oxidation-reduction; VEGFR2, vascular endothelial growth factor receptor-2.
Although proangiogenic activities are well documented in the literature, physiological normoxia- or hypoxia-induced antiangiogenic activities have also been described in a few human endothelial cell cultures (Table 1, in boldface; Refs. 15, 53, 69, 105). These contradictory effects on angiogenic activities do not appear to depend on the origin of endothelial cells since either pro- (41, 43, 67, 102) or antiangiogenic (15) activities induced by physiological normoxia or hypoxia have been reported in HUVECs. To date, what causes these discrepancies remains unknown; however, different cell preparation procedures (e.g., cell isolation procedure, cell passage number, and/or culture condition) could be contributing factors.
It is noteworthy that the majority of primary human endothelial cells listed in Table 1 are originally derived under atmospheric O2 (~21% O2; hyperoxia, also termed standard culture condition). Since atmospheric O2 is a hyperoxic condition that most endothelial cells might never be exposed to in vivo, the phenotypes of these cells obviously do not fully represent their in vivo states. To address this issue, we have isolated paired HUVECs and HUAECs from the same umbilical cord and steadily cultured them under CPN (3% O2, ≤25 days) or chronic hyperoxia (CH; 21% O2, ≤25 days) immediately after cell isolation (42, 43). Like CH cells, these CPN cells maintain classic endothelial phenotypes (e.g., a cobblestone morphology, positive 1,1′-dioctadecyl-1,3,3,3′,3′-tetramethylindocarbocyanine perchlorate acetylated-LDL uptake, CD31 positive, and formation of capillary-like tube structures). However, compared with CH, CPN decreases cell surface areas (~25% in HUVECs and HUAECs) after attachment, indicating that CPN cells are less spread out in culture, which may be a morphological adaptation of fetal endothelial cells to different O2 levels without known function and mechanisms.
Beside its actions on morphological adaptations of HUVECs and HUAECs, CPN also alters cellular functional phenotypes. In this regard, CPN further promotes FGF2- and VEGFA-stimulated cell proliferation (approximately 3- to 4-fold) and migration (approximately 1- to 2-fold) in HUVECs and HUAECs compared with CH, whereas CPN does not significantly affect the formation of capillary-like tube structures (42, 43). These data suggest that CPN and CH may program specific but not all behaviors of human fetoplacental endothelial cells. In addition, these CPN- and CH-programmed cellular responses to FGF2 and VEGFA are relatively stable, as altering culture O2 levels even up to 5 days fails to completely diminish these chronic O2 programmed cell responses in HUVECs (43). This supports the notion that acutely changing O2 levels is not sufficient to fully reverse cellular responses. Similar responses to reversing O2 levels are also observed in HUAECs (42). With the observation that it takes much less time to alter the CPN-enhanced cell migration than the CPN-enhanced cell proliferation (43), fetoplacental endothelial migration responses appear to be more sensitive to O2 changes than proliferation. Further studies are needed to dissect the mechanisms underlying such disparities.
Transcriptional Regulation
Chi et al. (18) have used human microarrays to analyze global gene expression in 53 human endothelial cell cultures (presumably all established under atmospheric O2) including 2 HUVEC preparations. They revealed that endothelial cells of different origins (e.g., microvasculature, arteries, and veins) have distinct and characteristic gene expression profiles. However, little is known about whether and how different O2 levels differentially regulate the gene expression profiles and function between fetoplacental artery and vein endothelial cells (1, 22, 80).
Several studies have analyzed physiological normoxia- or hypoxia-regulated gene profiles in endothelial cells using human microarrays. All of these studies using cells derived under hyperoxia suggest that acute hypoxic exposure alters gene expression profiles, which are capable of altering endothelial angiogenic activities. Acute hypoxia (1% O2 for 3 h) robustly induces ~112 transcripts in HUVECs, and many transcripts are involved in biological processes, such as proliferation, angiogenesis, and vasodilation (56). Exposure of primary HUVECs to 1% O2 for ≤48 h also upregulates expression of diverse genes (e.g., VEGFA, VEGFR1, and IGF-binding protein 3) in HUVECs (83). Acute hypoxia (1% O2, 8 and/or 24 h) elevates expression of VEGFA and placental growth factor (PlGF) in hPAECs (57), whereas it induces transcripts including heat shock factors, glycolytic enzymes, extracellular matrix factors, cytoskeletal factors, apoptotic factors, cell cycle regulators, and angiogenic factors in human aortic endothelial cells (68). Unlike those previous studies on acute hypoxic exposure (56, 68, 83), we have focused on determining the CPN-induced transcriptional profile changes in HUVECs and HUAECs (42, 43) using paired CPN and CH cell preparations that derived from the same vein or artery. Results from these studies revealed that CPN induces dramatic changes in cell cycle, cell death, cellular movement, and cellular development-related genes in HUVECs and HUVECs in association with stimulation of fetal angiogenesis (42, 43). Importantly, these CPN-altered gene expression profiles in HUVECs diverge from those induced by acute physiological normoxia (2% O2, ≤48 h) in HUVECs that are derived under atmospheric O2 (83). In particular, out of 62 differentially expressed (DE) genes identified in CPN HUVECs (43), only 6 are identical to those induced by the acute 2% O2 treatment (83). Moreover, in contrast to this previous report (83), CPN does not promote mRNA expression of FGF2, VEGFA, FGFR1, VEGFR1, and VEGFR2 in HUVECs (43). Similarly, CPN fails to alter the FGF2, FGFR1, VEGFR1, or VEGFR2 mRNA levels in HUAECs, whereas it only slightly increases VEGFA mRNA expression (42). Although caution should be taken when comparing microarray data among studies using different sample preparations, microarray platforms, and data analysis methods, it is clear that CPN induces distinctive transcriptional profiles in endothelial cells, suggesting that HUVECs and HUAECs generated under CPN are unique cell models for studies of fetoplacental endothelial cell function. These studies also suggest that the CPN-enhanced angiogenic responses in human placental endothelial cells are not mediated primarily via elevating expression of FGF2 and VEGFA and their major receptors.
