

Summary
CD38 is expressed on Waldenström macroglobulinaemia (WM) cells, but its role as a therapeutic target remains undefined. With recent approval of the anti-CD38 monoclonal antibody, daratumumab (Dara), we hypothesized that blocking CD38 would be lethal to WM cells. In vitro Dara treatment of WM cells (including ibrutinib-resistant lines) elicited antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cell phagocytosis (ADCP) and direct apoptosis. In vivo, Dara treatment was well tolerated and delayed tumour growth in RPCI-WM1-xenografted mice. CD38 is reported to augment B-cell receptor (BCR) signalling; we noted that Dara significantly attenuated phosphorylated SYK, LYN, BTK, PLCγ2, ERK1/2, AKT, mTOR, and S6 levels, and this effect was augmented by cotreatment with ibrutinib. Indeed, WM cells, including ibrutinib-resistant WM cell lines treated with the ibrutinib + Dara combination, showed significantly more cell death through ADCC, CDC, ADCP and apoptosis relative to single-agent Dara or ibrutinib. In summary, we are the first to report the in vitro and in vivo anti-WM activity of Dara. Furthermore, we show a close connection between BCR and CD38 signalling, which can be co-targeted with ibrutinib + Dara to induce marked WM cell death, irrespective of acquired resistance to ibrutinib.
Keywords: Bruton tyrosine kinase, CD38, daratumumab, ibrutinib, Waldenström macroglobulinaemia
Waldenström macroglobulinaemia (WM) is characterized by immunoglobulin (Ig) M-secreting lymphoplasmacytic lymphoma cells that arise from within the bone marrow but then infiltrate systemic lymphoid tissues and other organs. Clinical management includes the use of targeted agents, such as bortezomib-carfilzomib (proteasome inhibitors) and rituximab (anti-CD20 monoclonal antibody [mAb]), alone or in combination with chemotherapy and corticosteroids (Kasi et al, 2015; Leblond et al, 2016; Kapoor et al, 2017). In 2015, the Bruton tyrosine kinase (BTK) inhibitor ibrutinib became the first drug approved to treat WM (Treon et al, 2015). This treatment ushered in a new era of scientific investigations that delivered insight about the role of the B-cell receptor (BCR) and associated signalling pathways in WM (Argyropoulos et al, 2016; Koehrer & Burger, 2016; Paulus et al, 2016a; de Rooij et al, 2016). Currently, novel therapeutic strategies that exploit BCR-associated pathways at various nodes within the signalling complex are being intensely investigated (Paulus et al, 2016a).
CD38 is a cell surface receptor that is expressed on B cells at various stages of maturation and is also commonly present on WM cells (Malavasi et al, 2008). One function of CD38 is to amplify signalling through the BCR complex and enhance B cell proliferation (Funaro et al, 1993; Deaglio et al, 2003). Although this particular role of CD38 (as a coreceptor) has been studied in chronic lymphocytic leukaemia cells, it has not been investigated in WM cells (Funaro et al, 1993; Malavasi et al, 2008). Given the biological function of CD38 and its expression on WM cells, we hypothesized that targeting CD38 with the anti-CD38 mAb daratumumab (Dara) would be lethal to WM cells. Furthermore, because of CD38’s interconnecting role with BCRs, we postulated that the anti-WM activity of Dara could be enhanced by pairing it with ibrutinib. Our investigations herein describe the effects of targeting CD38 in WM cells in vitro and in a WM xenograft model in vivo.
Materials and methods
Cell lines, primary tumor cells, and reagents
The WM cell lines BCWM.1 and RPCI-WM1 and their isogenic ibrutinib-resistant (IR) subclones BCWM.1/IR and RPCI-WM1/IR were used in all experiments. Of note, IR subclones do not harbour BTKC481S or CXCR4WHIM-like mutations but are MYD88L265P positive, as previously described (Paulus et al, 2017). In some experiments, CD19+ cells from a patient with relapsed and refractory disease and a confirmed diagnosis of WM (WM Patient 11) were used. Cells from the patient were collected with approval by the Mayo Clinic Institutional Review Board, in accordance with the Declaration of Helsinki.
Cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS), penicillin (100 u/ml) and streptomycin (100 μg/ml). Cell viability was always maintained at >90% and was measured with a trypan blue exclusion assay and a ViCell-XR viability counter (Beckman Coulter, Brea, CA USA). RPMI medium, penicillin, streptomycin, tetramethylrhodamine methyl ester, and FBS were purchased from Life Technologies (Waltham, MA, USA). Ibrutinib was purchased from Sellekhem (Houston, TX, USA); Dara was obtained from clinical sources.
CD38 expression analysis
CD38 expression and mean fluorescence intensity (MFI) on WM cell lines was determined by using a phycoerythrin-conjugated anti-CD38 antibody and an Attune NxT flow cytometer (Invitrogen, Waltham, MA, USA). Quantification of CD38 cell surface molecules was determined by using Quantibrite beads (BD Biosciences, San Jose, CA, USA), followed by analysis with QuantiCALC software (BD Biosciences); data are expressed as the specific antibody-binding capacity (sABC), defined as the number of antibodies bound per cell. The utility of this approach has been previously described (Moulard & Ozoux, 2016) and its use in malignant B cells (treated with Dara) has also been reported (Matas-Cespedes et al, 2017).
Cell death assays
Antibody-dependent cellular cytotoxicity.
To measure antibody-dependent cellular cytotoxicity (ADCC), target cells (WM cell lines) were labelled with calcein-AM (1 μmol/l, 30 min, 37°C, in the dark (ThermoFisher Scientific, Waltham, MA, USA), washed thrice with phosphate-buffered saline (PBS) and plated in triplicate at 1 × 104 cells/well in 96-well, round-bottom plates. Cells were pre-incubated (15 min, 37°C, in the dark) with an isotype control (IgG1-b12, 0.1 μg/ml), vehicle control (dimethyl sulphoxide [DMSO], 0.001%), ibrutinib (1 μmol/l), Dara (0.1 μg/ml), or the combination of ibrutinib and Dara; doses for in vitro studies were previously established (de Weers et al, 2011; Ponader et al, 2012; Matas-Cespedes et al, 2017). Culture medium was added instead of mAb to determine the spontaneous calcein release and Triton X-100 (1%) was used to determine maximal calcein release. Peripheral blood mononuclear cells (PBMCs; also termed effectors), freshly isolated from healthy human donors, were added at an effector:target (E:T) ratio of 50:1 (optimized previously [de Weers et al, 2011]), and cells were incubated for 6 h at 37°C. Plates were centrifuged, the supernatant was transferred into black plates (ThermoFisher Scientific), and fluorescence was measured in a Synergy spectrophotometer (Scientific Bio-Tek, Winooski, VT, USA) with the excitation filter at 485 ± 20 nm and the band-pass filter at 530 ± 20 nm. The percentage of cellular cytotoxicity was calculated by using the following formula:
where RFU is the relative fluorescent unit.
Complement-dependent cytotoxicity.
Complement-dependent cytotoxicity (CDC) was determined in a manner similar to that of ADCC. Calcein-AM-labelled WM cell lines were pre-incubated (15 min, 37°C, in the dark) with IgG1-b12 (0.1 μg/ml), DMSO (0.001%), ibrutinib (1 μmol/l), Dara (0.1 μg/ml), or ibrutinib + Dara. Similarly, culture medium was added instead of mAb to determine the spontaneous calcein release and Triton X-100 (1%) was used to determine the maximal calcein release. Thereafter, 10% normal human AB serum was added, and cells were incubated for 1 h at 37°C. Cell viability and specific lysis were determined by using the same protocol used for ADCC.
Antibody-dependent cellular phagocytosis.
Macrophages were generated from peripheral blood monocytes isolated from healthy human donors. Monocytes were cultured for 7 days in Dulbecco’s modified Eagle medium supplemented with 10% FBS, 2 mmol/l L-glutamine, 50 μg/ml and 50 u/ml penicillin/streptomycin, and granulocyte-macrophage colony-stimulating factor (10 ng/ml); the culture medium was renewed every 3 days. Macrophages were detached on day 7 with 0.1% trypsin-EDTA and characterized by flow cytometry (CD11b+; BD Biosciences). Macrophages were seeded at 2 × 105 cells/well into non-tissue culture-treated 24-well plates and allowed to adhere overnight at 37°C.
