

Abstract
Emerging evidence has demonstrated that (pro)renin receptor (PRR)-mediated activation of intrarenal renin-angiotensin system (RAS) plays an essential role in renal handling of Na+ and water balance and blood pressure. The present study tested the possibility that the intrarenal RAS served as a molecular target for the protective action of ELABELA (ELA), a novel endogenous ligand of apelin receptor, in the distal nephron. By RNAscope and immunofluorescence, mRNA and protein expression of endogenous ELA was consistently localized to the collecting duct (CD). Apelin was also found in the medullary CDs as assessed by immunofluorescence. In cultured CD-derived M1 cells, exogenous ELA induced parallel decreases of full-length PRR (fPRR), soluble PRR (sPRR), and prorenin/renin protein expression as assessed by immunoblotting and medium sPRR and prorenin/renin levels by ELISA, all of which were reversed by 8-bromoadenosine 3′,5′-cyclic monophosphate. Conversely, deletion of PRR in the CD or nephron in mice elevated Apela and Apln mRNA levels as well as urinary ELA and apelin excretion, supporting the antagonistic relationship between the two systems. Administration of exogenous ELA-32 infusion (1.5 mg·kg−1·day−1, minipump) to high salt (HS)-loaded Dahl salt-sensitive (SS) rats significantly lowered mean arterial pressure, systolic blood pressure, diastolic blood pressure, and albuminuria, accompanied with a reduction of urinary sPRR, angiotensin II, and prorenin/renin excretion. HS upregulated renal medullary protein expression of fPRR, sPRR, prorenin, and renin in Dahl SS rats, all of which were significantly blunted by exogenous ELA-32 infusion. Additionally, HS-induced upregulation of inflammatory cytokines (IL-1β, IL-2, IL-6, IL-17A, IFN-γ, VCAM-1, ICAM-1, and MCP-1), fibrosis markers (TGF-β1, FN, Col1A1, PAI-1, and TIMP-1), and kidney injury markers (NGAL, Kim-1, albuminuria, and urinary NGAL excretion) were markedly blocked by exogenous ELA infusion. Together, these results support the antagonistic interaction between ELA and intrarenal RAS in the distal nephron that appears to exert a major impact on blood pressure regulation.
Keywords: (pro)renin receptor, ELABELA, intrarenal renin-angiotensin system, kidney injury, salt-sensitive hypertension
INTRODUCTION
Hypertension is the most prevalent chronic disease and the major risk factor for the high morbidity and mortality of cardiovascular disease and stroke, which have resulted in overwhelming health and economic problems nationally and internationally, as evidenced by the phenomenon that the number of adults with elevated blood pressure has increased dramatically from 594 million in 1975 to 1.13 billion in 2015 (1a, 57). More importantly, enhanced sensitivity of blood pressure to salt intake is present in nearly half of Americans who are afflicted with hypertension, including ~75% of African-American patients with hypertension (1a, 57). The positive correlation relationship between dietary salt intake and blood pressure has been well characterized (1, 10a), but the precise mechanisms of salt-sensitive hypertension are poorly understood. A better understanding of the mechanisms of salt-sensitive hypertension is important for the prevention and control of hypertension and improving mortality and healthcare costs worldwide.
The apelinergic system has emerged as a key signaling pathway in regulating cardiovascular homeostasis, since the discovery of the G protein-coupled receptor in 1993, which shares significant sequence homology and similar patterns of tissue expression with the angiotensin II type 1 receptor (AT1R) called apelin peptide jejunum (APJ, apelin receptor/Aplnr; Ref. 30) and its endogenous ligand, apelin (46). The beneficial effects of apelin-APJ system on hypertension, vasodilation, fluid and cardiovascular homeostasis, and energy metabolism, et cetera, are well established (19). ELABELA (ELA, also known as Apela/Toddler; http://www.elabela.com/), a newly discovered 32-residue peptide hormone, has been identified as an early endogenous ligand for APJ and is highly conserved among vertebrates (6, 31). Similar to the functions of apelin, ELA has also been shown to regulate fluid homeostasis (10), lower blood pressure (52, 53), promote angiogenesis and relaxation of blood vessels (10), and protect against heart failure (40) and renal injury (4), et cetera. Additionally, both apelin and ELA protect against pressure overload- and angiotensin II (ANG II)-induced heart failure and cardiac damage and regulate renin-angiotensin system (RAS) with a subtle difference, apelin negatively regulates ANG II-AT1R signaling through heterodimeric interaction of APJ and AT1R in atherosclerosis (7, 43) or upregulation of angiotensin-converting enzyme 2 (ACE2) expression in pressure overload heart failure (41) and ANG II-mediated heart disease (39, 56), whereas ELA downregulates angiotensin-converting enzyme (ACE) expression with little effect on ACE2 expression in the stressed hearts (40). These results implicate fine-tuned mechanisms for a ligand-induced APJ activation on RAS regulation.
The RAS has been known for over a century and has a pivotal role in the maintenance of extracellular volume homeostasis and blood pressure through complex mechanisms. Aberrant activation of intrarenal RAS has been recognized as an important mechanism for hypertension as well as cardiovascular and renal diseases (55). (Pro)renin receptor (PRR), a new component of the RAS, was first cloned as a specific receptor for prorenin and renin (26). Increasing evidence has demonstrated that PRR-mediated activation of intrarenal RAS plays an essential role in renal handling of Na+ and water balance and blood pressure (33, 35, 37, 51, 55). In particular, Kobori et al. (18) and Zhu et al. (58), respectively, reported that high salt (HS) intake suppressed circulating RAS but enhanced intrarenal RAS as reflected by increases of renal PRR, angiotensinogen (AGT), ACE, AT1R, and ANG II levels in Dahl salt-sensitive (SS) rats. Recently, Schreiber et al. (42) showed that long-term Apela gene delivery by adeno-associated virus serotype 9 vectors effectively lowered hypertension, preserved glomerular architecture, and decreased renal fibrosis in HS-loaded Dahl SS rats. However, the mechanisms on the antihypertensive and renoprotective effects of ELA remain largely unknown. The present study tested the possibility of the mutual interaction between the intrarenal RAS and apelin/ELA, with particular emphasis on the intrarenal RAS as a molecular target for the protective action of ELA in the distal nephron in Dahl SS rats.
MATERIALS AND METHODS
Animal care.
All rats and mice were given free access to tap water and were fed the standard diet (Na+: 0.3% and K+: 1%). Rats and mice were housed in a temperature- and humidity-controlled room with a 12:12-h light-dark cycle. All animal studies were conducted with the approval of the University of Utah Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Generation of cell-specific PRR KO mice.
Mice with conditional deletion of PRR in the collecting duct (termed CD PRR KO) were generated by genetic crosses between PRR-floxed mice and aquaporin 2 (AQP2)-Cre mice (48). Mice homozygous for the floxed PRR gene but lacking the AQP2-Cre transgene were used as controls (termed Floxed). Mice with nephron-specific PRR KO (termed Neph PRR KO) were generated by genetic crosses between PRR-floxed mice and mice transgenic for Pax8-rtTA transgene (which contains the reverse tetracycline transactivator under control of the paired box gene 8 promoter) and the LC-1 transgene (which encodes tetracycline-inducible bicistronic Cre recombinase and luciferase; Ref. 36). To induce nephron PRR KO, at 1 mo of age, mice were given 2 mg/mL doxycycline in 5% sucrose drinking water for 14 days followed by 4 wk of doxycycline. PRR-floxed mice without Pax8-rtTA or LC-1 transgenes were given doxycycline treatment at 1 mo of age and were used as controls (termed Floxed).
Animal treatment.
Male Dahl SS rats (10–12 wk old, purchased from Charles River) were fed a normal-Na+ (NS) diet (Na+: 0.3%), a high-salt (HS) diet (Na+: 4%), or a HS diet in combination with exogenous ELA-32 treatment (1.5 mg·kg−1·day−1 subcutaneous minipump infusion; Refs. 9, 40) for 14 days. The NS and HS groups received 0.9% NaCl (subcutaneous minipump infusion). Rat ELA-32 peptide (>98% purity), EKSVNFPRRRKLYRHNCFRRRCISLHSRVPFP, was purchased from GenScript (Piscataway, NJ). Rats were housed in metabolic cages in a temperature- and humidity-controlled room with a 12:12-h light-dark cycle for 24-h urine collection and to measure body weight and 24-h food and water intake. At the end of the treatment, rats were euthanized and blood and kidneys were harvested.
Measurement of blood pressure.
Blood pressure was assessed by using radiotelemetry. Dahl SS rats were instrumented with radiotelemetric devices. The radiotelemetric device was implanted via catheterization of carotid artery and was turned on for 4 h/day from 5:00 to 9:00 PM.
Enzyme immunoassay.
To detect urinary, plasma, and cell culture medium soluble PRR (sPRR), total renin/prorenin, and cAMP, urinary and plasma AGT and ANG II, urinary ELA, apelin, PGE2, neutrophil gelatinase-associated lipocalin (NGAL), and albumin, we used the commercially available enzyme immunoassay (EIA) kits according to the manufacturers’ instructions: soluble (Pro) renin Receptor Assay Kit (cat. no. 27782; Immuno-Biological, Gunma, Japan), Rat Prorenin and Renin Total Antigen ELISA Kit (cat. no. RPRENKT-TOT; Molecular Innovations), Cyclic AMP ELISA Kit (cat. no. 581001; Cayman Chemical), Rat Angiotensinogen ELISA Kit (cat. no. MBS762162; MyBioSource), ANG II ELISA kit (cat. no. ADI-900-204; Enzo Life Sciences, Farmingdale, NY), [pGlu1]-ELA-32 (Human) EIA Kit (cat. no. EK-007-19; Phoenix Pharmaceuticals), Mouse Apelin EIA Kit (cat. no. Q9R0R4; RayBiotech Life), Prostaglandin E2 ELISA Kit (cat. no. 514010; Cayman Chemical), and Rat NGAL ELISA Kit (cat. no. 501530; Cayman Chemical), respectively.