Recently, we have re-analyzed the previously published microarray data set on CPN and CH HUVECs and HUAECs (National Center for Biotechnology Information Gene Expression Omnibus: GSE49958) based on the sexes and origins (vein or artery) of cells (Fig. 2; Supplemental Table S1; all supplemental material is available at https://doi.org/10.6084/m9.figshare.10325657). Despite the small sample size (n = 2–3 for each sex of CPN and CH HUVECs), 1 DE gene [potassium voltage-gated channel subfamily A member regulatory β-subunit 1 (KCNAB1); ~20-fold increases] was identified between female and male HUVECs under CPN. KCNAB1 is a hypertension-associated gene (19, 63) that may play a critical role in human placental vascular (50) and cardiac function (25). Additionally, sex-specific gene expression in response to different O2 levels was detected between HUVECs and HUAECs (Fig. 2). Specifically, CPN dramatically shifted the gene expression differences between HUVECs and HUAECs in a sex-specific manner. There were more CPN-specific DE genes between HUVECs and HUAECs in female (185 genes) than male (21 genes) cells (Fig. 2A), whereas there were fewer CH-specific DE genes between HUVECs and HUAECs in female (40 genes) than male (110 genes) cells (Fig. 2B). The number of DE genes between HUVECs and HUAECs that were not affected by different culture O2 levels was larger in female (131 genes) versus male (50 genes) cells. Endothelin-1 and nitric oxide, both of which can be released by vascular endothelium, are important regulators of blood pressure and vascular tone (34, 58). The canonical pathway enrichment analysis reveals that genes in endothelin-1 signaling pathway are more significantly differentially expressed between HUVECs and HUVECs under CPN in male than in female cells (Fig. 2, C and D). In contrast, genes in nitric oxide signaling pathway are significantly differentially expressed between HUVECs and HUAECs under CPN in male but not in female cells (Fig. 2, C and E). Moreover, as female and male HUVECs cultured under CH exhibit considerably distinct transcriptomic profiles and cellular responses to cytokines and growth factors (103), it is expected that CPN should have similarly sexually dimorphic actions on HUVECs and HUAECs.
Fig. 2.

Effects of chronic physiological normoxia (CPN) on differentially expressed genes between human umbilical vein endothelial cells (HUVECs) and human umbilical arterial endothelial cells (HUAECs) from male and female fetuses. Venn diagrams show differentially expressed genes between HUVECs and HUAECs in female (A) and male (B) cells cultured under CPN and chronic hyperoxia (CH); bar chart (C) shows canonical pathways that are enriched in differentially expressed (DE) genes between HUVECs and HUAECs in female and male cells cultured under CPN and CH; heat map shows endothelin-1 (D) and nitric oxide signaling cardiovascular system (E) pathway-associated DE genes between HUVECs and HUAECs in female and male cells cultured under CPN and CH. Data set from the previously published microarray analysis (National Center for Biotechnology Information Gene Expression Omnibus: GSE49958) was re-analyzed based on sex and vessel origins of cells. n = 2 For CPN female HUVECs; n = 3 for all other groups per sex.
Collectively, these data further support the importance of culturing cells under chronic physiological O2 levels, although different physiological O2 levels may differentially regulate endothelial responses in transcriptome and function, especially when one considers the influence of cell sex.
Signaling Regulation
Among numerous signaling pathways that mediate angiogenic responses to FGF2 and VEGFA, MEK1/2/ERK1/2 and PI3K/Akt1 are two major placental endothelial function-associated pathways (16, 99). Accumulating evidence indicates that acute physiological normoxia and hypoxia alone can elevate basal activation of ERK1/2 and Akt in different cell types, including endothelial cells (13, 14, 21, 44, 66, 82). Specifically, acute hypoxic exposure (1% O2, ≤6 h) rapidly (≤30 min) activates the Akt/mammalian target of rapamycin pathway in rat aortic endothelial cells (54). Additionally, acute physiological normoxic exposure (<5% O2, within 1 h) increases the phosphorylation and activity of ERK1/2 in HUVECs, human microvascular endothelial cells, and porcine aorta endothelial cells (14, 32, 66, 82). Acute hypoxia exposure (<0.1% O2, 24 h) could induce the basal activation of ERK1/2 via a hypoxia-driven VEGF/VEGFR1 and FGF2 autocrine loop that interacts with HIF-1α (21).
