

Abstract
With the development of growth factors and growth factor modulators as therapeutics for a range of disorders, it is prudent to consider whether modulating the growth factor profile in a tissue can influence tumour initiation or progression. As recombinant human TGF-β3 (avotermin) is being developed for the improvement of scarring in the skin it is important to understand the role, if any, of this cytokine in tumour progression.
Elevated levels of TGF-β3 expression detected in late-stage tumours have linked this cytokine with tumourigenesis, although functional data to support a causative role are lacking. While it has proved tempting for researchers to interpret a ‘correlation’ as a ‘cause’ of disease, what has often been overlooked is the normal biological role of TGF-β3 in processes that are often subverted in tumourigenesis. Clarifying the role of this cytokine is complicated by inappropriate extrapolation of the data relating to TGF-β1 in tumourigenesis, despite marked differences in biology between the TGF-β isoforms. Indeed, published studies have indicated that TGF-β3 may actually play a protective role against tumourigenesis in a range of tissues including the skin, breast, oral and gastric mucosa. Based on currently available data it is reasonable to hypothesize that administration of acute low doses of exogenous TGF-β3 is unlikely to influence tumour initiation or progression.
Keywords: Transforming growth factor β, Tumourigenesis, Cytokines, Avotermin
1. Introduction
The role of TGF-β1 in tumourigenesis has been extensively studied and reviewed in some depth in a number of publications [1–5]. However, the data are often contradictory, reflecting the complexity of the biological role of this cytokine. Data from a number of studies have helped to determine which molecular and cellular mechanisms involving TGF-β1 play a causative role in tumourigenesis [6–8]. In contrast to the TGF-β1 isoform, the role of the TGF-β3 isoform in tumourigenesis has been considerably less well studied, with assumptions regarding its role in tumourigenesis often being made based on observations reported for TGF-β1. While there is overlap in some of the biological roles of these isoforms, it has been demonstrated in a number of systems that TGF-β3 and -β1 display clear isoform-specific biology, such that extrapolation of data generated for TGF-β1 to the other TGF-β isoforms is inappropriate and may be misleading.
This review summarises published studies investigating the expression and potential role of TGF-β3 in tumourigenesis and attempts to place this information in the context of the unique isoform-specific biology of TGF-β3, the normal biological role of TGF-β3 in development and healthy tissues, and the potential role of TGF-β3 in tumour suppression or protection from carcinogenesis.
2. TGF-β3 is structurally and biologically unique compared to TGF-β1 and -β2
2.1. Sequence and structural differences between the TGF-β isoforms
Alignment of the amino acid sequences of the three mammalian TGF-β isoforms reveals that the different isoforms share a high level of similarity between the active domains; TGF-β3 is 86% similar to that of TGF-β1 while it shares 91% similarity with that of TGF-β2 (Fig. 1). However, despite TGF-β2 and -β3 sharing the highest level of sequence similarity of the three isoforms, TGF-β2 binds to the TGF-β receptor II (TβRII) in a different way from TGF-β1 and -β3, through different residues [9]. Furthermore, while TGF-β1 and -β3 are both capable of binding directly to the type II receptor (TβRII), presentation of TGF-β2 to the receptor requires the presence of a co-receptor (beta glycan or endoglin), which may explain the differences in activities of TGF-β2 and -β1 [10–12].
Fig. 1.

Amino acid sequence alignments of human TGF-β isoforms. Comparison of the amino acid sequence of the active domains of the three human isoforms of TGF-β reveals that the active domain of TGF-β3 shares 76% identity (86/112) and 86% similarity (97/112) with TGF-β1; 79% identity (89/112) and 91% similarity (103/112) with TGF-β2; and 76% identity (86/112). TGF-β isoform sequences were sourced from NCBI (TGF-β3: accession number P10600; TGF-β1: accession number P01137; TGF-β2: accession number P61812). (*) Indicates positions of identical residues; (:) indicates positions of conserved substitution; ( ) indicates positions of semi-conserved substitutions.
Despite homology in amino acid sequence, it is known that TGF-β3 differs significantly from TGF-β1 and -β2 in its detailed tertiary structure of the active domain (Fig. 2A). Nuclear Magnetic Resonance (NMR) data show that the alpha3 helical region of TGF-β1 is structurally ordered [13], while the alpha3 helical region of TGF-β3 is structurally disordered [14,15]. This indicates that TGF-β3 can adopt a more flexible “open” state, which is observed in both the crystal structure of free TGF-β3 [16] and in its complex with TβRII [17]. One consequence of this difference in structural flexibility is that TGF-β1 may lock the receptor complex in a closed tight conformation, while TGF-β3 may allow a more open conformation of the receptor complex due to the greater flexibility of the TGF-β3 dimer (Fig. 2B). The implications of these observations are that the structure of the ligand/receptor complexes for TGF-β1 and -β3 may be significantly different and may engage the downstream signaling pathways in different ways, thus leading to qualitatively and quantitatively different biological outcomes. Furthermore, the temporal-spatial expression of the TGF-β isoforms in embryogenesis is very different, indicating uncompensated non-overlapping functions throughout development [18].
Fig. 2.
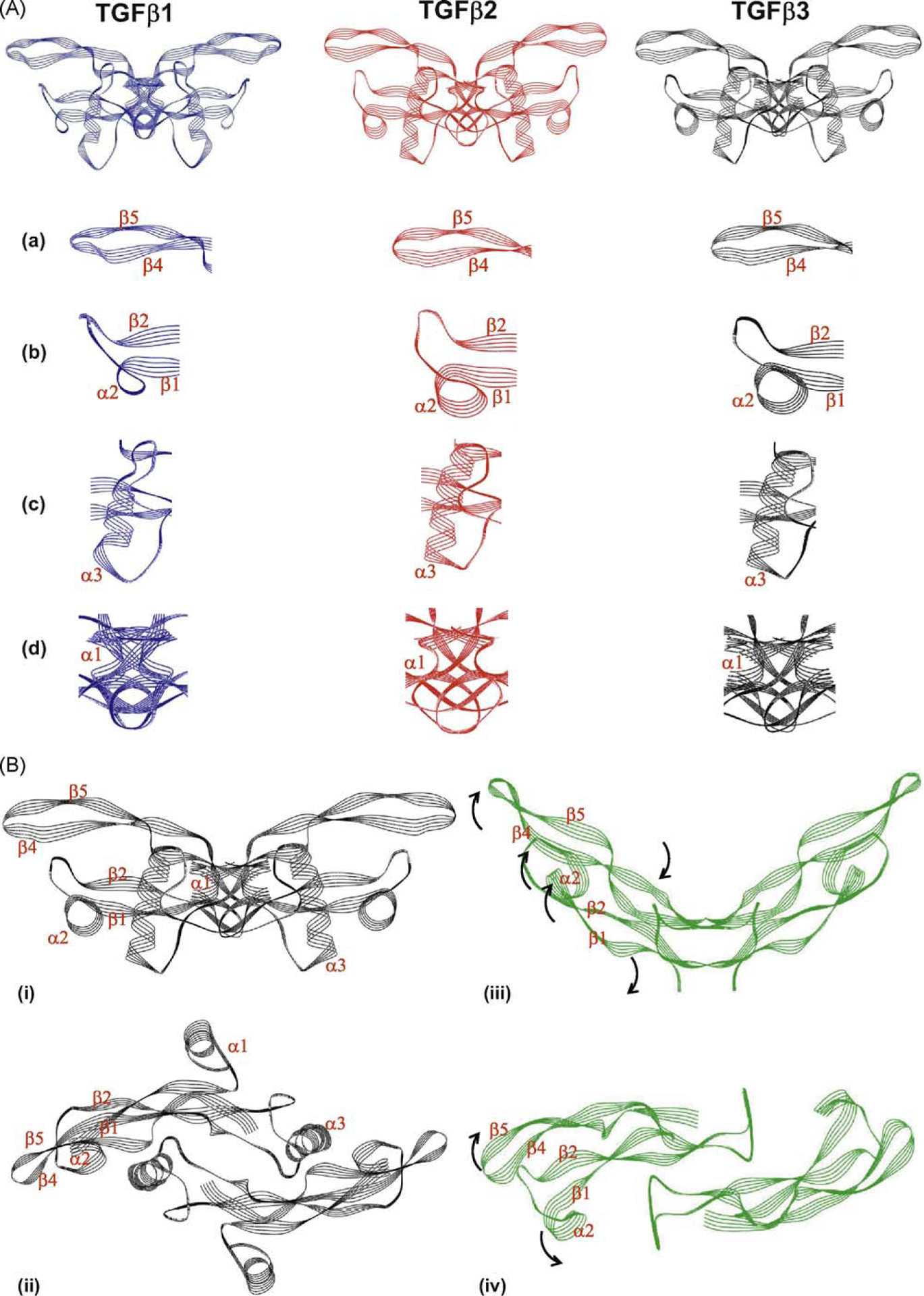
The structure of TGF-β3 differs from that of the other TGF-β isoforms and is capable of undergoing extensive rearrangement. (A) The structures of TGF-β1 (blue), TGF-β2 (red) and TGF-β3 (black) in their ‘closed’ form are shown (the dimeric forms of each isoform consisting of two monomers are represented). Domains of the protein which differ with the other isoforms are highlighted in the panels beneath each isoform; (a) β-strand β4 and β5; (b) α-helix α2; (c) a-helix α3 (d) monomer interface comprising N-terminal, a-helix a1 and the C-terminal domain. (B) Structural differences between ‘closed’ and ‘open’ forms of TGF-β3. The ‘closed’ structure of TGF-β3 is shown in black in two orientations (i) side-on and (ii) from above; while the ‘open’ structure of TGF-β3 is represented in green in two orientations (iii) side-on and (iv) from above. Individual peptide motifs of the TGF-β3 protein that undergo steric rearrangement are denoted on the individual representations. The direction of the steric rearrangement of peptide motifs between the ‘closed and ‘open’ forms is denoted by black arrows on the open form. Note that in the ‘open’ form the structure of the α-helices α1 and α3 is not represented as these motifs are disordered and therefore co-ordinates are not available. The structures of the individual isoforms are derived from co-ordinates previously published [12,17,146–148]. Images were generated using Weblab Viewer Lite (Molecular Simulations Inc. (now Accelrys)); superpositions of the structures were performed with a previously written software [149]. The structural alignments were based on the carbon a-atoms of the similar residues (as indicated by the sequential alignments) of the individual TGF-β isoforms. Other proteins that were reported in the published structures were disregarded for this analysis.
