

Abstract
Replication protein A (RPA) is the major human single stranded DNA (ssDNA)-binding protein, playing essential roles in DNA replication, repair, recombination, and DNA-damage response (DDR). Inhibition of RPA–DNA interactions represents a therapeutic strategy for cancer drug discovery and has great potential to provide single agent anticancer activity and to synergize with both common DNA damaging chemotherapeutics and newer targeted anticancer agents. In this letter, a new series of analogues based on our previously reported TDRL-551 (4) compound were designed to improve potency and physicochemical properties. Molecular docking studies guided molecular insights, and further SAR exploration led to the identification of a series of novel compounds with low micromolar RPA inhibitory activity, increased solubility, and excellent cellular up-take. Among a series of analogues, compounds 43, 44, 45, and 46 hold promise for further development of novel anticancer agents.
Keywords: Replication protein A, DNA damage and repair, reversible inhibitors, SAR, medicinal chemistry
The clinical efficacy of many DNA damaging cancer chemotherapeutics involves inducing DNA damage to push cancer cells into apoptosis.1,2 DNA damage is repaired by intrinsic repair pathways and is coordinated by the DNA damage response (DDR) pathway. One mechanism to enhance the therapeutic window associated with these therapies is to direct treatment to those cancers that harbor intrinsic DNA repair deficiencies.3−5 This approach has been effectively exploited in synthetic lethal strategies to develop safe and effective treatments.1 With an appropriately selected drug target, there is the potential to enhance both single agent activity and synergistic anticancer effects of the same chemical entity. Therefore, targeting DNA repair and the DDR deficiencies has the potential for more selective, better tolerated therapies to improve cancer patient survival.6,7
Replication protein A (RPA) is the major human ssDNA binding protein and plays an integral role in both nucleotide excision repair (NER) and homologous recombination (HR) DNA repair pathways in addition to its essential role in DNA replication and DDR.8−10 Each of these critical roles of RPA make the RPA–DNA interaction a promising target to develop novel anticancer therapeutics. RPA is also overexpressed in a number of cancers including lung, ovarian, breast, colon, and esophageal.11,12 One rationale for targeting RPA is based on RPA exhaustion which can lead to replication catastrophe. By extension, one can envision that cancer cells exhibiting replication stress require more RPA for survival compared to noncancer cells to provide a therapeutic treatment window.
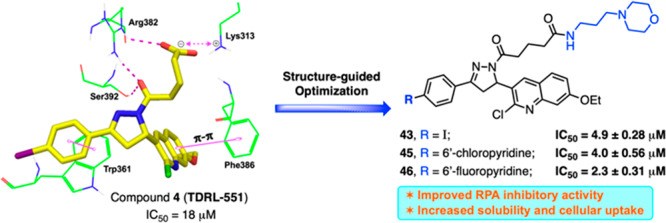
Over the past few years, several RPA inhibitors have been identified and have focused on blocking either the protein–protein13,15−22 or the protein–DNA interaction14,23−26 (Figure 1A). Targeting the F-domain has proven effective for disrupting protein–protein interactions and the A and B domains of RPA70 for protein–DNA inhibitors.27−29 Previously, we have successfully discovered and developed compounds 1–4 which block the RPA–DNA interaction.23−26 Particularly, compounds 3 (TDRL-505) and 4 (TDRL-551) display single agent activity in lung and ovarian cancer cell lines and also synergize with cisplatin and etoposide.25,26 The mechanism of action of both compounds is via a reversible interaction with the central OB-folds in DNA binding domains A and B of RPA70. In vivo analysis of compound 4 (TDRL-551) revealed minimal cytotoxicity alone and in combination with platinum was able to significantly delay tumor growth in a NSCLC xenograft model.26 Both compounds 3 and 4, however, had limited solubility and cell permeability. In this Letter, we further expanded our structure-guided drug design efforts by exploiting the compound 4 (TDRL-551) scaffold with the aim to improve RPA inhibitory potency, solubility, and cellular uptake.
Figure 1.