Bioinformatics analysis (Ingenuity Pathway Analysis tools) of CPN-induced DE genes in HUVECs also predicts that the MEK1/2/ERK1/2 and PI3K/Akt1 signaling pathways are located in the centers of gene interaction networks (43), implicating the importance of these two signaling pathways in CPN-programmed cellular function in fetoplacental endothelial cells. Biochemical analysis further confirms that CPN indeed enhances FGF2- and VEGFA-induced ERK1/2 activation and VEGFA-induced Akt1 activation in HUVECs in association with CPN-enhanced FGF2- and VEGFA-stimulated cell proliferation and VEGFA-stimulated migration (43). This CPN-enlarged activation of ERK1/2 and Akt1 observed in HUVECs is in contrast to the previous report that acute exposure (24 h) of CH hPAECs to 3% O2 does not affect either FGF2- and VEGFA-induced ERK1/2 or Akt1 activation (98). These observations further confirm the unique characteristics of CPN cells and imply that the MEK1/2/ERK1/2 and PI3K/Akt1 pathways mediate the CPN-enhanced fetoplacental endothelial angiogenic activities. In addition, recent evidence has also shown that G proteins may mediate FGF2- and VEGFA-stimulated cell function in HUVECs under CPN via phospholipase C-3β (PLC-3β; Refs. 106, 107).
O2 Regulation of Activation of VEGF Receptors and FGF Receptors
Little is known about effects of different O2 levels on expression and activation of receptors for VEGFA and FGF2 in endothelial cells. Acute hypoxia (≤2% O2 for ≤48 h) has been shown to upregulate expression of VEGFR1, but not VEGFR2 (30, 83), whereas it increases heparin sulfate proteoglycans-FGF2 binding sites, but not FGFR1 in HUVECs (52). Similarly, CPN also does not upregulate VEGFR2 and FGFR1 in HUVECs and HUAECs (42, 43). Hence, CPN-enhanced VEGFA- and FGF2-stimulated angiogenic responses do not appear to be mediated via increasing expression of these major receptors for FGF2 and VEGFA. However, CPN may modulate activation of these receptors on stimulation by their ligands as CPN can significantly enhance VEGFR2 activation and moderately enhance the FGFR1 activation as suggested (43).
HIF-1α Transcriptional Activity and Upregulation of Proangiogenic Genes
Numerous studies have demonstrated the importance of HIFs in regulating cell function in different types of cells after exposure to acute low O2 (2–5% O2; 4–120 h; Refs. 47, 75, 84). One function of HIFs is to upregulate an array of proangiogenic genes, such as FGF2, VEGFA, PlGF, angiopoietins 1 and 2, and VEGFR1, in endothelial cells and other nonendothelial types of cells (84, 85). Specifically, both constitutive expression of active HIF-1α and hypoxia treatment (1% O2, 24 h) upregulate many cell growth-associated genes, including VEGFA and PlGF, but without altering expression of major receptors for VEGF and FGF2 in hPAECs (57). However, little is known about the role of HIFs in FGF2- and VEGFA-regulated endothelial function under CPN.
Recently, it has been reported that knockdown of HIF-1α in CPN HUVECs does not affect FGF2- and VEGFA-stimulated angiogenic responses (proliferation and migration), whereas HIF-1α overexpression in CH HUVECs enhances these angiogenic responses (41). These data indicate that HIF-1α alone may not significantly regulate angiogenic responses in CPN endothelial cells. This conclusion is supported by our re-examination of the CPN-induced DE genes in HUVECs (43) in which only a few (~9.7%) of the DE genes are found to be direct targets of HIF-1α (41). Together with the previous study reporting that mice with endothelial cell-specific HIF-1α deletion develop normally and have no significant defects in angiogenesis (90), endothelial HIF-1α seems not to be an essential regulator for fetal endothelial growth during embryonic vasculogenesis and angiogenesis.
CONCLUSIONS
In conclusion, convincing evidence has shown that chronic physiological normoxia further enhances fetoplacental endothelial angiogenic activity in association with extremely different gene profiles (Fig. 3). Thus chronic physiological normoxia instead of acute low-O2 exposure should be recognized as a critical factor when culturing and studying endothelial cells and perhaps most other types of cells (e.g., vascular smooth muscle cells, trophoblast cells, and embryonic stem cells) as well. Chronic physiological normoxia-derived endothelial cells represent a unique endothelial cell model that more closely mimics the in vivo microenvironment under physiological states. To date, although much progress has been made, we still face many tough challenges in deciphering cellular and molecular mechanisms underlying fetoplacental endothelial function under chronic physiological normoxia and possibly more importantly under chronic hypoxia occurring in pregnancy complications such as preeclampsia and intrauterine growth restriction. In particular, since O2 levels vary dynamically in vivo, exactly what O2 levels should we consider physiological normoxia/hypoxia and use to establish and maintain endothelial cell culture in vitro? Whether differentially expressed genes identified in chronic physiological normoxia-derived cells affect cellular responses to angiogenic stimuli? What other signaling pathways are involved in mediating fetoplacental endothelial function under chronic physiological normoxia? Does and how the sex of cells affect cellular behaviors under chronic physiological normoxia? Additionally, although being the most popular cell model for studying human endothelial cell biology, most HUVECs reported in the literature are isolated from term deliveries. Thus it is likely that the cellular response of these cells might only reflect the end point of fetal endothelial adaptations to either normal or complicated pregnancies. These unanswered questions warrant further investigations. Better understanding of the mechanisms controlling endothelial function under chronic physiological normoxia will aid us to develop optimal diagnostic or intervention strategies for endothelial dysfunction-associated cardiovascular disorders such as preeclampsia, hypertension, and stroke.