2.2. Individual TGF-β isoform null animals display unique phenotypes
The distinct, uncompensated non-overlapping functions of the TGF-β isoforms are most clearly defined in transgenic knockout mice. Targeted disruption of the TGF-β1 gene leads to hematopoietic and vasculogenic defects that result in death of about half of null embryos by 10 days gestation [19]. The embryos that survive succumb to a wasting syndrome and multi-organ failure due to inflammation after weaning [20]. TGF-β2 knockout mice exhibit perinatal mortality as a result of multiple developmental abnormalities affecting the cardiopulmonary, urogenital, visual, auditory, neural and skeletal systems [21]. These phenotypes contrast to that of mice lacking TGF-β3 which exhibit cleft palate with 100% penetration and die immediately after birth due to an inability to suckle effectively [22,23]. The unique feature of TGF-β3 knockout mice compared with knockout mice deficient in either of the other TGF isoforms is that no other morphological anomalies occur in the craniofacial region or in other organs with the exception of the lung [22,23].
2.3. TGF-β3 isoform-specific biology
TGF-β3 has been found to have an important role in normal developmental biology including systems such as the heart, lung and breast and to display isoform-specific biology at both the in vivo and in vitro level. Understanding the unique biology of TGF-β3 is important to understanding the role it may have in tumourigenesis. However, this is not the primary focus of this review and a summary of the major TGF-β3 isoform-specific biological differences is given in Table 1. Two of the models where the differences in TGF-β biology have been studied in some depth are in the palate and in cutaneous wound healing.
Table 1.
TGF-β isoform specific differences.
| References | ||
|---|---|---|
| Structural differences | TGF-β3 shares 79% identity with TGF-β32 (see Fig. 1) | |
| TGF-β3 shares 76% identity with TGF- β1 (see Fig. 1) | ||
| TGF-β1 and TGF-β3 bind TβRII to initiate signal transduction | ||
| cascades whereas TGF-β2 requires binding of TβRIII prior to binding TβRII | ||
| The reported structures of TGF-β1 and TGF-β2 indicate that they adopt a rigid ‘closed’ structure. The structure for TGF-J33 indicates that its structure is flexible and capable of adopting either ‘open’ or ‘closed’ structures (see Fig. 2) | ||
| Differences observed in vitro | TGF-β1 induces migration of THP1 monocytes, whereas TGF-β3 does not | Unpublished |
| At low doses TGF-β1 induces collagen production by human dermal fibroblasts, whereas TGF-β3 does not | Unpublished | |
| TGF-β3 activates signal transduction pathways differently to TGF-β1, e.g., Smad | Unpublished | |
| TGF-β3 is a more potent inhibitor of DNA synthesis in human keratinocytes compared to TGF-β1 and TGF-β2 | [150] | |
| TGF-β3 is a more potent stimulator of neovascularisation compared to TGF-β1 and TGF-β2 as measured by the chicken chorio-allantoic membrane assay | [151] | |
| Whereas TGF-β1 and TGF-β2 have dose-dependent effects on the stimulation of granulocyte-macrophage colony stimulating factor from human bone marrow hematopoietic progenitor cells, TGF-β3 produced only an inhibitory effect and was more potent than other isoforms | [152] | |
| TGF-β3 is a more potent inhibitor of both interleukin-3-induced colony formation and IL-3 receptor expression compared to TGF-β1 | [153] | |
| TGF-β3 significantly increases cellular proliferation, whereas TGF-β1 induced precartilage condensation in posterfrontal suture-derived mesenchymal cells | [154] | |
| Tissue specific expression of TGF-β3 | TGF-β1, −β2 and −β3 are present in independent localised patterns during the process of secondary palate formation | [24] |
| TGF-β3 is the only isoform constitutively expressed in intact human epidermis | [70] | |
| TGF-β3 is an essential mediator of EMT in cardiac morphogenesis | [155,156] | |
| TGF-β3, but not TGF-β1 or TGF-β2, is up-regulated by milk stasis and induces apoptosis in mammary gland epithelium during involution | [157] | |
| TGF-β3 is a more potent regulator of functions associated with bone formation than TGF-β1 | [158] | |
| TGF-β3 mRNA expressed in most regions of the brain whereas TGF-β1 expression is induced following injury | [159,160] | |
| TGF-β3 but not TGF-β1 induces the expression of presenilin in post-mitotic neurons and astrocytes | [161] | |
| Transgenic phenotypes | TGF-β1 null animals display defects of hematopoiesis and vasculogenesis in development. | [19,20] |
| Those that survive to term present with a wasting syndrome and multi-organ failure due to inflammation after weaning | ||
| TGF-β2 null animals exhibit perinatal lethality as a result of developmental abnormalities | [21] | |
| TGF-β3 null animals die immediately after birth due to cleft palate and a failure to suckle | [22,23] | |
| Scarring and fibrosis | Exogenous addition of TGF-β3 to dermal wounds reduces scarring; neutralisation of TGF-β1 and TGF-β2 reduces scarring; exogenous addition of TGF-β1 and TGF-β2 has no effect on scarring | [31–33] |
| Neutralisation of TGF-β1 and TGF-β2 reduces adhesions; addition of exogenous TGF-β3 increases adhesions | [162] | |
| Exogenous addition of TGF-β3 to wounds changes the inflammatory and granulation cell profile | Unpublished | |
| TGF-β3 induces differential gene expression in wounds compared to TGF-β1 | Unpublished | |
| Progressive pulmonary fibrosis is mediated by TGF-β1 but not by TGF-β3 | [163] |
TGF-β1, -β2 and -β3 are present in independent localised patterns during the process of secondary palate formation in the mouse embryo [24]. Subsequent functional studies demonstrated that while inhibition of TGF-β1 or TGF-β2 activity did not prevent normal mouse embryonic palate fusion, inhibition of TGF-β3 (using either antisense oligonucleotides or neutralising antibodies) resulted in an inability of palatal shelves to fuse. Palatal fusion was restored by the exogenous addition of TGF-β3 in ex vivo studies[25]. TGF-β3 induces filopodia, a phenotype strongly associated with palatal shelf fusion, on the surface of medial edge epithelial cells whereas TGF-β1 and -β2 do not [26]. It could be argued that some of the differences observed in vivo are due to differences in the temporal-spatial expression of individual isoforms rather than different biological activities. However, the intrinsic differences in the biological activities of different isoforms have been elegantly demonstrated in vivo where TGF-β1 has been knocked into the TGF-β3 locus. In these studies TGF-β1 could only partially rescue the cleft palate phenotype [27].
One of the most dramatic examples of TGF-β isoform-specific differences is in cutaneous scarring [28]. In contrast to adults, mammalian embryos were found to heal with no signs of scarring and complete restitution of the normal skin architecture [29] and exhibit expression of high levels of TGF-β3 and low levels of TGF-β1 and -β2 [30]. Further scarring studies in adult rodents demonstrated that exogenous addition of recombinant TGF-β3 or neutralisation of TGF-β1 and/or TGF-β2 in cutaneous wounds reduced scarring and markedly improved the architecture of the neodermis; in contrast, control wounds healed with a scar [31–33]. TGF-β3 also improves scarring in man [34].
Taken together, these studies demonstrate different and specific roles for the isoforms of TGF-β in palate development and cutaneous scarring. These roles are elicited by isoform-specific effects on the modulation of several cellular processes such as cell migration, actin cytoskeleton organisation, angiogenesis and epithelial-mesenchymal transformation.
3. The role of TGF-β3 in tumourigenesis
At the time of its discovery, TGF-β1 was found to induce malignant behaviour of normal fibroblasts, leading to the hypothesis that TGF-β1 may be involved in uncoupling a cell from normal growth control [35]. This presumed malignant function was difficult to reconcile with the ubiquitous pattern of expression of TGF-β1 in normal tissues, including its prevalence in human platelets. Subsequently TGF-β1 was found to have a profound growth suppressive effect on many cells, including epithelial cells and lymphoid cells indicating that TGF-β1 may be a potent tumour suppressor [36]. Data from experimental systems and human cancers clearly show that, in addition to the TGF-β ligands, the TGF-β receptors and their primary cytoplasmic signal transducers all play an important role in suppressing primary tumourigenesis in many organs [37,38]. However, observations made in the later stages of disease suggest that increased TGF-β1 expression is required for disease progression [37,39] indicating a duality for TGF-β1 in terms of tumourigenesis. It has since become clear that TGF-β1 maintains tissue homeostasis and prevents incipient tumours from progressing to a malignant phenotype by regulating not only cellular proliferation, differentiation, survival and adhesion, but also the cellular microenvironment. However, many cancer cells have the capacity to evade the suppressive influence of the TGF-β pathway, leading to dominance of the pro-progression activities of the pathway, such as promoting tumour growth and invasion, evasion of immune surveillance, and cancer cell metastasis. This subversion of the tumour suppressive effects of TGF-β can be achieved by many different mechanisms and at different points in the TGF-β signaling pathway. Importantly any role that TGF-β may have in promoting tumourigenesis is dependent on the context in which these disruptions occur [5].