Antibody-dependent cellular phagocytosis (ADCP) was assessed similarly to ADCC and CDC assays. Calcein-AM-labelled WM cell lines were pre-incubated with IgG1-b12 (0.1 μg/ml), DMSO (0.001%), ibrutinib (1 μmol/l), Dara (0.1 μg/ml), or ibrutinib + Dara. After 6 h of incubation, the non-phagocytosed target cells were collected. Macrophages were detached with 0.1% trypsin-EDTA, added to the non-phagocytosed target cells, and stained for CD11b-phycoerythrin expression. The amount of doubly positive target cells (CD11b-phycoerythrin and calcein) was determined on an Attune NxT flow cytometer (Invitrogen).
Determination of apoptosis
Apoptosis was conducted as previously described (Manna et al, 2015; Paulus et al, 2016b). Briefly, 5 × 105 cells were incubated with IgG1-b12 (0.1 μg/ml), DMSO (0.001%), ibrutinib (1 μmol/l), Dara (0.1 μg/ml), or ibrutinib + Dara at 37°C and 5% CO2 for 24 h. After 2 washes, cells were resuspended in 100 μl annexin V binding buffer, incubated for 5 min at room temperature for equilibration with buffer, and annexin V-fluorescein isothiocyanate was added for 15 min at 37°C, per the manufacturer’s instructions (BD Pharmingen, San Jose, CA, USA). Before data acquisition, propidium iodide (PI; 0.1 μg/ml) was added and fluorescence was quantified in an Attune NxT flow cytometer (Invitrogen).
Immunoblotting
Immunoblotting was performed with WM cell lysates, as described previously (Manna et al, 2015; Paulus et al, 2017). Antibodies (Cell Signaling Technology, Danvers, MA, USA) against LYN, SYK, BTK, PLCγ2, ERK1/2, AKT, mTOR, S6, and their phosphorylated isoforms were used. An irrelevant IgG1 antibody (BioLegend, San Diego, CA, USA) was used as an isotype control in drug treatment experiments. Glycer-aldehyde-3-phosphate dehydrogenase was used as a loading control in protein analysis and Western blot experiments. Densitometry analysis was conducted by using GelQuant software (biochemlabsolutions.com).
In vivo xenograft model of WM
A partially humanized mouse model of human WM was established in a manner similar to that previously reported (Paulus et al, 2016c) and as per Mayo Clinic Institutional Animal Care and Use Committee approval and institutional guidelines. Briefly, luciferase-labelled RPCI-WM1 cells (2 × 106 cells) were subcutaneously injected into the right flanks of NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) mice (Taconic Biosciences, Rensselaer, NY, USA). After 10 days, when a bioluminescent signal on a IVIS® Spectrum imaging machine (PerkinElmer, Waltham, MA USA) was noticeable, mice were randomized to receive vehicle (PBS, 0.9%), 8 × 106 PBMCs from healthy human donors (via tail vein [intravenous] injection), or Dara (10 mg/kg, via intraperitoneal injection) + PBMCs; this treatment schema was consistent with prior investigations of Dara (de Weers et al, 2011; Nijhof et al, 2015). An initial saturating dose of Dara (20 mg/kg) was administered to mice on day 11 after tumour implantation, thereafter followed by a 10 mg/kg dose on days 14, 17 and 20. In the Dara-receiving study arm, PBMCs were given 1 day before the Dara injection. Tumour burden was quantified by bioluminescent signal intensity on an IVIS imaging machine and by calliper measurements every 4 days; body weight was also calculated on the same days. Mice were euthanized on day 36 to avoid any possibility of graft-versus-host disease (GVHD)-like symptoms, which could impact the results of the study, particularly in mice that received healthy donor PBMCs. This phenomenon is commonly observed in partially humanized mouse models (Herman & Wiestner, 2016).
Statistical analysis
ADCC, CDC and ADCP experiments were performed in WM cell lines; cells were plated in triplicate and assays were performed at least twice. Results are expressed as mean ± standard error of the mean (SEM) because of the low number of biological samples tested. Statistical analysis was performed with the nonparametric Mann–Whitney test or nonparametric Kruskal–Wallis multiple comparison test (as applicable), using GraphPad Prism software, version 7 (GraphPad Software Inc., La Jolla, CA, USA). P values less than 0.05 were considered statistically significant. Associations of CD38 sABC expression with each cell death measure were evaluated by using the Spearman test of correlation. The Spearman correlation coefficient r was estimated, and all statistical tests were 2-sided. Statistical analyses also were performed with R statistical software (version 3.1.1; R Foundation for Statistical Computing, Vienna, Austria).
Results
CD38 expression in WM cells
CD38 is expressed at varying intensity, ranging from dim to bright by immunohistochemistry analysis, in approximately 70% of patients with WM (Morice et al, 2009). We have previously shown a similar pattern of expression in WM cell lines and primary patient WM cells (Paulus et al, 2015), with CD38 sABC in BCWM.1, RPCI-WM1 and WM Patient 11 being 4.86 × 103, 3.59 × 104 and 3.80 × 103 units, respectively (Fig 1A–C). The differentially increased expression in RPCI-WM1 cells appeared to arise at the gene level, with RNA-Seq analysis showing ~10-fold increase in CD38 mRNA expression in RPCI-WM1 versus BCWM.1 (reads per kilobase per million mapped reads of 84.1 vs. 8.4, respectively) (Fig 1D). Indeed, these findings were also echoed in primary WM cells. Gene expression analysis from a publicly-available dataset (series GSE9656; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9656), examining CD19+ B cells from healthy donors (n = 7) versus patients with WM (n = 12) showed a 5.72-fold upregulation of CD38 in the latter (P < 0.001) (Fig 1D).
Fig 1.
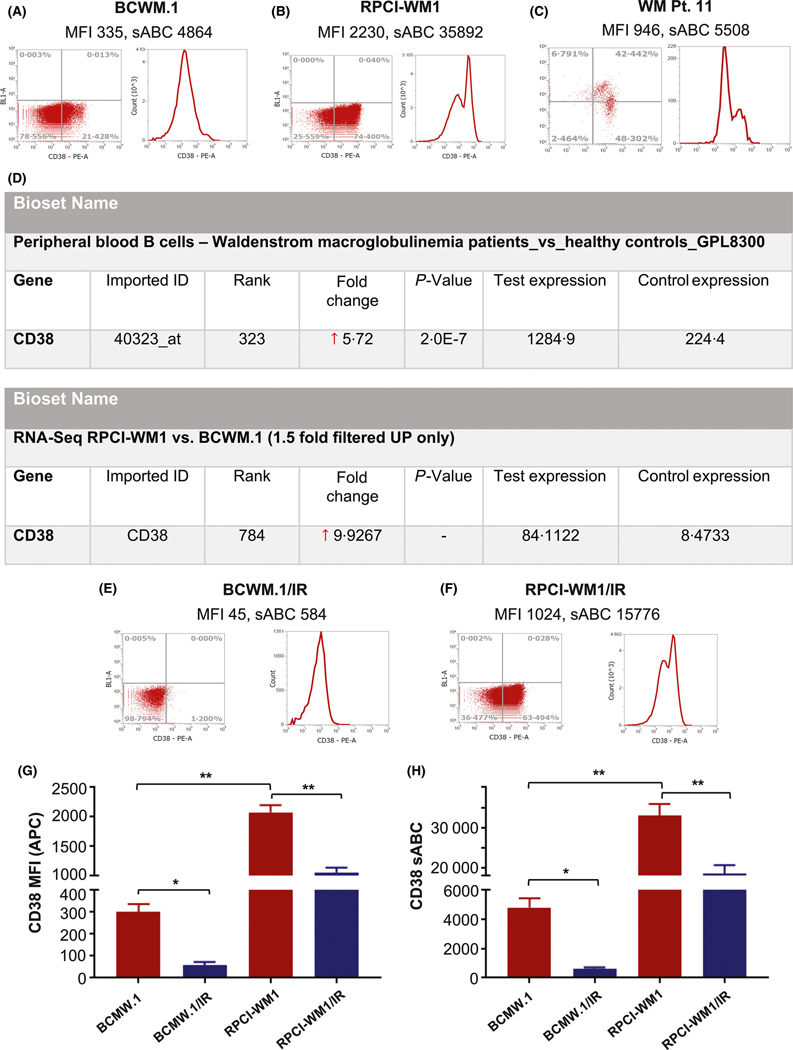
CD38 expression in wild-type Waldenström macroglobulinaemia cell lines. (A–C) CD38 receptor expression and mean fluorescence intensity (MFI) in BCWM.1 cells (A), RPCI-WM1 cells (B) and CD19+/CD138+ tumour cells from a patient with relapsed and refractory WM patient (WM Pt. 11) (C). Expression was determined by flow cytometry with a phycoerythrin-conjugated anti-CD38 antibody. Quantification of surface expression of CD38 molecules per cell was also determined by using BD Quantibrite beads; data are represented as mean expression of CD38 ± SEM or the number of antibodies bound per ± cell SEM (sABC). (D) CD38 gene expression in CD19+ peripheral blood mononuclear cells from healthy donors (n = 7, control expression) and CD19+ bone marrow B cells from patients with WM (n = 12, test expression) from a previously published dataset (series GSE9656) was analysed in the Illumina BaseSpace Correlation Engine suite (Illumina, San Diego, CA, USA). Relative to healthy PBMCs, CD38 expression was 5.72-fold higher in WM cells. Likewise, CD38 expression from RPCI-WM1 (test expression) and BCWM.1 (control expression) cell lines was analysed. Relative to BCWM.1 cells, CD38 was expressed 9.9-fold higher in RPCI-WM1 cells. (E–F) CD38 receptor expression, MFI and sABC were analysed in ibrutinib-resistant (IR) BCWM.1/IR and RPCI-WM1/IR cell lines. (G–H) Comparison and statistical difference in CD38 MFI and sABC between BCWM.1, ibrutinib-resistant BCWM.1/IR, RPCI-WM1, and RPCI-WM1/IR cell lines. *P value <0.05; **P value <0.005 (both considered statistically significant).