Fig. 3.
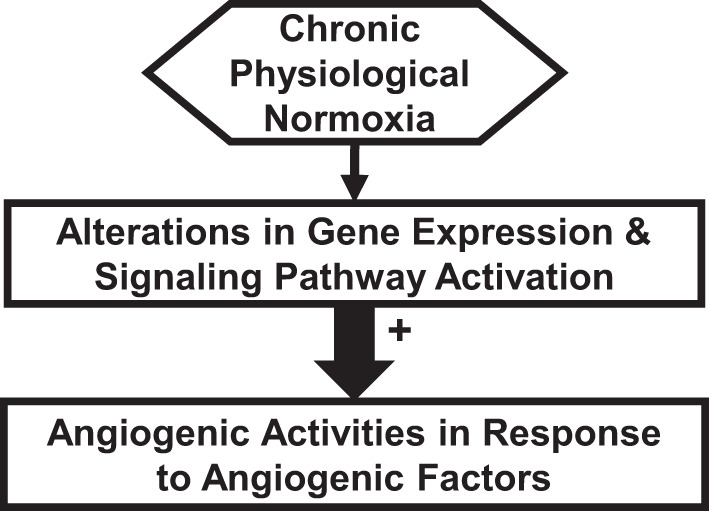
Schematic of proposed mechanisms underlying chronic physiological normoxia (CPN) regulation in fetoplacental endothelial cells. In these mechanisms, we propose that CPN induces significantly distinct gene expression profiles and activation of diverse signaling pathways (e.g., mitogen-activated protein kinase kinase/ERK1/2 and phosphatidylinositol-3 kinase/v-akt murine thymoma viral oncogene homolog 1) compared with chronic hyperoxia. These changes further promote angiogenic activities of fetoplacental endothelial cells in response to angiogenic factors (e.g., VEGFA and FGF2) under CPN.
GRANTS
This study was supported by the US National Institute of Child Health and Human Development Grants P01-HD-38843 (to J. Zheng), P01-HD-038843-14S1 (to J. Zheng), and R03-HD-100778 (to C. Zhou) as well as American Heart Association Awards 17POST33670283 and 19CDA34660348 (to C. Zhou). The project was also supported by a Translational Basic & Clinical Research Pilot Award (to J. Zheng and C. Zhou) from the University of Wisconsin Institute for Clinical and Translational Research and the Clinical and Translational Science Award program, through the NIH National Center for Advancing Translational Sciences Grant UL1TR002373.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.Z. and Q.-Y.Z. prepared figures; C.Z., Q.-Y.Z., Y.-Z.J., and J.Z. drafted manuscript; C.Z., Q.-Y.Z., Y.-Z.J., and J.Z. edited and revised manuscript; C.Z., Q.-Y.Z., Y.-Z.J., and J.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank our collaborators and colleagues (Drs. Kai Wang, Dong-bao Chen, Ian M. Bird, Ronald R. Magness, Christina Kendziorski, Shi-An Huang, Wei Lei, Yan Li, and Cai-feng Dai) for their valuable contributions to this work. We thank Lori Uttech-Hanson, a grant writer from the Office of Research Administration and Proposal Development, University of Wisconsin-Madison, for English editing.