This paradigm of duality in tumourigenesis established for TGF-β1 is now widely accepted for all TGF-β isoforms, but there is a paucity of functional data specifically relating to the role of TGF-β3 in disease progression. The few observations that have been reported for TGF-β3 expression in disease tissues have been limited by the low number of samples analysed and the scope of the study, e.g., immunohistochemical analysis of late-stage tumour biopsies. While these observations are interesting, they provide only correlative data from which a causative role in disease progression cannot be robustly determined. Observations based on transcript profiling or immunohistochemical data do not take account of the multiple levels of control of TGF-β activity. All TGF-β isoforms are expressed as latent complexes that have to be activated and, in order to propagate a signal, the activated ligand must complex with cell surface receptors [40–42]. Hence most immunohistochemical techniques measure total and not biologically active ligand. It is, therefore, difficult to determine the amount of active ligand present from isolated observations at the gene expression or protein level. Often overlooked in the interpretation of these observations is the normal role of TGF-β3 expression in tissue homeostasis and response to injury. If a tumour is recognised as an insult to tissue homeostasis then elevated TGF-β3 levels could be interpreted as a tissue response to injury. In an effort to further define the specific role of TGF-β3 in tumourigenesis, we have reviewed data published on the expression of TGF-β3 in different tumour types and attempted to place these observations in the context of the unique biology of TGF-β3.
3.1. Skin neoplasms
3.1.1. Melanocytic neoplasia
Cutaneous melanoma is an often aggressively metastatic and fatal neoplasm that accounts for most skin cancer deaths [43,44]. The early stages of melanoma are characterised as being susceptible to TGF-β-mediated growth inhibition, while later stages are increasingly less affected [45]. Several studies show that melanomas resistant to TGF-β-mediated growth inhibition are more aggressively invasive and metastatic than variants that retain the ability to respond to growth inhibitory signals [46–48].
3.1.2. Animal skin carcinogenesis models
Understanding the role of TGF-β1 in skin carcinogenesis has been greatly helped by the use of the mouse skin model of chemical carcinogenesis [49–51]. In this model cells are initiated with the mutation and activation of the Ha-ras oncogene by a single treatment with 7,12-dimethylbenz(a)anthracene (DMBA), followed by repeated doses of phorbo 12-myristate 13-acetate (TPA) to induce a hyperproliferative response in keratinocytes [50].
Using the skin chemical carcinogenesis model, transgenic mice over-expressing TGF-β1 were found to be more resistant to the induction of benign skin tumours than controls, but the malignant conversion rate was vastly increased [52] with a higher incidence of spindle cell carcinomas that were found to express high levels of TGF-β3. The data suggest that TGF-β1 elicits an epithelial-mesenchymal transition in vivo and that TGF-β3 might be involved in the maintenance of the spindle cell phenotype [52]. Gold et al.[53] investigated the introduction of keratinocytes of different tumourigenic phenotypes in vivo, into surface transplants and subcutaneous tumours in nude mice, and examined what effect this had on metastasis of the cell lines and the expression of TGF-β isoforms. In keratinized epithelia TGF-β1 was localised to the upper differentiated layers, the stratum granulosum and the corneum in a perimembranous pattern, whereas TGF-β2 and -β3 were detected in all suprabasal layers of normal keratinizing epithelia. Immunohistochemical staining for TGF-β3 was much lower than that for TGF-β1 and -β2. In contrast, non-keratinizing transplants of non-tumourigenic or highly aggressive cells showed little to no immunoreactivity for TGF-β1. TGF-β2 staining was moderate in the upper layers of non-tumourigenic epithelia, whereas large tumour cells of malignant HaCaT-ras clones were strongly positive for TGF-β2, particularly at the invasion front. Interestingly TGF-β3 immunostaining was most pronounced in the stroma of malignant tumours, implying its paracrine induction by the malignant tumour transplants. These results suggest differential functions for each of the TGF-β isoforms in epidermal carcinogenesis, such that TGF-β1 is associated with the more differentiated state; TGF-β2 with highly malignant and invading cells; and TGF-β3 is induced in cells surrounding the tumour by factors produced by the tumour itself. These data indicate that rather than being causative of non-melanoma skin cancer progression, TGF-β3 expression in the surrounding tissue is being induced by the tumour. However the precise role of TGF-β3 remains unclear due to a lack of in vivo studies investigating the experimental manipulation of TGF-β3 in appropriate animal models.
3.1.3. Transcriptomic analysis of melanoma progression
In order to identify the transcriptional changes that underpin changes in metastatic potential of melanomas, several analyses have been undertaken. A comparison of the gene expression profile of an early melanoma cell type with a variety of isogenic derivative cell lines of increasing aggressiveness identified 66 genes whose expression correlated with melanoma progression [54]. Interestingly, none of the identified genes included TGF-β3 or any members of the TGF-β superfamily. Detailed examination of the 66-gene cohort suggested that DNA methylation had an important role in mediating gene expression alterations between parental and derivative cell lines.
Uveal melanoma is the most common form of eye cancer and the second most common site for melanoma [55]. Using microarray-based gene expression profiling of human biopsies it was shown that uveal melanoma clusters into two distinct groups [56,57]. Class 1 tumours are associated with an excellent prognosis, whereas class 2 tumours are associated with epithelioid cytology, looping matrix patterns, monosomy 3 and metastasis [56–58]. TGF-β3 expression was not associated with either of the two classes reported. Indeed the profiling data suggest that a global down-regulation of neural crest/melanocyte genes and an up-regulation of epithelial genes are associated with metastatic disease [59].
Hoek et al. [60] carried out three separate DNA microarray analyses on a total of 86 cultures of melanoma-derived cells. By combining the transcriptional profiles of three panels of melanoma cell cultures the transcriptional variations responsible for intra-panel sample differences could be excluded allowing the identification of co-regulating gene sets. Cohort differentiating genes were split between only two expression patterns: Motif 1 contained genes critical to neural crest differentiation and cell cycle control, whereas Motif 2 genes were up-regulated in melanoma of high metastatic potential. Many of the genes in Motif 2 are regulated by TGF-β-type signaling, however there was no cohort specific-expression of TGF-β receptors or Smad genes, or any change in the expression pattern of the three TGF-β isoforms (TGF-β1, TGF-β2 or TGF-β3). This suggests that the progression of tumourigenesis observed in metastatic melanoma is associated with a subversion of TGF-β responsive genes but not the TGF-β signal transduction pathway [60]. This observation is supported by the results of a study that used genetics to map the genetic architecture of mouse skin inflammation, tumour susceptibility and pigmentation [61]. Interestingly, the TGF-β pathway does not appear anywhere in the tumour susceptibility or genetic hierarchy.
3.1.4. The expression of TGF-β3 in skin neoplasms
Early in vitro studies found that normal melanocytes and some melanoma-derived cells express TGF-β [62], although melanoma cells have shown varying degrees of TGF-β resistance [63,64]. In vivo studies using in situ hybridisation analysis of primary melanomas revealed that TβRII was heterogeneously distributed when compared to benign melanocytic nevi, suggesting variable degrees of TGF-β resistance among melanoma cells within individual lesions [65]. Of the three TGF-β isoforms that were studied (TGF-β1, -β2 and -β3), TGF-β3 was the only isoform identified as being consistently expressed in skin metastases at both the mRNA and protein level [65]. However, this reported pattern of expression for TGF-β3 was not supported by observations made by Van Belle et al. [66] who identified significant linear trends of expression for TGF-β1, -β2 and -β3 associated with the progression from melanocytes through nevi to primary metastatic melanomas. TGF-β1 was expressed by some melanocytes and almost uniformly by nevi and melanomas. TGF-β2 and -β3 were not expressed in normal melanocytes but were expressed in nevi, early, advanced primary and metastatic melanomas in a tumour-progression-related manner. The expression of TGF-β3 increased with progression from melanocytes to nevi and again from nevi to the more biologically advanced vertical growth phase (VGP) primary melanoma [66]. Extramammary Paget’s Disease (EPD) and Bowen’s Disease are skin cancers of unknown histogenesis and TGF-β3 over-expression was detected in all EPD patients and down-regulated TGF-β3 expression was detected in all Bowen’s Disease patients [67]. The data, from this comparatively small study, suggest that TGF-β3 levels may be higher or lower depending on the context and not necessarily related to, or indeed causative of, tumour progression.
3.1.5. TGF-β signaling is intact in melanoma
In keeping with the failure to identify a TGF-β transcriptional signature in melanoma progression [54,59,60], genetic studies have shown no inactivating mutations in the TGF-β receptor system or in the Smad signaling cascade, suggesting other mechanisms are involved in resistance to the growth-inhibitory effects of TGF-β in melanoma [68]. The level and activity of the V-Ski avian sarcoma viral oncogene homologue (SKI), rather than the levels of individual TGF-β ligands, may be a critical determinant in melanoma progression by activating b-catenin signaling and repressing TGF-β activity [69]. Elevated levels of SKI repress TGF-β signaling in the absence of inactivating mutations to the TGF-β signal transduction pathway, highlighting the importance of mutations in other pathways modulating TGF-β responsiveness and the dangers of interpreting isolated observations of TGF-β ligand levels.