(A) Structure of previously reported RPA inhibitors targeting RPA–DNA interaction.23−26 (B) Schematic representation of SAR design rationale: a large space-filling pocket surrounding Ring A of compound 4 and amino acid residues surrounding the alkyl carboxylic acid side chain which can be extended toward the solvent-exposed region for further structural optimization. [Compound 4 hydrogen bond interaction with Ser392 and Arg382 is indicated with the dashed magenta line, π–π stacking interactions with Trp361 and Phe386 are shown in solid magenta dumbbell, and salt-bridge interactions with Lys313 are shown in dashed two-sided magenta arrow.]
Initial molecular docking studies with compounds 3 and 4 revealed that both compounds have a high predicted affinity for DNA binding domain B. We performed molecular docking studies mainly focusing on the central DNA binding domains A and B of RPA70 by using RPA70181–422 X-ray crystal structure (PDB code: 1FGU).29 The interaction presented in Figure 1B reveals the stability is driven via hydrophobic and π–π interactions and that the conserved alkyl carboxylic acid side chain is stabilized via interactions with basic amino acid residues which are critical for inhibitory activity. In addition, docking studies revealed the extended terminal carboxylic acid side chain appeared to orient toward the solvent-exposed region of the protein and provides a potential site for modulating the physicochemical properties of these compounds. We have exploited compound 4’s interaction with domain B to outline a SAR design for further structural optimization (Figure 1B). We first pursued optimizing aromatic Ring A exploiting the large pocket around it and also simultaneously optimizing the alkyl carboxylic acid side chain to improve the druglike properties.
The synthesis of target compound 4 and its analogs 16–25 is depicted in Scheme 1. We have obtained compounds 4 and 16–25 from starting material 3-ethoxyaniline by slightly modifying our previous synthetic protocol.26 The requisite quinoline carbaldehyde 6 was prepared by acylation of 3-ethoxyaniline with acetic anhydride, followed by multicomponent reaction that involves the process of Vilsmeier–Haack chlorination, formylation, and cyclization of acetanilide using DMF and POCl3. Claisen–Schmidt condensation of the corresponding substituted acetophenones 7–10 with quinoline carbaldehyde 6 in the presence of aqueous NaOH in ethanol yielded the corresponding 1,3-diarypropenones 11–14 which on further treatment with hydrazine hydrate in ethanol under reflux afforded the corresponding 2-pyrazolines 15a–d in good yields. Compounds 4 and 16–18 were obtained in good yields by acylation at N1 of the pyrazoline ring of compounds 15a–d with glutaric anhydride in chloroform. Upon reduction of the nitro group of 18 by stannous chloride, the desired amine product 19 was obtained in an acceptable yield. The synthesis of compounds 20 and 21 was accomplished by acylation of the hydroxyl group of compound 17 using acryloyl chloride and morpholinecarbonyl chloride, respectively, in basic conditions. To further extend the substitution on the para-position at ring A, we have synthesized target compounds 22–29 as depicted in Scheme 1 and Supporting Information Scheme S1. Compound 30 was prepared from an above synthesized precursor 15a by N1-acylation of the pyrazoline ring using ethyl glutaryl chloride in basic condition. Intermediates 31a–d were prepared by optimizing Pd-catalyzed Suzuki coupling of compound 30 with corresponding boronic acids/esters using nonaqueous CsF in a dimethoxyethane protocol. Final target compounds 22–25 were obtained from compounds 31a–d by hydrolysis of the corresponding alkyl esters using 10 N NaOH at room temperature. Our initial attempt utilizing Scheme 1 to synthesize halopyridine containing compounds 26–28 had limited efficiency as a function of the late stage Suzuki coupling reaction. Therefore, we have achieved efficient synthesis of target compounds 26–29 as outlined in Supporting Information Scheme S1. Early stage Suzuki coupling of 4′-iodoacetophenone 7 with commercially available boronic acids/esters 32a–d catalyzed by tetrakis(triphenylphosphine)palladium(0) in the presence of aqueous potassium carbonate base provided precursors 33a–d. 1,3-Diarypropenones 34a–d were synthesized using an above-described Claisen–Schmidt condensation of the corresponding substituted methyl ketones 33a–d with quinoline carbaldehyde 6 and then 1,3-diarypropenones 34a–d refluxed with hydrazine hydrate in ethanol to afford the corresponding 2-pyrazolines 35a–d in good yields. Finally, acylation at N1 of the pyrazoline ring of compounds 35a–d with glutaric anhydride in chloroform under reflux provided target compounds 26–29 in moderate to good yields after recrystallization in ethanol.