REFERENCES
- 1.Aitsebaomo J, Portbury AL, Schisler JC, Patterson C. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circ Res 103: 929–939, 2008. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akazawa S, Unterman T, Metzger BE. Glucose metabolism in separated embryos and investing membranes during organogenesis in the rat. Metabolism 43: 830–835, 1994. doi: 10.1016/0026-0495(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 3.Bennett A, Sain SR, Vargas E, Moore LG. Evidence that parent-of-origin affects birth-weight reductions at high altitude. Am J Hum Biol 20: 592–597, 2008. doi: 10.1002/ajhb.20784. [DOI] [PubMed] [Google Scholar]
- 4.Benzing H, Lösse B, Schuchhardt S, Niederle N. Simultaneous measurement of regional blood flow and oxygen pressure in the dog myocardium during coronary occlusion or hypoxic hypoxia. Adv Exp Med Biol 37A: 541–546, 1973. doi: 10.1007/978-1-4684-3288-6_69. [DOI] [PubMed] [Google Scholar]
- 5.Berk BC, Haendeler J, Sottile J. Angiotensin II, atherosclerosis, and aortic aneurysms. J Clin Invest 105: 1525–1526, 2000. doi: 10.1172/JCI9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernardi ML, Fléchon JE, Delouis C. Influence of culture system and oxygen tension on the development of ovine zygotes matured and fertilized in vitro. J Reprod Fertil 106: 161–167, 1996. doi: 10.1530/jrf.0.1060161. [DOI] [PubMed] [Google Scholar]
- 7.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer 8: 967–975, 2008. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boeldt DS, Bird IM. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J Endocrinol 232: R27–R44, 2017. doi: 10.1530/JOE-16-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton GJ. Oxygen, the Janus gas; its effects on human placental development and function. J Anat 215: 27–35, 2009. doi: 10.1111/j.1469-7580.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev 96: 1509–1565, 2016. doi: 10.1152/physrev.00029.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caniggia I, Winter J, Lye SJ, Post M. Oxygen and placental development during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta 21, Suppl A: S25–S30, 2000. doi: 10.1053/plac.1999.0522. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 6: 389–395, 2000. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 13.Carrera S, de Verdier PJ, Khan Z, Zhao B, Mahale A, Bowman KJ, Zainol M, Jones GD, Lee SW, Aaronson SA, Macip S. Protection of cells in physiological oxygen tensions against DNA damage-induced apoptosis. J Biol Chem 285: 13658–13665, 2010. doi: 10.1074/jbc.M109.062562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casanello P, Torres A, Sanhueza F, González M, Farías M, Gallardo V, Pastor-Anglada M, San Martín R, Sobrevia L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ Res 97: 16–24, 2005. doi: 10.1161/01.RES.0000172568.49367.f8. [DOI] [PubMed] [Google Scholar]
- 15.Chapple SJ, Keeley TP, Mastronicola D, Arno M, Vizcay-Barrena G, Fleck R, Siow RCM, Mann GE. Bach1 differentially regulates distinct Nrf2-dependent genes in human venous and coronary artery endothelial cells adapted to physiological oxygen levels. Free Radic Biol Med 92: 152–162, 2016. doi: 10.1016/j.freeradbiomed.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Chen DB, Zheng J. Regulation of placental angiogenesis. Microcirculation 21: 15–25, 2014. doi: 10.1111/micc.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR. Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Front Cell Dev Biol 6: 132, 2018. doi: 10.3389/fcell.2018.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA 100: 10623–10628, 2003. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu YF, Chung RH, Lee CY, Kao HY, Hou L, Hsu FC. Identification of rare variants for hypertension with incorporation of linkage information. BMC Proc 8, Suppl 1: S109, 2014. doi: 10.1186/1753-6561-8-S1-S109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danilov CA, Fiskum G. Hyperoxia promotes astrocyte cell death after oxygen and glucose deprivation. Glia 56: 801–808, 2008. doi: 10.1002/glia.20655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das B, Yeger H, Tsuchida R, Torkin R, Gee MF, Thorner PS, Shibuya M, Malkin D, Baruchel S. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1α through mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 pathway in neuroblastoma. Cancer Res 65: 7267–7275, 2005. doi: 10.1158/0008-5472.CAN-04-4575. [DOI] [PubMed] [Google Scholar]
- 22.dela Paz NG, D’Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res 335: 5–16, 2009. doi: 10.1007/s00441-008-0706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8: 425–437, 2008. [Erratum in Nat Rev Cancer 8: 654, 2008.] doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ducsay CA, Goyal R, Pearce WJ, Wilson S, Hu XQ, Zhang L. Gestational hypoxia and developmental plasticity. Physiol Rev 98: 1241–1334, 2018. doi: 10.1152/physrev.00043.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.England SK, Uebele VN, Shear H, Kodali J, Bennett PB, Tamkun MM. Characterization of a voltage-gated K+ channel β subunit expressed in human heart. Proc Natl Acad Sci USA 92: 6309–6313, 1995. doi: 10.1073/pnas.92.14.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 102: 4783–4788, 2005. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 9: 669–676, 2003. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 28.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376: 66–70, 1995. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 29.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 277: 1669–1672, 1997. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 30.Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 272: 23659–23667, 1997. doi: 10.1074/jbc.272.38.23659. [DOI] [PubMed] [Google Scholar]