3.1.6. The role of TGF-β3 in suppressing melanoma progression
In contrast to the previously discussed reports linking TGF-β3 expression with the progression of melanoma, one report identified a strong tumour suppressive role for TGF-β3 expression in vivo. TGF-β3 is reported as being the predominant TGF-β isoform expressed in human epidermis [70]. It has been shown that TGF-β3 mRNA expression is decreased in the epidermis overlaying primary melanomas, when compared to normal epidermis, suggesting that melanoma cells themselves may suppress keratinocyte TGF-β3 expression via a paracrine mechanism [63]. The authors suggest that a lack of TGF-β3 expression in the epidermis could precede melanoma development and promote clonal expansion of transformed melanocytes. Since TGF-β1 is a potent inhibitor of melanocyte proliferation in vitro [63], epidermal TGF-β3 may have a physiological function to control cell division and/or the differentiation of melanocytes. Consequently, melanomas may arise predominantly at sites of low epidermal TGF-β3 expression [63]. In this context exogenous TGF-β3 applied to the dermis may produce the same paracrine suppressive effect on any activated melanocytes that may be present.
3.2. Breast carcinoma
Interpretation of data from clinical samples has indicated a potential role for TGF-β ligands in breast carcinoma metastasis. Data from histological analysis of human tumour biopsies, although an important source of information, are limited by the fact that clinical samples are frequently derived from late-stage disease tissues when the events that result in disease initiation and progression have already occurred. Furthermore, specific samples may potentially be unrepresentative of the overall heterogeneity of the tumour. Unfortunately very few pre-clinical animal studies have specifically investigated the role of TGF-β3 in breast carcinoma, with the majority of studies concentrating solely on TGF-β1.
3.2.1. Animal breast carcinogenesis models
Using four cell lines that spanned the spectrum of tumour progression previously described [71,72], Tang et al. [6] introduced a dominant negative TβRII receptor into all four cell lines and tested for effects on tumourigenicity in vivo. Decreased TGF-β responsiveness alone was demonstrated not to initiate tumourigenesis. Another initiating oncogenic lesion was required to make a premalignant breast cell (and a low grade tumourigenic cell line) tumourigenic. However, although reduced TGF-β responsiveness had no effect on primary tumourigenesis, it significantly decreased metastasis. Results of a more recent publication suggest that TGF-β signaling is not required for the metastasis of breast cancer cells[73]. When mice carrying a conditional deletion of TβRII in the mammary epithelium (Tgfbr2MGKO) were mated with the mouse mammary tumour virus-polyomavirus middle T antigen (PyVmT) transgenic mouse model of metastatic breast cancer, loss of TβRII in the mammary epithelium resulted in formation of mammary tumours with a significantly shortened median latency when compared with PyVmT mice. Contrary to expectation, the mice in which the TβRII had been conditionally knocked out displayed significantly more pulmonary metastases compared with wild type mice on the PyVmT background, consistent with TβRII acting as a tumour suppressor in the mammary gland. These findings are in agreement with other studies using colon and prostate mouse models in which disruption of TGF-β signaling, by introduction of a mutant TβRII, results in enhanced tumour progression [74,75].
Support for the hypothesis that normal TGF-β signaling is affected by mutation of its receptors is provided by in vitro observations using a variety of cell lines, some of which express the estrogen receptor (ER). The data indicate that, not only is TGF-β signaling reduced by receptor mutation, but also that mutation of other signaling pathways is required. ER-positive MCF7 cells transfected with the mouse oncogene Ha-Ras show increased growth despite a resultant rise in constitutive TGF-β1 [76]. Human breast cancer cells stably transfected with the TGF-β1 gene have an unaltered response to estrogen and anti-estrogens despite greatly increased expression of TGF-β1. These transfected cells have increased tumourigenicity and show estrogen independence when grown in athymic mice [77]. The high production of TGF-β1 by these cell lines would not be expected if this growth factor was acting as a primary epithelial inhibitor, rather it might indicate disruption of a negative feedback loop or a switch of function from growth suppression to growth promotion [77,78]. Fibroblasts from benign and malignant breast tumours produce and secrete TGF-β1 and -β2, and there is increased synthesis of TGF-β1 in primary cultures of breast-tumour fibroblasts after tamoxifen exposure[79]. Other studies have confirmed that primary cultures of breast-tumour fibroblasts secrete TGF-β1 and -β2 [80].
Studies that have reported an involvement of TGF-β in metastases have also shown the requirement for other signal transduction pathways in order for cells to metastasise. For example TGF-β/Smad and p38 signaling pathways have been shown to co-operate to promote metastasis of human breast cancer cell lines to bone by inducing expression of the osteolytic factor, Parathyroid hormone-related protein (PTHrP; [81,82]), supporting the hypothesis that mutations in other pathways may lead to the increased TGF-β expression levels observed in tumours. However, Lebrecht et al. have reported that there is no correlation between serum TGF-β1 levels and the clinicopatho-logical parameters of breast diseases including carcinoma [83].
It appears that breast cancer cell lines express more TGF-β than less tumourigenic cell lines in an attempt to inhibit their own proliferation via both autocrine and paracrine inhibitory loops. However these cell lines are unable to respond to the inhibitory effects of TGF-β via as yet unidentified molecular mechanisms.
3.2.2. Gene expression associated with human breast carcinoma metastases and prognosis
Given that some clinical observations have reported an association between TGF-β expression and disease progression it would be expected that a screen for genes important in the metastasis of breast carcinoma would identify one or more of the TGF-β isoforms or TGF-β signaling pathways. van de Vijver et al.[84] used microarray data derived from clinical breast-tumour biopsies to define a 70 gene signature that was strongly predictive of a short interval to distant metastases (poor prognosis). TGF-β3 was identified as one of the genes in this signature but with reduced expression associated with poor prognosis. Increased expression of TGF-β3 mRNA was associated with a good prognosis. The prognostic value of this gene expression profile was subsequently validated in independent clinical data sets [85,86]. Minn et al. [87] had previously derived a similar gene expression profile for breast cancers that metastasise to the lung. Of the 95 genes identified in the metastatic gene expression signature, none were TGF-β superfamily ligands, TGF-β superfamily receptors or downstream signaling components. The only gene related to the TGF-β superfamily was Latent TGF-β Binding Protein (LTBP1), a molecule that sequesters TGF-β and limits its activity [88]. In experiments to validate the role of these genes in the metastasis of cancer cell lines it was found that single gene expression changes alone were not capable of conferring metastatic potential, indicating that multiple gene expression changes are characteristic of metastasis [87].
More recently, a TGF-β gene response signature was defined using human epithelial cell lines and turned into a bioinformatics classifier tool [89]. In different clinical cohorts approximately 40% of human breast tumours showed a TGF-β gene response signature and this status coincided with a high expression of TGF-β1, -β2, LTBP1, Smad3 and Smad4. Interestingly, high TGF-β3 expression was not found to be part of this gene expression signature. A review of the Oncomine database (www.oncomine.org) identified six independent breast cancer studies that showed an inverse correlation between TGF-β3 mRNA levels and increasing tumour grade i.e., lower TGF-β3 expression correlated with higher tumour grade ((using a P value threshold of 1E-4); [84,90–94]). A similar trend between increasing tumour grade and decreasing TGF-β3 expression has been reported for prostate cancer studies (discussed later).
The studies described above highlight the importance of context in determining any role that TGF-β3 might have. Although all studies investigated breast cancer, each attempted to measure something different: the first study was investigating a signal of prognostic value; the second a signal associated with metastases; and the third study tried to define a TGF-β gene response signature. Each produced a different result because of the context and this highlights the care that should be used when interpreting and comparing data. Although each study investigated a different aspect of breast tumourigenesis the only TGF-β3 specific data showed that increased TGF-β3 expression is associated with good prognosis.
3.2.3. The role of TGF-β3 in breast carcinogenesis
Although animal studies and transcriptional analysis have not identified changes in TGF-β3 expression as causative of breast cancer progression, several clinical studies have reported an increase in TGF-β3 expression associated with breast tumourigenesis. Li et al. [95] describe an assessment of plasma levels of TGF-β1 and -β3 (using chemiluminescence Enzyme Linked Immunosorbent Assay, ELISA) in 80 patients with untreated breast cancer, 14 of whom had lymph node metastases. Enhanced levels of TGF-β3 and TGF-β3/receptor complexes (as measured by ELISA for receptor/ligand complexes) were found to correlate with the presence of lymph node metastases. This correlation was supported by a subsequent study of 153 invasive breast cancer tissue samples [96] that found intense immunostaining for TGF-β3 in breast cancer samples. The increased expression of TGF-β3 correlated significantly with a decrease in overall survival (p = 0.0204), with elevated TGF-β3 expression becoming a more significant prognostic factor when linked to lymph node metastasis. Additional support for a role of TGF-β3 in breast carcinoma metastasis is provided by a study performed by Amatschek et al.[97], who found that the expression of the TGF-β3 gene was elevated in patients with short survival time from breast cancer.
In an analysis of 25 breast carcinomas and adjacent normal tissue specimens Soufla et al. [98] found that TGF-β3 transcript levels were significantly elevated in cancer specimens compared to normal tissues (in contradiction of gene expression data described previously). In this study they evaluated the mRNA expression profile of Vascular Endothelial Growth Factor (VEGF), Fibroblast Growth Factor 2 (FGF2), TGF-β1, -β2 and -β3 and TβRI, TβRII and TβRIII in 25 breast cancer samples. The majority of samples were infiltrating ductal carcinoma and the most frequent histological grade was G3 [98]. Increased levels of all three TGF-β isoforms were associated with cancer specimens compared to controls whereas all TβRs displayed lower levels of expression. However, only levels of FGF2 and TGF-β3 were statistically significantly elevated in tumours compared with adjacent normal breast tissue (p = 0.031 and p = 0.043, respectively). Importantly, even though the elevated level of TGF-β3 mRNA expression was significant between tumour tissue and adjacent normal tissue no correlation was established between TGF-β3 transcript levels in normal or malignant breast tissue and the grade of the tumour [98].
