

Abstract
Loss-of-function mutations in phospholipase C-ε1 (PLCE1) have been detected in patients with nephrotic syndrome, but other family members with the same mutation were asymptomatic, suggesting additional stressor are required to cause the full phenotype. Consistent with these observations, we determined that global Plce1-deficient mice have histologically normal glomeruli and no albuminuria at baseline. Angiotensin II (ANG II) is known to induce glomerular damage in genetically susceptible individuals. Therefore, we tested whether ANG II enhances glomerular damage in Plce1-deficient mice. ANG II increased blood pressure equally in Plce1-deficient and wild-type littermates. Additionally, it led to 20-fold increased albuminuria and significantly more sclerotic glomeruli in Plce1-deficient mice compared with wild-type littermates. Furthermore, Plce1-deficient mice demonstrated diffuse mesangial expansion, podocyte loss, and focal podocyte foot process effacement. To determine whether these effects are mediated by hypertension and hyperfiltration, rather than directly through ANG II, we raised blood pressure to a similar level using DOCA + salt + uninephrectomy and norepinephrine. This caused a fivefold increase in albuminuria in Plce1-deficient mice and a significant increase in the number of sclerotic glomeruli. Consistent with previous findings in mice, we detected strong PLCE1 transcript expression in podocytes using single cell sequencing of human kidney tissue. In hemagglutinin-tagged Plce1 transgenic mice, Plce1 was detected in podocytes and also in glomerular arterioles using immunohistochemistry. Our data demonstrate that Plce1 deficiency in mice predisposes to glomerular damage secondary to hypertensive insults.
Keywords: diffuse mesangial sclerosis, glomerulosclerosis, hypertension, phospholipase C-ε1
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is a common cause of nephrotic syndrome. Loss of visceral glomerular epithelial cell (podocytes) is thought to be one of the major pathophysiological processes driving the development of FSGS (9, 10). Supporting this, it has been shown that isolated loss of podocytes using transgenic animal models is sufficient for the development of glomerulosclerosis (33). Moreover, loss-of-function mutations in proteins that are integral to podocyte and slit diaphragm function and homeostasis, such as nephrin, podocin, Wilms’ tumor-1 (WT1), and laminin-β2, result in FSGS or diffuse mesangial sclerosis (DMS) (1).
Phospholipase C-ε1 (PLCE1) is an isoform of phospholipase C that was discovered to be a candidate gene for FSGS and DMS via positional cloning (12, 17). Plce1 expression has been localized to podocytes in rats, where it interacts with v-Raf murine sarcoma viral oncogene homolog B and Ras-GTPase activating-like protein (IQGAP1) (6, 17). Initial studies have suggested that ~8–12% of infantile, familial, steroid-resistant FSGS and ~33–50% of DMS cases are secondary to missense or nonsense mutations in PLCE1 (5, 12). However, after establishing cohorts of patients with mutations in PLCE1, it was discovered that multiple parents of affected children also carried homozygous missense or nonsense PLCE1 mutations but did not develop FSGS or DMS (14). This suggests that the absence of PLCE1 is not sufficient for the development of the full nephrotic phenotype and that additional environmental stressors are needed. This “two-hit” hypothesis has been demonstrated for other models of nephrotic syndrome. Most notably, patients with high-risk apolipoprotein L1 variants are at higher risk of developing nephropathy if exposed to human immunodeficiency virus (HIV) (21). Moreover, mice deficient in receptor-type protein-tyrosine phosphatase O (ptpro, GLEPP1), an important podocyte protein-tyrosine phosphatase, have grossly normal renal histology and no proteinuria at baseline but do develop accelerated kidney disease and hypertension when they undergo nephrectomy (32).
Angiotensin II (ANG II) is the major effector molecule of the renin-angiotensin system. It has been implicated in the development and progression of glomerulosclerosis by multiple mechanisms including selective glomerular hyperfiltration and direct podocyte damage (4, 18). ANG II also acts as a “second hit” for the development of glomerulosclerosis in genetically susceptible humans and mice. Patients with activating mutations in transient receptor potential cation channel subfamily C member 6 (TRPC6) are prone to develop FSGS in the fourth decade of life (35). In contrast, however, Trpc6+/+ mice are not phenotypically different from Trpc6−/− mice at baseline but do develop more proteinuria and have accelerated glomerulosclerosis when they develop ANG II-mediated hypertension (11). Similarly, mice that express an accessory protein of HIV in a podocyte-specific manner develop accelerated glomerulosclerosis and proteinuria in response to ANG II-induced hypertension (19). Similarly, global Plce1 knockout mice (Plce1−/− mice) also exhibit normal kidney structure and function (17). Whether these mice develop glomerulosclerosis and proteinuria in response to ANG II-mediated hypertension is unknown.
In the present study, we examine whether Plce1 deficiency increases susceptibility to ANG II-mediated glomerular damage and whether the effects of ANG II are directly mediated by ANG II or secondary to hypertension.
METHODS
Mice.
Global Plce1−/− mice on a C57BL/6J background were used (31). Mice had previously been backcrossed at least 10 generations onto a C57BL/6 background. F0 Plce1−/− mice were bred with Plce1+/+ mice to generate F1 Plce1+/− mice. F1 heterzygotes were bred to generate F2 littermates on which all experiments were performed. Male and female mice at a age of 10–12 wk old were used, with the exception of mice used for DOCA + salt + uninephrectomy (UNx)-induced hypertension, in which only male mice were used because blood pressure elevation is more severe in male mice (see below) (13) (Sex-specific results are provided in the Supplemental Data.) Mice that died before the scheduled euthanization were excluded from the analysis.
Plce1+/+, Plce1+/−, and Plce1−/− mice were genotyped as previously described (31) using GoTaq Green Master Mix (Promega, Madison, WI) as per the manufacturer’s instructions. Genomic DNA (50–200 ng) was combined with 0.4 µM of a common 5′ primer (GCGTATTTCCAGAGTTAGAACAAGG) and 0.2 µM of a 3′ wild-type Plce1+/+ primer (CCACAACCAGGACCAGAGATG) and 0.2 µM of a 3′ Lac Z primer from Plce1−/− construct (CTGCAAAGGGTCGCTACAGA). Using a Bio-Rad C1000 Touch cycler, samples underwent 60 s of initial denaturation at 97°C followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 63.5°C for 30 s, and extension at 72°C for 90 s. Cycler products were run on a 1.5% agarose gel and stained with 0.5 µg ethidium bromide. The reaction generates a 550-bp amplicon product for Plce1+/+ mice, a 740-bp amplicon product for Plce1−/− mice, and both amplicon products for Plce1+/− mice.