Quantitative RT-PCR.
Snap-frozen renal samples were homogenized in TRIzol reagent (cat. no. 15596018; Life Technologies). Total RNA isolation and reverse transcription (RT) were performed according to the manufacturer’s recommendations. We used 1 μg of total RNA as a template for RT by using the Transcriptor First Strand cDNA Synthesis Kit (cat. no. 4368813; Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using the ABI PRISM StepOnePlus System (Applied Biosystems, Life Technologies) and the SYBR Premix Ex Taq kit (Tli RNaseH Plus, cat. no. 1803132; Thermo Fisher Scientific) according to the manufacturers’ instructions. The primer sequences used are shown in Table 1. All reactions were run in duplicate. The data are shown as relative values normalized by GAPDH.
Table 1.
Sequences of oligonucleotides used for quantitative RT-PCR
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Product Size, bp |
|---|---|---|---|
| R PRR | ATCCTTGAGACGAAACAAGA | AGCCAGTCATAATCCACAGT | 109 |
| R Renin | GATCACCATGAAGGGGGTCTCTGT | GTTCCTGAAGGGATTCTTTTGCAC | 274 |
| R AGT | AGCATCCTCCTTGAACTCCA | TGATTTTTGCCCAGGATAGC | 244 |
| R ACE | GAGCCATCCTTCCCTTTTTC | CCACATGTTCCCTAGCAGGT | 266 |
| R AT1R | TGAGCACGCTTTCTTACC | CTGCTTAGATCCTGAGGC | 189 |
| R AT2R | CAATCTGGCTGTGGCTGACTT | TGCACATCACAGGTCCAAAGA | 101 |
| R ACE2 | GAATGCGACCATCAAGCG | CAAGCCCAGAGCCTACGA | 228 |
| R COX-2 | TCCTCAGAAGAACCTTTTCC | GGAGTCTGGAACATTGTGAA | 265 |
| R TNF-α | AGTGACAAGCCCGTAGCC | GGGTGAGGAGCACGTAGTC | 205 |
| R IL-1β | CGACAGTGAGGAGAATGACC | TCCACGGGCAAGACATAG | 206 |
| R IL-2 | AGCGTGTGTTGGATTTGACTC | ATGATGCTTTGACAGATGGCTA | 190 |
| R IL-6 | GCCAGAGTCATTCAGAGCAATA | GTTGGATGGTCTTGGTCCTTAG | 160 |
| R IL-17A | GGGAAGTTGGACCACCACCT | TTCTCCACCCGGAAAGTGAA | 101 |
| R IFN-γ | AGGCCATCAGCAACAACATAAGTG | GACAGCTTTGTGCTGGATCTGTG | 140 |
| R Apela | ATTCTCGAGTGCCCTTCCCATG | TCCGAAAAGCCATCCAAGGTAC | 174 |
| R Apln | GACCCATGCCTTTCTAAAGCAG | TAACAGGTGCAAGATGAGAGCC | 131 |
| R Aplnr | CAGGGTGCTTGCTGAGTTGCTG | GGAAAACCAGAATGTAGATGGC | 194 |
| R TGF-β1 | CACGATCATGTTGGACAACTGCTCC | CTTCAGCTCCACAGAGAAGAACTGC | 298 |
| R Fibronectin | AGACCATACCTGCCGAATGTAG | GAGAGCTTCTTGTCCTGTAGAG | 129 |
| R Collagen1a | ACGCATGGCCAAGAAGACATCCC | TTGCATAGCACGCCATCGCACAC | 143 |
| R PAI-1 | TGGTGAACGCCCTCTATTTC | GAGGGGCACATCTTTTTCAA | 248 |
| R NGAL | TCTGGGCCTCAAGGATAACAAC | AGACAGGTGGGACCTGACCA | 127 |
| R TIMP-1 | GACCACCTTATACCAGCGTT | GTCACTCTCCAGTTTGCAAG | 334 |
| R KIM-1 | CTCCAGGAAGCCGAGCAAAC | AAGCACTGGGTACAGATCCAAA | 124 |
| R MCP-1 | GCCCCACTCACCTGCTGCTACT | CCTGCTGCTGGTGATTCTCTTGT | 87 |
| R ICAM-1 | TACAAGTGCCGTGCCTTTAG | CATGGTACAGCACTGTCAGGT | 70 |
| R VCAM-1 | GCTACATCCACACTGACGCT | CAGGGAATGAGTAGACCTCCA | 124 |
| R GAPDH | GTCTTCACTACCATGGAGAAGG | TCATGGATGACCTTGGCCAG | 197 |
| M PRR | TCTCCGAACTGCAAGTGCTA | CTGCAAACTTTTGGAGAGCA | 195 |
| M Apela | TTTGCAGAGACTTCCCGCTT | GCTCACCCCACATCCTATGG | 93 |
| M Apln | GCTGCTGCTGCTCTGGCTCT | GGGGGCGCTGTCTGCGAAAT | 176 |
| M Aplnr | GCCTGTCATGGTGTTCCG | CTCAATGCGCTCCTTTCGG | 229 |
| M GAPDH | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA | 123 |
M, Mus musculus; R, Rattus norvegicus.
Immunoblot analysis.
Tissue samples from the renal cortex and medulla were lysed and subsequently sonicated in homogenization buffer (0.3 M sucrose, 50 mM Tris·HCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, pH 7.5) with protease inhibitor cocktail (Roche, Berlin, Germany). Cell samples were homogenized in cell lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM DTT, 1% Nonidet P-40, 1% protease inhibitor cocktail, pH 7.5). Protein concentrations were determined with the Pierce BCA Protein Assay Kit (cat. no. NCI3225CH; Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s instructions. Samples were resolved by SDS-PAGE and transferred onto polyvinylidene fluoride membrane (Immobilon-P; Millipore, Bedford, MA). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline-Tween 20 (TBST) for 1 h at room temperature, followed by incubation with primary antibodies [PRR, 1:1,000 dilution, HPA003156, Sigma; renin, 1:1,000 dilution, sc-133145, Santa Cruz Biotechnology; APJ, 1:1,000 dilution, ab214369, Abcam; cyclooxygenase-2 (COX-2), 1:1,000 dilution, cat. no. 160126, Cayman Chemical; phosphorylated CRE-binding protein (CREB; Ser133), 1:1,000 dilution, cat. no. 9198, Cell Signaling Technology; CREB (48H2), 1:1,000 dilution, cat. no. 9197, Cell Signaling Technology; β-actin, 1:10,000 dilution, A2066, Sigma-Aldrich] diluted in antibody dilution buffer [1.5 g of bovine serum albumin (BSA), 0.1 g of NaN3, 50 mL of TBST] overnight at 4°C. After membranes were washed with TBST, they were incubated with secondary antibodies (goat anti-rabbit/mouse horseradish peroxidase-conjugated secondary antibody; Thermo Fisher Scientific) for 1 h at room temperature and visualized with enhanced chemiluminescence (EMD Millipore). Signals on immunoblots were detected using ChemiDoc XRS+ System (Bio-Rad) and quantitated using Image-Pro Plus version 6.0 software. The expression of protein was calculated in relation to β-actin.
Antibody for ELA peptide.
Rabbit polyclonal antibody against ELA (AB1095) was raised from a mouse COOH-terminal epitope (K33LYRHNCFRRRCIPLHSRVPFP54). This antibody was used for immunofluorescence staining in this study.
ELA RNAscope assay.
RNAscope assay was performed on paraffin-embedded tissue using RNAscope 2.5 HD Reagent Kit-RED (cat. no. 322350; Advanced Cell Diagnostics, Hayward, CA) according to the manufacturer’s recommendations. Briefly, tissue sections were deparaffinized with xylene and 100% ethanol and incubated with pretreat-1 solution for 10 min, pretreat-2 for 15 min, and pretreat-3 for 30 min (Pretreatment Reagents, cat. no. 322330). The slides were then hybridized with a custom mApela (m, Mus musculus; cat. no. 416811) C2 probe at 40°C for 2 h. After hybridizations, slides were subjected to signal amplification using RNAscope 2.5 HD Detection Kit (cat. no. 322360), and hybridization signal was detected using a mixture of Fast-RED solutions A and B (1:60). After slides were counterstained with Gill hematoxylin, they were dried in a 60°C dry oven for 15 min and mounted with Glycergel Mounting Medium. Images were captured using a Leica DMI4000B fluorescence microscope (Wetzlar, Germany).
Immunofluorescence staining.
Kidneys were fixed overnight at 4°C in 3% paraformaldehyde-0.1 M phosphate salt buffer and processed for immunofluorescence analysis. Five-micrometer cryosections were cut after cryoprotection with 30% sucrose in phosphate salt buffer for ≥12 h. Slides were preincubated with 3% BSA to block nonspecific reactions and were then incubated with anti-ELA antibody (1:100 dilution; AB1095) or ELA peptide (KLYRHNCFRRRCIPLHSRVPFP), anti-apelin antibody (1:100 dilution; ab59469; Abcam) or apelin peptide (ab141010; Abcam), anti-APJ antibody (1:100 dilution; ab214369; Abcam), anti-Na/K/2Cl cotransporter (NKCC2) antibody (1:100 dilution; SPC-401; StressMarq), and anti-AQP2 antibody (1:1,000 dilution; sc-9882; Santa Cruz Biotechnology). Sections were incubated with primary antibodies at 4°C overnight and with fluorescent dye-conjugated secondary antibodies at room temperature for 1 h. Images were captured using a Leica DMI4000B fluorescence microscope.
Cell culture.