Scheme 1. Synthesis of Compound 4 and Analogs 16–25.

Reagents and conditions: (a) acetic anhydride, DIPEA, DMAP, DCM, rt for 2 h, 88%; (b) (i) DMF, POCl3, 0 °C for 25 min, (ii) acetanilide 5, 110 °C for 3 h, 74%; (c) 2.5 M NaOH, EtOH, 45 °C for 45 min to 1 h, 53–74%; (d) hydrazine hydrate, EtOH, reflux for 2–3 h, 70–83%; (e) glutaric anhydride, CHCl3, reflux for 2 h, 58–71% (after recrystallization); (f) SnCl2, EtOH:THF (1:1), reflux for 2 h, 48% (after recrystallization); (g) acryloyl chloride, 2 N NaOH, THF, 0 °C to rt for 2 h, 34%; (h) 4-morpholinecarbonyl chloride, TEA, DMAP, THF, 0 °C to rt for 12 h, 63%. (i) ethyl glutaryl chloride, DIPEA, DCM, rt for 12 h, 78%; (j) boronic acid/ester, Pd(PPh3)4, CsF, DME, 90 °C for 15–18 h, 63–69%; (k) 10 N NaOH, THF:MeOH (1:2), rt for 6–8 h, 75–87%.
To optimize the alkyl carboxylic acid side chain of compound 4, we have synthesized compounds 37–47 as depicted in Supporting Information Schemes S2 and S3. The N1-acylation of the pyrazoline ring of precursor 15a using alkyl acyl chloride in basic conditions afforded ester derivatives 36a–b, and subsequent ester hydrolysis using NaOH provided analogs 37–38 in excellent yields (Supporting Information Scheme S2). To introduce rigidity in the RPA binding pocket, we have synthesized compounds 39–40 from precursor 11 and using corresponding substituted hydrazinobenzoic acid under reflux condition in AcOH and n-butanol. Furthermore, the morpholine/morpholinoalkylamine group was attached to the oxopentanoic acid side chain of corresponding compound 4 or 16 or 26 or 28 or 29 by utilizing the EDCI/HOBt amide synthetic protocol to achieve final compounds 41–47 in good yields (Supporting Information Scheme S3).
The assessment of RPA inhibitory activity was performed using a highly sensitive and quantitative electrophoretic mobility shift assay (EMSA). The assay employs purified heterotrimeric human RPA devoid of any tags or modifications and measures the impact of the compounds on direct RPA binding to a ss-DNA substrate.23,26,30 We also have confirmed that the compounds specifically target OB-folds A and B using an RPA construct consisting of this minimal DNA binding domain (See Supporting Information Table S2), as we have previously described.25 RPA can also interact with damage duplex DNA via binding to the undamaged ssDNA regions opposite the damage.30 The RPA inhibitory compounds also, as expected, block this interaction. For the purpose of quantifying RPA inhibitory activity we used gold-standard, full length RPA binding to single stranded DNA to calculate IC50 values of newly synthesized analogs and provide insight into the structure–activity relationships (Table 1 and 2). The replacement of the iodo group of compound 4 (IC50 = 15.3 ± 1.42 μM) with hydroxyl or nitro groups resulted in a near complete loss of RPA inhibitory activity while changing to an amine resulted in a 7-fold increase in potency (IC50 = 2.3 ± 0.22 μM). Acrylate containing compound 20 exhibited a 2-fold increase in activity compared to 4; surprisingly, the inclusion of morpholinecarbonyl group in compound 21 resulted in poor RPA inhibition. Introduction of a heteroaromatic ring on Ring A such as 3′-furan, 2′-furan, and 4′-isoxazole boosted potency drastically as 2′-substituted furan at Ring A (compound 23, IC50 = 2.1 ± 0.48 μM) exhibited almost 3.2-fold increase in activity compared to 3′-substituted furan containing compound 22 (IC50 = 6.7 ± 0.64 μM). Insertion of another heteroatom such as nitrogen into the furan ring in the form of an isoxazole displayed even more potent RPA inhibitory activity (compound 24, IC50 = 1.7 ± 0.28 μM). The difference in activity with the small changes of heteroatom within the 5-membered ring may be due to the orientation of the ring or the ability of the heteroatom to interact with RPA backbone binding pocket amino acid residues. Further extension of compound 4 Ring A in the space-filling binding cleft with a substituted bulky biphenyl heteroaromatic ring such as halogenated pyridinyl compounds (26–29) exhibited even more potent RPA inhibitory activities than parent compound with an increased potency between 4- and 15-fold. Among all the Ring A modifications, chloro-pyridinyl compound 26 and bromo-pyridinyl compound 27 showed the most potent RPA inhibitory activity in the EMSA assay (26, 1.6 ± 0.04 μM and 27, IC50 = 1.0 ± 0.60 μM). Further, we have included fluorine and trifluoromethyl substituents in pyridinyl compounds 28–29. However, both compounds displayed slightly weaker activity compared to chloro- and bromo-pyridinyl containing compounds 26–27. These data revealed the importance of bulky hydrophobic substituents on Ring A of compound 4 to occupy the space-filling binding pocket of RPA and are consistent with our postulated molecular docking binding pockets outlined in an above SAR design section.