Hemagglutinin-tagged (HA) Plce1 mice were generated by the University of Rochester Mouse Genome Editing Resource using the CRISPR/Cas9 approach. Briefly, a 49-bp 5′ arm-MluI-3xHA stop-49-bp 3′ arm, silent mutation for sgB PAM single-strand DNA template containing Plce1-HA single-stranded donor oligonucleotide (ssODN) was synthesized and purified by Integrated DNA Technologies. The sequence of the oligonucleotide was as follows: 5′-AAACCTGTGGGTGCCTTGTCCTCTAGTGACACAGTGGGTTACCAGCAGACGCGTTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTGATTAGGCAAGTGCGGCATATTTGTTCCAGGtATATGAAGGTACTGTAGCATTCT-3′. In vitro transcription for sgRNA was performed with the MEGAshortscript T7 kit (Life Technologies) using sgRNA template cloned in px330 plasmid (Addgene plasmid 42230); the IVT product was purified using the MEGAclear kit (Life Technologies). The sequence of the sgRNA template was as follows: 5′-AGTGCGGCATATTTGTTCCA(GGG)-3′. A mixture of Cas9 mRNA (TriLink Biotechnologies, 100 ng/μL), sgRNA (50 ng/μL), and ssODN (100 ng/μL) was injected into fertilized eggs from C57BL/6J mice (Stock no. 000664, Jackson Laboratory, Bar Harbor, ME). Viable two-cell stage embryos were transferred to pseudo-pregnant ICR female mice to generate founder mice, which were subsequently bred with C57BL/6J mice for germline transmission to generate F1 mice. Pups with the desired mutation were identified by PCR and were further sequencing confirmed. The primer sequences used for HA-tagged mice were as follows: Plce1-HA wild-type forward, 5′-GGAGTGCATACTACCTCTAAGTATGACATG-3′; Plce1-HA wild-type reverse, 5′-CCTGTCTTCAGAGGACAAGCCTCTTGA-3′; Plce1-HA mutant forward, 5′-CACAGTGGGTTACCAGCAGACGCG T-3′; and Plce1-HA mutant reverse, 5′-ACTTGCCTAATCAGCGTAATCTGGAAC-3′. Primer sequences for WT mice were as follows: Plce1-HA wild-type forward, 5′-GGAGTGCATACTACCTCTAAGTATGACATG-3′; and Plce1-HA wild-type reverse, 5′-CCTGTCTTCAGAGGACAAGCCTCTTGA-3′. Amplicon lengths were as follows: 392 bp for wild-type, 491 bp for HA-tagged Plce1, and 276+215 bp for cut HA-tagged Plce1.
Surgical procedures and euthanasia.
For the implantation of subcutaneous, osmotic mini-pumps, or DOCA pellets, mice were anesthetized with 2–5% inhaled isofluorane. Perioperative analgesia was provided with 0.1 mg/kg subcutaneous buprenorphine before surgery and every 12 h for 72 h postoperatively. Ophthalmic ointment was applied. Body temperature was maintained using a heating pad for the duration of the surgery. The nape of the neck was shaved and cleaned with 70% ethanol and betadine. Under sterile conditions, the mini-pump or implant was implanted in the subcutaneous tissue through a small incision. The wound was closed with 2-0 silk suture (Ethicon, Somerville, NJ). Mice were monitored for recovery and then returned to their cages. Osmotic mini-pumps (models 1002 and 1004) were purchased from Alzet (Durect, Cupertino, Ca) for 2- and 4-wk infusions, respectively. Osmotic mini-pumps were primed overnight in sterile PBS at 37°C before implantation.
For UNx, mice were anesthetized, and the left flank was shaved and cleaned as above. Under sterile conditions, a subcostal incision was made through the skin and fascia, and the left kidney was exposed. The renal vasculature and ureter were exposed and ligated with 4-0 silk suture (Ethicon), and the kidney was removed. The fascia was closed with 4-0 silk suture and the skin with 2-0 silk suture.
Mice were euthanized under 5% inhaled isoflurane and via bilateral pneumothorax and aortic transection. Their kidneys were flushed with PBS via cardiac perfusion and placed in various fixatives for further processing as listed below.
Drugs and experimental groups.
ANG II (Sigma, St. Louis, MO) was delivered via osmotic mini-pump at a dose of 1.5 mg·kg−1·day−1 (19). DOCA (Sigma) was delivered at a dose of 50 mg/20 g body wt (3). Implants were made by combining DOCA with Silastic Paste no. RTV-3110 (Dow Corning, Midland, MI) and no. 4 Catalyst (Dow Corning) before being allowed to cure overnight. Appropriate-sized implants were then cut and implanted as above. Mice treated with DOCA also received 1.0% NaCl and 0.2% KCl in their drinking water. Norepinephrine (NE; Sigma) was infused at 3.8 mg·kg−1·day−1 (19), as a bitartrate, monohydrate salt.
Baseline kidney structure and function were assessed in 10- to 12-wk-old Plce1+/+ (n = 5), Plce1+/− (n = 5), and Plce1−/− (n = 6) mice. ANG II pumps were inserted in 10- to 12-wk-old mice. Mice were euthanized 2 wk [Plce1+/+ (n = 9), Plce1+/− (n = 7), and Plce1−/− (n = 7)] and 4 wk [Plce1+/+ (n = 10), Plce1+/− (n = 8), and Plce1−/− (n = 8)] later. Similarly, mice were treated for 4 wk with DOCA + salt + UNx and an additional NE mini-pump at nephrectomy to achieve equivalent blood pressure as ANG II. Plce1+/+ (n = 7) and Plce1−/− (n = 5) mice were treated for 4 wk before euthanasia.
Blood pressure measurements.
Systolic blood pressure was measured noninvasively using the CODA tail-cuff system (Kent Scientific, Torrington, CT) (2). Each data point is the mean of 10 blood pressure measurements.
Urine collection.
Urine samples were collected in metabolic caging for 4 h, and urine albumin and creatinine concentration were determined as previously described (22)
Immunohistochemistry and podometric analyses.
Immunohistochemistry was performed on 3-μm sections as previously described (22). Primary antibodies were incubated overnight at 4°C, and a Vectastain anti-rabibit HRP kit (Vector Laboratories, Burlingame, CA) with diaminobenzidine (Sigma) as per the manufacturer’s instructions were used. All slides were stained with 0.5% periodic acid (Electron Microscopy Sciences) for 5 min and Schiff’s reagent (Sigma) for 15 min before being counterstained by hematoxylin for 90 s.
The podocyte index was determined for at least 30 glomeruli from each sample with a primary antibody for GLEPP1 as previously described (9, 10). The total GLEPP1 area was normalized to glomerular tuft area measurements. To determine podocyte volume, the GLEPP1 area was normalized to the glomerular tuft area using the average of Eq. 9 in Venkatareddy et al. (30). Podocyte density was determined for at least 30 glomeruli using a primary antibody against WT1 (Abcam, Cambridge, UK). Podocyte nucleus counts were normalized to glomerular tuft area and glomerular volume as above (9, 10, 22). The mesangial index was determined from at least 30 glomeruli/sample as previously described (22). In brief, the minimum hue, intensity, and saturation (HIS) threshold according to periodic acid-Schiff (PAS)-positive material was set, which, if exceeded, would count as the mesangial matrix. The PAS-positive area was normalized to the glomerular tuft area. Glomeruli that were noted to be segmentally or globally sclerosed by PAS staining were tabulated as previously described (3) and normalized to the total number of glomeruli. Plce1 staining was determined using rabbit monoclonal antibody against HA (Cell Signaling Technology, Danvers, MA). Staining for renin was performed using a rabbit monoclonal antibody (Abcam). All analyses were performed with ImageJ software (National Institutes of Health).
Transmission electron microscopy.
The kidney cortex was immersion fixed in 2.5% glutaraldehyde overnight at 4°C. Tissue processing and imaging was performed by the University of Michigan Microscope and Image Analysis core facility. Kidney sections were examined using a JEOL USAJEM-1400Plus electron microscope. Two to three glomeruli per sample were visualized.
Quantitative PCR.