In addition to wild-type WM cell lines, we also examined ibrutinib-resistant (IR) isogenic subclones of BCWM.1 and RPCI-WM1 (termed BCWM.1/IR and RPCI-WM1/IR). Intriguingly, we found CD38 expression was significantly lower in the IR variants compared with the non-IR wild-type parent cells (P < 0.05). Mean CD38 sABC in these drug-resistant cells was 8400 units (BCWM.1/IR: 584 units; RPCI-WM1/IR: 15 776 units) (Fig 1E–H). Overall, our analysis shows that although CD38 is expressed on WM cells, receptor density and expression of the molecule per cell varies considerably, and this variability may further be affected by resistance to therapeutic agents (e.g., ibrutinib).
Dara induces immune effector-mediated death and direct programmed cell death in WM cells with and without ibrutinib resistance
We used BCWM.1, RPCI-WM1 and WM Patient 11 cells, as well as subclones BCWM.1/IR and RPCI-WM1/IR, to investigate the anti-tumour activity profile of Dara in vitro and ex vivo. We first assessed ADCC induced by immune effector cells from healthy donor PBMCs in BCWM.1, RPCI-WM1 and WM Patient 11 cells treated with single-agent Dara.
ADCC is triggered when natural killer or T effector cells (or both) engage antibody-bound tumour cells, resulting in specific lysis of the tumour cell (Neri et al, 2001; Strome et al, 2007; Boyerinas et al, 2015). ADCC (lysis) in BCWM.1, RPCI-WM1 and WM Patient 11 cells was 23.86 ± 0.02%, 24.46 ± 0.10% and 30.24 ± 0.66%, respectively (Fig 2A). ADCC was lower in the IR variants; being 14.68 ± 2.56% in BCWM.1/IR cells and 24.09 ± 3.71% in RPCI-WM1/IR cells (Fig 2B).
Fig 2.
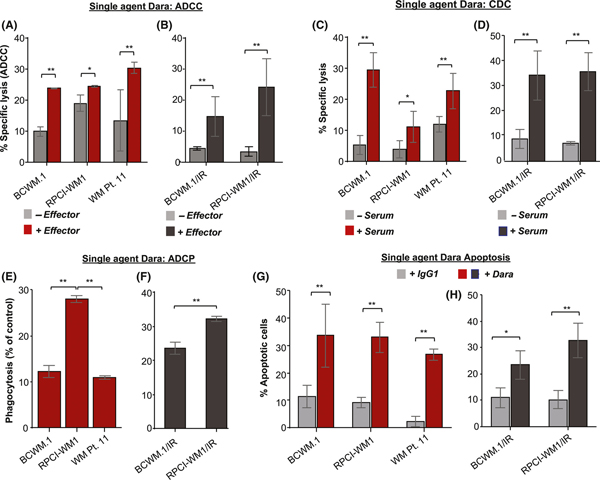
Dara induced immune-mediated and direct cytotoxicity in Waldenström macroglobulinaemia cell lines in vitro. All results are expressed as mean ± SEM. Comparative significance analyses between the groups (brackets) show P values. *P value <0.05; **P value <0.005 (both considered statistically significant). (A–B) Antibody-dependent cell cytotoxicity (ADCC) induced by single-agent daratumumab (Dara; 0.1 μg/ml) was assessed in calcein-AM labelled BCWM.1, RPCI-WM1, primary WM Patient 11 (WM Pt. 11) cells, as well as isogenic ibrutinib-resistant BCWM.1/IR and RPCI-WM1/IR target cells, in the absence or presence of PBMCs from healthy donors (effectors) at an effector:target ratio of 50:1 for 4 h. Spontaneous release and maximum release were determined by using an irrelevant IgG1-b12 isotype antibody (0.1 μg/ml). (C–D) Specific lysis from complement-dependent cell cytotoxicity (CDC) was measured in wild-type and ibrutinib-resistant WM cells in the presence of 10% human serum from a healthy donor for 1 h. (E–F) Cell death through phagocytosis (ADCP) was assessed in calcein-AM-labelled WM cell lines, primary WM Pt. 11and BCWM.1/IR and RPCI-WM1/IR cells, using CD11b+ macrophages derived from healthy human donor monocytes at an effector:target ratio of 2:1 for 2 h. G-H, Apoptosis was assessed in WM cell lines, WM Pt. 11 cells, as well as BCWM.1/IR and RPCI-WM1/IR cells, treated with Dara or IgG1-b12 isotype antibody for 24 h. Cells were stained with annexin-V and propidium iodide and were analysed by flow cytometry.
CDC has not been reported as a major mechanism of cell death induced by mAbs, such as rituximab; however, Dara can induce CDC in various B-lymphoid cell types (de Weers et al, 2011; Nijhof et al, 2016). In WM cells, we noted specific lysis from CDC in BCWM.1, RPCI-WM1 and WM Patient 11 cells (29.49 ± 2.04%, 11.10 ± 1.82%, and 22.66 ± 2.06%, respectively) (Fig 2C). In contrast, CDC was marginally higher in IR cells: 34.48 ± 5.70% in BCWM.1/IR and 35.91 ± 4.34% in RPCI-WM1/IR (Fig 2D).
Another manner by which Dara can trigger cell death is through macrophage-mediated phagocytosis (ADCP) of antibody-coated tumour cells (Overdijk et al, 2015). Phagocytic cell death in BCWM.1 and WM Patient 11 cells was 12.18 ± 0.54% and 9.73 ± 0.51%, respectively; however, ADCP was significantly higher in RPCI-WM1 cells (28.07 ± 0.28%) (Fig 2E). In IR isogenic subclones, ADCP was 23.59 ± 1.24% and 32.16 ± 0.51% in BCWM.1/IR and RPCI-WM1/IR cells, respectively (Fig 2F).
Finally, as Dara has been reported to directly induce apoptosis (Overdijk et al, 2016), we measured annexin-V/PI positivity in WM cells treated with Dara. Mean annexin-V/PI positivity across WM cell lines and primary WM cells was 33.35%; individually, values were 33.64 ± 6.61%, 33.06 ± 3.21% and 26.78 ± 1.46% in BCWM.1, RPCI-WM1 and WM Patient 11 cells, respectively (Fig 2G). In BCWM.1/IR and RPCI-WM1/IR cells, apoptosis was largely preserved (23.43 ± 3.18% and 32.75 ± 3.81%, respectively) (Fig 2H).
Altogether, although ADCC was similar across wild-type WM cell lines and primary patient cells, it was significantly lower because of lower CD38 sABC in the IR cells. Yet despite this difference, CDC, ADCP and apoptosis were generally similar for wild-type and IR cells, with more variation in CDC and ADCP noted between BCWM.1, RPCI-WM1 and WM Patient 11 cells.