Table 1. SAR of Ring A with RPA IC50 Values of Analogs 16–29a.


Determined using EMSA; binding of full length human RPA to DNA was assessed.
Compounds that displayed greater than 80% inhibition at 25 μM were analyzed in titration experiments.
IC50 values are a mean of minimum of triplicate independent experiments and data are presented as the mean ± SD.
Table 2. SAR of Side Chain Modifications with RPA IC50 Values of Analogs 41–47a.


Determined using EMSA; binding of full length human RPA to DNA was assessed.
Compounds that displayed greater than 80% inhibition at 25 μM were analyzed in titration experiments.
IC50 values are a mean of minimum of triplicate independent experiments, and data are presented as the mean ± SD.
The next aim was to explore SAR on the alkyl carboxylic acid side chain of compound 4, with a focus on enhancing physicochemical properties while retaining or improving potency. Modification of the alkyl side chain of compound 4 revealed a series of interesting activity against RPA (Supporting Information Table S1). The conversion to an oxopentanoic ethyl ester (compound 30) completely abrogated RPA inhibitory activity. A similar trend was observed with compounds 39–40 when we replaced oxopentanoic acids with 2′- or 3′-subsitituted benzoylic carboxylic acids. The intramolecular distances between the benzoylic carboxylic acids and the core dihydropyrazole were very similar compared to the oxopentanoic acids. The dramatic reduction in activity of the benzoylic carboxylic acid derivatives is likely a function of the rigidity induced restricting motion of the molecule within the RPA-DNA binding pocket independent of the position of the carboxylic acid. Increasing the length of the aliphatic side chain by 1 and 2 carbons, in compounds 37 and 38, respectively, had only modest effects on potency, and similar inhibition was observed by direct addition of an N-morpholino group on the alkyl carboxylic acid side chain depicted in compound 41.
As discussed earlier, initial docking studies positioned the terminal carboxylic group toward the solvent-exposed region of the protein and could provide a good potential for modulating the physicochemical properties of our compounds. In addition, the carboxylic acid functional group often has limited utility in drug discovery due to limited passive diffusion across biological membranes (cellular permeability) and metabolic instability.31 Therefore, we articulated that RPA inhibitors could be improved by introducing an additional solubilizing group into the terminal carboxylic acid group without significantly influencing the RPA inhibitory activities. To this end, the water-soluble morpholine moiety was employed to enhance the physicochemical properties. Consistent with our docking analysis, replacement of the terminal carboxylic group with morpholinoethane (compound 42) and morpholinopropane (compound 43) retained the inhibitory activity while compound 43 showed an almost 3-fold increase in potency (IC50 = 4.9 ± 0.28 μM) compared to our parent compound 4. Encouraged by these results, a series of new analogs (Table 2, compounds 44–47) were further designed and synthesized by incorporating morpholinopropane at the terminal carboxylic group of compounds 16, 26, and 28–29. All these compounds exhibited modest to potent RPA inhibitory activity compared to the parent compound 4 and were anticipated to improve physiochemical properties for better cellular and in vivo activities.