Thirty grams of the mouse kidney were stored in RNAlater (Invitrogen, Carlsbad, CA) at −80°C. RNA was extracted using TRIzol (Ambion, Carlsbad, CA) and chloroform using the RNeasy kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. Extracted RNA was treated with DNAse. One microgram of DNAse-treated RNA was reverse transcribed using a high-capacity cDNA kit (Applied Biosystems, Foster, CA) as per the manufacturer’s instructions. For quantitative PCR, 1 μl of cDNA was then amplified using 200 nM of specific primers in a thermal cycler and power SYBR green master mix (Applied Biosystems). The primer sequences used were as follows: collagen type I-α1 (Col1a1), forward 5′-GACGCCATCAAGGTCTACTG-3′ and reverse 5′-ACGGGAATCCATCGGTCA-3′; Gapdh, forward 5′-GCATTGTGGAAGGGCTCA-3′ and reverse 5′-GGGTAGGAACACGGAAGG-3′; collagen type III-α1 (Col3a1), forward 5′-AATTTGGTGTGGACGTTGGC-3′ and reverse 5′-TTGTCGGTCACTTGCACTGG-3′; and fibronectin (Fn1), forward 5′-TGATCACATGGACGCCTGC-3′ and reverse 5′-GAGTCAAGCCGGACACAACG-3′ (16, 27). Quantification of relative RNA abundance was performed using the ΔΔCt method (where Ct is threshold cycle).
Single cell RNA sequencing.
Single cell RNA sequencing (scRNA-Seq) was performed as previously described (15, 24). Briefly, the single cell data were generated from the combined analysis of three scRNA-Seq data sets generated from healthy portions of tumor nephrectomy samples specifically harvested for single cell analysis using 10x Genomics methodology. The sequencing data were first analyzed using Cell Ranger software (10x Genomics) to extract the gene expression data matrix. Downstream analysis steps including unsupervised clustering and identification of cluster-specific markers were performed using the Seurat R package. From a total of 4,734 cells, 14 clusters of specific cell types as defined by differentially expressed cell type-specific genes were identified. The data for the gene expression matrix are provided in the Supplemental Data (https://doi.org/10.6084/m9.figshare.10262909.v1).
Statistics.
Comparisons of the means between two groups was performed with Student’s t test. Comparisons of the mean between three or more groups was performed with one-way ANOVA and Tukey’s post hoc test. A P value of <0.05 was considered significant for all tests. All figures are presented as means ± SE unless otherwise specified. All individual data points are provided in the Supplemental Tables.
Research assurances.
All procedures were approved by the University of Michigan Institutional Animal Care and Use Committee and adhered to the guiding principles in the care and use of experimental animals in accordance with National Institutes of Health guidelines. The University of Michigan operates an Association for Assessment and Accreditation of Laboratory Animal Care-certified animal facility. All experiments using human kidney tissue were approved by the University of Michigan Institutional Review Board. Experiments adhered to the requirements of the United States Federal Policy for the Protection of Human Subjects (45 CFR, Part 46).
RESULTS
To determine whether Plce1 deficiency is sufficient to cause abnormalities in the kidney, we evaluated the kidneys of 10- to 12-wk-old Plce1+/+, Plce1+/−, and Plce1−/− mice. Kidneys of Plce1−/− mice did not show sclerotic glomeruli or foot process effacement or basement membrane thickening (Fig. 1). Furthermore, podocyte number and density and mesangial area were not different between unchallenged Plce1+/+, Plce1+/−, and Plce1−/− mice (Tables 1 and 2 and Supplemental Table S1, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). Glomerular volume was slightly enlarged in untreated Plce1−/− mice relative to Plce1+/+ mice; there was no difference between untreated Plce1−/− and Plce1+/− mice (Tables 1 and 2 and Supplemental Table S1). No difference was detected in cortical glomerular density among the three groups. Besides mildly enlarged glomeruli, no other glomerular parameter was different in untreated Plce1−/− mice. Albuminuria was not significantly different between untreated Plce1+/+, Plce1+/−, and Plce1−/− mice (40.2 ± 15.3, 30.7 ± 8.4, and 27.7 ± 5.9 µg/mg creatinine; n = 15, 19, and 15, respectively).
Fig. 1.

Representative light microscopy images of glomeruli and transmission electron microscopy (TEM) of podocyte foot processes from untreated phospholipase C-ε1 (Plce1)+/+, Plce1+/−, and Plce1−/− mice. Podocyte nuclei and cytoplasm are stained with Wilms’ tumor-1 (WT1) and receptor-type protein-tyrosine phosphatase O (GLEPP1), respectively. No significant abnormalities were detected in Plce1−/− glomeruli. Magnification: ×40 and ×8,000 for light microscopy and TEM, respectively.
Table 1.
Glomerular parameters in untreated Plce1+/+, Plce1+/−, and Plce1−/− mice
| Groups |
|||
|---|---|---|---|
| Parameter | Plce1+/+ | Plce1+/− | Plce1−/− |
| Number of mice/group | 5 | 5 | 6 |
| Mesangial index, % | 4.5 ± 2.0 | 5.1 ± 0.9 | 3.0 ± 1.1 |
| Podocytes/glomerulus | 83.4 ± 6.8 | 84.1 ± 2.2 | 90.7 ± 5.3 |
| Individual podocyte volume, μm3 | 1,164 ± 135 | 1,195 ± 115 | 1,171 ± 108 |
| Glomerular volume, μm3 | 165,780 ± 11,316 | 175,115 ± 12,103 | 203,949 ± 6,533 * |
| Glomeruli per mm2 | 9.91 ± 0.45 | 10.36 ± 0.87 | 10.10 ± 0.79 |
Data are presented as means ± SE. Glomerular volume was significantly elevated in phospholipase C-ε1 (Plce1)−/− mice versus Plce1+/+ mice but did not significantly differ from Plce1+/− mice.
P < 0.05 vs. Plce1+/+ mice.
Table 2.
Sexes and body weights of untreated mice
| Groups |
|||
|---|---|---|---|
| Parameter | Plce1+/+ | Plce1+/− | Plce1+/+ |
| Male/female mice | 2/3 | 2/3 | 3/3 |
| Body weight, g | 22.0 ± 1.0 | 21.9 ± 1.9 | 22.9 ± 1.3 |
Plce1, phospholipase C-ε1.
To determine whether Plce1 deficiency confers greater susceptibility to ANG II-mediated glomerular damage, we infused Plce1+/+, Plce1+/−, and Plce1−/− mice with ANG II for 2 or 4 wk. ANG II increased blood pressure after 1 and 4 wk independent of genotype (Fig. 2A and Supplemental Table S2A, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). While blood pressure was elevated above baseline in all three groups at week 2, the values in the Plce1−/− group were lower than in the Plce1+/+ group. Thus, our data demonstrate that the ANG II infusion was successful at inducing hypertension.
Fig. 2.

A: effect of ANG II on systolic blood pressure in phospholipase C-ε1 (Plce1) deficiency. Systolic blood pressure rose equally in all three genotypes after 1 wk of treatment with ANG II. Blood pressure was significantly lower in Plce1−/− mice at week 2 compared with Plce1+/+ mice. Blood pressure in Plce1−/− mice was equivalent to the other two genotypes at week 4. Group sizes for Plce1+/+, Plce1+/−, and Plce1−/− mice were as follows n = 19, 15, and 15 at baseline, n = 18, 11, and 14 at week 1, n = 19, 15, and 15 at week 2, and n = 10, 8, and 8 at week 4, respectively. †P < 0.05 vs. baseline for all three genotypes; ‡P < 0.05 vs. Plce1+/+ mice at week 2. B: effect of ANG II on albuminuria in Plce1 deficiency. The urine albumin-to-creatinine ratio was significantly higher in Plce1−/− mice compared with the other two genotypes at weeks 1, 2, and 4 of ANG II infusion. Albuminuria levels were lower at week 2 compared with week 1 in Plce1−/− mice but were still greater than levels experienced by the other two genotypes. Group sizes for Plce1+/+, Plce1+/−, and Plce1−/− mice were as follows: n = 19, 15, and 15 at baseline, week 1, and week 2, respectively, and n = 10, 8, and 8 at week 4, respectively. *P < 0.05 vs. Plce1+/+ and Plce1+/− mice; #P < 0.05 vs. Plce1−/− mice at week 1.