- 31.Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension 38: 718–722, 2001. doi: 10.1161/01.HYP.38.3.718. [DOI] [PubMed] [Google Scholar]
- 32.Härtel FV, Holl M, Arshad M, Aslam M, Gündüz D, Weyand M, Micoogullari M, Abdallah Y, Piper HM, Noll T. Transient hypoxia induces ERK-dependent anti-apoptotic cell survival in endothelial cells. Am J Physiol Cell Physiol 298: C1501–C1509, 2010. doi: 10.1152/ajpcell.00333.2009. [DOI] [PubMed] [Google Scholar]
- 33.Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med 24: 165–169, 2014. doi: 10.1016/j.tcm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Houde M, Desbiens L, D’Orléans-Juste P. Endothelin-1: biosynthesis, signaling and vasoreactivity. Adv Pharmacol 77: 143–175, 2016. doi: 10.1016/bs.apha.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem 45: 1005–1019, 1997. doi: 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- 36.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J 16: 771–780, 2002. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 37.Hustin J, Schaaps JP. Echographic and anatomic studies of the maternotrophoblastic border during the first trimester of pregnancy. Am J Obstet Gynecol 157: 162–168, 1987. doi: 10.1016/S0002-9378(87)80371-X. [DOI] [PubMed] [Google Scholar]
- 38.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12: 149–162, 1998. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James JL, Stone PR, Chamley LW. The effects of oxygen concentration and gestational age on extravillous trophoblast outgrowth in a human first trimester villous explant model. Hum Reprod 21: 2699–2705, 2006. doi: 10.1093/humrep/del212. [DOI] [PubMed] [Google Scholar]
- 40.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol 157: 2111–2122, 2000. doi: 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang YZ, Li Y, Wang K, Dai CF, Huang SA, Chen DB, Zheng J. Distinct roles of HIF1A in endothelial adaptations to physiological and ambient oxygen. Mol Cell Endocrinol 391: 60–67, 2014. doi: 10.1016/j.mce.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Enhanced cellular responses and distinct gene profiles in human fetoplacental artery endothelial cells under chronic low oxygen. Biol Reprod 89: 133, 2013. doi: 10.1095/biolreprod.113.110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Transcriptional and functional adaptations of human endothelial cells to physiological chronic low oxygen. Biol Reprod 88: 114, 2013. doi: 10.1095/biolreprod.113.108225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin N, Hatton N, Swartz DR, Xia X, Harrington MA, Larsen SH, Rhoades RA. Hypoxia activates jun-N-terminal kinase, extracellular signal-regulated protein kinase, and p38 kinase in pulmonary arteries. Am J Respir Cell Mol Biol 23: 593–601, 2000. doi: 10.1165/ajrcmb.23.5.3921. [DOI] [PubMed] [Google Scholar]
- 45.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL‐1β mediated up‐regulation of HIF‐lα via an NFκB/COX‐2 pathway identifies HIF‐1 as a critical link between inflammation and oncogenesis. FASEB J 17: 1–22, 2003. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 46.Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402, 2008. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Keeley TP, Mann GE. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol Rev 99: 161–234, 2019. doi: 10.1152/physrev.00041.2017. [DOI] [PubMed] [Google Scholar]
- 48.Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol 209: 254–267, 1999. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 49.Kroon ME, Koolwijk P, van der Vecht B, van Hinsbergh VW. Urokinase receptor expression on human microvascular endothelial cells is increased by hypoxia: implications for capillary-like tube formation in a fibrin matrix. Blood 96: 2775–2783, 2000. doi: 10.1182/blood.V96.8.2775. [DOI] [PubMed] [Google Scholar]
- 50.Lang I, Schweizer A, Hiden U, Ghaffari-Tabrizi N, Hagendorfer G, Bilban M, Pabst MA, Korgun ET, Dohr G, Desoye G. Human fetal placental endothelial cells have a mature arterial and a juvenile venous phenotype with adipogenic and osteogenic differentiation potential. Differentiation 76: 1031–1043, 2008. doi: 10.1111/j.1432-0436.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- 51.Li J, Foote RH. Culture of rabbit zygotes into blastocysts in protein-free medium with one to twenty per cent oxygen. J Reprod Fertil 98: 163–167, 1993. doi: 10.1530/jrf.0.0980163. [DOI] [PubMed] [Google Scholar]
- 52.Li J, Shworak NW, Simons M. Increased responsiveness of hypoxic endothelial cells to FGF2 is mediated by HIF-1alpha-dependent regulation of enzymes involved in synthesis of heparan sulfate FGF2-binding sites. J Cell Sci 115: 1951–1959, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Li RK, Shaikh N, Weisel R, Tumiati L, Wu TW, Mickle D. Cultured vascular endothelial cell susceptibility to extracellularly generated oxidant injury. J Mol Cell Cardiol 24: 595–604, 1992. doi: 10.1016/0022-2828(92)91044-6. [DOI] [PubMed] [Google Scholar]
- 54.Li W, Petrimpol M, Molle KD, Hall MN, Battegay EJ, Humar R. Hypoxia-induced endothelial proliferation requires both mTORC1 and mTORC2. Circ Res 100: 79–87, 2007. doi: 10.1161/01.RES.0000253094.03023.3f. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Zhao YJ, Zou QY, Zhang K, Wu YM, Zhou C, Wang K, Zheng J. Preeclampsia does not alter vascular growth and expression of CD31 and vascular endothelial cadherin in human placentas. J Histochem Cytochem 63: 22–31, 2015. doi: 10.1369/0022155414558063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang GP, Su YY, Chen J, Yang ZC, Liu YS, Luo XD. Analysis of the early adaptive response of endothelial cells to hypoxia via a long serial analysis of gene expression. Biochem Biophys Res Commun 384: 415–419, 2009. doi: 10.1016/j.bbrc.2009.04.160. [DOI] [PubMed] [Google Scholar]