Targeting CD38 with dara delays in vivo tumor growth in WM-bearing mice
To determine whether in vitro WM cell death, induced by Dara, can be recapitulated in an in vivo setting, we used our previously reported WM xenograft mouse model (Chitta et al, 2013, 2014; Ailawadhi et al, 2016; Paulus et al, 2016b). This model relies on implantation of RPCI-WM1 cells, which were developed from a patient with highly drug-resistant terminal-stage WM, into immunocompromised mice (Drexler et al, 2013; Ailawadhi et al, 2016). Thus, we xenografted luciferase-labelled RPCI-WM1 cells into mice and began drug treatment on day 10 (when tumour growth was detected by bioluminescent signal intensity); mice were randomized to receive either vehicle, healthy donor PBMCs (containing natural killer and T cells) or Dara + PBMCs. Indeed, mice receiving Dara showed reduced tumour growth compared with controls (Fig 3A–B). Bioluminescent intensity analysis on day 34 showed tumour burden to be lower by 77% and 54% in Dara-treated versus vehicle- or PBMC-administered mice, respectively. Calliper-based manual measurements of maximal tumour diameter were relatively similar (data not shown). Notably, we did not observe any significant weight loss up to day 34 (Fig 3C). Mice were euthanized after day 34 because of development of GVHD-like symptoms, which is common after administration of human PBMCs (Herman & Wiestner, 2016). Altogether, our data demonstrate that Dara significantly prohibited WM cell growth in mice and was well tolerated, with no observable drug-related toxicities.
Fig 3.
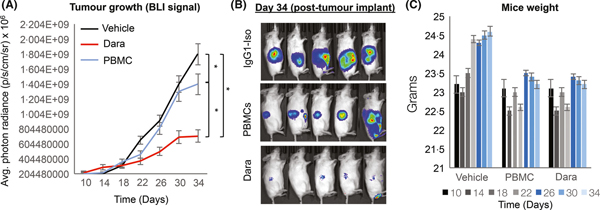
Targeting CD38 with dara delayed in vivo tumour growth in WM-bearing mice. Luciferase-labelled RPCI-WM1 cells were subcutaneously injected into the right flanks of NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) and randomized to receive vehicle (phosphate-buffered saline, 0.9%), healthy donor peripheral blood mononuclear cells (PBMCs; 8 × 106 cells) or Dara + PBMCs. After a bioluminescent signal was detected on post-implantation day 10, mice received the assigned treatments. In the Dara group, an initial saturating dose of Dara (20 mg/kg) was administered to mice on day 11 after tumour implantation, followed by a 10 mg/kg dose on days 14, 17 and 20; PBMCs were given 1 day before Dara injection. Data are represented as the mean ± SEM. (A–B) Tumour burden was quantified by bioluminescent signal intensity (BLI) every 4 days. Comparative significance analyses between the groups (brackets) show P values. *P value <0.05; **P value <0.005 (both considered statistically significant). (C) Weight was measured every 4 days.
Ibrutinib-enhances therapeutic disruption of CD38 modulates BCR-associated signalling components
Functionally, CD38 acts as a molecular hub, integrating signals from the BCR complex and the microenvironment to drive B cell proliferation (Funaro et al, 1993; Deaglio et al, 2003). The intracellular impact on protein signalling after blocking CD38 in WM cells is not known. Thus, we probed wild-type and IR-WM cells treated with Dara for BCR-associated proteins, including phosphorylated (p) isoforms (Fig 4). As anticipated, we observed significant downregulation of LYN, pLYN, pSYK, pBTK, PLCγ2 and pERK1/2 after a 1-h exposure to Dara in wild-type BCWM.1 (Fig 4A–E, red-outlined bars indicate Dara-treated cells) and RPCI-WM1 cells (Fig 4I–M). In addition to canonical BCR signalling, CD19-associated signalling (facilitated through phosphoinositide 3-kinase, PI3K) can also be initiated in parallel during BCR activation (Otero et al, 2001; Depoil et al, 2008). With this in mind, we probed for AKT, mTOR and S6 (including phosphorylated isoforms). Indeed, pAKT, pmTOR and pS6 protein levels were downregulated in BCWM.1 (Fig 4F–H) and RPCI-WM1 cells (Fig 4N–P). Of note, WM Patient 11 cells could not be examined because of low tumour yield, and most of the cells were used in the other cell death assays.
Fig 4.
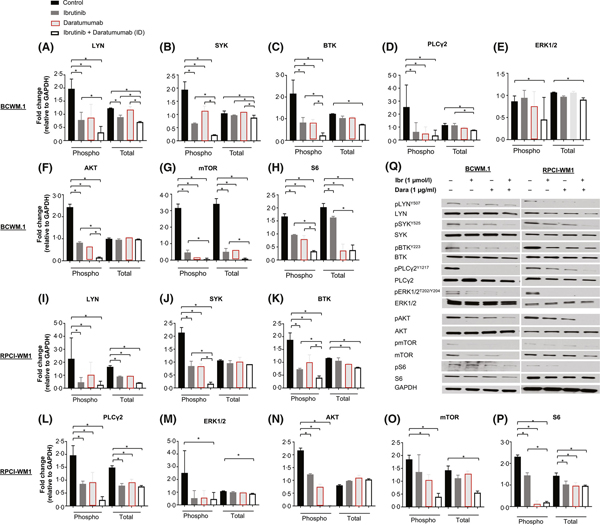
Dara modulates B-cell receptor and AKT signalling proteins in wild-type Waldenström macroglobulinaemia cells and its effect is augmented by ibrutinib. Proteins associated with B-cell receptor (BCR) signalling were examined by Western blot in wild-type cell lines BCWM.1 and RPCI-WM1, treated with isotype IgG1-b12, ibrutinib (Ibr), daratumumab (Dara), or their combination (ID; ibrutinib + Dara) for 2 h. Experiments were conducted at least twice. Protein levels were quantified by densitometry analysis, and fold change in expression was normalized to GAPDH. Data are represented as the mean ± SEM. A comparative significance analysis between the groups show P values <0.05 (denoted by *), which were considered statistically significant. (A–P) Phosphorylated isoforms (Phospho, p) and total protein levels for LYN, SYK, BTK, PLCγ2, ERK1/2, AKT, mTOR and S6 were measured in BCWM.1 cells (A–H) and RPCI-WM1 cells (I–P). Q, A representative Western blot protein expression profile of each protein from BCWM.1 and RPCI-WM1 cells treated under different experimental conditions (Ibr, Dara, or Ibr + Dara).
Next, we profiled the same proteins in IR WM cells in response to Dara (Fig 5, blue-outlined bars indicate Dara-treated cells). Across both IR cell lines, the effects of Dara on LYN/pLYN, SYK/pSYK, mTOR/pmTOR were different from those observed in wild-type cells (Fig 5A, B, G, I, J and O), but downregulation of pBTK, PLCG2/PLCγ2, pERK1/2 and pS6 was comparable (albeit in a cell line-specific manner) (Fig 5C–E, H, K-M, and P). pAKT was decreased only in RPCI-WM1/IR cells and not in BCWM.1/IR cells (Fig 5F, N).
Fig 5.
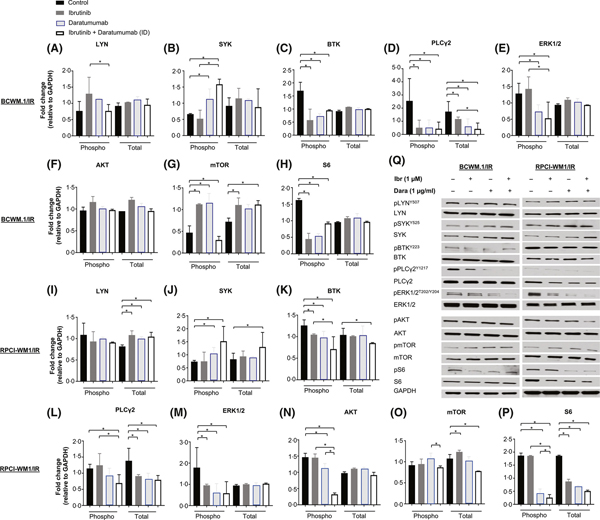
B-cell receptor and AKT Signalling-associated proteins in ibrutinib-resistant Waldenström macroglobulinaemia cells were variably altered after exposure to daratumumab alone or in combination with ibrutinib. Immunoblot analysis probing for proteins associated with B-cell receptor (BCR) signalling was performed in ibrutinib (Ibr)-resistant WM cell lines (BCWM.1/IR and RPCI-WM1/IR) following the procedure described in the legend of Fig 4. Proteins associated with BCR signalling were assessed in ibrutinib-resistant WM cells treated with isotype IgG1-b12, Ibr, Dara, or Ibr + Dara for 2 h. Experiments were conducted at least twice; protein levels were quantified by densitometry analysis, and fold change in expression was normalized to GAPDH. Data are represented as the mean ± SEM. A comparative significance analysis between the groups show P values <0.05 (denoted by *), which were considered statistically significant. (A–P) Phosphorylated isoforms (Phospho, p) and total protein levels for LYN, SYK, BTK, PLCγ2, ERK1/2, AKT, mTOR and S6 were measured in BCWM.1/IR cells (A–H) and RPCI-WM1/IR cells (I–P). Q, A representative Western blot protein expression profile of each protein from BCWM.1/IR and RPCI-WM1/IR cells treated under different experimental conditions (Ibr, Dara, or Ibr + Dara).