To determine whether ANG II-induced hypertension caused a functional glomerular defect, we measured albuminuria. One, two, and four weeks after the initiation of ANG II infusion, albuminuria was increased in Plce1−/− mice compared with Plce1+/+ and Plce1+/− mice (Fig. 2B and Supplemental Table S2B, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). In Plce1−/− mice, albuminuria was less at week 2 than week 1, although it still greater than in untreated Plce1+/+ and Plce1+/− mice. This corresponded with the lower levels of hypertension experienced by Plce1−/− mice at week 2 of ANG II infusion. Our data demonstrate that ANG II-induced hypertension causes a functional glomerular defect in Plce1−/− mice.
We evaluated kidneys for evidence of histological damage in response to ANG II-induced hypertension. After 2 wk of ANG II infusion, Plce1−/− mice were noted to have focal glomerulosclerosis and focal loss of GLEPP1 and WT1 expression (Fig. 3) and had significantly more sclerotic glomeruli at weeks 2 and 4 compared with Plce1+/+ and Plce1−/− mice (Tables 3 and 4 and Supplemental Table S3, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). Furthermore, Plce1−/− glomeruli showed evidence of focal foot process effacement by electron microscopic examination at week 4 (Fig. 3), whereas foot processes of Plce1+/+ and Plce1+/− glomeruli were normal. In addition to focal sclerosis, Plce1−/− glomeruli showed evidence of mesangial matrix expansion and podocyte depletion at 4 wk compared with both Plce1+/+ and Plce1+/− glomeruli (Tables 3 and 4 and Supplemental Table S3). No differences were detected at week 2. Our data demonstrate that Plce1−/− mice develop structural glomerular damage consistent with increased susceptibility to ANG II-mediated hypertension.
Fig. 3.

Representative light microscopy images of glomeruli and transmission electron microscopy (TEM) of podocyte foot processes from phospholipase C-ε1 (Plce1)+/+, Plce1+/−, and Plce1−/− mice treated with 4 wk of ANG II. Focal glomerulosclerosis was noted in Plce1−/− mice treated with ANG II. Note the complete loss of Wilms’ tumor-1 (WT1) and receptor-type protein-tyrosine phosphatase O (GLEPP1) staining within the sclerotic Plce1−/− glomerulus consistent with podocyte loss. TEM demonstrated focal foot process effacement and basement membrane thickening in Plce1−/− glomeruli. Magnification: ×40 and ×8,000 for light microscopy and TEM, respectively.
Table 3.
Glomerular parameters in mice treated with 2 or 4 wk of ANG II
|
Week 2 |
Week 4 |
|||||
|---|---|---|---|---|---|---|
| Parameter | Plce1+/+ | Plce1+/− | Plce1−/− | Plce1+/+ | Plce1+/− | Plce1−/− |
| Number of mice/group | 9 | 7 | 7 | 10 | 8 | 8 |
| Sclerotic glomeruli, % | 0 ± 0 | 0 ± 0 | 0.46 ± 0.19* | 0.03 ± 0.03 | 0 ± 0 | 0.87 ± 0.39* |
| Mesangial index, % | 7.2 ± 1.4 | 5.1 ± 1.4 | 9.9 ± 1.6 | 6.0 ± 0.8 | 6.1 ± 1.2 | 11.2 ± 1.5* |
| Podocytes/glomerulus | 90.9 ± 3.2 | 89.7 ± 4.2 | 91.2 ± 3.6 | 111.6 ± 7.6 | 83.9 ± 8.0 | 77.4 ± 7.1* |
| Individual podocyte volume, µm3 | 1,336 ± 83 | 1,307 ± 99 | 1,314 ± 107 | 942 ± 71 | 1,313 ± 188 | 1,280 ± 68 |
| Glomerular volume, µm3 | 234,076 ±16,351 | 230,259 ±12,093 | 226,521 ±13,209 | 191,111 ±6,535 | 192,807 ±13,911 | 185,884 ±12,047 |
Data are presented as means ± SE. More sclerotic glomeruli were noted at week 2 in phospholipase C-ε1 (Plce1)−/− mice treated with ANG II. Plce1−/− mice had more sclerotic glomeruli and mesangial expansion and fewer podocytes per glomerulus after 4 wk of ANG II treatment.
P < 0.05 vs. Plce1+/+ and Plce1+/− mice.
Table 4.
Sexes as well as starting and ending body weights of ANG II-treated mice
|
Week 2 |
Week 4 |
|||||
|---|---|---|---|---|---|---|
| Parameter | Plce1+/+ | Plce1+/− | Plce1−/− | Plce1+/+ | Plce1+/− | Plce1−/− |
| Number of mice/group | 9 | 7 | 7 | 10 | 8 | 8 |
| Male/female | 7/2 | 4/3 | 3/4 | 2/8 | 6/2 | 3/5 |
| Start weight, g | 23.9 ± 1.4 | 22.4 ± 1.4 | 22.1 ± 0.9 | 23.2 ± 1.1 | 24.3 ± 1.1 | 22.5 ± 0.9 |
| End weight, g | 23.3 ± 1.2 | 21.9 ± 2.4 | 21.3 ± 0.8 | 22.7 ± 1.0 | 23.0 ± 1.0 | 22.5 ± 0.8 |
Data are presented as mean ± SE. Plce1, phospholipase C-ε1.
ANG II is also known induce renal interstitial fibrosis. We assessed for this by performing quantitative PCR for Col1a1, Col3a1, and Fn1 using RNA isolated from whole kidneys of mice treated with ANG II for 4 wk. While no statistically significant difference in Col1a1 and Fn1 mRNA expression was detected between any of the three groups at week 4, there was a trend toward higher expression in kidneys of Plce1−/− mice. Similarly, Col3a1 mRNA expression was highest in Plce1−/− mice, reaching statistical significance compared with Plce1+/− mice (Supplemental Figure S1, available online at https://doi.org/10.6084/m9.figshare.11858031.v1). Our data suggest that expression of extracellular matrix genes is increased in Plce1−/− mice after 4 wk of ANG II exposure.
Because ANG II can cause glomerular damage through hemodynamic effects as well as directly causing podocyte damage (4, 18), we sought to determine whether Plce1−/− mice develop glomerular damage after induction of hypertension through an ANG II-independent mechanism. Therefore, we used the DOCA + salt + UNx model of hypertension in Plce1−/− and Plce1+/+ mice, as this has been previously shown to induce glomerulosclerosis in genetically susceptible mice (3). However, our initial dose-finding experiments demonstrated that DOCA + salt + UNx was unable to raise blood pressure to a similar level as ANG II (Supplemental Table S4C, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). To account for this, we added additional NE via mini-pump to the DOCA + salt + UNx model. As shown in Fig. 4A (and Supplemental Table 4A, available online at https://doi.org/10.6084/m9.figshare.11858022.v1), DOCA + salt + UNx + NE significantly increased blood pressure in Plce1+/+ and Plce1−/− mice at both 2 and 4 wk but was significantly less in Plce1−/− mice compared with Plce1+/+ mice.