Collecting duct-derived M1 cells or mouse inner medullary collecting duct (IMCD) cell line (IMCD3; ATCC CRL-2123; American Type Culture Collection, Manassas, VA) were grown to 95% confluence in Dulbecco’s modified Eagle medium/Ham’s F-12 medium supplemented with 10% fetal bovine serum and 10,000 U/mL penicillin-streptomycin. M1 cells were treated with 100 μM 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP), 30 nM ELA (4), 10 nM sPRR-His (23), or 30 nM ELA in combination with 100 μM 8-Br-cAMP or 10 nM sPRR-His for 24 h. The cells were then harvested and homogenized in cell lysis buffer for protein expression analysis, and the cell culture medium was harvested for sPRR, prorenin/renin, and cAMP ELISA assay. IMCD3 cells were treated with 10 nM sPRR-His or ATP6AP2 siRNA (SR426490; OriGene, Rockville, MD) transfection for various hours, and the cells were harvested for Apela and Apln mRNA analysis.
Statistical analysis.
Data are summarized as means ± SE. Statistical analysis was performed by one-way analysis of variance with the Bonferroni test for multiple comparisons or by unpaired Student’s t test for two comparisons using IBM SPSS 19 software. P < 0.05 was considered statistically significant.
RESULTS
Localization of ELA and apelin in the kidney.
A clue indicating a potential biological function of ELA came from the cellular distribution in mouse kidneys using RNAscope and immunofluorescence. By RNAscope, clear, punctate, and strong Apela (encoding ELA) labeling was present in the CDs, especially in the outer and inner medullary CDs (Fig. 1A). Consistently, immunofluorescence showed predicted predominant labeling of ELA mainly on the apical membrane of CD principal cells in normal mice, overlapping with AQP2, the marker of CD principal cells, confirming the localization in principal cells but not in intercalated cells. We performed a competition assay to validate the specificity of anti-ELA antibody. The signal from this antibody disappeared after preincubation with the immunizing peptide (Fig. 1B). Similarly, the labeling with anti-apelin antibody was found predominantly on the apical membrane of CD principal cells, and the specificity of the apelin labeling was validated similarly by using the immunizing peptide (Fig. 2).
Fig. 1.

ELABELA (ELA) expression in the adult mouse kidney. A: RNAscope with mApela (m, Mus musculus) probe in the mouse kidney. Clear and punctate Apela labeling is present in the collecting ducts (CDs), especially in the outer medulla CDs and inner medulla CDs. B: immunofluorescence staining of ELA in the mouse kidney. To validate the specificity of the labeling of renal ELA in mice, the competition with the immunizing peptide in the consecutive sections from the same tissue samples was performed, and the consecutive sections were stained with ELA antibody preincubated with or without its immunizing peptides. Male mice were used in these studies. AQP2, aquaporin 2.
Fig. 2.

Apelin expression in the adult mouse kidney. Immunofluorescence staining of apelin in the mouse kidney was performed. To validate the specificity of the labeling of renal apelin in mice, the competition with the immunizing peptide in the consecutive sections from the same tissue samples was performed, and the consecutive sections were stained with ELABELA (ELA) antibody preincubated with or without its immunizing peptides. Male mice were used in these studies. AQP2, aquaporin 2.
PRR regulates the apelinergic system in the kidney.
In CD PRR KO mice, medullary fPRR and sPRR protein expressions were significantly reduced (Fig. 3A), along with an increase of Apela and Apln (encoding apelin) mRNA expression as assessed by quantitative RT-PCR (Fig. 3B), consistent with the increased urinary ELA and apelin excretion as assessed by ELISA (Fig. 3C). However, CD PRR KO did not affect APJ protein and Aplnr (encoding APJ) mRNA levels. Similarly, nephron PRR KO dramatically decreased fPRR and sPRR protein expression in renal cortical and medullary regions (Fig. 3D) but significantly upregulated Apela and Apln mRNA expression (Fig. 3E) and urinary ELA and apelin excretion (Fig. 3F). In contrast with the effect of CD PRR KO on APJ expression, nephron PRR KO markedly downregulated medullary APJ protein and Aplnr mRNA expression (Fig. 3, D and E). Colabeling with AQP2 and labeling of consecutive sections for the electroneutral NKCC2, a marker of thick ascending limb (TAL) cells, confirmed APJ expression in the TALs (Fig. 4). Immunofluorescence for APJ also showed that nephron PRR KO dramatically decreased the immunofluorescence staining of APJ in the renal medullary TAL compared with PRR-Floxed control; the labeling in nephron PRR KO mice almost disappeared (Fig. 4). In cultured IMCD3 cells, PRR knockdown by ATP6AP2 siRNA significantly increased (Fig. 3G), whereas exogenous recombinant sPRR-His treatment remarkably decreased (Fig. 3H), Apela and Apln mRNA expression.
Fig. 3.

ELABELA (ELA), apelin, and apelin peptide jejunum (APJ) expression in the kidneys of mice with conditional deletion of (pro)renin receptor in the collecting duct (CD PRR KO) and nephron (Neph) PRR KO mice. A and D: representative immunoblotting and densitometric analysis of renal PRR and APJ expression in the CD PRR KO mice (A) and nephron PRR KO mice (D); expression was normalized by β-actin. B and E: quantitative RT-PCR analysis of medullary Apela, Apln, and Aplnr mRNA expression in the CD PRR KO mice (B) and nephron PRR KO mice (E); expression was normalized by GAPDH. C and F: urinary ELA and apelin excretion in the CD PRR KO mice (C) and nephron PRR KO mice (F). Male floxed and PRR KO mice were used in these studies. n = 5–10 Mice per group. Data are means ± SE. *P < 0.05, **P < 0.01, ***P < 0.001 vs. floxed. G and H: siRNA-mediated PRR knockdown increased (G) but exogenous sPRR-His (H) decreased Apela and Apln mRNA expression in cultured IMCD3 cells. **P < 0.01, ***P < 0.001 vs. negative control (NC) or vehicle. Statistical analysis was performed by using unpaired Student’s t test (A–G) or 1-way analysis of variance with the Bonferroni test (H) using IBM SPSS 19 software. fPRR, full-length PRR; IMCD, inner medullary collecting duct; sPRR, soluble PRR.
Fig. 4.

Representative immunofluorescence images of apelin peptide jejunum (APJ) in floxed and nephron (pro)renin receptor knockout (Neph PRR KO) mice. The images shown are representative of 5 animals per group. Male floxed and nephron PRR KO mice were used in these studies. AQP2, aquaporin 2; NKCC2, Na/K/2Cl cotransporter.
ELA-32 suppresses HS-induced hypertension, fibrosis, and inflammation in Dahl SS rats.
Compared with the Dahl salt-resistant (SR) rats, Dahl SS rats exhibited slightly lower expression of Apela, Apln, and Aplnr mRNA in the renal medulla (Fig. 5A). To assess whether the renal apelinergic system is involved in HS-induced hypertension in Dahl SS rats, the mRNA levels of Apela, Apln, and Aplnr were examined by quantitative RT-PCR, and markedly decreased Apela, Apln, and Aplnr expression was observed in the renal medulla of HS-loaded Dahl SS rats (Fig. 5B).
Fig. 5.

ELA-32 suppresses high salt (HS)-induced hypertension, fibrosis, inflammation, and oxidative stress in Dahl salt-sensitive (SS) rats. A: quantitative RT-PCR results of medullary Apela, Apln, and Aplnr mRNA expression in the kidneys of Dahl salt-resistant (SR) rats and Dahl SS rats; expression was normalized by GAPDH. B: quantitative RT-PCR results of medullary Apela, Apln, and Aplnr mRNA expression in the kidneys of normal Na+ (NS)- and HS-loaded Dahl SS rats; expression was normalized by GAPDH. C: effect of ELA-32 on HS-induced hypertension in Dahl SS rats. All rats were subjected to a high-salt diet for 14 days. Radiotelemetry was performed to record mean arterial pressure (MAP), systolic blood pressure (SBP), and diastolic blood pressure (DBP). D: urinary albumin excretion. E: urinary neutrophil gelatinase-associated lipocalin (NGAL) excretion. F and G: quantitative RT-PCR analysis of fibrosis genes, including TIMP-1, NGAL, Col1A1, FN, PAI-1, TGF-β1, and Kim-1 mRNA expression in the kidney cortical (F) and medullary (G); expression was normalized by GAPDH. H and I: quantitative RT-PCR analysis of inflammatory genes, including TNF-α, IL-1β, IL-2, IL-6, IL-17A, IFN-γ, COX-2, VCAM-1, ICAM-1, and MCP-1 mRNA expression in the kidney cortical (H) and medullary (I); expression was normalized by GAPDH. J: urinary PGE2 excretion. Male Dahl SR rats and Dahl SS rats were used in these studies. n = 5 Rats per group. Data are means ± SE. $P < 0.05, $$P < 0.01 vs. Dahl SR; *P < 0.05, **P < 0.01, ***P < 0.001 vs. NS; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. HS. Statistical analysis was performed by using unpaired Student’s t test (A–C) or 1-way analysis of variance with the Bonferroni test (D–J) using IBM SPSS 19 software. ELA, ELABELA.
Radiotelemetry was performed to measure blood pressure in Dahl SS rats after a HS diet. Exogenous ELA-32 infusion significantly suppressed HS-induced hypertension in Dahl SS rats [mean arterial pressure (MAP): 113.0 ± 1.7 vs. 133.8 ± 1.5 mmHg, P < 0.001; systolic blood pressure (SBP): 122.5 ± 1.8 vs. 142.9 ± 2.6 mmHg, P < 0.01; and diastolic blood pressure (DBP): 105.8 ± 2.3 vs. 127.4 ± 0.9 mmHg, P < 0.001; Fig. 5C].