To confirm the mechanism of inhibition via compound binding to the target protein and not via binding to the DNA substrates, we conducted a fluorescent intercalator displacement (FID) assay as described previously.32 The displacement of a DNA binding dye and decrease in fluorescence is indicative of the compound’s ability to bind DNA. In order to probe the potential role of DNA intercalation as a mechanism for RPA inhibition, most potent compounds 19, 23, 26, 27, 43, and 45 were analyzed using FID assay along with doxorubicin (Dox) as a positive control. The results presented in Figure 2 demonstrate as expected that positive control, doxorubicin, a known noncovalent DNA binding chemotherapeutic, resulted in a concentration dependent reduction in fluorescence. As expected, minimal DNA binding activity was observed for any of the novel RPA inhibitors (19, 23, 26, 27, 43, and 45). The slight reduction in signal cannot account for the high potency of the compounds. These data suggest that the mechanism of action involves targeting the protein–DNA interaction by directly binding to the RPA protein. Consistent with this conclusion is the lack of inhibition against other OB-fold containing proteins.
Figure 2.
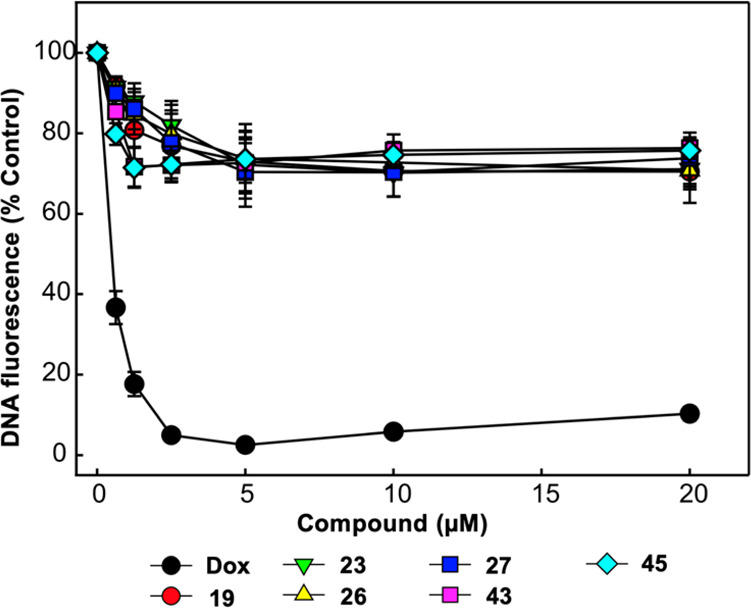
Analysis of compound interactions with DNA using fluorescent intercalator displacement (FID) assay. The indicated concentrations of doxorubicin (Dox), compounds 19, 23, 26, 27, 43, and 45 were analyzed for the ability to displace a fluorescent Sybr-green DNA intercalator as a measure of compound DNA interactions. The data represent the average and SD of three independent experimental determinations performed in duplicate.
To delineate the key interactions and to understand the SAR of newly synthesized compounds, these compounds were flexibly docked mainly focusing on the central DNA binding domains A and B of RPA70 as discussed above.29 All newly developed inhibitors docked well at this site. Figure 3 shows the binding orientation and molecular interactions of representative compounds 26, 42, and 45 within the RPA domain B region. The molecular interactions of 26, 42, and 45 (Figures 3A–C, respectively) are largely ascribed to various electrostatic interactions, including (i) compounds 42 and 45 amide carbonyls making hydrogen bond contacts with the amine of Lys313, while the compound 26 terminal carboxylic acid makes salt-bridge interactions with the Lys313; (ii) the amide carbonyl (attached to pyrazole ring) of all three compounds making strong hydrogen bond contacts with the hydroxyl group of Ser392 as well as with the amine group of Arg382; (iii) the π–π stacking interactions between the phenyl moiety (Ring A) and the aromatic ring of Trp361 in all three compounds. In addition, all three compounds’ quinoline moiety is also well positioned to make π–π stacking interactions with Phe386; (iv) The terminal alkyl morpholino side chain is well fitted and located as extending out of the RPA binding region into a solvent exposed region. Docking studies predicted a stronger affinity of compound 26 than other series of compounds including compounds 42 and 45; in fact, compound 26 also showed potent RPA inhibition compared to other series of compounds in in vitro EMSA assay (Table 1).
Figure 3.