Fig. 4.

A: effect of DOCA + salt + uninephrectomy (UNx) + norepinephrine (NE) on blood pressure in phospholipase C-ε1 (Plce1) deficiency. DOCA + salt + UNx + NE increased blood pressure similarly in both groups by week 2 of the protocol compared with baseline. Blood pressure remained elevated in both groups but was significantly lower in the Plce1−/− group after 4 wk. †P < 0.05 for both genotypes vs. baseline; *P < 0.05 vs. Plce1+/+ mice. B: effect of DOCA + salt + UNx + NE on albuminuria in Plce1 deficiency. Urine albumin-to-creatinine ratios were significantly elevated in Plce1−/− mice treated with DOCA + salt + UNx + NE compared with Plce1+/+ mice at weeks 2 and 4 of treatment. *P < 0.05 vs. Plce1+/+ mice.
To determine whether DOCA + salt + UNx + NE caused accelerated glomerular damage in Plce1−/− mice, we measured the effect on albuminuria. Urine albumin-to-creatinine ratios were significantly higher in Plce1−/− mice compared with Plce1+/+ mice at both week 2 and week 4 (Fig. 4B and Supplemental Table S4B, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). Plce1−/− mice also showed evidence of focal glomerulosclerosis after treatment with DOCA + salt + UNx + NE (Fig. 5). Again, there was significant focal loss of GLEPP1 and WT1 staining within sclerosed glomeruli, consistent with focal podocyte loss. Quantification of the number of sclerotic glomeruli showed significantly more in Plce1−/− mice compared with Plce1+/+ mice (Tables 5 and 6 and Supplemental Table S5A, available online at https://doi.org/10.6084/m9.figshare.11858022.v1). In contrast to ANG II, however, there was no diffuse mesangial matrix expansion or podocyte loss noted in Plce1−/− mice compared with Plce1+/+ mice (Tables 5 and 6 and Supplemental Table S5). Our data demonstrate that Plce1−/− mice develop increased glomerular damage as evidenced by their higher levels of albuminuria and higher rates of focal glomerulosclerosis. In conjunction with our ANG II data, these data suggest that Plce1−/− mice are more susceptible to glomerular damage from hypertensive and hyperfiltering injuries.
Fig. 5.
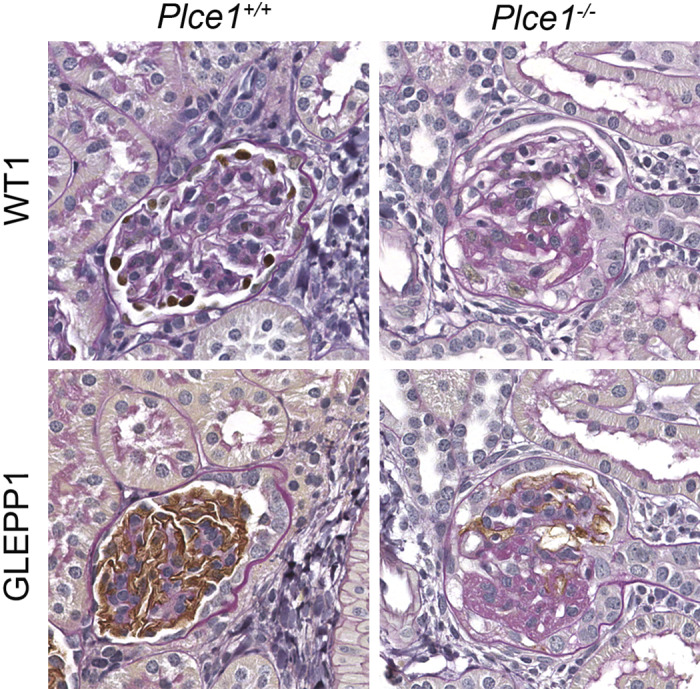
Representative light microscopy images of glomeruli from phospholipase C-ε1 (Plce1)+/+ and Plce1−/−mice treated with DOCA + salt + uninephrectomy + norepinephrine. Focal glomerulosclerosis was noted within Plce1−/− mice treated with this protocol. Note the lack of Wilms’ tumor-1 (WT1) and receptor-type protein-tyrosine phosphatase O (GLEPP1) staining within the Plce1−/− glomerulus, consistent with focal podocyte depletion. Magnification: ×40.
Table 5.
Glomerular parameters in mice treated with 4 wk of DOCA + salt + uninephrectomy + norepinephrine
| Parameter | Plce1+/+ | Plce1−/− |
|---|---|---|
| Number of mice/group | 7 | 5 |
| Sclerotic glomeruli, % | 0 ± 0 | 2.30 ± 1.07* |
| Mesangial index, % | 7.1 ± 1.0 | 8.0 ± 4.6 |
| Podocytes/glomerulus | 76.0 ± 3.2 | 78.0 ± 1.7 |
| Individual podocyte volume, µm3 | 1,629 ± 98 | 1,699 ± 143 |
| Glomerular volume, µm3 | 240,022 ± 7,257 | 252,218 ± 11,126 |
Data are presented as means ± SE. More sclerotic glomeruli were noted after 4 wk of treatment in phospholipase C-ε1 (Plce1)−/− mice. No other significant differences were noted.
P < 0.05 vs. Plce1+/+ mice.
Table 6.
Sexes and starting and ending body weights of DOCA + salt + uninephrectomy + norepinephrine-treated mice
| Parameter | Plce1+/+ | Plce1−/− |
|---|---|---|
| Number of mice/group | 7 | 5 |
| Male/female | 7/0 | 5/0 |
| Start weight, g | 21.5 ± 0.6 | 21.2 ± 1.1 |
| End weight, g | 22.8 ± 0.5 | 22.4 ± 1.4 |
Data are presented as means ± SE. Plce1, phospholipase C-ε1.
PLCE1 has previously been localized to podocytes (6, 17). However, whether PLCE1 is expressed within other cells in the kidney is unknown. To determine PLCE1 expression in human kidneys, we performed scRNA-Seq of human kidney tissue obtained from patients undergoing nephrectomy. Fourteen different cell populations were identified based on their distinct molecular markers (Fig. 6A). As demonstrated in the violin plots, PLCE1 was found to be highly enriched in podocytes, consistent with previous data. However, PLCE1 mRNA was also detected in cells of vascular smooth muscle and/or mesangial origin as well as certain endothelial cell populations. To validate these data, we performed immunohistochemical staining of kidneys from mice expressing HA-tagged Plce1. As shown in Fig. 6B, Plce1 was found to be abundantly expressed in podocytes and in vascular smooth muscle cells of glomerular arterioles, consistent with the scRNA-Seq data. No significant endothelial staining of HA-tagged Plce1 was observed. Wild-type mice stained with HA antibody did not show any nonspecific staining (Fig. 6C). To determine Plce1 protein expression within glomerular arterioles, we performed immunohistochemistry for HA and renin in serial sections of glomeruli of HA-tagged Plce1 mice. As shown in Fig. 7, A and B, HA-tagged Plce1-positive glomerular arterioles express renin in close proximity, suggesting that Plce1 is expressed in the afferent arteriole. Additionally, HA-tagged Plce1 staining was also detected in interlobular arteries (Fig. 7, C and D). No significant staining was noted within segmental branches of the main renal arteries (Supplemental Figure S2, available online at https://doi.org/10.6084/m9.figshare.11858034.v1). Our data suggest that Plce1 is predominantly expressed in podocytes and resistance vessels in the kidneys.