HS-induced upregulation of urinary albumin (Fig. 5D) and NGAL (Fig. 5E) excretion was also significantly suppressed by ELA-32 treatment. Consistently, the mRNA levels of various fibrosis markers (TGF-β1, FN, Col1A1, PAI-1, and TIMP-1) and kidney injury markers (NGAL and Kim-1) were significantly increased in the kidneys of HS-loaded Dahl SS rats (Fig. 5F), ELA-32 markedly inhibited these markers in the cortex (Fig. 5F), whereas HS-induced Col1A1, NGAL, and Kim-1 mRNA levels were also inhibited by ELA-32 in the medulla (Fig. 5G).
Inflammatory genes, such as TNF-α, IL-1β, IL-2, IL-6, IL-17A, IFN-γ, ICAM-1, VCAM-1, and MCP-1, were further analyzed by using quantitative RT-PCR. The mRNA levels of IL-1β, IL-2, IL-6, IL-17A, IFN-γ, VCAM-1, ICAM-1, and MCP-1 in the cortex (Fig. 5H) and TNF-α, IL-1β, and IL-6 in the medulla (Fig. 5I) were markedly elevated after HS intake and blocked by ELA-32 treatment.
Consistent with the previous reports (5, 16, 54), renal medullary COX-2 mRNA (Fig. 5I) and protein (Fig. 6A) expression as well as 24-h urinary PGE2 excretion (Fig. 5J) were markedly stimulated by a high-salt diet in Dahl SS rats, whereas renal cortical COX-2 expression was unaffected (Fig. 5H). Remarkably, HS-induced renal medullary COX-2 expression and urinary PGE2 excretion were partially abolished by ELA-32 treatment (Fig. 5, I and J, and Fig. 6A).
Fig. 6.

ELA-32 treatment inhibits the activation of intrarenal renin-angiotensin system in high salt (HS)-loaded Dahl salt-sensitive (SS) rats. A: representative immunoblotting and densitometric analysis of renal (pro)renin receptor (PRR) and cyclooxygenase-2 (COX-2) expression in different experimental groups; expression was normalized by β-actin. B: quantitative RT-PCR analysis of PRR, renin, AGT, ACE, AT1R, AT2R, and ACE2 mRNA expression in the kidney cortical and medullary; expression was normalized by GAPDH. C: urinary and plasma soluble PRR (sPRR) levels. D: urinary and plasma prorenin/renin levels. E: urinary and plasma angiotensinogen (AGT) levels. F: urinary and plasma angiotensin II (ANG II) levels. Male Dahl SS rats were used in these studies. n = 5 Rats per group. Data are means ± SE. *P < 0.05, **P < 0.01, ***P < 0.001 vs. normal Na+ (NS); #P < 0.05, ##P < 0.01, ###P < 0.001 vs. HS. Statistical analysis was performed by using 1-way analysis of variance with the Bonferroni test using IBM SPSS 19 software. ELA, ELABELA; fPRR, full-length PRR.
ELA-32 suppresses the activation of intrarenal RAS in HS-loaded Dahl SS rats.
To investigate whether ELA-32 lowers blood pressure and protects renal injury via the inhibition of intrarenal RAS, we examined the levels of RAS components in the kidney, urine, and plasma. By Western blotting, HS-induced fPRR, sPRR, prorenin, and renin protein expression in kidney medulla of Dahl SS rats were significantly blunted by ELA-32 infusion (Fig. 6A). The mRNA levels of AT1R and AT2R in the cortex as well as renin and AGT in the medulla were all elevated in HS-loaded Dahl SS rats and suppressed by ELA-32 treatment (Fig. 6B). ELA-32 also reversed HS-induced downregulation of ACE2 mRNA expression in the medulla but not that in the cortex (Fig. 6B). Consistently, as assessed by ELISA, urinary but not plasma sPRR (Fig. 6C), prorenin/renin (Fig. 6D), AGT (Fig. 6E), and ANG II (Fig. 6F) levels were significantly increased in HS-loaded Dahl SS rats, all of which were blunted by ELA-32 infusion.
ELA-32 inhibits PRR/sPRR and prorenin/renin expression in M1 cells.
To further validate the regulation of PRR and renin expression by ELA, we determined the abundance of PRR and renin expression in ELA-treated M1 cells. As shown in Fig. 7, ELA treatment decreased fPRR, sPRR, prorenin, renin, and phosphorylated (p) CREB (pCREB)/cAMP-dependent transcription factor-1 (pATF-1) protein expression as assessed by immunoblotting (Fig. 7A), accompanied with the inhibition of cell medium sPRR levels (Fig. 7B) and prorenin/renin levels (Fig. 7C), all of which were blocked by 8-Br-cAMP treatment. What is more, ELA-induced inhibition of prorenin, renin, and pCREB/pATF-1 protein expression (Fig. 7D), medium prorenin/renin levels (Fig. 7C), as well as cAMP levels in the cell lysis and medium (Fig. 7E) were reversed by sPRR-His treatment.
Fig. 7.

ELA-32 treatment suppresses (pro)renin receptor (PRR)/soluble PRR (sPRR) and prorenin/renin expression in cultured M1 cells. A and D: representative immunoblotting and densitometric analysis of full-length PRR (fPRR), sPRR, prorenin, renin, phosphorylated CRE-binding protein (pCREB), and total CREB (tCREB) protein expression in different experimental groups; expression was normalized by β-actin. B: sPRR levels in the cell medium. C: prorenin/renin levels in the cell medium. E: cAMP levels in cell lysis and cell medium. n = 6 Cells per group. Data are means ± SE. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. ELABELA (ELA). Statistical analysis was performed by using 1-way analysis of variance with the Bonferroni test using IBM SPSS 19 software. 8-Br-cAMP, 8-bromoadenosine 3′,5′-cyclic monophosphate; pATF-1, phosphorylated cAMP-dependent transcription factor-1.
DISCUSSION
Our hypothesis was that the antagonist interaction between ELA and intrarenal RAS in the distal nephron appears to exert a major impact on blood pressure regulation. Herein, we first characterized the localization of the components of the apelinergic system (ELA, apelin, and APJ) and directly tested the effects of exogenous ELA-32 on HS-induced hypertension, renal injury, inflammation, and intrarenal RAS activation in Dahl SS rats. In addition, we studied the regulation of PRR on the apelinergic system using the cell-specific PRR KO mice. In this report, we provide evidence that 1) there is antagonistic interaction between ELA and intrarenal RAS in the distal nephron and 2) exogenous ELA-32 infusion offers a potential therapy for hypertension and chronic kidney disease, which may be partially via the inhibition of intrarenal RAS.
Both Apln and Aplnr mRNA transcripts are widely expressed in various organs in adults, such as heart, lung, kidney, liver, stomach, adipose tissue, anterior pituitary, neuronal system, and mammary gland (27). Of note, the precise cellular distribution of Apln and Aplnr in the kidney is still unclear since the current evidence for them is equivocal as well as less convincing and remains controversial (14, 28, 29, 34, 38), which may be caused by the limitations and detection sensitivity of the methods. An early study employing immunostaining for the first time demonstrated that apelin expression is mainly detected in tubular epithelial cells and sparsely distributed in tissue endothelial cells, including vascular epithelial cells and glomeruli (38). By RT-PCR, Aplnr mRNA in the rat and mouse kidney is mainly expressed in vascular elements (vasa recta) of the outer medulla and glomeruli with lower levels in the CDs (14, 29, 34). A recent study using in situ hybridization histochemistry (ISHH) with radiolabeled oligonucleotides, branched-chain ISHH, and RNA-Seq showed that Apln mRNA was largely restricted to isolated cells mainly in the vicinity of Aplnr cells in the medulla and is absent from the tubules, vascular endothelial cells, and glomeruli and that Aplnr mRNA was highly expressed in nontubular structures in the outer medulla and glomeruli with low levels in the distal convoluted tubule and cortical TAL (28). In contrast to these previous reports, in the present study, we found that apelin protein expression is exclusively present in the apical membrane of medullary CD principal cells, whereas APJ protein expression is highly detected in TAL in the outer medulla, as demonstrated by immunofluorescence staining. The reason for the discrepancy is unclear but could be related to the differences in experimental reagents and protocols.
Compared with Apln and Aplnr, Apela mRNA expression as assessed by RT-PCR appears to be more prevalent during development with high expression in the heart and kidney of embryonic rodents and restricted to adult rat and mouse kidney (4, 10). However, by branched-chain ISHH, Apela mRNA exists in isolated cells in other tissues such as the heart (28). Within the kidney, ISHH and RNA-Seq results consistently showed that Apela mRNA expression is throughout the tubule with strongest expression in the inner medullary CDs (28). Recently, by immunostaining of paraffin kidney sections, Chen et al. (4) reported that ELA protein expression was predominantly found in renal tubular epithelial cells, especially on the luminal side of the tubules, as well as in the CDs, with very little glomerular staining, whereas by immunofluorescence staining of frozen kidney sections, Schreiber et al. (42) showed that ELA protein expression is largely restricted to the CDs, since the endogenous ELA signals frequently colocalized with Dolichos biflorus agglutinin (a marker of CD) but not biotinylated Lotus tetragonolobus lectin (a marker of proximal tubule) or biotinylated Lycopersicon esculentum (tomato) lectin (a marker of TAL). Herein, we have utilized RNAscope and immunofluorescence staining to reveal strong endogenous Apela mRNA and ELA protein expression in the medullary CDs, similar to renal apelin protein expression overlapping with AQP2 expression in the apical membrane of CD principal cells. Taken together, these results indicate that ELA is constitutively expressed in renal medullary CDs in rodents.