3.2.4. Parity-induced protection from breast cancer
Women who give birth to a child when they are younger than 24 years of age exhibit a decrease in their lifetime risk of developing breast cancer, and additional pregnancies increase their protection [99]. The protective effect of full-term pregnancy is a well established concept not only in humans, but also in experimental rodent models [100–107]. In rodents, maximal incidence of DMBA-induced mammary cancer occurs when the carcinogen is administered to young, virgin cycling rats. However the same carcinogen fails to induce tumours when administered to rats that have had a full-term pregnancy [100–107]. The high susceptibility of the young, virgin rat mammary gland to develop malignancies is the result of the interaction of the carcinogen with the rapidly dividing epithelium composed of undifferentiated terminal end buds (TEBs), which are contained in the ductal structure of the mammary parenchyma. Both the binding of the carcinogen DMBA to the DNA of rapidly proliferating epithelial cells and a low DNA repair capacity result in fixation of transformation, leading to the initiation of cancer [108–112].
D’Cruz et al. [113] used high density oligonucleotide microarrays to analyse the impact of early, first, full-term pregnancy on global gene expression profiles within the murine mammary gland. A panel of genes was subsequently identified whose expression in the mammary gland is persistently altered as a consequence of parity. These observations were replicated in three strains of mice, as well as in two widely used rat models for parity-induced protection against breast cancer. The findings demonstrated that parity induces the persistent up-regulation of TGF-β3, and several of its downstream targets e.g., Eta-1, Clusterin and Id2. In addition, their findings indicate that parity results in a persistent increase in the differentiated state of the mammary gland as well as permanent changes in the hematopoietic cell type’s resident within the gland and that the expression of TGF-β3 is of central importance to the parity-induced protection against breast cancer [113].
3.3. Other tumour types
TGF-β3 expression levels have been reported for several other tumour types. Key data are summarised below to provide an overview of the field.
3.3.1. Colon carcinoma
3.3.1.1. Animal model of colon carcinoma.
Placement of a null TGF-β1 locus onto the immunodeficient background of (129S6 X CF-1) Rag2−/− mice, which lack both B- and T-cells, permits the double knockout animals to survive to adulthood. However, by five months of age the TGF-β1−/− Rag2−/− mice exhibit carcinomas in caecum the and the multiple colon [113]. In contrast the caecums and colons from nearly all TGF-β1+/+Rag2−/− and TGF-β1+/+ Rag2−/− mice remain hyperplastic [114]. TGF-β2 and TGF-β3 continue to be expressed in the caecum and colon of TGF-β1 −/− Rag2−/− mice. Interestingly, when the TGF-β signal transduction molecule Smad3 was mutated, Smad3-deficient mice (which in contrast to Smad4 knockout mice are viable) become moribund at 4–6 months of age, manifesting highly invasive and metastatic colorectal adenocarcinoma [115]. This is in contrast to the TGF-β1−/− Rag2−/− mice which develop only non-metastatic colon cancer. As Smad3 has been shown to transduce the signals of all three TGF-β ligands it is reasonable to hypothesize that in TGF-β1−/− Rag2−/− mice TGF-β2 and TGF-β3 inhibit metastasis at late stages of colon tumourigenesis via Smad3.
3.3.1.2. Clinical studies of colon carcinoma.
Biopsies from 39 colon carcinoma patients who underwent surgical resections were examined for the expression of all three TGF-β isoforms at both the protein and mRNA level [116]. The data from this study indicate that the pattern of TGF-β3 expression in colonic mucosa is distinct from that observed for the other two TGF-β isoforms. Immunoreactivity for TGF-β3 was high in normal mucosa and not statistically different between normal mucosa and tumour sections, whereas a linear relationship was found between elevated TGF-β1 expression and colon cancer progression. In addition tumour-associated expression of TGF-β3 was uniform across successive stages of tumour progression and TGF-β3 mRNA levels were similar between normal and tumour tissues. Taken together the clinical observations and pre-clinical data suggest that it is unlikely that changes in the expression of TGF-β3 at both the protein and mRNA level play a role in the initiation and progression of colon cancer in humans.
3.3.2. Gastric carcinoma
In a study of 110 gastrectomy specimens, of which 95 were low grade and 15 were high grade malignancies [117], TGF-β3 protein was detected in both normal mucosa and neoplastic tissue although there was no statistically significant difference in TGF-β3 protein levels between the normal and neoplastic cells [117]. Whereas TGF-βb1 protein expression correlated with better patient survival and longer disease-free survival time, TGF-β2 protein expression correlated with worse survival, and no such correlation was found between TGF-β3 expression and patient survival or disease-free survival time. Interestingly, TGF-β3 expression has been associated with protection from mucosal injury. In a study in which mice were irradiated with X-rays (15.8-Gy) almost 100% of animals pre-treated with TGF-β3 survived to 30 days but only approximately 35% of vehicle control animals survived to the same time point [118]. Following injury to the oral mucosa, the levels of TGF-β3 are elevated three-fold. In contrast, TGF-β1 levels fall following injury indicating that there is a shift in the ratio of these isoforms that protects the oral mucosa and reduces subsequent scarring [118,119]. Therefore it is possible that elevated levels of TGF-β3 reflect a protective, rather than a causative, role in disease biopsies.
3.3.3. Ovarian carcinoma
Two studies have reported contradictory levels of TGF-β3 expression in ovarian carcinoma biopsies. Bristow et al. [120] found that enhanced TGF-β1 and -β3 expression, as well as loss of expression of TβRI and TβRII, may contribute to ovarian carcinogenesis and/or tumour progression. However in a separate analysis of angiogenesis markers in biopsies of ovarian tumours from 40 patients, no change in the expression of TGF-β3 was found between normal and disease tissue [121]. Evidence is available that increased TGF-β3 expression protects against the development of ovarian cancer. Routine use of oral contraceptives (especially the estrogen-progestin combination oral contraceptive) for as little as 3 years confers as much as a 50% reduction in the risk of ovarian cancer and this protective association increases with duration of use and lasts for as long as 20 years after discontinuation of use [122]. In young adult female cynomolgus macaques progestin treatment, either alone or when combined with estrogen, was associated with a highly statistically significant decrease in the expression of TGF-β1 in ovarian epithelium (p < 0.001) and a moderate decrease in the expression of TGF-β1 in the oocyte cytoplasm (p = 0.002) [123]. In contrast, progestin treatment was associated with a marked increase in the expression of TGF-β2 and -β3 in the ovarian epithelium (p < 0.001). Analysis of the ovarian epithelium revealed that the progestin induced changes in TGF-β-isoform expression correlated with an increase in apoptosis. Taken together these data demonstrate that progestin-induced apoptosis in the ovarian epithelium is associated with an isoform switch in expression from TGF-β1 to the protective expression of TGF-β2 and -β3 [123]. The observation of increased TGF-β3 expression associated with a reduction in the risk of developing ovarian cancer [123] is similar to the role identified for TGF-β3 in parity-induced protection from breast cancer described earlier [113].
3.3.4. Prostate cancer
In an analysis of 14 prostate adenocarcinomas using immunohistochemistry only three displayed TGF-β3 expression [124]. Other studies have reported the loss of TGF-β3 expression from basal epithelial cells of prostate carcinoma [125]. Gene expression analyses of epithelial tissues found TGF-β3 expression to be increased two-fold in one study [126], but down-regulated in another study [127]. A review of four independent datasets for prostate cancer in the Oncomine database (www.oncomine.org) revealed that TGF-β3 expression was lower in diseased tissue compared with healthy tissue, indicating that elevated TGF-β3 expression levels are associated with suppression of prostate cancer [128–131].
3.3.5. Osteosar
Immunohistochemical analysis for TGF-β3 staining of 25 high grade osteosarcoma samples identified minimal to moderate staining in 11 of 25 tumours (44%) and strong to intense immunoreactivity was observed in the remaining 14 tumours (56%) [132]. Immunoreactivity for TGF-β3 was found to be related to disease progression (p = 0.027) whereas immunoreactivity for TGF-β1 and -b2 was not. Although the level of TGF-β3 expression was found to correlate with disease progression, it was not significantly related to patient survival (p = 0.39) [132]. In an independent study of 16 human osteosarcoma biopsies, expression of the TGF-β1 or TGF-β3 isoforms was associated with a higher rate of subsequent lung metastases (p < 0.05; [133]). Although Kleon et al. [132] reported that there was no link between TGF-β expression and metastases, in this study 5 of the 10 patients whose tumour displayed immunoreactivity for TGF-β3 later developed lung metastases. However the small sample size of these studies makes interpretation of the data difficult.
Increased levels of TGF-β3 detection in osteosarcoma can be explained when the normal biological role of TGF-β3 in bone is taken into consideration. High levels of TGF-β3 are present in the bone matrix and in instances of bone damage, such as during osteoporosis, high levels of TGF-β3 appear to be released [134]. TGF-β3 is considered a more potent regulator of functions associated with osteogenesis and angiogenesis compared with the other TGF-β isoforms [135]. Indeed, TGF-β3 has been found to stimulate remarkable regeneration of alveolar bone, periodontal ligament and cementum within exposed furcations in non-human primate periodontal tissue [136]. It is possible that elevated levels of TGF-β3 observed may be associated with attempts by the tissue to repair rather than disease progression.