Fig. 6.

A and B: UMAP (A) and violin plots (B) demonstrating 14 separate cell populations noted from single cell RNA profiling of surgically removed human kidney tissue. The violin plots demonstrate podocyte-specific marker podocin (NPHS2). Phospholipase C-ε1 (PLCE1) transcript was selectively elevated in podocytes, although lower-level RNA expression was noted in other tissues as well. C: staining of Plce1 within the glomerulus from a hemagglutinin (HA)-tagged Plce1-expressing mouse. Plce1 was noted to be expressed in podocytes, consistent with previous data. Plce1 expression was also noted within smooth muscle cells of the glomerular arteriole. D: negative control. A wild-type mouse glomerulus stained with anti-HA antibody is shown. No staining was noted. NKC, natural killer cell; aLOH, ascending loop of Henle (LOH); dLOH, descending LOH; VSMC, vascular smooth muscle cell.
Fig. 7.
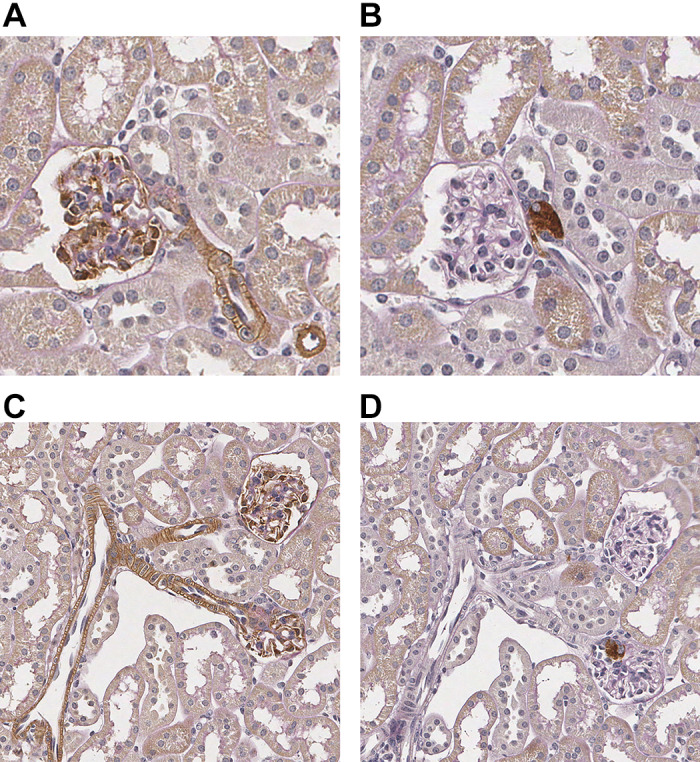
A: staining of phospholipase C-ε1 (Plce1) within the glomerulus and afferent arteriole from a hemagglutinin (HA)-tagged Plce1-expressing mouse at ×40 magnification. B: staining of renin within the juxtaglomerular apparatus of the afferent arteriole in the same glomerulus as shown in A. Magnification: ×40. C: staining of Plce1 in the afferent arterioles and interlobular artery in a HA-tagged Plce1-expressing mouse at ×20 magnification. D: staining of renin in juxtaglomerular cells from the afferent arterioles from the same glomeruli as shown in C. Magnification: ×20.
DISCUSSION
In the present study, we demonstrated that Plce1−/− mice have largely normal kidney structure and function at baseline but develop albuminuria and glomerular damage when exposed to hypertension induced by ANG II and DOCA + salt + UNx + NE. These findings support the hypothesis that Plce1 deficiency predisposes to glomerular damage after injury or when exposed to stressors including hypertension.
Loss-of-function mutations in PLCE1 are thought to predispose to the development of juvenile FSGS or DMS (5, 12). However, these mutations are insufficient for the development of either of these syndromes, as persons who express identical loss-of-function mutations have no detectable kidney disease (14). We did find that untreated Plce1−/− mice have modestly enlarged glomeruli at baseline. However, none of the other structural parameters, in particular cortical glomerular density, mesangial index, podocyte volume and density, were significantly different from wild-type or heterozygous mice. Additionally, Plce1−/− mice do not have any proteinuria, all of which is consistent with a relatively normal baseline kidney structure. The significance of the mild glomerulomegaly at baseline is unclear; it is not related to decreased glomerular number. We speculate that it may be the sequela of mild hyperfiltration at baseline in Plce1−/− mice (34). Overall, our findings are consistent with a previously reported absence of pathological kidney phenotype in unchallenged global Plce1−/− mice (17). As such, it has been postulated that a pathophysiological stressor is needed to induce the nephrotic phenotype in the setting of Plce1 deficiency. This requirement for a physiological stressor has also been noted in other murine models that mimic human glomerular disease: ANG II is necessary for the development of the full nephrotic phenotype in Trpc6+/+ and podocin/Vpr mice, which are models of primary FSGS and HIV-associated nephropathy, respectively (11, 19). Thus, ANG II-induced hypertension appears to provoke clinically meaningful phenotypes in mice with genetic predisposition to glomerular disease.
ANG II-induced hypertension in Plce1−/− mice recapitulated many of the clinical aspects of PLCE1 deficiency in patients with FSGS and DMS. Focal glomerulosclerosis and diffuse mesangial matrix expansion were noted in Plce1−/− mice after treatment with ANG II but not in wild-type or heterozygous mice. These patterns of injury mirror their human counterparts with loss-of-function mutations in PLCE1, namely, FSGS and DMS (5, 12, 17). Additionally, our murine model of Plce1 deficiency shared a similar inheritance pattern to patients with PLCE1 mutations. Loss-of-function PLCE1 mutations have been proposed to be inherited in a homozygous recessive manner (5). Consistent with this, the heterozygous Plce1+/− mice in our study did not develop a detectable glomerular phenotype when exposed to ANG II-mediated hypertension.
High levels of PLCE1 expression have been preferentially localized to podocytes in past studies (6, 17). Our scRNA and immunohistochemistry experiments are consistent with these data, as we demonstrated robust Plce1 protein expression in podocytes via both immunohistochemical staining and mRNA expression via scRNA-Sequencing of human kidney tissue. The function of PLCE1 within podocytes is not fully understood. In all cell types, PLCE1 is involved in the generation of diacylglycerol and release of Ca2+ from intracellular stores (28). PLCE1 is located downstream of a multitude of different G protein-coupled receptors, serving as an intracellular integrator for extracellular signals (28). PLCE1 is known to regulate certain signaling cascades within podocytes. PLCE1 is known to directly bind to TRPC6 (20). While PLCE1 does not directly interact with nephrin at the slit diaphragm, PLCE1 has been found to directly interact with both v-Raf murine sarcoma viral oncogene homolog B and IQGAP1 within podocytes (6, 25). Given the importance of IQGAP1 in the regulation of the actin cytoskeleton, it has been suggested that PLCE1 likely is involved in the regulation of the podocyte’s actin cytoskeleton. Consistent with this, PLCE1 is known to directly interact with Advillin (26), a regulator of the actin cytoskeleton (23). In the same set of experiments, PLCE1 knockdown impaired the migratory activity of cultured podocytes, further supporting a role for PLCE1 in the regulation of actin dynamics. Our findings also suggest that PLCE1 plays an important role in the regulation of podocyte homeostasis. In Plce1−/− mice, ANG II-mediated hypertension caused significant focal podocyte foot process effacement as well as diffuse podocyte loss after 4 wk of treatment. Additionally, we noted focal podocyte loss as manifested by loss of GLEPP1 and WT1 staining within focally sclerosed glomeruli. Since the presence of Plce1 abrogated these effects, it implies that Plce1 may protect podocytes from hyperfiltration-mediated injury. Further studies are needed to precisely define the molecular pathways through which PLCE1 regulates these effects.