A functional antagonism between apelin-APJ and ANG II-AT1R signaling has been suggested by the evidence that apelin-APJ inhibits ANG II-AT1R signaling via the inhibition of AT1R activity by the heterodimer formation of APJ and AT1R or via converting ANG II to ANG 1–7 by apelin-upregulated ACE2; in turn, ANG II downregulates apelin expression (19). Interestingly, increasing evidence has demonstrated that PRR/sPRR and ELA/apelin have opposite functions in the regulation of renal handling of Na+ and blood pressure and urine concentrating ability (7, 10, 23, 40, 42, 48, 55). In light of the colocalization of PRR with ELA/apelin in the CDs, our hypothesis was that PRR/sPRR and ELA/apelin, with the tangled and intimate relationship, may be recognized as two opposite “Yin” and “Yang” players for fluid and cardiovascular homeostasis as well as blood pressure regulation. In the present study, we found that renal cell-specific PRR KO mice, including CD PRR KO and nephron PRR KO mice, consistently exhibited increased medullary Apela and Apln mRNA levels, accompanied with enhanced 24-h urinary ELA and apelin excretion; these results were consistent with our in vitro results that PRR knockdown by ATP6AP2 siRNA promoted but exogenous sPRR-His treatment inhibited Apela and Apln mRNA expression in cultured IMCD3 cells. Whereas nephron PRR KO but not CD PRR KO dramatically downregulated Aplnr mRNA and APJ protein expression, the expression of APJ protein in nephron PRR KO mice nearly disappeared. Of note, nephron PRR KO mice exhibited larger elevation in Apela/Apln expression compared with that in CD PRR KO mice. We suspect that the downregulation of APJ expression may be secondary to increases of its ligands ELA and apelin, which were indirectly regulated by PRR. In turn, in cultured CD-derived M1 cells, exogenous ELA-32 inhibited fPRR/sPRR, prorenin/renin, and pCREB expression, accompanied with decreased medium cAMP, sPRR, and prorenin/renin levels, all of which were reversed by 8-Br-cAMP, suggesting that inhibition of PRR and renin expression by ELA may be partially via cAMP signaling. ELA binds to APJ with an affinity similar to apelin to activate the Gi signaling pathway, including inhibition of adenylate cyclase and stimulation of MAP kinase phosphorylation (10). However, as discussed above, APJ is absent from the CDs, indicating that novel receptors for ELA may exist in the CDs. Indeed, the APJ-independent role of ELA has already been reported previously (4, 12, 20). In addition, ELA-inhibited prorenin/renin expression as well as cAMP production was also reversed by exogenous recombinant sPRR-His protein treatment, suggesting that ELA-induced inhibition of sPRR expression may lead to a decrease in cAMP generation, then inhibiting prorenin/renin expression. Overall, these results indicate that there is antagonistic interaction between ELA/apelin and PRR in the distal nephron. It is thus of great interest to understand the precise mechanisms for the antagonistic interaction between ELA/apelin and intrarenal RAS.
There is little evidence for the regulation of renal ELA expression under pathophysiological conditions. In this regard, the expression was found to be unaffected in spontaneously hypertensive rats, Dahl SS rats, and rats with polycystic kidney disease (42). In the present study, we reported that compared with Dahl SR rats, the renal expression of Apela, Apln, and Aplnr mRNA in the renal medulla of Dahl SS rats was slightly lower during NS loading. After HS diet treatment, Apela, Apln, and Aplnr mRNA expression were significantly reduced in the renal medulla of Dahl SS rats. These results are compatible with the notion that the defective apelinergic system may play a causal role in the pathogenesis of salt-sensitive hypertension.
By using tail-cuff plethysmography, Schreiber et al. (42) made an initial observation that Apela gene delivery by adeno-associated virus serotype 9 vectors induced a transient antihypertensive effect of ELA in HS-loaded Dahl SS rats. The present study has significantly extended this observation in many aspects. First, the Journal of the American Heart Association published a position paper on the use of radiotelemetry versus tail cuff plethysmography for blood pressure measurement in chronic studies and stated that tail cuff plethysmography is unable to offer accurate determination of blood pressure variance, especially with the small magnitude of the apparent blood pressure change (50). With the use of radiotelemetry, we were able to observe ~20 mmHg decreases of MAP, SBP, and DBP in HS-treated Dahl SS rats following ELA-32 treatment for 14 days. These results are in line with the reported vasodilatory effects of ELA (49). Second, the present study revealed translational potential of ELA in management of hypertension. Currently, clinical trials with recombinant adenovirus are only limited to fatal genetic diseases due to the safety concerns. Finally, we provide a novel mechanism of antihypertensive action of ELA concerning its interaction with PRR and intrarenal RAS.
In the present study, we found ELA-32 peptide treatment not only significantly suppressed inflammatory molecules (IL-1β, IL-2, IL-6, IL-17A, IFN-γ, VCAM-1, ICAM-1, and MCP-1 mainly in the renal cortex), but also inhibited various fibrosis markers (TGF-β1, FN, Col1A1, PAI-1, and TIMP-1 mainly in the renal cortex) and kidney injury markers (NGAL, Kim-1, and urinary albumin) in HS-loaded Dahl SS rats. Interestingly, HS diet increases TGF-β1, FN, and PAI-1 mRNA expression in the renal cortex but not in the medulla in Dahl SS rats (45), suggesting differences in severity of renal fibrosis in the two kidney regions. It has been reported that the abundance of TNF-α mRNA was significantly decreased in the renal cortex but markedly increased in the renal medulla in HS-loaded Dahl SS rats (13, 47) and that etanercept (an inhibitor of TNF-α) attenuated salt-induced hypertension (13), suggesting a particular role of renal medullary TNF-α in pathogenesis of salt-sensitive hypertension. We also found that HS intake increased mRNA levels of IL-2, IL-17A, IFN-γ, VCAM-1, and ICAM-1 in the renal cortex but decreased mRNA expression of IL-2, IL-17A, and IFN-γ without affecting VCAM-1 and ICAM-1 levels in the renal medulla. These results may suggest functionally distinct roles of various inflammatory cytokines in the kidney regions of salt-loaded Dahl SS rats. Of note, IL-2 (15) and IFN-γ (17) have antihypertensive effect, whereas IL-17A (11) has prohypertensive effect.
Increasing evidence from pharmacological and genetic studies has established the important role of intrarenal RAS in hypertension and renal diseases (22, 55). Herein, our hypothesis was that the intrarenal RAS served as a molecular target for the antihypertensive and renoprotective action of ELA in HS-induced hypertensive Dahl SS rats. Hypertensive Dahl SS rat is a well-known low-systemic-renin hypertension model with the overactivation of intrarenal RAS, as reflected by decreased plasma renin activity/concentration and plasma ANG II concentration, but increased renal PRR, prorenin/renin, AGT, ACE, AT1R, and ANG II levels (2, 3, 18, 22, 58). Suppression of plasma renin may delay the salt-induced blood pressure rise (2), whereas activation of local renin in the kidney may play a major role to promote the elevation of salt-induced blood pressure in Dahl SS rats (3). Consistently, in the present study, the expression of medullary PRR and renin, as well as urinary sPRR, prorenin/renin, AGT, and ANG II excretion, were markedly elevated in HS-loaded Dahl SS rats, all of which were consistently blocked by ELA-32 treatment, accompanied with lower blood pressure and improved renal injury. However, these parameters in plasma were relatively inconsistent. HS intake significantly downregulated plasma prorenin/renin and ANG II concentration without affecting the levels of plasma sPRR and AGT. What is more, renin-deficient Dahl SS rats exhibited undetectable plasma renin activity/concentration and lower blood pressure as well as underdeveloped kidneys as reflected by the displaced medulla, incomplete formation of the medullary rays, the presence of large central lesions, and cortical thinning (25, 32), suggesting that renin plays an essential role to maintain normal blood pressure and renal functions in Dahl SS rats on NS diet. Overall, ELA functions as an antihypertensive and renoprotective factor during HS intake in Dahl SS rats, at least in part, depending on the inhibition of intrarenal RAS. Furthermore, it is well addressed that ACE2 signaling protects against hypertension and kidney injury, thus opposing the action of the classic RAS (21). ELA restored the level of renal medullary but not renal cortical ACE2 expression, which may contribute to ELA-induced suppression of intrarenal RAS.
In summary, the present study revealed a novel mechanism underlying the antihypertensive, antifibrotic, and anti-inflammatory actions of ELA, involving the inhibition of intrarenal RAS (Fig. 8). Additionally, we for the first time demonstrated the antagonistic interaction between ELA and intrarenal RAS in the distal nephron. All of these suggest that ELA may be considered as a potential candidate to treat hypertension and renal injury in salt-induced hypertension.
Fig. 8.
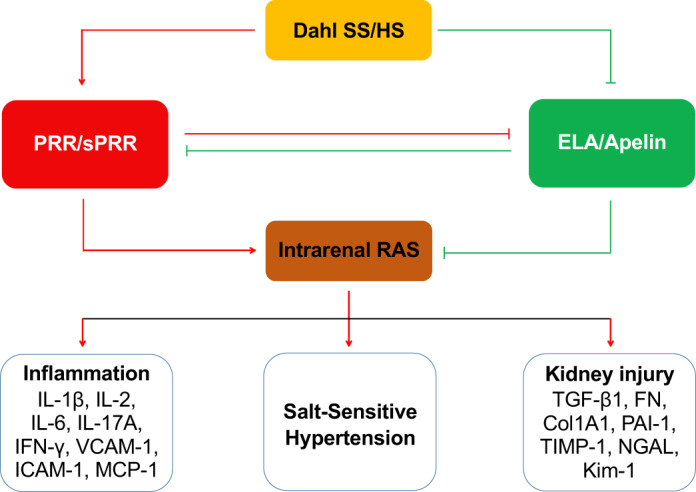
The proposed renal mechanisms of high salt (HS)-induced salt-sensitive hypertension in Dahl salt-sensitive (SS) rats. Experimental evidence is available to demonstrate that HS intake causes activation of (pro)renin receptor (PRR) and intrarenal renin-angiotensin system (RAS), along with the inhibition of ELABELA (ELA)/apelin system, ultimately resulting in elevated blood pressure, inflammation, and kidney injury. PRR/soluble PRR (sPRR) and ELABELA (ELA)/apelin, with the tangled and intimate relationship, may be recognized as 2 opposite “Yin” and “Yang” players: PRR/sPRR inhibits ELA/apelin expression; in turn, ELA/apelin blocks PRR/sPRR expression.