3.3.6. Uterine leiomyomas
Comparison of TGF-β1 and -β3 mRNA levels in matched pairs of myometria and leiomyoma specimens showed that leiomyomas had a five-fold higher level of expression of TGF-β3 mRNA than did the corresponding myometria ((p < 0.05); [137]), whereas levels of TGF-β1 mRNA were similar among all biopsies. Furthermore, immunohistochemical analysis of TGF-β3 protein expression showed an increase in TGF-β3 levels in leiomyoma tissues compared with normal myometrial tissues. Unfortunately the majority of data relating to uterine leiomyomas and TGF-β3 are based on expression levels determined by immunohistochemistry and, with a lack of supporting animal model data, are only correlative. Based on the available data it is difficult to interpret the observed increases in TGF-β3 expression and whether they are related to leiomyoma progression or simply to the extensive changes observed in the myometrium given the important role for TGF-β3 in maintenance and turnover of the endometrium [138].
3.3.7. Acute lymphoblastic leukaemia, malignant fibrous histiocytoma and desmoplastic small round cell tumour
Quantification of the plasma levels of TGF-β3 in 60 children with acute lymphoblastic leukaemia (ALL) using a specific enzyme linked immunosorbent assay (ELISA) found that TGF-β3 levels were significantly lower in patients with ALL compared to controls [139]. In a major subgroup of this study 45 children with common acute lymphoblastic leukaemia (cALL) had significantly lower TGF-β3 levels compared with controls (p = 0.0006).
Immunostaining of samples from 43 malignant fibrous histiocytoma biopsies identified TGF-β3 expression in 38 (88%) of the biopsies. Due to the nature of the study control specimens were not examined [140].
In an analysis of 24 desmoplastic small round cell tumours that carried the EWS-WT1 fusion product variable immunoreactivity was reported for TGF-β3 with expression detected in 21 of 24 biopsies examined. However, no correlation between tumour-associated desmoplasia and TGF-β3 expression was found [141].
4. Summary and perspective
Compared with the wealth of data available for TGF-β1 it is apparent that there is a lack of functional data demonstrating a causative role for TGF-β3 in tumourigenesis. The majority of data that have been published in respect of TGF-β3 are correlative, generated by either analysis of protein or mRNA expression in tumour biopsies. In addition, the small sample sizes analysed in the majority of studies may explain the often contradictory data generated. Consequently, the interpretation of reported observations is not only very difficult but potentially confusing. Of particular note is the widespread extrapolation by researchers of data generated for the TGF-β1 isoform to the TGF-β3 isoform. As isoform specific roles have been identified in normal biological processes and in tumourigenesis, these extrapolations are both incorrect and misleading; TGF-β3 is not the same as TGF-β1. Clearly there is a need to further define the relevance of the correlative clinical observations with data from robust animal models that address the functional role of TGF-β3 in tumourigenesis.
Interestingly, TGF-β3 expression has been associated with protection against the onset of disease in several situations. TGF-β3 is expressed at high levels in the epidermis where it is reported to have a protective effect against the onset of melanoma. Although increased expression of TGF-β3 has been observed in breast cancer biopsies, elevated levels of TGF-β3 have been associated with good prognosis in breast cancer. In large epidemiological studies it has been found that women who give birth when they are younger than 24 years of age exhibit a decrease in their lifetime risk of developing breast cancer, and additional pregnancies increase the level of protection. Gene expression studies have identified an association between TGF-β3 expression and parity-induced protection against breast cancer. Additionally, the use of oral contraception is associated with a 50% reduction in the risk of developing ovarian cancer and this protection has been correlated with an increased expression of TGF-β3 (Table 2).
Table 2.
Summary of data indicating a protective role for TGF-β3 in tumourigenesis.
|
While it has proved tempting for researchers to report a ‘correlation’ as a ‘cause’ of disease, what has often been overlooked is the normal biological role of the TGF-β3 molecule. TGF-β3 is important in embryonic development, scarless repair of injury in the embryo, adult wound healing and tissue homeostasis. It has an important role in regulating cell migration, angiogenesis, epithelial-mesenchymal transition, apoptosis, modulation of immune function, extracellular matrix (ECM) production and the regulation of ECM remodelling; biological processes that are often required for tumour growth and maintenance. Therefore the expression patterns of TGF-β3 observed in some tumours may directly reflect the subversion of these biological processes in tumour progression rather than a specific role for TGF-β3 in driving tumour progression. Again, the functional relevance of these observations remains to be tested in appropriate models. It has also been proposed that tumour stroma is ‘normal wound healing gone awry’ [142] and that many of the normal reparative processes are active in the tumour milieu [143,144]. Indeed a similar gene expression signature has been identified that is shared between fibroblasts responding to serum and that of some tumours [145]. In light of the protective role that TGF-β3 has in many tissues including the breast, skin, oral and gastric mucosa, and given its prominent role in scar-free healing, it is reasonable to hypothesize that increased TGF-β3 expression in tumours is a tissue protective response to tumour ‘injury’.
Clearly, the administration of biologicals such as growth factors and antibodies to patients for therapeutic benefit raises questions about the safety and the potential risk of tumourigenicity with these agents (e.g., becaplermin, recombinant human PDGF; and ustekinumab, human monoclonal antibody against interleukin-12 and interleukin-23; both of which are applied to the skin). Avotermin (human recombinant TGF-β3) is in clinical development for the improvement of scar appearance in the skin. To date, there are no data from studies of avotermin applied to wounds that raise any safety concerns. Indeed pre-clinical data showing the suppressor effects of TGF-β3 in the early stages of tumourigenesis, coupled with the acute, low doses applied and the low systemic bioavailability all give reassurance. Clinical trial subjects exposed to avotermin are closely monitored and appropriate supportive animal studies are planned in order to confirm the safety profile of avotermin and to better understand the isoform-specific biology of TGF-β3.
Acknowledgements
Preparation of the manuscript was funded by Renovo. The authors would like to thank Dawn Lobban for editorial assistance.
Biographies

Hugh Laverty initially studied for his degree at The Queen’s University, Belfast and after that, he studied for his doctorate at The University of Glasgow (UK). He then followed on from this with post-doctoral research in palate development. He is currently working as a Discovery Manager at Renovo Ltd. Manchester (UK), with a focus on investigating the basic biology of wound healing and scarring; the identification of new therapeutics for heal-ingand scarring, along with elucidating the mechanism of action of Renovo’s lead compounds. His main research interests include, understanding the molecular and cellular basis of embryonic development along with applying this to regenerative medicine and disease processes.

Lalage Wakefield DPhil, initially trained in biochemistry at Oxford University (UK) before moving to the National Cancer Institute (USA) in 1983 to work on the newly discovered transforming growth factor TGF-β for her postdoctoral fellowship. Currently she is working as a senior investigator in the Lab of Cancer Biology and Genetics at the National Cancer Institute. Her main research focuses on the complex role of the TGF-β pathway in breast cancer progression, and also on the pre-clinical development of TGF-β antagonists.

Nick Occleston is currently working as the senior vice president of discovery at Renovo Ltd. Manchester (UK). He received his PhD from the University of London in the molecular and cellular biology of ocular tissue repair. He has previously held positions at Pfizer Global Research and Development and also at the Institute of Ophthalmology at Moorfields Eye Hospital (UK). His main research interests include mechanisms of tissue regeneration, repair and fibrosis at multiple body sites as well as the utility of human experimental medicine in reverse discovery for the identification and development of new therapeutics.

Sharon O’Kane has a PhD and BSc (first class) honours degree from the University of Ulster, a diploma in industrial science, and a diploma in company direction from the Institute of Directors. Sharon sits on Lord Drayson’s Government Office for Life Sciences and on the Government’s Bioscience Innovation and Growth Team Steering Group, is co-chair of the biomedical sector of the ‘Innovation Manchester’ initiative of Manchester City Council/Manchester Knowledge Capital, is a member of AGMA’s Business Leadership Council and is a member of the North West Science Council Biohealth team. Sharon is the co-founder and chief scientific officer at Renovo: a biotechnology company developing drugs to improve scarring and enhance tissue regeneration. Her research interests are in wound healing, scarring and fibrosis, cleft palate, craniofacial development and ‘cosmeceuticals’.

Professor Mark Ferguson graduated from the Queens University of Belfast with degrees in dentistry (BDS), anatomy and embryology (BSC, PhD) and medical sciences (DMedSc), holds Fellowships from the Royal Colleges of Surgeons in Ireland (FFD) and Edinburgh (FDS), is a founding fellow of the UK Academy of Medical Sciences (FMed Sci) and was made a “Commander of the British Empire” (CBE) by the Queen in 1999 for services to health and life sciences. He is a professor in the faculty of life sciences at the University of Manchester and the Co-founder and CEO of Renovo: a biotechnology company developing drugs to improve scarring and enhance tissue regeneration. His research interests are in wound healing, scarring and fibrosis, cleft palate, craniofacial development and alligator and crocodile biology.