Next, we tested whether the effects of ANG II on glomerular damage in Plce1-deficient mice were specific to ANG II or could be caused by hypertension per se. To address this question, we used a second model of hypertension, the DOCA + salt + UNx model, which can also lead to albuminuria and glomerulosclerosis in genetically susceptible mice (3) and also rats through its effects on the development of glomerular capillary hypertension (8). In our preliminary dose-finding experiments, DOCA + salt + UNx was unable to raise blood pressure, and, as such, we added NE via mini-pump, which significantly increased blood pressure. We determined that Plce1−/− mice develop similar levels of albuminuria and glomerulosclerosis when treated with DOCA + salt + UNx + NE, suggesting that Plce1 deficiency predisposes to glomerular injury from multiple different types of hypertension.
One unexpected finding in our study was differences in blood pressure experienced by Plce1−/− mice in response to both models of hypertension. Plce1−/− mice had lower levels of hypertension at week 2 of ANG II infusion and week 4 of the DOCA + salt + UNx + NE protocol. To our knowledge, Plce1 has not been previously shown to affect blood pressure. In addition, albuminuria was decreased in Plce1−/− mice at the time of lower blood pressure and increased again at week 4 when blood pressure had increased again. This is consistent with the hypothesis that changes in glomerular perfusion pressure are the driving force behind glomerular damage in Plce1 deficiency. We do not suspect that these differences in blood pressure confound our results: Plce1−/− mice developed worse glomerular damage in spite of a lower blood pressure.
Our scRNA and immunohistochemical staining data demonstrate a wider expression of PLCE1 than had previously been reported. Consistent with previous experiments (6, 17), we confirmed abundant PLCE1 expression in podocytes. However, we also detected enhanced PLCE1 expression within a population of vascular smooth muscle cells. To our knowledge, PLCE1 expression has not been previously demonstrated within vascular smooth muscle cells. Our data suggest that Plce1 is predominantly found in the afferent and interlobular vessels. No significant expression was detected in main segment branches of the main renal arteries. This would suggest that Plce1 is predominantly expressed in resistance vessels in the kidney. We speculate that the expression of Plce1 within arteriolar smooth muscle cells may play a role in the differences in blood pressure in Plce1−/− mice. ANG II-mediated hypertension is known to be predominantly mediated by the kidneys. This has been demonstrated by the abrogation of ANG II-mediated hypertension via renal cross-transplantation of ANG II type 1 receptor knockout kidneys into wild-type mice during ANG II infusions (7). Furthermore, this effect is likely in part due to the deletion of the ANG II type 1 receptor from resistance arteries in the kidney leading to changes in natriuresis (29). Further experiments are needed to clarify the role of Plce1 in the vasculature.
We did not find significant expression of Plce1 within the tubulointerstitium of HA-tagged Plce1 mice. Additionally, we only detected minimal amounts of PLCE1 mRNA within tubuli of our human samples. In kidneys of Plce1−/− mice exposed to ANG II for 4 wk, we detected a trend toward higher mRNA expression of Col1a1, Col3a1, and Fn1. At a time point when glomerular damage is already advanced, these very mild changes likely reflect early stages of interstitial fibrosis secondary to glomerular damage rather than a direct effect of Plce1 in the tubule interstitium consistent with the observed predominant expression of Plce1 within the podocytes and arterioles.
In conclusion, we demonstrated that Plce1 deficiency causes an increased susceptibility to glomerular damage from hypertensive stressors. Further experiments are needed to elucidate the molecular pathways and cellular localization by which Plce1 exerts its protective effects in the glomerulus against hypertensive stressors.
GRANTS
D. K. Atchison received funding from The Division of Nephrology, University of Michigan [National Institutes of Health (NIH) Training Grant T32-DK-007378]. M. Bitzer received funding from NIH Grant R01-DK-100449 and the George O’Brien Kidney Translational Core Center Small Grants Program (NIH Grant P30-DK-081943). R. C. Wiggins received funding from NIH Grant R01-DK-102643. A. V. Smrcka received funding from NIH Grant R35-GM-127303. S. K. Ganesh received funding from NIH Grants R01-HL-122684 and R01-HL-139672.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.K.A., C.L.O., R.C.W., A.V.S., and M.B. conceived and designed research; D.K.A., C.L.O., R.M., E.A.O., and S.K.G. performed experiments; D.K.A., C.L.O., R.M., E.A.O., S.K.G., R.C.W., and M.B. analyzed data; D.K.A., C.L.O., S.K.G., R.C.W., A.V.S., and M.B. interpreted results of experiments; D.K.A., C.L.O., R.M., and E.A.O. prepared figures; D.K.A. and C.L.O. drafted manuscript; D.K.A., C.L.O., S.K.G., R.C.W., A.V.S., and M.B. edited and revised manuscript; M.B. approved final version of manuscript.