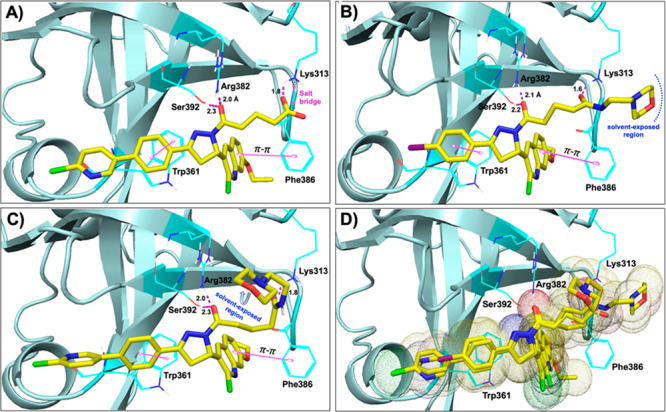
Molecular docking studies (PDB code: 1FGU):29 (A–C) Molecular interactions of compounds 26 (A), 42 (B), and 45 (C) (all in yellow carbon) with hRPA (key amino acids are shown in dark cyan carbon and cartoon is shown in pale cyan color). Interaction with amino acid side chains is indicated with the dashed magenta lines, π–π stacking interactions are shown in solid magenta dumbbell, and salt-bridge interactions are shown in dashed two-sided magenta arrow. Interaction distances indicated in Å. (D) Molecular overlay (superimposition) of compounds 26, 42, and 45 (all in yellow carbon) in the RPA binding site.
Our previous data demonstrated that compounds 3 and 4 displayed a significantly higher cellular IC50 compared to the biochemical in vitro IC50.25,26 This suggested cellular uptake could be limiting cellular activity. The charge on the carboxylic acid, while potentially increasing aqueous solubility, was also anticipated to negatively impact cellular uptake. We therefore assessed aqueous solubility of a series of the most biochemically active compound 4 derivatives. Figure 4A shows data obtained with assessing aqueous solubility in unbuffered H2O at pH 7. The data reveal a general trend of the morpholino modified compounds displaying increased solubility compared to their carboxylic acid counterparts (compounds 4, 16, 26, 28, 29 vs 43, 44, 45, 46, 47, respectively).
Figure 4.

Solubility analysis and cellular uptake of RPA inhibitors. (A) Aqueous solubility and (B) Cellular uptake of representative compounds were determined as described in the Supporting Information. Individual data points are plotted, and bars represent the mean and SD of three independent experimental determinations.
The cellular uptake or intracellular metabolism of the compounds plays an important role in cellular efficacy and it allows proper rationalization of the structure–activity relationships to enhance the sustained therapeutic effect. Therefore, we evaluated cellular uptake of our lead compounds in H460 NSCLC cells (Figure 4B). The data demonstrate that as expected, compound 4 has relatively poor uptake while compound 26 showed increased uptake even with the carboxylic acid moiety. The morpholino derivative of both these compounds demonstrated considerably superior uptake observed with compounds 43 and 45. We expect the increased uptake will increase cellular RPA inhibition and anticancer activity. A similar trend of increased cellular uptake was also observed in morpholino modified compound 46 and 47 compared to their carboxylic acid counterparts (Data not shown).
In conclusion, we have extended SAR study of our previously reported RPA inhibitor by utilizing a structure-based drug design strategy. We have identified a series of novel chemical inhibitors that interact directly with RPA to block its interaction with single-stranded DNA, and most importantly these inhibitors do not bind directly with DNA. The systematic SAR exploration had revealed that a heteroaromatic ring or reasonably a larger lipophilic biphenyl ring at Ring A and an extension of the terminal carboxylic acid side chain are well tolerated. Particularly, the introduction of a morpholino group at the terminal alkyl carboxylic acid side chain resulted in the enhanced potency, solubility, and cellular up-take. Compounds 43, 45, and 46 represent excellent lead compounds with drug-like properties suitable for future cellular and in vivo studies. These efforts will be addressed in a subsequent manuscript.
Acknowledgments
We thank Dr. Lifan Zeng and Erica Woodall for technical assistance with HPLC and mass spectrometry (LCMS and HRMS).
Glossary
Abbreviations
- DDR
DNA damage response
- NER
nucleotide excision repair
- RPA
replication protein A
- DBD
DNA binding domain
- HOBt
hydroxybenzotriazole
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- TEA
triethylamine
- DME
dimethoxyethane
- EMSA
electrophoretic mobility shift assay
- SAR
structure activity relationship
- FID
fluorescent intercalator displacement
- Dox
doxorubicin
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00440.