References
- [1].Wakefield LM, Letterio JJ, Geiser AG, Flanders KC, O’Shaughnessy J, Roberts AB, et al. Transforming growth factor-beta s in mammary tumorigenesis: promoters or antipromoters? Prog Clin Biol Res 1995;391:133–48. [PubMed] [Google Scholar]
- [2].Gold LI. The role for transforming growth factor-beta (TGF-βeta) in human cancer. Crit Rev Oncog 1999;10:303–60. [PubMed] [Google Scholar]
- [3].Padgett RW. TGFbeta signaling pathways and human diseases. Cancer Metastasis Rev 1999;18:247–59. [DOI] [PubMed] [Google Scholar]
- [4].Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest 2007;87:1077–91. [DOI] [PubMed] [Google Scholar]
- [5].Massague J TGFb in cancer. Cell 2008;134:215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-βeta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 2003;112:1116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tian F, DaCosta Byfield S, Parks WT, Yoo S, Felici A, Tang B, et al. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res 2003;63:8284–92. [PubMed] [Google Scholar]
- [8].Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci USA 2003;100:8621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Crescenzo G, Hinck CS, Shu Z, Zúñiga J, Yang J, Tang Y, et al. Three key residues underlie the differential affinity of the TGFbeta isoforms for the TGFbeta type II receptor. J Mol Biol 2006;355:47–62. [DOI] [PubMed] [Google Scholar]
- [10].Lyons RM, Miller DA, Graycar JL, Moses HL, Derynck R. Differential binding of transforming growth factor-beta 1, -beta 2, and -beta 3 by fibroblasts and epithelial cells measured by affinity cross-linking of cell surface receptors. Mol Endocrinol 1991;5:1887–96. [DOI] [PubMed] [Google Scholar]
- [11].Mitchell EJ, O’Connor-McCourt MD. A transforming growth factor beta (TGF-beta) receptor from human placenta exhibits a greater affinity for TGF-βeta 2 than for TGF-βeta 1. Biochemistry 1991;30:4350–6. [DOI] [PubMed] [Google Scholar]
- [12].Lopez-Casillas F, Wrana JL, Massagué J. b-glycan presents ligand to the TGFb signaling receptor. Cell 1993;73:1435–44. [DOI] [PubMed] [Google Scholar]
- [13].Hinck AP, Archer SJ, Qian SW, Roberts AB, Sporn MB, Weatherbee JA, et al. Transforming growth factor beta 1: three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor beta2. Biochemistry 1996;35:8517–34. [DOI] [PubMed] [Google Scholar]
- [14].Bocharov EV, Blommers MJ, Kuhla J, Arvinte T, Bürgi R, Arseniev AS. Sequence-specific 1H and 15N assignment and secondary structure of transforming growth factor beta3. J Biomol NMR 2000;16:179–80. [DOI] [PubMed] [Google Scholar]
- [15].Bocharov EV, Korzhnev DM, Blommers MJ, Arvinte T, Orekhov VY, Billeter M, et al. Dynamics-modulated biological activity of transforming growth factor beta3. J Biol Chem 2002;277:46273–9. [DOI] [PubMed] [Google Scholar]
- [16].Grütter C, Wilkinson T, Turner R, Podichetty S, Finch D, McCourt M, et al. A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions. Proc Natl Acad Sci USA 2008;105:20251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP. Crystal structure of the human TbetaR2 ectodomain–TGF-βeta3 complex. Nat Struct Biol 2002;9:203–8. [DOI] [PubMed] [Google Scholar]
- [18].Akhurst RJ, Lehnert SA, Faissner A, Duffie E. TGF beta in murine morphogenetic processes: the early embryo and cardiogenesis. Development 1990;108:645–56. [DOI] [PubMed] [Google Scholar]
- [19].Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995;121:1845–54. [DOI] [PubMed] [Google Scholar]
- [20].Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992;359:693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997;124:2659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 1995;11:409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-βeta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 1995;11: 415–21. [DOI] [PubMed] [Google Scholar]
- [24].Fitzpatrick DR, Denhez F, Kondaiah P, Akhurst RJ. Differential expression of TGF beta isoforms in murine palatogenesis. Development 1990;109: 585–95. [DOI] [PubMed] [Google Scholar]
- [25].Brunet CL, Sharpe PM, Ferguson MW. Inhibition of TGF-βeta 3 (but not TGF-beta 1 or TGF-βeta 2) activity prevents normal mouse embryonic palate fusion. Int J Dev Biol 1995;39:345–55. [PubMed] [Google Scholar]
- [26].Taya Y, O’Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-βeta3 knockout mice. Development 1999;126:3869–79. [DOI] [PubMed] [Google Scholar]
- [27].Yang LT, Kaartinen V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev Biol 2007;312:384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ferguson MW, O’Kane S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos Trans R Soc Lond B Biol Sci 2004;359:839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Whitby DJ, Ferguson MW. Immunohistochemical localization of growth factors in fetal wound healing. Dev Biol 1991;147:207–15. [DOI] [PubMed] [Google Scholar]
- [30].Whitby DJ, Ferguson MW. The extracellular matrix of lip wounds in fetal, neonatal and adult mice. Development 1991;112:651–68. [DOI] [PubMed] [Google Scholar]
- [31].Shah M, Foreman DM, Ferguson MW. Control of scarring in adult wounds by neutralising antibody to transforming growth factor beta. Lancet 1992;339:213–4. [DOI] [PubMed] [Google Scholar]
- [32].Shah M, Foreman DM, Ferguson MW. Neutralising antibody to TGF-βeta 1,2 reduces cutaneous scarring in adult rodents. J Cell Sci 1994;107: 1137–57. [DOI] [PubMed] [Google Scholar]
- [33].Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-βeta 1 and TGF-beta 2 or exogenous addition of TGF-βeta 3 to cutaneous rat wounds reduces scarring. J Cell Sci 1995;108:985–1002. [DOI] [PubMed] [Google Scholar]
- [34].Ferguson MW, Duncan J, Bond J, Bush J, Durani P, So K, et al. Prophylactic administration of avotermin for improvement of skin scarring: three double-blind, placebo-controlled, phase I/II studies. Lancet 2009;373:1264–74. [DOI] [PubMed] [Google Scholar]
- [35].Roberts AB, Sporn MB. Transforming growth factors. Cancer Surv 1985;4: 683–705. [PubMed] [Google Scholar]
- [36].Wakefield LM, Sporn MB. Suppression of carcinogenesis: a role for TGF-βeta and related molecules in prevention of cancer. Immunol Ser 1990;51: 217–43. [PubMed] [Google Scholar]
- [37].Wakefield LM, Roberts AB. TGF-βeta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 2002;12:22–9. [DOI] [PubMed] [Google Scholar]
- [38].de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst 2000;92:1388–402. [DOI] [PubMed] [Google Scholar]
- [39].Akhurst RJ, Derynck R. TGF-βeta signaling in cancer—a double-edged sword. Trends Cell Biol 2001;11:S44–51. [DOI] [PubMed] [Google Scholar]
- [40].Miyazono K, Heldin CH. Latent forms of TGF-βeta: molecular structure and mechanisms of activation. Ciba Found Symp 1991;157:81–9. [DOI] [PubMed] [Google Scholar]
- [41].Lawrence DA. Latent-TGF-βeta: an overview. Mol Cell Biochem 2001;219: 163–70. [DOI] [PubMed] [Google Scholar]
- [42].Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003;116:217–24. [DOI] [PubMed] [Google Scholar]
- [43].Chin L, Merlino G, DePinho RA. Malignant melanoma: modern black plague and genetic black box. Genes Dev 1998;12:3467–81. [DOI] [PubMed] [Google Scholar]
- [44].Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DG, et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol 2001;19:3635–48. [DOI] [PubMed] [Google Scholar]
- [45].Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol 2005;23:2078–93. [DOI] [PubMed] [Google Scholar]
- [46].Heredia A, Villena J, Romarís M, Molist A, Bassols A. The effect of TGF-βeta 1 on cell proliferation and proteoglycan production in human melanoma cells depends on the degree of cell differentiation. Cancer Lett 1996;109: 39–47. [DOI] [PubMed] [Google Scholar]
- [47].Krasagakis K, Garbe C, Schrier PI, Orfanos CE. Paracrine and autocrine regulation of human melanocyte and melanoma cell growth by transforming growth factor beta in vitro. Anticancer Res 1994;14:2565–71. [PubMed] [Google Scholar]
- [48].Roberts AB, Anzano MA, Wakefield LM, Roche NS, Stern DF, Sporn MB. Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci USA 1985;82:119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hennings H, Shores R, Mitchell P, Spangler EF, Yuspa SH. Induction of papillomas with a high probability of conversion to malignancy. Carcinogenesis 1985;6:1607–10. [DOI] [PubMed] [Google Scholar]
- [50].Akhurst RJ, Balmain A. Genetic events and the role of TGF beta in epithelial tumour progression. J Pathol 1999;187:82–90. [DOI] [PubMed] [Google Scholar]
- [51].Derynck R, Akhurst RJ, Balmain A. TGF-βeta signaling in tumor suppression and cancer progression. Nat Genet 2001;29:117–29. [DOI] [PubMed] [Google Scholar]
- [52].Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, et al. TGF-βeta 1 inhibits the formation of benign skin tumours but enhances progression to invasive spindle cell carcinomas in transgenic mice. Cell 1996;86: 531–42. [DOI] [PubMed] [Google Scholar]
- [53].Gold LI, Jussila T, Fusenig NE, Stenback F. TGF-βeta isoforms are differentially expressed in increasing malignant grades of HaCaT keratinocytes, suggesting separate roles in skin carcinogenesis. J Pathol 2000;190:579–88. [DOI] [PubMed] [Google Scholar]
- [54].Gallagher WM, Bergin OE, Rafferty M, Kelly ZD, Nolan IM, Fox EJ, et al. Multiple markers for melanoma progression regulated by DNA methylation: insights from transcriptomic studies. Carcinogenesis 2005;26: 1856–67. [DOI] [PubMed] [Google Scholar]
- [55].Egan KM, Seddon JM, Glynn RJ, Gragoudas ES, Albert DM. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol 1988;32:239–51. [DOI] [PubMed] [Google Scholar]
- [56].Tschentscher F, Hüsing J, Hölter T, Kruse E, Dresen IG, Jöckel KH, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res 2003;63:2578–84. [PubMed] [Google Scholar]