- 57.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669, 2005. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 58.Martínez-Ruiz A, Lamas S. Two decades of new concepts in nitric oxide signaling: from the discovery of a gas messenger to the mediation of nonenzymatic posttranslational modifications. IUBMB Life 61: 91–98, 2009. doi: 10.1002/iub.144. [DOI] [PubMed] [Google Scholar]
- 59.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE 2001: re21, 2001. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 60.Matsuo K, Malinow AM, Harman CR, Baschat AA. Decreased placental oxygenation capacity in pre-eclampsia: clinical application of a novel index of placental function preformed at the time of delivery. J Perinat Med 37: 657–661, 2009. doi: 10.1515/JPM.2009.121. [DOI] [PubMed] [Google Scholar]
- 61.Mayhew TM. Changes in fetal capillaries during preplacental hypoxia: growth, shape remodelling and villous capillarization in placentae from high-altitude pregnancies. Placenta 24: 191–198, 2003. doi: 10.1053/plac.2002.0895. [DOI] [PubMed] [Google Scholar]
- 62.Mayhew TM, Charnock-Jones DS, Kaufmann P. Aspects of human fetoplacental vasculogenesis and angiogenesis. III. Changes in complicated pregnancies. Placenta 25: 127–139, 2004. doi: 10.1016/j.placenta.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 63.McCarthy NS, Vangjeli C, Cavalleri GL, Delanty N, Shianna KV, Surendran P, O’Brien E, Munroe PB, Masca N, Tomaszewski M, Samani NJ, Stanton AV. Two further blood pressure loci identified in ion channel genes with a gene-centric approach. Circ Cardiovasc Genet 7: 873–879, 2014. doi: 10.1161/CIRCGENETICS.113.000190. [DOI] [PubMed] [Google Scholar]
- 64.Meininger CJ, Schelling ME, Granger HJ. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. Am J Physiol Heart Circ Physiol 255: H554–H562, 1988. doi: 10.1152/ajpheart.1988.255.3.H554. [DOI] [PubMed] [Google Scholar]
- 65.Meschia G. Placental respiratory gas and exchange and fetal oxygenation. Creasy and Resnik’s Maternal-Fetal Medicine: Principles and Practice, edited by Resnik R, Lockwood CJ, Moore TR, Greene MF, Copel JA, Silver RM. Philadephia, PA: Elsevier, 2014, p. 210–222. [Google Scholar]
- 66.Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, Remacle J, Michiels C. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett 468: 53–58, 2000. doi: 10.1016/S0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- 67.Namiki A, Brogi E, Kearney M, Kim EA, Wu T, Couffinhal T, Varticovski L, Isner JM. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem 270: 31189–31195, 1995. doi: 10.1074/jbc.270.52.31189. [DOI] [PubMed] [Google Scholar]
- 68.Ning W, Chu TJ, Li CJ, Choi AM, Peters DG. Genome-wide analysis of the endothelial transcriptome under short-term chronic hypoxia. Physiol Genomics 18: 70–78, 2004. doi: 10.1152/physiolgenomics.00221.2003. [DOI] [PubMed] [Google Scholar]
- 69.Olszewska-Pazdrak B, Hein TW, Olszewska P, Carney DH. Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells. Am J Physiol Cell Physiol 296: C1162–C1170, 2009. doi: 10.1152/ajpcell.00533.2008. [DOI] [PubMed] [Google Scholar]
- 70.Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays 22: 108–112, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 71.Patel J, Landers K, Mortimer RH, Richard K. Regulation of hypoxia inducible factors (HIF) in hypoxia and normoxia during placental development. Placenta 31: 951–957, 2010. doi: 10.1016/j.placenta.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 72.Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc Natl Acad Sci USA 97: 8386–8391, 2000. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phillips PG, Birnby LM, Narendran A. Hypoxia induces capillary network formation in cultured bovine pulmonary microvessel endothelial cells. Am J Physiol Lung Cell Mol Physiol 268: L789–L800, 1995. doi: 10.1152/ajplung.1995.268.5.L789. [DOI] [PubMed] [Google Scholar]
- 74.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 7: 165–197, 2000. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 75.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9: 677–684, 2003. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 76.Rajakumar A, Brandon HM, Daftary A, Ness R, Conrad KP. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 25: 763–769, 2004. doi: 10.1016/j.placenta.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 77.Rajakumar A, Conrad KP. Expression, ontogeny, and regulation of hypoxia-inducible transcription factors in the human placenta. Biol Reprod 63: 559–569, 2000. doi: 10.1095/biolreprod63.2.559. [DOI] [PubMed] [Google Scholar]
- 78.Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, Fisher SJ. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest 114: 744–754, 2004. doi: 10.1172/JCI200422991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reynolds LP, Redmer DA. Utero-placental vascular development and placental function. J Anim Sci 73: 1839–1851, 1995. doi: 10.2527/1995.7361839x. [DOI] [PubMed] [Google Scholar]
- 80.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis 12: 139–147, 2009. doi: 10.1007/s10456-009-9132-x. [DOI] [PubMed] [Google Scholar]