Supplementary Figures S1, synthetic Schemes S1–S3, Table S1 and Table S2, synthetic experimental procedures along with characterization data, biological and physiochemical experimental procedures, copies of 1H NMR spectra of final compounds, molecular docking overlays, and 2D interactions of RPA inhibitors with RPA protein (PDF)
Author Contributions
The manuscript was written by N.S.G. and J.J.T. with editing support from P.S.V., K.S.P., M.R.J., and T.L.V. All authors have given approval to the final version of the manuscript.
This work is supported by NIH grants R01-CA180710 and R41-CA162648 and the Tom and Julie Wood Family Foundation.
The authors declare the following competing financial interest(s): J. Turchi is a co-founder and CSO of NERx Biosciences and co-inventor on patents covering the compounds described in this Letter.
Supplementary Material
References
- Gavande N. S.; VanderVere-Carozza P. S.; Hinshaw H. D.; Jalal S. I.; Sears C. R.; Pawelczak K. S.; Turchi J. J. DNA repair targeted therapy: The past or future of cancer treatment?. Pharmacol. Ther. 2016, 160, 65–83. 10.1016/j.pharmthera.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willers H.; Azzoli C. G.; Santivasi W. L.; Xia F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. 10.1097/PPO.0b013e318292e4e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford A. N.; Jackson S. P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Brown J. S.; O’Carrigan B.; Jackson S. P.; Yap T. A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery 2017, 7, 20–37. 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. P.; Helleday T. Drugging DNA repair. Science 2016, 352, 1178–1179. 10.1126/science.aab0958. [DOI] [PubMed] [Google Scholar]
- Hosoya N.; Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor M. J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- Oakley G. G.; Patrick S. M. Replication Protein A: directing traffic at the intersection of replication and repair. Front. Biosci., Landmark Ed. 2010, 15, 883–900. 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz S. K.; Sheehan A. M.; Wold M. S. Replication Protein A phosphorylation and the cellular response to DNA damage. DNA Repair 2004, 3, 1015–1024. 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- Marechal A.; Zou L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. 10.1038/cr.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D.; Domont J.; Taranchon E.; Monnet I.; Tredaniel J.; Caliandro R.; Validire P.; Besse B.; Soria J.-C.; Fouret P. The NER proteins are differentially expressed in ever smokers and in never smokers with lung adenocarcinoma. Ann. Oncol. 2009, 20, 1257–1263. 10.1093/annonc/mdn785. [DOI] [PubMed] [Google Scholar]
- Jekimovs C.; Bolderson E.; Suraweera A.; Adams M.; O’Byrne K. J.; Richard D. J. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Front. Oncol. 2014, 4, 86. 10.3389/fonc.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone J. D.; Waterson A. G.; Fesik S. W. Recent advancements in the discovery of protein–protein interaction inhibitors of replication protein A. MedChemComm 2017, 8, 259–267. 10.1039/C6MD00460A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavande N. S.; VanderVere-Carozza P. S.; Pawelczak K. S.; Turchi J. J.. Targeting the nucleotide excision repair pathway for therapeutic applications. In DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications; Kelley M., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2016; pp 135–150. [Google Scholar]
- Patrone J. D.; Kennedy J. P.; Frank A. O.; Feldkamp M. D.; Vangamudi B.; Pelz N. F.; Rossanese O. W.; Waterson A. G.; Chazin W. J.; Fesik S. W. Discovery of protein-protein interaction inhibitors of replication protein A. ACS Med. Chem. Lett. 2013, 4, 601–605. 10.1021/ml400032y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkamp M. D.; Frank A. O.; Kennedy J. P.; Patrone J. D.; Vangamudi B.; Waterson A. G.; Fesik S. W.; Chazin W. J. Surface reengineering enables co-crystallization with an inhibitor of the RPA interaction motif of ATRIP. Biochemistry 2013, 52, 6515–6524. 10.1021/bi400542z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J. G.; Carnes K. A.; Soto P.; Liu S.; Parkhurst L. J.; Oakley G. G. A Small Molecule Directly Inhibits the p53 Transactivation Domain from Binding to Replication Protein A. Nucleic Acids Res. 2013, 41, 2047–2059. 