GRANTS
This work was supported by NIH Grants HL-135851, HL-139689, and DK-104072, Veterans Affairs Merit Review from the Department of Veterans Affairs, and Postdoctoral Fellowship Award 19POST34410068 from the American Heart Association. T. Yang is a Research Career Scientist in the Department of Veterans Affairs.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.X. and T.Y. conceived and designed research; C.X., F.W., Y.C., S.X., and D.S. performed experiments; C.X., F.W., Y.C., S.X., and D.S. analyzed data; T.Y. interpreted results of experiments; C.X. prepared figures; C.X. and T.Y. drafted manuscript; C.X., B.R., and T.Y. edited and revised manuscript; T.Y. approved final version of manuscript.
REFERENCES
- 1.Alderman MH. Salt, blood pressure, and human health. Hypertension 36: 890–893, 2000. doi: 10.1161/01.HYP.36.5.890. [DOI] [PubMed] [Google Scholar]
- 1a.Benjamin EJ, Montaner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics–2019 update: a report from the American Heart Association. Circulation 139: e56–e528, 2019. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Campbell WG Jr, Gahnem F, Catanzaro DF, James GD, Camargo MJ, Laragh JH, Sealey JE. Plasma and renal prorenin/renin, renin mRNA, and blood pressure in Dahl salt-sensitive and salt-resistant rats. Hypertension 27: 1121–1133, 1996. doi: 10.1161/01.HYP.27.5.1121. [DOI] [PubMed] [Google Scholar]
- 3.Chandramohan G, Bai Y, Norris K, Rodriguez-Iturbe B, Vaziri ND. Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-κB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol 28: 158–167, 2008. doi: 10.1159/000110021. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Wang L, Wang W, Cheng C, Zhang Y, Zhou Y, Wang C, Miao X, Wang J, Wang C, Li J, Zheng L, Huang K. ELABELA and an ELABELA fragment protect against AKI. J Am Soc Nephrol 28: 2694–2707, 2017. doi: 10.1681/ASN.2016111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hébert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol 295: F818–F825, 2008. doi: 10.1152/ajprenal.90253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell 27: 672–680, 2013. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, Zheng L, Leeper NJ, Pearl NE, Patterson AJ, Anderson JP, Tsao PS, Lenardo MJ, Ashley EA, Quertermous T. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest 118: 3343–3354, 2008. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coquerel D, Chagnon F, Sainsily X, Dumont L, Murza A, Côté J, Dumaine R, Sarret P, Marsault É, Salvail D, Auger-Messier M, Lesur O. ELABELA improves cardio-renal outcome in fatal experimental septic shock. Crit Care Med 45: e1139–e1148, 2017. doi: 10.1097/CCM.0000000000002639. [DOI] [PubMed] [Google Scholar]
- 10.Deng C, Chen H, Yang N, Feng Y, Hsueh AJ. Apela regulates fluid homeostasis by binding to the APJ receptor to activate Gi signaling. J Biol Chem 290: 18261–18268, 2015. doi: 10.1074/jbc.M115.648238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10a.Ha SK. Dietary salt intake and hypertension. Electrolyte Blood Press 12: 7–18, 2014. doi: 10.5049/EBP.2014.12.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc 125: 130–138, 2014. [PMC free article] [PubMed] [Google Scholar]
- 12.Ho L, Tan SY, Wee S, Wu Y, Tan SJ, Ramakrishna NB, Chng SC, Nama S, Szczerbinska I, Chan YS, Avery S, Tsuneyoshi N, Ng HH, Gunaratne J, Dunn NR, Reversade B. ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 17: 435–447, 2015. [Erratum in Cell Stem Cell 17: 635, 2015.] doi: 10.1016/j.stem.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Huang B, Cheng Y, Usa K, Liu Y, Baker MA, Mattson DL, He Y, Wang N, Liang M. Renal tumor necrosis factor α contributes to hypertension in Dahl salt-sensitive rats. Sci Rep 6: 21960, 2016. doi: 10.1038/srep21960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hus-Citharel A, Bouby N, Frugière A, Bodineau L, Gasc JM, Llorens-Cortes C. Effect of apelin on glomerular hemodynamic function in the rat kidney. Kidney Int 74: 486–494, 2008. doi: 10.1038/ki.2008.199. [DOI] [PubMed] [Google Scholar]
- 15.Ishimitsu T, Uehara Y, Numabe A, Tsukada H, Ogawa Y, Yagi S. Antihypertensive effect of interleukin-2 in salt-sensitive Dahl rats. Hypertension 23: 68–73, 1994. doi: 10.1161/01.HYP.23.1.68. [DOI] [PubMed] [Google Scholar]
- 16.Jensen BL, Kurtz A. Differential regulation of renal cyclooxygenase mRNA by dietary salt intake. Kidney Int 52: 1242–1249, 1997. doi: 10.1038/ki.1997.449. [DOI] [PubMed] [Google Scholar]
- 17.Jiang L, He P, Liu Y, Chen J, Wei X, Tan N. Mechanism of IFN-γ in regulating OPN/Th17 pathway during vascular collagen remodeling of hypertension induced by ANG II. Int J Clin Exp Pathol 8: 14433–14440, 2015. [PMC free article] [PubMed] [Google Scholar]
- 18.Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuba K, Sato T, Imai Y, Yamaguchi T. Apelin and Elabela/Toddler; double ligands for APJ/Apelin receptor in heart development, physiology, and pathology. Peptides 111: 62–70, 2019. doi: 10.1016/j.peptides.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 20.Li M, Gou H, Tripathi BK, Huang J, Jiang S, Dubois W, Waybright T, Lei M, Shi J, Zhou M, Huang J. An Apela RNA-containing negative feedback loop regulates p53-mediated apoptosis in embryonic stem cells. Cell Stem Cell 16: 669–683, 2015. doi: 10.1016/j.stem.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li XC, Zhang J, Zhuo JL. The vasoprotective axes of the renin-angiotensin system: physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol Res 125: 21–38, 2017. doi: 10.1016/j.phrs.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin L, Phillips WE, Manning RD Jr. Intrarenal Angiotensin II is associated with inflammation, renal damage and dysfunction in Dahl salt-sensitive hypertension. J Am Soc Hypertens 3: 306–314, 2009. doi: 10.1016/j.jash.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X, Wang F, Xu C, Soodvilai S, Peng K, Su J, Zhao L, Yang KT, Feng Y, Zhou SF, Gustafsson JÅ, Yang T. Soluble (pro)renin receptor via β-catenin enhances urine concentration capability as a target of liver X receptor. Proc Natl Acad Sci USA 113: E1898–E1906, 2016. doi: 10.1073/pnas.1602397113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno C, Hoffman M, Stodola TJ, Didier DN, Lazar J, Geurts AM, North PE, Jacob HJ, Greene AS. Creation and characterization of a renin knockout rat. Hypertension 57: 614–619, 2011. doi: 10.1161/HYPERTENSIONAHA.110.163840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002. doi: 10.1172/JCI0214276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol 219: R13–R35, 2013. doi: 10.1530/JOE-13-0227. [DOI] [PubMed] [Google Scholar]
- 28.O’Carroll AM, Salih S, Griffiths PR, Bijabhai A, Knepper MA, Lolait SJ. Expression and functional implications of the renal apelinergic system in rodents. PLoS One 12: e0183094, 2017. doi: 10.1371/journal.pone.0183094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Carroll AM, Selby TL, Palkovits M, Lolait SJ. Distribution of mRNA encoding B78/apj, the rat homologue of the human APJ receptor, and its endogenous ligand apelin in brain and peripheral tissues. Biochim Biophys Acta 1492: 72–80, 2000. doi: 10.1016/S0167-4781(00)00072-5. [DOI] [PubMed] [Google Scholar]
- 30.O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136: 355–360, 1993. doi: 10.1016/0378-1119(93)90495-O. [DOI] [PubMed] [Google Scholar]