Given the putative function and interplay of CD38 with the BCR, we hypothesized that the combination of Dara with ibrutinib would further downregulate BCR and associated proteins. Thus, we probed the aforementioned proteins in wild-type and IR WM cell lines exposed to ibrutinib, with and without Dara. Protein modulation in wild-type BCWM.1 and RPCI-WM1 cells was relatively similar in Dara versus ibrutinib-treated cells (Fig 4). In ibrutinib + Dara-treated cells, however, pLYN, pSYK, pBTK, PLCγ2, pERK1/2, pAKT, pmTOR, and pS6 generally were markedly reduced compared with single-agent ibrutinib- or Dara-treated cells (Fig 4A–P, open white bars indicate combination ibrutinib + Dara-treated cells). As expected, cell line-specific variability between BCWM.1 and RPCI-WM1 cells, the expression levels and the resultant change in proteins were evident.
In IR cells, we anticipated that BCR protein changes in response to ibrutinib with or without Dara would be some-what different from that observed in wild-type cells because of variances in basal protein expression and potential reliance on these proteins. Although ibrutinib increased pLYN levels in BCWM.1/IR cells, this change was not observed in RPCI-WM1/IR cells, with the ibrutinib + Dara combination appearing to normalize pLYN levels back to baseline (Fig 5A, I). Intriguingly, although ibrutinib had no effect on pSYK expression in either IR cell line, a significant increase of pSYK (in both IR cell lines) was observed when comparing with control, single-agent ibrutinib, or Dara-treated counter-parts; with total SYK also increasing in RPCI-WM1/IR cells (Fig 5B, J). However, pBTK and PLCγ2 were reduced in RPCI-WM1/IR cells (Fig 5K, L) and pERK1/2 was lower in BCWM.1/IR cells exposed to the ibrutinib + Dara combination, compared with single-agent ibrutinib (Fig 5E, M). We and others have previously shown that IR cells preferentially utilize PI3K/AKT-associated pathways via transcriptional reprogramming to safeguard against neoplastic growth and survival (Chiron et al, 2014; Paulus et al, 2017). Indeed, a significant decrease in pAKT and its downstream effector pS6/S6 was seen in RPCI-WM1/IR cells treated with ibrutinib + Dara (Fig 5N, P), but this effect was not evident in BCWM.1/IR cells (Fig 5F, H). Notably, basal mTOR levels appeared lower in BCWM.1/IR cells compared with RPCI-WM1/IR clones and, in general, the effects of ibrutinib + Dara treatment on mTOR were variable between the 2 models (Fig 5G, O).
The combination of ibrutinib and dara is highly lethal to WM cells that are sensitive or resistant to ibrutinib
With modulation of BCR and parallel pathway signalling proteins noted across wild-type and IR WM cell lines, albeit in a cell type-specific manner, we questioned whether the combined effect of ibrutinib + Dara would translate into enhanced lethality in WM cells. We assessed ADCC in BCWM.1 cells and WM Patient 11 cells (Fig 6A–D). Specific lysis from ibrutinib alone was 11.70 ± 0.70% and 16.70 ± 1.88%, respectively, but it was not observed in RPCI-WM cells (compared with conditions lacking PBMC effector cells). However, in ibrutinib + Dara-treated BCWM.1, RPCI-WM1 and WM Patient 11 cells, ADCC was significantly increased to 55.67 ± 2.73%, 49.74 ± 3.89% and 65.59 ± 3.28%, respectively (Fig 6A–C). The average ADCC in wild-type WM cells treated across conditions is shown in Fig 6D.
Fig 6.
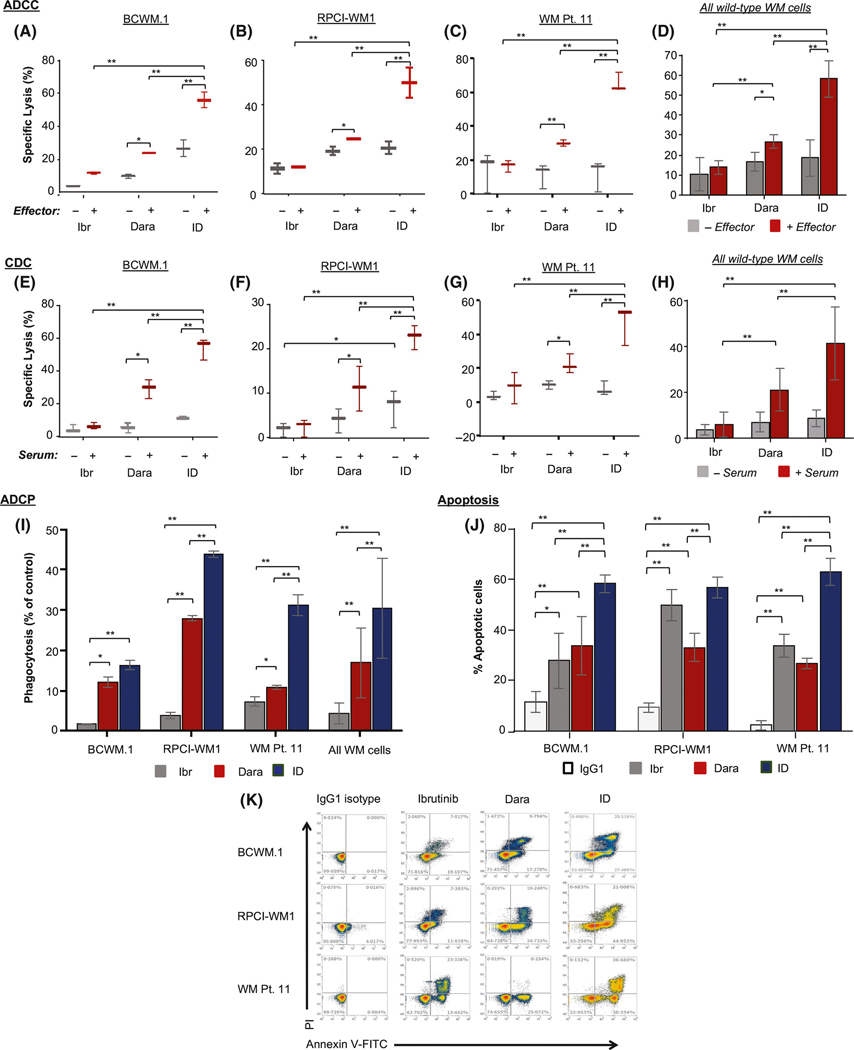
Combination Ibrutinib + Dara is significantly more lethal to wild-type WM cells compared with single-agent treatment. Cells were treated with single-agent ibrutinib (Ibr; 1 μmol/l), daratumumab (Dara; 0.1 μg/ml), or combination Ibr + Dara. (A–C) Antibody-dependent cell cytotoxicity (ADCC) of calcein-AM labelled BCWM.1 (A), RPCI-WM1 (B), and primary WM cells (WM Pt. 11) (C) target cells, with or without peripheral blood mononuclear cells (PBMCs, effectors) from healthy donors at an effector:target ratio of 50:1 for 4 h. D, Specific lysis from ADCC averaged across all cell lines. (E–G) Complement-dependent cell cytotoxicity (CDC) was measured in BCWM.1 (E), RPCI-WM1 (F) and WM Pt. 11 (G) tumour cells treated with the different agents for 1 h in the presence of 10% human serum from a healthy donor. (H) Specific lysis from CDC averaged across all cell lines. (I) Antibody-dependent cellular phagocytosis (ADCP) was assessed in calcein-AM labelled WM cell lines and WM Pt. 11 cells treated under the indicated conditions, using CD11b+ macrophages derived from healthy human donor monocytes (effectors) at an effector:target ratio of 2:1 for 2 h. All data are expressed as mean SEM. (J) Apoptosis was assessed in WM cells treated with IgG1-b12 isotype antibody, Ibr, Dara or Ibr + Dara for 24 h, followed by staining with annexin-V and propidium iodide (PI) and flow cytometry analysis. Results are expressed as mean SEM of the percent of cells that underwent apoptosis. K, Scatterplot showing apoptosis in WM cell lines and primary WM cells treated with various agents. *P value <0.05; **P value <0.005 (both considered statistically significant).