10.1093/nar/gks1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. O.; Feldkamp M. D.; Kennedy J. P.; Waterson A. G.; Pelz N. F.; Patrone J. D.; Vangamudi B.; Camper D. V.; Rossanese O. W.; Chazin W. J.; Fesik S. W. Discovery of a Potent Inhibitor of Replication Protein A Protein–Protein Interactions Using a Fragment-Linking Approach. J. Med. Chem. 2013, 56, 9242–9250. 10.1021/jm401333u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. O.; Vangamudi B.; Feldkamp M. D.; Souza-Fagundes E. M.; Luzwick J. W.; Cortez D.; Olejniczak E. T.; Waterson A. G.; Rossanese O. W.; Chazin W. J.; Fesik S. W. Discovery of a potent stapled helix peptide that binds to the 70N domain of replication protein A. J. Med. Chem. 2014, 57, 2455–2461. 10.1021/jm401730y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J. G.; Liu S.; Wang L.; Mosel A.; Peng A.; Oakley G. G. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. 10.1158/0008-5472.CAN-14-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterson A. G.; Kennedy J. P.; Patrone J. D.; Pelz N. F.; Feldkamp M. D.; Frank A. O.; Vangamudi B.; Souza-Fagundes E. M.; Rossanese O. W.; Chazin W. J.; Fesik S. W. Diphenylpyrazoles as replication protein a inhibitors. ACS Med. Chem. Lett. 2015, 6, 140–145. 10.1021/ml5003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone J. D.; Pelz N. F.; Bates B. S.; Souza-Fagundes E. M.; Vangamudi B.; Camper D. V.; Kuznetsov A. G.; Browning C. F.; Feldkamp M. D.; Frank A. O.; Gilston B. A.; Olejniczak E. T.; Rossanese O. W.; Waterson A. G.; Chazin W. J.; Fesik S. W. Identification and optimization of anthranilic acid-based Inhibitors of replication protein A. ChemMedChem 2016, 11, 893–899. 10.1002/cmdc.201500479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews B. J.; Turchi J. J. Development of a high-throughput screen for inhibitors of replication protein A and its role in nucleotide excision repair. Mol. Cancer Ther. 2004, 3, 385–391. [PubMed] [Google Scholar]
- Neher T. M.; Bodenmiller D.; Fitch R. W.; Jalal S. I.; Turchi J. J. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol. Cancer Ther. 2011, 10, 1796–1806. 10.1158/1535-7163.MCT-11-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck S. C.; Turchi J. J. Targeted inhibition of replication protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. 10.1158/0008-5472.CAN-09-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A. K.; Dormi S. S.; Turchi A. M.; Woods D. S.; Turchi J. J. Chemical inhibitor targeting the replication protein A–DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 2015, 93, 25–33. 10.1016/j.bcp.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey C. A.; Soss S. E.; Brooks S.; Yan C.; Ivanov I.; Dorai K.; Chazin W. J. Functional dynamics in replication protein a DNA binding and protein recruitment domains. Structure 2015, 23, 1028–1038. 10.1016/j.str.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Pavletich N. P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26, 2337–2347. 10.1101/gad.194787.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva E.; Belegu V.; Korolev S.; Bochkarev A. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J. 2001, 20, 612–618. 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick S. M.; Turchi J. J. Replication protein A (RPA) binding to duplex cisplatin damaged DNA is mediated through the generation of single-stranded DNA. J. Biol. Chem. 1999, 274, 14972–14978. 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- Lassalas P.; Gay B.; Lasfargeas C.; James M. J.; Tran V.; Vijayendran K. G.; Brunden K. R.; Kozlowski M. C.; Thomas C. J.; Smith A. B. III; Huryn D. M.; Ballatore C. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59, 3183–3203. 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavande N. S.; VanderVere-Carozza P.; Mishra A. K.; Vernon T. L.; Pawelczak K. S.; Turchi J. J. Design and Structure-Guided Development of Novel Inhibitors of the Xeroderma Pigmentosum Group A (XPA) Protein-DNA Interaction. J. Med. Chem. 2017, 60, 8055–8070. 10.1021/acs.jmedchem.7b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.