- 31.Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science 343: 1248636, 2014. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavlov TS, Levchenko V, Ilatovskaya DV, Moreno C, Staruschenko A. Renal sodium transport in renin-deficient Dahl salt-sensitive rats. J Renin Angiotensin Aldosterone Syst 17: 1470320316653858, 2016. doi: 10.1177/1470320316653858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Renal Physiol 312: F245–F253, 2017. doi: 10.1152/ajprenal.00178.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pope GR, Roberts EM, Lolait SJ, O’Carroll AM. Central and peripheral apelin receptor distribution in the mouse: species differences with rat. Peptides 33: 139–148, 2012. doi: 10.1016/j.peptides.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prieto MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, McLellan M, Gentile O, Jensen VB, Ichihara A, McDonough AA, Pochynyuk OM, Gonzalez AA. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol 313: F1243–F1253, 2017. doi: 10.1152/ajprenal.00152.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, Siragy HM, Ichihara A, Kohan DE. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Renal Physiol 309: F48–F56, 2015. doi: 10.1152/ajprenal.00126.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramkumar N, Stuart D, Mironova E, Bugay V, Wang S, Abraham N, Ichihara A, Stockand JD, Kohan DE. Renal tubular epithelial cell prorenin receptor regulates blood pressure and sodium transport. Am J Physiol Renal Physiol 311: F186–F194, 2016. doi: 10.1152/ajprenal.00088.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ripoll E, Pluvinet R, Torras J, Olivar R, Vidal A, Franquesa M, Cassis L, Cruzado JM, Bestard O, Grinyó JM, Aran JM, Herrero-Fresneda I. In vivo therapeutic efficacy of intra-renal CD40 silencing in a model of humoral acute rejection. Gene Ther 18: 945–952, 2011. doi: 10.1038/gt.2011.39. [DOI] [PubMed] [Google Scholar]
- 39.Sato T, Kadowaki A, Suzuki T, Ito H, Watanabe H, Imai Y, Kuba K. Loss of apelin augments angiotensin II-induced cardiac dysfunction and pathological remodeling. Int J Mol Sci 20: 239, 2019. doi: 10.3390/ijms20020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato T, Sato C, Kadowaki A, Watanabe H, Ho L, Ishida J, Yamaguchi T, Kimura A, Fukamizu A, Penninger JM, Reversade B, Ito H, Imai Y, Kuba K. ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc Res 113: 760–769, 2017. doi: 10.1093/cvr/cvx061. [DOI] [PubMed] [Google Scholar]
- 41.Sato T, Suzuki T, Watanabe H, Kadowaki A, Fukamizu A, Liu PP, Kimura A, Ito H, Penninger JM, Imai Y, Kuba K. Apelin is a positive regulator of ACE2 in failing hearts. J Clin Invest 123: 5203–5211, 2013. doi: 10.1172/JCI69608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreiber CA, Holditch SJ, Generous A, Ikeda Y. Sustained ELABELA gene therapy in high-salt diet-induced hypertensive rats. Curr Gene Ther 16: 349–360, 2017. doi: 10.2174/1566523217666161121111906. [DOI] [PubMed] [Google Scholar]
- 43.Siddiquee K, Hampton J, McAnally D, May L, Smith L. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol 168: 1104–1117, 2013. doi: 10.1111/j.1476-5381.2012.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takizawa T, Takasaki I, Shionoiri H, Ishii M. Progression of glomerulosclerosis, renal hypertrophy, and an increased expression of fibronectin in the renal cortex associated with aging and salt-induced hypertension in Dahl salt-sensitive rats. Life Sci 61: 1553–1558, 1997. doi: 10.1016/S0024-3205(97)00734-0. [DOI] [PubMed] [Google Scholar]
- 46.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251: 471–476, 1998. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 47.Tian N, Moore RS, Braddy S, Rose RA, Gu JW, Hughson MD, Manning RD Jr. Interactions between oxidative stress and inflammation in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 293: H3388–H3395, 2007. doi: 10.1152/ajpheart.00981.2007. [DOI] [PubMed] [Google Scholar]
- 48.Wang F, Lu X, Peng K, Fang H, Zhou L, Su J, Nau A, Yang KT, Ichihara A, Lu A, Zhou SF, Yang T. Antidiuretic action of collecting duct (pro)renin receptor downstream of vasopressin and PGE2 receptor EP4. J Am Soc Nephrol 27: 3022–3034, 2016. doi: 10.1681/ASN.2015050592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z, Yu D, Wang M, Wang Q, Kouznetsova J, Yang R, Qian K, Wu W, Shuldiner A, Sztalryd C, Zou M, Zheng W, Gong DW. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci Rep 5: 8170, 2015. doi: 10.1038/srep08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilde E, Aubdool AA, Thakore P, Baldissera L Jr, Alawi KM, Keeble J, Nandi M, Brain SD. Tail-cuff technique and its influence on central blood pressure in the mouse. J Am Heart Assoc 6: e005204, 2017. doi: 10.1161/JAHA.116.005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu C, Lu A, Lu X, Zhang L, Fang H, Zhou L, Yang T. Activation of renal (pro)renin receptor contributes to high fructose-induced salt sensitivity. Hypertension 69: 339–348, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang P, Maguire JJ, Kuc RE, Southwood M, Morrell NW, Davenport A. ELABELA/Toddler (Apela), is an endogenous ligand of the human apelin receptor and is reduced in pulmonary arterial hypertension (Abstract). Circulation 132, suppl_3: A13911, 2015. [Google Scholar]
- 53.Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R, Upton PD, Crosby A, Sawiak SJ, Carpenter TA, Glen RC, Morrell NW, Maguire JJ, Davenport AP. Elabela/Toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135: 1160–1173, 2017. doi: 10.1161/CIRCULATIONAHA.116.023218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang T, Singh I, Pham H, Sun D, Smart A, Schnermann JB, Briggs JP. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am J Physiol Renal Physiol 274: F481–F489, 1998. doi: 10.1152/ajprenal.1998.274.3.F481. [DOI] [PubMed] [Google Scholar]
- 55.Yang T, Xu C. Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J Am Soc Nephrol 28: 1040–1049, 2017. doi: 10.1681/ASN.2016070734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang ZZ, Wang W, Jin HY, Chen X, Cheng YW, Xu YL, Song B, Penninger JM, Oudit GY, Zhong JC. Apelin is a negative regulator of angiotensin II-mediated adverse myocardial remodeling and dysfunction. Hypertension 70: 1165–1175, 2017. doi: 10.1161/HYPERTENSIONAHA.117.10156. [DOI] [PubMed] [Google Scholar]
- 57.Zhou B, Bentham J, Di Cesare M, Bixby H, Danaei G, Cowan MJ, Paciorek CJ, Singh G, Hajifathalian K, Bennett JE, Taddei C, Bilano V, Carrillo-Larco RM, Djalalinia S, Khatibzadeh S, Lugero C, Peykari N, Zhang WZ, Lu Y, Stevens GA, Riley LM, Bovet P, Elliott P, Gu D, Ikeda N, Jackson RT, Joffres M, Kengne AP, Laatikainen T, Lam TH, Laxmaiah A, Liu J, Miranda JJ, Mondo CK, Neuhauser HK, Sundström J, Smeeth L, Soric M, Woodward M, Ezzati M, Abarca-Gómez L, Abdeen ZA, Rahim HA, Abu-Rmeileh NM, Acosta-Cazares B, Adams R, Aekplakorn W, Afsana K, Aguilar-Salinas CA, Agyemang C, Ahmadvand A, Ahrens W, Al Raddadi R, Al Woyatan R, Ali MM, Alkerwi A, Aly E, Amouyel P, Amuzu A, Andersen LB, Anderssen SA, Ängquist L, Anjana RM, Ansong D, Aounallah-Skhiri H, Araújo J, Ariansen I, Aris T, Arlappa N, Aryal K, Arveiler D, Assah FK, Assunção MCF, Avdicová M, Azevedo A, Azizi F, Babu BV, Bahijri S, Balakrishna N, Bandosz P, Banegas JR, Barbagallo CM, Barceló A, Barkat A, Barros AJD, Barros MV, Bata I, Batieha AM, Baur LA, Beaglehole R, Romdhane HB, Benet M, Benson LS, Bernabe-Ortiz A, Bernotiene G, Bettiol H, Bhagyalaxmi A, Bharadwaj S, Bhargava SK, Bi Y, Bikbov M, Bjerregaard P, Bjertness E, Björkelund C, Blokstra A, Bo S, Bobak M, Boeing H, Boggia JG, Boissonnet CP, Bongard V, Braeckman L, Brajkovich I, Branca F, Breckenkamp J, Brenner H, Brewster LM, Bruno G, Bueno-de-Mesquita HB, Bugge A, Burns C, Bursztyn M, de León AC, Cacciottolo J, Cameron C, Can G, Cândido APC, Capuano V, Cardoso VC, Carlsson AC, Carvalho MJ, Casanueva FF, Casas JP, Caserta CA, Chamukuttan S, Chan AW, Chan Q, Chaturvedi HK, Chaturvedi N, Chen CJ, Chen F, Chen H, Chen S, Chen Z, Cheng CY, Dekkaki IC, Chetrit A, Chiolero A, Chiou ST, Chirita-Emandi A, Cho