REFERENCES
- 1.Akchurin O, Reidy KJ. Genetic causes of proteinuria and nephrotic syndrome: impact on podocyte pathobiology. Pediatr Nephrol 30: 221–233, 2015. doi: 10.1007/s00467-014-2753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atchison DK, Harding P, Beierwaltes WH. Vitamin D increases plasma renin activity independently of plasma Ca2+ via hypovolemia and β-adrenergic activity. Am J Physiol Renal Physiol 305: F1109–F1117, 2013. doi: 10.1152/ajprenal.00010.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, Vining C, Randolph A, Herbach N, Wanke R, Atkins KB, Gyung Kang H, Henger A, Brakebusch C, Holzman LB, Kretzler M. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 84: 920–930, 2013. doi: 10.1038/ki.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohrer MP, Deen WM, Robertson CR, Brenner BM. Mechanism of angiotensin II-induced proteinuria in the rat. Am J Physiol 233: F13–F21, 1977. doi: 10.1152/ajprenal.1977.233.1.F13. [DOI] [PubMed] [Google Scholar]
- 5.Boyer O, Benoit G, Gribouval O, Nevo F, Pawtowski A, Bilge I, Bircan Z, Deschênes G, Guay-Woodford LM, Hall M, Macher MA, Soulami K, Stefanidis CJ, Weiss R, Loirat C, Gubler MC, Antignac C. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J Med Genet 47: 445–452, 2010. doi: 10.1136/jmg.2009.076166. [DOI] [PubMed] [Google Scholar]
- 6.Chaib H, Hoskins BE, Ashraf S, Goyal M, Wiggins RC, Hildebrandt F. Identification of BRAF as a new interactor of PLCε1, the protein mutated in nephrotic syndrome type 3. Am J Physiol Renal Physiol 294: F93–F99, 2008. doi: 10.1152/ajprenal.00345.2007. [DOI] [PubMed] [Google Scholar]
- 7.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dworkin LD, Hostetter TH, Rennke HG, Brenner BM. Hemodynamic basis for glomerular injury in rats with desoxycorticosterone-salt hypertension. J Clin Invest 73: 1448–1461, 1984. doi: 10.1172/JCI111349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, Wickman LT, Wiggins JE, Muchayi T, Fingar D, Shedden KA, Inoki K, Wiggins RC. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol 23: 1351–1363, 2012. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, Shedden KA, Dysko RC, Wiggins JE, Wiggins RC. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int 81: 40–55, 2012. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G, Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L, Birbaumer L, Rosenberg P, Winn MP. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol 22: 526–535, 2011. doi: 10.1681/ASN.2010050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gbadegesin R, Hinkes BG, Hoskins BE, Vlangos CN, Heeringa SF, Liu J, Loirat C, Ozaltin F, Hashmi S, Ulmer F, Cleper R, Ettenger R, Antignac C, Wiggins RC, Zenker M, Hildebrandt F. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 23: 1291–1297, 2008. doi: 10.1093/ndt/gfm759. [DOI] [PubMed] [Google Scholar]
- 13.Giachini FR, Sullivan JC, Lima VV, Carneiro FS, Fortes ZB, Pollock DM, Carvalho MH, Webb RC, Tostes RC. Extracellular signal-regulated kinase 1/2 activation, via downregulation of mitogen-activated protein kinase phosphatase 1, mediates sex differences in desoxycorticosterone acetate-salt hypertension vascular reactivity. Hypertension 55: 172−179, 2010. doi: 10.1161/HYPERTENSIONAHA.109.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert RD, Turner CL, Gibson J, Bass PS, Haq MR, Cross E, Bunyan DJ, Collins AR, Tapper WJ, Needell JC, Dell B, Morton NE, Temple IK, Robinson DO. Mutations in phospholipase C epsilon 1 are not sufficient to cause diffuse mesangial sclerosis. Kidney Int 75: 415–419, 2009. doi: 10.1038/ki.2008.573. [DOI] [PubMed] [Google Scholar]
- 15.Gillies CE, Putler R, Menon R, Yasutake K, Nair V, Hoover P, Lieb D, Li S, Eddy S, Fermin D, McNulty MT; Nephrotic Syndrome Study Network (NEPTUNE), Hacohen N, Kiryluk K, Kretzler M, Wen X, Sampson MG. An eQTL Lanscape of Kidney Tissue in Human Nephrotic Syndrome. Am J Hum Genet 103: 232–244, 2018. doi: 10.1016/j.ajhg.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Z, Feng L, Zhang X, Geng Y, Parodi DA, Suarez-Quian C, Dym M. Expression of Col1a1, Col1a2 and procollagen I in germ cells of immature and adult mouse testis. Reproduction 130: 333–341, 2005. doi: 10.1530/rep.1.00694. [DOI] [PubMed] [Google Scholar]
- 17.Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE, Ashraf S, Becker C, Hennies HC, Goyal M, Wharram BL, Schachter AD, Mudumana S, Drummond I, Kerjaschki D, Waldherr R, Dietrich A, Ozaltin F, Bakkaloglu A, Cleper R, Basel-Vanagaite L, Pohl M, Griebel M, Tsygin AN, Soylu A, Müller D, Sorli CS, Bunney TD, Katan M, Liu J, Attanasio M, O’toole JF, Hasselbacher K, Mucha B, Otto EA, Airik R, Kispert A, Kelley GG, Smrcka AV, Gudermann T, Holzman LB, Nürnberg P, Hildebrandt F. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 38: 1397–1405, 2006. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann S, Podlich D, Hähnel B, Kriz W, Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol 15: 1475–1487, 2004. doi: 10.1097/01.ASN.0000127988.42710.A7. [DOI] [PubMed] [Google Scholar]
- 19.Ideura H, Hiromura K, Hiramatsu N, Shigehara T, Takeuchi S, Tomioka M, Sakairi T, Yamashita S, Maeshima A, Kaneko Y, Kuroiwa T, Kopp JB, Nojima Y. Angiotensin II provokes podocyte injury in murine model of HIV-associated nephropathy. Am J Physiol Renal Physiol 293: F1214–F1221, 2007. doi: 10.1152/ajprenal.00162.2007. [DOI] [PubMed] [Google Scholar]
- 20.Kalwa H, Storch U, Demeletiner J, Fiedler S, Mayer T, Kanneler M, Fahlbusch M, Barth H, Smrcka A, Hildebrandt F, Gudermann T, Dietrich A, Phospholipase C. Epsilon (PLCε) induced TRPC6 activation: a common but redundant mechanism in primary pdodcytes. J Cell Physiol 230: 1389–1399, 2015. doi: 10.1002/jcp.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, Mokrzycki MH, Kimmel PL, Limou S, Ahuja TS, Berns JS, Fryc J, Simon EE, Smith MC, Trachtman H, Michel DM, Schelling JR, Vlahov D, Pollak M, Winkler CA. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22: 2129–2137, 2011. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai JY, Luo J, O’Connor C, Jing X, Nair V, Ju W, Randolph A, Ben-Dov IZ, Matar RN, Briskin D, Zavadil J, Nelson RG, Tuschl T, Brosius FC III, Kretzler M, Bitzer M. MicroRNA-21 in glomerular injury. J Am Soc Nephrol 26: 805–816, 2015. doi: 10.1681/ASN.2013121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Liang W, Yang Y, Pan Y, Yang Q, Chen X, Singhal PC, Ding G. IQGAP1 regulates actin cytoskeleton organization in podocytes through interaction with nephrin. Cell Signal 27: 867–877, 2015. doi: 10.1016/j.cellsig.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menon R, Otto EA, Hoover P, Eddy S, Mariani L, Godfrey B, Berthier CC, Eichinger F, Subramaniam L, Harder J, Ju W, Nair V, Larkina M, Naik AS, Luo J, Sealfon R, Troyanskaya O, Hacohen N, Hodgin JB, Kretzler M; Kidney Precision Medicine Project (KPMP), Nephrotic Syndrome Study Network (NEPTUNE) . Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perico L, Mandalà M, Schieppati A, Carrara C, Rizzo P, Conti S, Longaretti L, Benigni A, Remuzzi G. BRAF signaling pathway inhibition, podocyte injury, and nephrotic syndrome. Am J Kidney Dis 70: 145–150, 2017. doi: 10.1053/j.ajkd.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Rao J, Ashraf S, Tan W, van der Ven AT, Gee HY, Braun DA, Fehér K, George SP, Esmaeilniakooshkghazi A, Choi WI, Jobst-Schwan T, Schneider R, Schmidt JM, Widmeier E, Warejko JK, Hermle T, Schapiro D, Lovric S, Shril S, Daga A, Nayir A, Shenoy M, Tse Y, Bald M, Helmchen U, Mir S, Berdeli A, Kari JA, El Desoky S, Soliman NA, Bagga A, Mane S, Jairajpuri MA, Lifton RP, Khurana S, Martins JC, Hildebrandt F. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J Clin Invest 127: 4257–4269, 2017. doi: 10.1172/JCI94138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, Rauterberg J, Lorkowski S. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol 15: 5707–5719, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal 24: 1333–1343, 2012. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol 26: 2953–2962, 2015. doi: 10.1681/ASN.2014080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, Wiggins RC. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol 25: 1118–1129, 2014. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 97: 1305–1313, 2005. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]
- 32.Wharram BL, Goyal M, Gillespie PJ, Wiggins JE, Kershaw DB, Holzman LB, Dysko RC, Saunders TL, Samuelson LC, Wiggins RC. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J Clin Invest 106: 1281–1290, 2000. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 34.Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, Kuick RD, Wiggins RC. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol 16: 2953–2966, 2005. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- 35.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]