Next CDC was surveyed (Fig 6E–H). Although ibrutinib induced no measurable specific lysis in any of the WM cell lines, relative to counterparts in which no complement-containing serum was added, CDC was significantly enhanced in ibrutinib + Dara-treated cells. CDC induction in combination-treated BCWM.1, RPCI-WM1 and WM Patient 11 cells was 54.21 ± 2.43%, 22.61 ± 0.99% and 47.03 ± 6.49%, respectively (Fig 6E–G). The average CDC in wild-type WM cells treated across conditions is shown in Fig 6H.
In a similar fashion, although phagocytic cell death was not induced beyond baseline in the WM cells, the ibrutinib + Dara combination treatment enhanced ADCP to 16.43 ± 0.49%, 42.87 ± 0.11% and 31.37 ± 1.83% in BCWM.1, RPCI-WM1 and WM Patient 11 cells, respectively (Fig 6I). Additionally, apoptosis was measured, and although ibrutinib alone induced 27.84 ± 6.28%, 49.72 ± 3.49% and 36.77 ± 3.69% annexin V/PI positivity in BCWM.1, RPCI-WM1 and WM Patient 11 cells, respectively, this effect was significantly increased to 58.31 ± 1.28% and 66.91 ± 5.21% in BCWM.1 and WM Patient 11 cells, respectively, after ibrutinib + Dara treatment for 24 h. In RPCI-WM1 cells, apoptosis increased to 56.75 ± 1.55%; this difference was not significant compared with apoptosis after treatment with ibrutinib alone, but it was significantly increased compared with Dara alone (Fig 6J; representative scatterplots are shown in Fig 6K).
We conducted a similar analysis in IR WM cell lines. In BCWM.1/IR and RPCI-WM1/IR cells, ADCC induction by single-agent ibrutinib was 11.37 ± 1.45% and 22.17 ± 2.41%, respectively. With the addition of Dara, ADCC increased significantly to 80.13 ± 10.49% and 47.82 ± 2.41% in BCWM.1/ IR and RPCI-WM1/IR clones, respectively (Fig 7A–B). The average specific lysis from ADCC across both IR WM cell lines treated across different conditions is shown in Fig 7C. Akin to their wild-type counterparts, CDC was not induced by ibrutinib; however, it was significantly increased in combination-treated BCWM.1/IR and RPCI-WM1/IR cells (52.76 ± 4.47% and 67.93 ± 2.46%, respectively; Fig 7D–E). The percent of lysis from CDC, averaged across both IR cell lines, is shown in Fig 7F.
Fig 7.

Addition of daratumumab to ibrutinib induces significant cell death in ibrutinib-resistant WM cells. Cells were treated with single-agent ibrutinib (Ibr; 1 μmol/l), daratumumab (Dara; 0.1 μg/ml) or combination Ibr + Dara (ID). (A–B) Antibody-dependent cell cytotoxicity (ADCC) was determined in calcein-AM labelled BCWM.1/IR (A) and RPCI-WM1/IR (B) target cells, with or without peripheral blood mononuclear cells (PBMCs, effectors) from healthy donors at an effector:target ratio of 50:1 for 4 h. (C) Specific lysis from ADCC averaged across BCWM.1/IR and RPCI-WM1/IR cell lines. (D–E) Complement-dependent cell cytotoxicity (CDC) was measured in BCWM.1/IR (D) or RPCI-WM1/IR (E) tumour cells treated with different agents for 1 h in the presence of 10% human serum from a healthy donor. (F) Specific lysis from CDC averaged across the ibrutinib-resistant WM cell lines. (G) Antibody-dependent cellular phagocytosis (ADCP) was assessed in calcein-AM labelled ibrutinib-resistant WM cell lines treated under the indicated conditions, using CD11b+ macrophages derived from healthy human donor monocytes (effectors) at an effector:target ratio of 2:1 for 2 h. All data are expressed as mean ± SEM. H, Apoptosis was assessed in BCWM.1/IR and RPCI-WM1/IR cells treated with IgG1-b12 isotype antibody, Ibr, Dara or Ibr + Dara for 24 h, followed by staining with annexin-V and propidium iodide (PI) and flow cytometry analysis. Results are expressed as mean ± SEM of the percentage of cells that underwent apoptosis. (I) Scatterplot showing apoptosis in ibrutinib-resistant WM cell lines treated with various agents is shown. *P value <0.05; **P value <0.005 (both considered statistically significant).
When measuring ADCP in BCWM.1/IR cells, we noted that nominal phagocytosis was induced by ibrutinib alone (10.28 ± 1.0%), but the combined effect of ibrutinib + Dara (24.36 ± 0.81%) was not superior to Dara alone. In RPCI-WM1/IR cells, ibrutinib alone did not induce ADCP: although the combination treatment (34.98 ± 0.37%) was superior to ibrutinib, ADCP was not significantly increased compared with single-agent Dara treatment (Fig 7G).
Finally, apoptosis was measured, and as expected, no significant change in annexin V/PI positivity was noted in IR WM cells treated with ibrutinib. However, combination ibrutinib + Dara treatment induced a significant degree of apoptosis in BCWM.1/IR and RPCI-WM1/IR cells (63.72 ± 1.92% and 72.75 ± 2.61%, respectively), as shown in Fig 7H (representative scatterplots are shown in Fig 7I). Altogether, these data affirm that combined targeting of BTK (ibrutinib) and CD38 (with Dara) is highly lethal to WM cells and that this effect is preserved in IR WM cells.
Discussion
WM cells express various surface antigens that are typically seen on monotypic B cells (CD19 and CD20) and plasma cells (CD38, CD138) (Paulus et al, 2015). As clinical treatment strategies have capitalized on CD20 expression with the use of rituximab, targeting CD38 in WM could be similarly equitable. The recent approval of Dara for treatment of patients with multiple myeloma and its examination in solid tumour malignancies, such as lung (Pillai et al, 2017) and brain tumours (Sonikpreet et al, 2018), provides a novel opportunity to investigate its potential as an anti-WM therapeutic (McKeage, 2016). As such, our investigations focused on understanding the anti-tumour activity profile of Dara in preclinical models of WM. We demonstrate for the first time that Dara induces WM cell death in vitro, ex vivo and in an in vivo model of disease.
Indeed, although single-agent ibrutinib is clinically effective in patients with WM, biologically, the sustained suppression of BTK can result in outgrowth of IR cells and lead to clinical disease progression (Woyach et al, 2014; Paulus et al, 2017). Thus, agents with anti-tumour activity in IR cells (and which have a potentially tractable path to clinical application) are of prime interest. Our investigations showed that single-agent Dara treatment induced death in WM cells through various mechanisms (ADCC, CDC, ADCP and apoptosis); this effect was equally conserved in isogenic IR WM cell lines. However, because healthy donor PBMCs were used for effector cells, it is possible that specific lysis carried out by WM PBMC (effectors) might be lower. Our group aims to examine this possibility in a future study, using primary patient WM cells and autologous PBMC effector cells. Collectively though, these cell death mechanisms contributed to the marked blunting of WM cell growth in WM-bearing mice and did not cause any unforeseen adverse effects.
CD38 is expressed at varying levels on WM cells, and its presence can dynamically change with several (cell-intrinsic or -extrinsic) factors (Konoplev et al, 2005; Patten et al, 2008). The first notable observation in our analysis was that chronic exposure of WM cell lines to ibrutinib downregulated cell surface expression of CD38. Although the precise cause of this change remains unclear, and it may be an in vitro phenomenon, this novel finding highlights the intimate link between CD38 and the BCR complex. Malignant CD38− cells are considered to be more anergic than CD38+ B cells and thus less reliant on canonical BCR signalling for their survival (Zupo et al, 1996; Lanham et al, 2003; Malavasi et al, 2011). Furthermore, we have previously shown that IR WM cells are less dependent on BCR signalling when selective pressure from ibrutinib is applied (Paulus et al, 2017); thus, we surmise that CD38 is reflexively downregulated because signalling through the BCR becomes of little use for cell survival. More intriguingly, however, we noted that in 2 of 4 assays (CDC, ADCP), cell death in IR WM cells was higher than that noted in wild-type (ibrutinib-sensitive) WM cells, with the percent average cell death being almost equivalent between wild-type and IR cells (24.6% vs. 27.6%, respectively). Perhaps there is a minimal CD38 sABC threshold at which Dara is capable of eliciting cytolysis through at least 1 or more of the cell death mechanisms demonstrated herein (and independent of the BCR pathway).