B, Cho Y, Chudek J, Cifkova R, Claessens F, Clays E, Concin H, Cooper C, Cooper R, Coppinger TC, Costanzo S, Cottel D, Cowell C, Craig CL, Crujeiras AB, Cruz JJ, D’Arrigo G, d’Orsi E, Dallongeville J, Damasceno A, Dankner R, Dantoft TM, Dauchet L, De Backer G, De Bacquer D, de Gaetano G, De Henauw S, De Smedt D, Deepa M, Dehghan A, Delisle H, Deschamps V, Dhana K, Di Castelnuovo AF, Dias-da-Costa JS, Diaz A, Dickerson TT, Do HTP, Dobson AJ, Donfrancesco C, Donoso SP, Döring A, Doua K, Drygas W, Dulskiene V, Džakula A, Dzerve V, Dziankowska-Zaborszczyk E, Eggertsen R, Ekelund U, El Ati J, Ellert U, Elliott P, Elosua R, Erasmus RT, Erem C, Eriksen L, de la Peña JE, Evans A, Faeh D, Fall CH, Farzadfar F, Felix-Redondo FJ, Ferguson TS, Fernández-Bergés D, Ferrante D, Ferrari M, Ferreccio C, Ferrieres J, Finn JD, Fischer K, Föger B, Foo LH, Forslund AS, Forsner M, Fortmann SP, Fouad HM, Francis DK, Franco MC, Franco OH, Frontera G, Fuchs FD, Fuchs SC, Fujita Y, Furusawa T, Gaciong Z, Gareta D, Garnett SP, Gaspoz JM, Gasull M, Gates L, Gavrila D, Geleijnse JM, Ghasemian A, Ghimire A, Giampaoli S, Gianfagna F, Giovannelli J, Goldsmith RA, Gonçalves H, Gross MG, Rivas JPG, Gottrand F, Graff-Iversen S, Grafnetter D, Grajda A, Gregor RD, Grodzicki T, Grøntved A, Gruden G, Grujic V, Gu D, Guan OP, Gudnason V, Guerrero R, Guessous I, Guimaraes AL, Gulliford MC, Gunnlaugsdottir J, Gunter M, Gupta PC, Gureje O, Gurzkowska B, Gutierrez L, Gutzwiller F, Hadaegh F, Halkjær J, Hambleton IR, Hardy R, Harikumar R, Hata J, Hayes AJ, He J, Hendriks ME, Henriques A, Cadena LH, Herrala S, Heshmat R, Hihtaniemi IT, Ho SY, Ho SC, Hobbs M, Hofman A, Dinc GH, Hormiga CM, Horta BL, Houti L, Howitt C, Htay TT, Htet AS, Hu Y, Huerta JM, Husseini AS, Huybrechts I, Hwalla N, Iacoviello L, Iannone AG, Ibrahim MM, Ikram MA, Irazola VE, Islam M, Ivkovic V, Iwasaki M, Jackson RT, Jacobs JM, Jafar T, Jamrozik K, Janszky I, Jasienska G, Jelakovic B, Jiang CQ, Joffres M, Johansson M, Jonas JB, Jørgensen T, Joshi P, Juolevi A, Jurak G, Jureša V, Kaaks R, Kafatos A, Kalter-Leibovici O, Kamaruddin NA, Kasaeian A, Katz J, Kauhanen J, Kaur P, Kavousi M, Kazakbaeva G, Keil U, Boker LK, Keinänen-Kiukaanniemi S, Kelishadi R, Kemper HCG, Kengne AP, Kersting M, Key T, Khader YS, Khalili D, Khang YH, Khaw KT, Kiechl S, Killewo J, Kim J, Klumbiene J, Kolle E, Kolsteren P, Korrovits P, Koskinen S, Kouda K, Koziel S, Kristensen PL, Krokstad S, Kromhout D, Kruger HS, Kubinova R, Kuciene R, Kuh D, Kujala UM, Kula K, Kulaga Z, Kumar RK, Kurjata P, Kusuma YS, Kuulasmaa K, Kyobutungi C, Laatikainen T, Lachat C, Lam TH, Landrove O, Lanska V, Lappas G, Larijani B, Laugsand LE, Laxmaiah A, Bao KLN, Le TD, Leclercq C, Lee J, Lee J, Lehtimäki T, Lekhraj R, León-Muñoz LM, Levitt NS, Li Y, Lilly CL, Lim WY, Lima-Costa MF, Lin HH, Lin X, Linneberg A, Lissner L, Litwin M, Lorbeer R, Lotufo PA, Lozano JE, Luksiene D, Lundqvist A, Lunet N, Lytsy P, Ma G, Ma J, Machado-Coelho GLL, Machi S, Maggi S, Magliano DJ, Majer M, Makdisse M, Malekzadeh R, Malhotra R, Rao KM, Malyutina S, Manios Y, Mann JI, Manzato E, Margozzini P, Marques-Vidal P, Marrugat J, Martorell R, Mathiesen EB, Matijasevich A, Matsha TE, Mbanya JCN, Posso AJMD, McFarlane SR, McGarvey ST, McLachlan S, McLean RM, McNulty BA, Khir ASM, Mediene-Benchekor S, Medzioniene J, Meirhaeghe A, Meisinger C, Menezes AMB, Menon GR, Meshram II, Metspalu A, Mi J, Mikkel K, Miller JC, Miquel JF, Mišigoj-Durakovic M, Mohamed MK, Mohammad K, Mohammadifard N, Mohan V, Yusoff MFM, Møller NC, Molnár D, Momenan A, Mondo CK, Monyeki KDK, Moreira LB, Morejon A, Moreno LA, Morgan K, Moschonis G, Mossakowska M, Mostafa A, Mota J, Motlagh ME, Motta J, Muiesan ML, Müller-Nurasyid M, Murphy N, Mursu J, Musil V, Nagel G, Naidu BM, Nakamura H, Námešná J, Nang EEK, Nangia VB, Narake S, Navarrete-Muñoz EM, Ndiaye NC, Neal WA, Nenko I, Nervi F, Nguyen ND, Nguyen QN, Nieto-Martínez RE, Niiranen TJ, Ning G, Ninomiya T, Nishtar S, Noale M, Noboa OA, Noorbala AA, Noorbala T, Noto D, Al Nsour M, O’Reilly D, Oh K, Olinto MTA, Oliveira IO, Omar MA, Onat A, Ordunez P, Osmond C, Ostojic SM, Otero JA, Overvad K, Owusu-Dabo E, Paccaud FM, Padez C, Pahomova E, Pajak A, Palli D, Palmieri L, Panda-Jonas S, Panza F, Papandreou D, Parnell WR, Parsaeian M, Pecin I, Pednekar MS, Peer N, Peeters PH, Peixoto SV, Pelletier C, Peltonen M, Pereira AC, Pérez RM, Peters A, Petkeviciene J, Pham ST, Pigeot I, Pikhart H, Pilav A, Pilotto L, Pitakaka F, Plans-Rubió P, Polakowska M, Polašek O, Porta M, Portegies MLP, Pourshams A, Pradeepa R, Prashant M, Price JF, Puiu M, Punab M, Qasrawi RF, Qorbani M, Radic I, Radisauskas R, Rahman M, Raitakari O, Raj M, Rao SR, Ramachandran A, Ramos E, Rampal S, Reina DAR, Rasmussen F, Redon J, Reganit PFM, Ribeiro R, Riboli E, Rigo F, de Wit TFR, Ritti-Dias RM, Robinson SM, Robitaille C, Rodríguez-Artalejo F, Rodriguez-Perez del Cristo M, Rodríguez-Villamizar LA, Rojas-Martinez R, Rosengren A, Rubinstein A, Rui O, Ruiz-Betancourt BS, Horimoto ARVR, Rutkowski M, Sabanayagam C, Sachdev HS, Saidi O, Sakarya S, Salanave B, Salazar Martinez E, Salmerón D, Salomaa V, Salonen JT, Salvetti M, Sánchez-Abanto J, Sans S, Santos D, Santos IS, dos Santos RN, Santos R, Saramies JL, Sardinha LB, Margolis GS, Sarrafzadegan N, Saum KU, Savva SC, Scazufca M, Schargrodsky H, Schneider IJ, Schultsz C, Schutte AE, Sen A, Senbanjo IO, Sepanlou SG, Sharma SK, Shaw JE, Shibuya K, Shin DW, Shin Y, Siantar R, Sibai AM, Silva DAS, Simon M, Simons J, Simons LA, Sjöström M, Skovbjerg S, Slowikowska-Hilczer J, Slusarczyk P, Smeeth L, Smith MC, Snijder MB, So HK, Sobngwi E, Söderberg S, Solfrizzi V, Sonestedt E, Song Y, Sørensen TIA, Jérome CS, Soumare A, Staessen JA, Starc G, Stathopoulou MG, Stavreski B, Steene-Johannessen J, Stehle P, Stein AD, Stergiou GS, Stessman J, Stieber J, Stöckl D, Stocks T, Stokwiszewski J, Stronks K, Strufaldi MW, Sun CA, Sundström J, Sung YT, Suriyawongpaisal P, Sy RG, Tai ES, Tammesoo ML, Tamosiunas A, Tang L, Tang X, Tanser F, Tao Y, Tarawneh MR, Tarqui-Mamani CB, Taylor A, Theobald H, Thijs L, Thuesen BH, Tjonneland A, Tolonen HK, Tolstrup JS, Topbas M, Topór-Madry R, Tormo MJ, Torrent M, Traissac P, Trichopoulos D, Trichopoulou A, Trinh OTH, Trivedi A, Tshepo L, Tulloch-Reid MK, Tuomainen TP, Tuomilehto J, Turley ML, Tynelius P, Tzourio C, Ueda P, Ugel E, Ulmer H, Uusitalo HMT, Valdivia G, Valvi D, van der Schouw YT, Van Herck K, van Rossem L, van Valkengoed IGM, Vanderschueren D, Vanuzzo D, Vatten L, Vega T, Velasquez-Melendez G, Veronesi G, Verschuren WMM, Verstraeten R, Victora CG, Viet L, Viikari-Juntura E, Vineis P, Vioque J, Virtanen JK, Visvikis-Siest S, Viswanathan B, Vollenweider P, Voutilainen S, Vrdoljak A, Vrijheid M, Wade AN, Wagner A, Walton J, Mohamud WNW, Wang MD, Wang Q, Wang YX, Wannamethee SG, Wareham N, Wederkopp N, Weerasekera D, Whincup PH, Widhalm K, Widyahening IS, Wiecek A, Wijga AH, Wilks RJ, Willeit J, Willeit P, Williams EA, Wilsgaard T, Wojtyniak B, Wong TY, Wong-McClure RA, Woo J, Woodward M, Wu AG, Wu FC, Wu SL, Xu H, Yan W, Yang X, Ye X, Yiallouros PK, Yoshihara A, Younger-Coleman NO, Yusoff AF, Yusoff MFM, Zambon S, Zdrojewski T, Zeng Y, Zhao D, Zhao W, Zheng Y, Zhu D, Zimmermann E, Zuñiga Cisneros J; NCD Risk Factor Collaboration (NCD-RisC) . Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet 389: 37–55, 2017. doi: 10.1016/S0140-6736(16)31919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu A, Yoneda T, Demura M, Karashima S, Usukura M, Yamagishi M, Takeda Y. Effect of mineralocorticoid receptor blockade on the renal renin-angiotensin system in Dahl salt-sensitive hypertensive rats. J Hypertens 27: 800–805, 2009. doi: 10.1097/HJH.0b013e328325d861. [DOI] [PubMed] [Google Scholar]