- 81.Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J 17: 3005–3015, 1998. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schäfer M, Ewald N, Schäfer C, Stapler A, Piper HM, Noll T. Signaling of hypoxia-induced autonomous proliferation of endothelial cells. FASEB J 17: 1–23, 2003. doi: 10.1096/fj.02-0398fje. [DOI] [PubMed] [Google Scholar]
- 83.Scheurer SB, Rybak JN, Rösli C, Neri D, Elia G. Modulation of gene expression by hypoxia in human umbilical cord vein endothelial cells: a transcriptomic and proteomic study. Proteomics 4: 1737–1760, 2004. doi: 10.1002/pmic.200300689. [DOI] [PubMed] [Google Scholar]
- 84.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24: 97–106, 2009. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 86.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66, 1995. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 87.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 9: 285–296, 2008. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 90: 4299–4308, 2005. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1α versus Hif-2α in regulation of the transcriptional response to hypoxia. Cancer Res 63: 6130–6134, 2003. [Erratum in Cancer Res 63: 8562, 2003.] [PubMed] [Google Scholar]
- 90.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 6: 485–495, 2004. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 91.Thompson JG, Simpson AC, Pugh PA, Donnelly PE, Tervit HR. Effect of oxygen concentration on in-vitro development of preimplantation sheep and cattle embryos. J Reprod Fertil 89: 573–578, 1990. doi: 10.1530/jrf.0.0890573. [DOI] [PubMed] [Google Scholar]
- 92.Tissot van Patot MC, Bendrick-Peart J, Beckey VE, Serkova N, Zwerdlinger L. Greater vascularity, lowered HIF-1/DNA binding, and elevated GSH as markers of adaptation to in vivo chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 287: L525–L532, 2004. doi: 10.1152/ajplung.00203.2003. [DOI] [PubMed] [Google Scholar]
- 93.Tissot van Patot MC, Murray AJ, Beckey V, Cindrova-Davies T, Johns J, Zwerdlinger L, Jauniaux E, Burton GJ, Serkova NJ. Human placental metabolic adaptation to chronic hypoxia, high altitude: hypoxic preconditioning. Am J Physiol Regul Integr Comp Physiol 298: R166–R172, 2010. doi: 10.1152/ajpregu.00383.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tucci M, Hammerman SI, Furfaro S, Saukonnen JJ, Conca TJ, Farber HW. Distinct effect of hypoxia on endothelial cell proliferation and cycling. Am J Physiol Cell Physiol 272: C1700–C1708, 1997. doi: 10.1152/ajpcell.1997.272.5.C1700. [DOI] [PubMed] [Google Scholar]
- 95.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10: 116–129, 2010. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 96.Umaoka Y, Noda Y, Narimoto K, Mori T. Effects of oxygen toxicity on early development of mouse embryos. Mol Reprod Dev 31: 28–33, 1992. doi: 10.1002/mrd.1080310106. [DOI] [PubMed] [Google Scholar]
- 97.Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal 9: 1221–1236, 2007. doi: 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- 98.Wang K, Jiang YZ, Chen DB, Zheng J. Hypoxia enhances FGF2- and VEGF-stimulated human placental artery endothelial cell proliferation: roles of MEK1/2/ERK1/2 and PI3K/AKT1 pathways. Placenta 30: 1045–1051, 2009. doi: 10.1016/j.placenta.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang K, Zheng J. Signaling regulation of fetoplacental angiogenesis. J Endocrinol 212: 243–255, 2012. doi: 10.1530/JOE-11-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zamudio S. The placenta at high altitude. High Alt Med Biol 4: 171–191, 2003. doi: 10.1089/152702903322022785. [DOI] [PubMed] [Google Scholar]
- 101.Zamudio S, Postigo L, Illsley NP, Rodriguez C, Heredia G, Brimacombe M, Echalar L, Torricos T, Tellez W, Maldonado I, Balanza E, Alvarez T, Ameller J, Vargas E. Maternal oxygen delivery is not related to altitude- and ancestry-associated differences in human fetal growth. J Physiol 582: 883–895, 2007. doi: 10.1113/jphysiol.2007.130708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao F, Sellgren K, Ma T. Low-oxygen pretreatment enhances endothelial cell growth and retention under shear stress. Tissue Eng Part C Methods 15: 135–146, 2009. doi: 10.1089/ten.tec.2008.0321. [DOI] [PubMed] [Google Scholar]
- 103.Zhou C, Yan Q, Zou QY, Zhong XQ, Tyler CT, Magness RR, Bird IM, Zheng J. Sexual dimorphisms of preeclampsia-dysregulated transcriptomic profiles and cell function in fetal endothelial cells. Hypertension 74: 154–163, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou J, Schmid T, Brüne B. Tumor necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol Biol Cell 14: 2216–2225, 2003. doi: 10.1091/mbc.e02-09-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou L, Dosanjh A, Chen H, Karasek M. Divergent effects of extracellular oxygen on the growth, morphology, and function of human skin microvascular endothelial cells. J Cell Physiol 182: 134–140, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 106.Zou QY, Zhao YJ, Li H, Wang XZ, Liu AX, Zhong XQ, Yan Q, Li Y, Zhou C, Zheng J. GNA11 differentially mediates fibroblast growth factor 2- and vascular endothelial growth factor A-induced cellular responses in human fetoplacental endothelial cells. J Physiol 596: 2333–2344, 2018. doi: 10.1113/JP275677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zou QY, Zhao YJ, Zhou C, Liu AX, Zhong XQ, Yan Q, Li Y, Yi FX, Bird IM, Zheng J. G protein α subunit 14 mediates fibroblast growth factor 2-induced cellular responses in human endothelial cells. J Cell Physiol 234: 10184–10195, 2019. doi: 10.1002/jcp.27688. [DOI] [PMC free article] [PubMed] [Google Scholar]