We acknowledge that our analysis was limited by low sample sizes and marked variance in CD38 sABC between the 2 IR WM cell lines and where CD38 expression was ~15-fold higher in RPCI-WM1/IR cells compared with BCWM.1/IR cells. Spearman correlation analysis did not show any significant association between CD38 sABC levels with cell death in any of the assays (ADCC, CDC, etc.). Indeed, increased sample sizes will be needed to fully investigate associations between CD38 expression and Dara-induced death in WM cells.
The most striking and novel observation in our study was that Dara significantly modulated BCR and AKT-associated signalling in WM cells. CD38 is an evolutionarily conserved protein expressed at varying quantity in various cell types and chiefly regulates nicotinamide adenine dinucleotide metabolism (Malavasi et al, 2008, 2010; Vaisitti et al, 2015). The net effect of CD38’s ecto-enzymatic hydrolase and cyclase activities is the mobilization of intracellular Ca+ from the endoplasmic reticulum, which in turn influences various cellular processes through modulation of target proteins (Vaisitti et al, 2011, 2015). The link between CD38 enzymatic and BCR coreceptor functions is unclear and, to our knowledge, alteration of BCR-associated signalling proteins upon disruption of CD38 via small molecule or mAb-based approaches has not been previously reported (in any cancer, let alone WM). Although BCR pathway proteins were downregulated in wild-type WM cell lines exposed to Dara, these changes were highly variable in IR WM clones (and more so when exposed to the combination treatment ibrutinib + Dara). We observed a sharp increase in SYK (Fig 5B, J) after a 1-h exposure to Dara in IR WM cells; this change also was observed at 2 and 24 h (data not shown). With precise mechanisms underlying this response being unknown, we speculate that Dara enhances binding of 1 or more transcription factors to the promoter region of the SYK gene, resulting in increased transcription and, ultimately, protein translation of SYK, perhaps as a compensatory survival mechanism. Also, although a Dara-mediated increase in mTOR was noted in the resistant cells, downstream signalling through S6 was inhibited. S6 kinase is a substrate of mTOR kinase; it can be phosphorylated at several sites in its Ser/Thr kinase catalytic domain, and its Ser411 residue can alternatively be phosphorylated by ERK1/2-dependent mechanisms (Hou et al, 2007). Thus, it is plausible that in IR-WM cells, the observed decrease in pS6 (Ser411) occurred independent of mTOR.
We posit that the Dara-induced apoptotic death of IR-WM cells must be due to mechanisms other than changes in the aforementioned proteins and possibly related to alterations in mitochondrial bioenergetics (Paulus et al, 2016d, 2017). Altogether, and considering the changes noted on the aforementioned immunoblot analyses in drug-treated WM cells, we hypothesized that combined inhibition of CD38 and BTK would increase the rate of cell death. Although we anticipated that this effect would be significantly evident in wild-type WM cells, we were surprised to observe that higher cell death (in ADCC, CDC and apoptosis assays) was observed in combination-treated IR WM cell lines compared with the wild-type WM cells. Further investigations are warranted with primary tumour cells from patients with IR WM, but our data show that the combination of ibrutinib + Dara induces greater lethality in WM cells (irrespective of ibrutinib resistance status) compared with the lethality observed with either single-agent Dara or single-agent ibrutinib treatment.
With a phase II clinical trial currently testing single-agent Dara in patients with WM (NCT03187262), our studies herein demonstrate that the anti-WM effects of targeting CD38 can be significantly enhanced with concomitant disruption of BTK. These findings help establish the basic framework for future clinical testing.
Acknowledgments
This work was supported by a grant from the Daniel Foundation of Alabama (A.A.C-K.). It also received support from the University of Iowa and Mayo Clinic Lymphoma SPORE, Developmental Research Program (P50 CA097274) (A.P.).We also acknowledge support from the Mayo Clinic Cancer Center (CA015083) (A.A.C-K.) and Predolin Foundation (Asher Chanan-Khan).
Footnotes
Competing interests
The authors declare no competing interests.
References
- Ailawadhi S, Paulus A & Chanan-Khan A (2016) Preclinical models of Waldenstrom’s macroglobulinemia and drug resistance. Best Practice & Research: Clinical Haematology, 29, 169–178. [DOI] [PubMed] [Google Scholar]
- Argyropoulos KV, Vogel R, Ziegler C, Altan-Bonnet G, Velardi E, Calafiore M, Dogan A, Arcila M, Patel M, Knapp K, Mallek C, Hunter ZR, Treon SP, van den Brink MR & Palomba ML (2016) Clonal B cells in Waldenstrom’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia, 30, 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY & Schlom J (2015) Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunology Research, 3, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP & Chen-Kiang S (2014) Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discovery, 4, 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitta KS, Paulus A, Ailawadhi S, Foster BA, Moser MT, Starostik P, Masood A, Sher T, Miller KC, Iancu DM, Conroy J, Nowak NJ, Sait SN, Personett DA, Coleman M, Furman RR, Martin P, Ansell SM, Lee K & Chanan-Khan AA (2013) Development and characterization of a novel human Waldenstrom macroglobulinemia cell line: RPCI-WM1, Roswell Park Cancer Institute - Waldenstrom Macroglobulinemia 1. Leukemia and Lymphoma, 54, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitta K, Paulus A, Caulfield TR, Akhtar S, Blake MK, Ailawadhi S, Knight J, Heckman MG, Pinkerton A & Chanan-Khan A (2014) Nimbolide targets BCL2 and induces apoptosis in preclinical models of Waldenstroms macroglobulinemia. Blood Cancer Journal, 4, e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Capobianco A, Bergui L, Durig J, Morabito F, Duhrsen U & Malavasi F (2003) CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood, 102, 2146–2155. [DOI] [PubMed] [Google Scholar]
- Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL & Batista FD (2008) CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nature Immunology, 9, 63–72. [DOI] [PubMed] [Google Scholar]
- Drexler HG, Chen S & Macleod RA (2013) Would the real Waldenstrom cell line please stand up? Leukemia and Lymphoma, 54, 224–226. [DOI] [PubMed] [Google Scholar]
- Funaro A, De Monte LB, Dianzani U, Forni M & Malavasi F (1993) Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. European Journal of Immunology, 23, 2407–2411. [DOI] [PubMed] [Google Scholar]
- Herman SE & Wiestner A (2016) Preclinical modeling of novel therapeutics in chronic lymphocytic leukemia: the tools of the trade. Seminars in Oncology, 43, 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, He L & Qi RZ (2007) Regulation of s6 kinase 1 activation by phosphorylation at ser-411. Journal of Biological Chemistry, 282, 6922–6928. [DOI] [PubMed] [Google Scholar]
- Kapoor P, Ansell SM, Fonseca R, Chanan-Khan A, Kyle RA, Kumar SK, Mikhael JR, Witzig TE, Mauermann M, Dispenzieri A, Ailawadhi S, Stewart AK, Lacy MQ, Thompson CA, Buadi FK, Dingli D, Morice WG, Go RS, Jevremovic D, Sher T, King RL, Braggio E, Novak A, Roy V, Ketterling RP, Greipp PT, Grogan M, Micallef IN, Bergsagel PL, Colgan JP, Leung N, Gonsalves WI, Lin Y, Inwards DJ, Hayman SR, Nowakowski GS, Johnston PB, Russell SJ, Markovic SN, Zeldenrust SR, Hwa YL, Lust JA, Porrata LF, Habermann TM, Rajkumar SV, Gertz MA & Reeder CB (2017) Diagnosis and management of Waldenstrom macroglobulinemia: mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncology, 3, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasi PM, Ansell SM & Gertz MA (2015) Waldenstrom macroglobulinemia. Clinical Advances in Hematology & Oncology, 13, 56–66. [PubMed] [Google Scholar]
- Koehrer S & Burger JA (2016) B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clinical Advances in Hematology & Oncology, 14, 55–65. [PubMed] [Google Scholar]
- Konoplev S, Medeiros LJ, Bueso-Ramos CE, Jorgensen JL & Lin P (2005) Immunophenotypic profile of lymphoplasmacytic lymphoma/ Waldenstrom macroglobulinemia. American Journal of Clinical Pathology, 124, 414–420. [DOI] [PubMed] [Google Scholar]
- Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F & Packham G (2003) Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood, 101, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Leblond V, Kastritis E, Advani R, Ansell SM, Buske C, Castillo JJ, Garcia-Sanz R, Gertz M, Kimby E, Kyriakou C, Merlini G, Minnema MC, Morel P, Morra E, Rummel M, Wechalekar A, Patterson CJ, Treon SP & Dimopoulos MA (2016) Treatment recommendations from the Eighth International Workshop on Waldenstrom’s Macroglobulinemia. Blood, 128, 1321–1328. [DOI] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T & Aydin S (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiological Reviews, 88, 841–886. [DOI] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Zaccarello G, Horenstein AL, Chillemi A, Audrito V, Serra S, Gandione M, Zitella A & Tizzani A (2010) The hidden life of NAD+-consuming ectoen- zymes in the endocrine system. Journal of Molecular Endocrinology, 45, 183–191. [DOI] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M & Chiorazzi N (2011) CD38 and chronic lymphocytic leukemia: a decade later. Blood, 118, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna A, De Sarkar S, De S, Bauri AK, Chattopadhyay S & Chatterjee M (2015) The variable chemotherapeutic response of Malabaricone-A in leukemic and solid tumor cell lines depends on the degree of redox imbalance. Phytomedicine, 22, 713–723. [DOI] [PubMed] [Google Scholar]
- Matas-Cespedes A, Vidal-Crespo A, Rodriguez V, Villamor N, Delgado J, Gine E, Roca-Ho H, Menendez P, Campo E, Lopez-Guillermo A, Colomer D, Roue G, Wiestner A, Parren PW, Doshi P, van Bueren JL & Perez-Galan P (2017) The human CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia-microenvironment inter-actions in chronic lymphocytic leukemia. Clinical Cancer Research, 23, 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage K (2016) Daratumumab: first global approval. Drugs, 76, 275–281. [DOI] [PubMed] [Google Scholar]
- Morice WG, Chen D, Kurtin PJ, Hanson CA & McPhail ED (2009) Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenstrom’s macroglobulinemia. Modern Pathology, 22, 807–816. [DOI] [PubMed] [Google Scholar]
- Moulard M & Ozoux ML (2016) How validated receptor occupancy flow cytometry assays can impact decisions and support drug development. Cytometry Part B: Clinical Cytometry, 90, 150–158. [DOI] [PubMed] [Google Scholar]
- Neri S, Mariani E, Meneghetti A, Cattini L & Facchini A (2001) Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clinical and Diagnostic Laboratory Immunology, 8, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, de Jong-Korlaar R, Yuan H, Noort WA, Klein SK, Martens AC, Doshi P, Sasser K, Mutis T & van de Donk NW (2015) Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia, 29, 2039–2049. [DOI] [PubMed] [Google Scholar]
- Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, Groen RW, van Duin M, Sonneveld P, Minnema MC, Zweegman S, Chiu C, Bloem AC, Mutis T, Lokhorst HM, Sasser AK & van de Donk NW (2016) CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood, 128, 959–970. [DOI] [PubMed] [Google Scholar]
- Otero DC, Omori SA & Rickert RC (2001) CD19-dependent activation of Akt kinase in B-lymphocytes. Journal of Biological Chemistry, 276, 1474–1478. [DOI] [PubMed] [Google Scholar]
- Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, Groen RW, Breij E, Martens AC, Bleeker WK & Parren PW (2015) Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs, 7, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overdijk MB, Jansen JH, Nederend M, Lammerts van Bueren JJ, Groen RW, Parren PW, Leusen JH & Boross P (2016) The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fc Gamma Receptor-Mediated Cross-Linking. Journal of Immunology, 197, 807–813. [DOI] [PubMed] [Google Scholar]
- Patten PE, Buggins AG, Richards J, Wother-spoon A, Salisbury J, Mufti GJ, Hamblin TJ & Devereux S (2008) CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood, 111, 5173–5181. [DOI] [PubMed] [Google Scholar]
- Paulus A, Chitta KS, Wallace PK, Advani PP, Akhtar S, Kuranz-Blake M, Ailawadhi S & Chanan-Khan AA (2015) Immunopheno-typing of Waldenstroms macroglobulinemia cell lines reveals distinct patterns of surface antigen expression: potential biological and therapeutic implications. PLoS ONE, 10, e0122338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus A, Ailawadhi S & Chanan-Khan A (2016a) Novel therapeutic targets in Waldenstrom macroglobulinemia. Best Practice & Research: Clinical Haematology, 29, 216–228. [DOI] [PubMed] [Google Scholar]
- Paulus A, Akhtar S, Caulfield TR, Samuel K, Yousaf H, Bashir Y, Paulus SM, Tran D, Hudec R, Cogen D, Jiang J, Edenfield B, Novak A, Ansell SM, Witzig T, Martin P, Coleman M, Roy V, Ailawadhi S, Chitta K, Linder S & Chanan-Khan A (2016b) Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer Journal, 6, e492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus A, Akhtar S, Bashir Y, Paulus SM, Yousaf H, Roy V, Ailawadhi S, Ansell S, Witzig TE & Chanan-Khan AA (2016c) Drug resistance alters CD38 expression and in vitro response to daratumumab in Waldenstrom macroglobulinemia cells. Blood, 128, 3018. [Google Scholar]
- Paulus A, Akhtar S, Hudec R, Paulus SM, Bashir Y, Witzig TE, Ansell S, Linder S, Ailawadhi S & Chanan-Khan AA (2016d) Waldenstrom macroglobulinemia cells modulate mitochondrial bioenergetics and induce a respiratory hyper-drive state upon acquisition of ibrutinib-resistance. Blood, 128, 2761. [Google Scholar]
- Paulus A, Akhtar S, Yousaf H, Manna A, Paulus SM, Bashir Y, Caulfield TR, Kuranz-Blake M, Chitta K, Wang X, Asmann Y, Hudec R, Springer W, Ailawadhi S & Chanan-Khan A (2017) Waldenstrom macroglobulinemia cells devoid of BTK(C481S) or CXCR4 (WHIM-like) mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer Journal, 7, e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai R, Ramalingam S, Paz-Ares L, Thayu M, Watson P & Reck M (2017) P3.04–008 A phase 1b/2 study of atezolizumab with or without daratumumab in advanced or metastatic non-small cell lung cancer (NSCLC). Journal of Thoracic Oncology, 12, S2288. [Google Scholar]
- Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N & Burger JA (2012) The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood, 119, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij MF, Kuil A, Kraan W, Kersten MJ, Treon SP, Pals ST & Spaargaren M (2016) Ibrutinib and idelalisib target B cell receptor-but not CXCL12/CXCR4-controlled integrin-mediated adhesion in Waldenstrom macroglobulinemia. Haematologica, 101, e111–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonikpreet D, Schiapparelli P, Manna A, Paulus A, Rosenfeld SS, Chanan-Khan A & Quinones-Hinojosa A (2018) CD38 targeted therapy in glioblastoma: a step forward. Neurology, 90, 160. [Google Scholar]
- Strome SE, Sausville EA & Mann D (2007) A mechanistic perspective of monoclonal antibodies in cancer therapy beyond target-related effects. Oncologist, 12, 1084–1095. [DOI] [PubMed] [Google Scholar]
- Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, Argyropoulos KV, Yang G, Cao Y, Xu L, Patterson CJ, Rodig S, Zehnder JL, Aster JC, Harris NL, Kanan S, Ghobrial I, Castillo JJ, Laubach JP, Hunter ZR, Salman Z, Li J, Cheng M, Clow F, Graef T, Palomba ML & Advani RH (2015) Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. New England Journal of Medicine, 372, 1430–1440. [DOI] [PubMed] [Google Scholar]
- Vaisitti T, Audrito V, Serra S, Bologna C, Brusa D, Malavasi F & Deaglio S (2011) NAD+-metabolizing ecto-enzymes shape tumor-host interactions: the chronic lymphocytic leukemia model. FEBS Letters, 585, 1514–1520. [DOI] [PubMed] [Google Scholar]
- Vaisitti T, Audrito V, Serra S, Buonincontri R, Sociali G, Mannino E, Pagnani A, Zucchetto A, Tissino E, Vitale C, Coscia M, Usai C, Pepper C, Gattei V, Bruzzone S & Deaglio S (2015) The enzymatic activities of CD38 enhance CLL growth and trafficking: implications for therapeutic targeting. Leukemia, 29, 356–368. [DOI] [PubMed] [Google Scholar]
- de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, Oomen LA, Peipp M, Valerius T, Slootstra JW, Mutis T, Bleeker WK, Anderson KC, Lokhorst HM, van de Winkel JG & Parren PW (2011) Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. Journal of Immunology, 186, 1840–1848. [DOI] [PubMed] [Google Scholar]
- Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ & Byrd JC (2014) Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. New England Journal of Medicine, 370, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupo S, Isnardi L, Megna M, Massara R, Malavasi F, Dono M, Cosulich E & Ferrarini M (1996) CD38 expression distinguishes two groups of B-cell chronic lymphocytic leukemias with different responses to anti-IgM antibodies and propensity to apoptosis. Blood, 88, 1365–1374. [PubMed] [Google Scholar]