- [57].Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res 2006;66:4602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Onken MD, Lin AY, Worley LA, Folberg R, Harbour JW. Association between microarray gene expression signature and extravascular matrix patterns in primary uveal melanomas. Am J Ophthalmol 2005;140:748–9. [DOI] [PubMed] [Google Scholar]
- [59].Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res 2004;64:7205–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 2006;19:290–302. [DOI] [PubMed] [Google Scholar]
- [61].Quigley DA, To MD, Pérez-Losada J, Pelorosso FG, Mao JH, Nagase H, et al. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature 2009;458:505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Albino AP, Davis BM, Nanus DM. Induction of growth factor RNA expression in human malignant melanoma: markers of transformation. Cancer Res 1991;51:4815–20. [PubMed] [Google Scholar]
- [63].Rodeck U, Bossler A, Graeven U, Fox FE, Nowell PC, Knabbe C, et al. Transforming growth factor beta production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res 1994;54:575–81. [PubMed] [Google Scholar]
- [64].MacDougall JR, Kobayashi H, Kerbel RS. Responsiveness of normal dysplastic melanocytes and melanoma cells from different lesional stages of disease progression to the growth inhibitory effects of TGF-β. Mol Cell Different 1993;1:21–40. [Google Scholar]
- [65].Schmid P, Itin P, Rufli T. In situ analysis of transforming growth factor-beta s (TGF-βeta 1, TGF-βeta 2, TGF-βeta 3), and TGF-βeta type II receptor expression in malignant melanoma. Carcinogenesis 1995;16:1499–503. [DOI] [PubMed] [Google Scholar]
- [66].Van Belle P, Rodeck U, Nuamah I, Halpern AC, Elder DE. Melanoma-associated expression of transforming growth factor-beta isoforms. Am J Pathol 1996;148:1887–94. [PMC free article] [PubMed] [Google Scholar]
- [67].Kawakami T, Soma Y, Mizoguchi M, Saito R. Overexpression of transforming growth factor-beta3 immunohistochemical staining in extramammary Paget’s disease, but downregulated expression in Bowen’s disease. Int J Dermatol 2001;40:262–7. [DOI] [PubMed] [Google Scholar]
- [68].Rodeck U, Nishiyama T, Mauviel A. Independent regulation of growth and SMAD-mediated transcription by transforming growth factor beta in human melanoma cells. Cancer Res 1999;59:547–50. [PubMed] [Google Scholar]
- [69].Chen D, Xu W, Bales E, Colmenares C, Conacci-Sorrell M, Ishii S, et al. SKI activates Wnt/beta-catenin signaling in human melanoma. Cancer Res 2003;63:6626–34. [PubMed] [Google Scholar]
- [70].Schmid P, Cox D, Bilbe G, McMaster G, Morrison C, Stähelin H, et al. TGF-βeta s and TGF-βeta type II receptor in human epidermis: differential expression in acute and chronic skin wounds. J Pathol 1993;171:191–7. [DOI] [PubMed] [Google Scholar]
- [71].Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol 1996;148:313–9. [PMC free article] [PubMed] [Google Scholar]
- [72].Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat 2001;65:101–10. [DOI] [PubMed] [Google Scholar]
- [73].Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, et al. Effect of conditional knockout of the type II TGF-βeta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res 2005;65:2296–302. [DOI] [PubMed] [Google Scholar]
- [74].Tu WH, Thomas TZ, Masumori N, Bhowmick NA, Gorska AE, Shyr Y, et al. The loss of TGF-βeta signaling promotes prostate cancer metastasis. Neoplasia 2003;5:267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska AE, Wirth PS, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res 2004;64: 4687–92. [DOI] [PubMed] [Google Scholar]
- [76].Dickson RB, Kasid A, Huff KK, Bates SE, Knabbe C, Bronzert D, et al. Activation of growth factor secretion in tumorigenic states of breast cancer induced by 17 beta-estradiol or v-Ha-ras oncogene. Proc Natl Acad Sci USA 1987;84: 837–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Arteaga CL, Hurd SD, Winnier AR, Johnson MD, Fendly BM, Forbes JT. Anti-transforming growth factor (TGF)-beta antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-βeta interactions in human breast cancer progression. J Clin Invest 1993;92:2569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Dumont N, Arteaga CL. Transforming growth factor-beta and breast cancer: tumor promoting effects of transforming growth factor-beta. Breast Cancer Res 2000;2:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Benson JR, Wakefield LM, Baum M, Colletta AA. Synthesis and secretion of transforming growth factor beta isoforms by primary cultures of human breast tumour fibroblasts in vitro and their modulation by tamoxifen. Br J Cancer 1996;74:352–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chakravarthy D, Green AR, Green VL, Kerin MJ, Speirs V. Expression and secretion of TGF-βeta isoforms and expression of TGF-βeta-receptors I, II and III in normal and neoplastic human breast. Int J Oncol 1999;15: 187–94. [DOI] [PubMed] [Google Scholar]
- [81].Kakonen SM, Selander KS, Chirgwin JM, Yin JJ, Burns S, Rankin WA, et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem 2002;277:24571–8. [DOI] [PubMed] [Google Scholar]
- [82].Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-βeta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999;103:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lebrecht A, Grimm C, Euller G, Ludwig E, Ulbrich E, Lantzsch T, et al. Transforming growth factor beta 1 serum levels in patients with preinvasive and invasive lesions of the breast. Int J Biol Markers 2004;19:236–9. [DOI] [PubMed] [Google Scholar]
- [84].van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009. [DOI] [PubMed] [Google Scholar]
- [85].Bueno-de-Mesquita JM, Linn SC, Keijzer R, Wesseling J, Nuyten DS, van Krimpen C, et al. Validation of 70-gene prognosis signature in node-negative breast cancer. Breast Cancer Res Treat )2008;(September). [DOI] [PubMed] [Google Scholar]
- [86].van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–6. [DOI] [PubMed] [Google Scholar]
- [87].Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-βeta. Microsc Res Tech 2001;52:354–62. [DOI] [PubMed] [Google Scholar]
- [89].Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like4. Cell 2008;133:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bittner M Expression project for oncology - breast samples. International Genomics Consortium, Phoeniz, AZ 85004. https://expo.intgen.org/expo/public/; 2005. [Google Scholar]
- [91].Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 2005;102:13550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res 2006;66:10292–301. [DOI] [PubMed] [Google Scholar]
- [93].Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 2006;98:262–72. [DOI] [PubMed] [Google Scholar]
- [94].Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, et al. TRANSBIG consortium. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res 2007;13: 3207–14. [DOI] [PubMed] [Google Scholar]
- [95].Li C, Wang J, Wilson PB, Kumar P, Levine E, Hunter RD, et al. Role of transforming growth factor beta3 in lymphatic metastasis in breast cancer. Int J Cancer 1998;79:455–9. [DOI] [PubMed] [Google Scholar]
- [96].Ghellal A, Hayes M, Byrne G, Bundred N, Kumar S. Prognostic significance of TGF beta 1 and TGF beta 3 in human breast carcinoma. Anticancer Res 2000;20:4413–8. [PubMed] [Google Scholar]
- [97].Amatschek S, Koenig U, Auer H, Steinlein P, Pacher M, Gruenfelder A, et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res 2004;64:844–56. [DOI] [PubMed] [Google Scholar]
- [98].Soufla G, Porichis F, Sourvinos G, Vassilaros S, Spandidos DA. Transcriptional deregulation of VEGF, FGF2, TGF-βeta1, 2, 3 and cognate receptors in breast tumorigenesis. Cancer Lett 2006;235:100–13. [DOI] [PubMed] [Google Scholar]
- [99].Lambe M, Hsieh CC, Chan HW, Ekbom A, Trichopoulos D, Adami HO. Parity, age at first and last birth, and risk of breast cancer: a population-based study in Sweden. Breast Cancer Res Treat 1996;38:305–11. [DOI] [PubMed] [Google Scholar]
- [100].Russo J, Russo IH. Influence of differentiation and cell kinetics on the susceptibility of the rat mammary gland to carcinogenesis. Cancer Res 1980;40:2677–87. [PubMed] [Google Scholar]
- [101].Russo J, Tay LK, Russo IH. Differentiation of the mammary gland and susceptibility to carcinogenesis. Breast Cancer Res Treat 1982;2:5–73. [DOI] [PubMed] [Google Scholar]
- [102].Welsch CW. Host factors affecting the growth of carcinogen-induced rat mammary carcinomas: a review and tribute to Charles Brenton Huggins. Cancer Res 1985;45:3415–43. [PubMed] [Google Scholar]
- [103].Sinha DK, Pazik JE, Dao TL. Prevention of mammary carcinogenesis in rats by pregnancy: effect of full-term and interrupted pregnancy. Br J Cancer 1988;57:390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev 1993;15:36–47. [DOI] [PubMed] [Google Scholar]
- [105].Swanson SM, Whitaker LM, Stockard CR, Myers RB, Oelschlager D, Grizzle WE, et al. Hormone levels and mammary epithelial cell proliferation in rats treated with a regimen of estradiol and progesterone that mimics the preventive effect of pregnancy against mammary cancer. Anticancer Res 1997;17:4639–45. [PubMed] [Google Scholar]
- [106].Yang J, Yoshizawa K, Nandi S, Tsubura A. Protective effects of pregnancy and lactation against N-methyl-N-nitrosourea-induced mammary carcinomas in female Lewis rats. Carcinogenesis 1999;20:623–8. [DOI] [PubMed] [Google Scholar]
- [107].Rajkumar L, Guzman RC, Yang J, Thordarson G, Talamantes F, Nandi S. Short-term exposure to pregnancy levels of estrogen prevents mammary carcinogenesis. Proc Natl Acad Sci USA 2001;98:11755–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Russo J, Russo IH. DNA labeling index and structure of the rat mammary gland as determinants of its susceptibility to carcinogenesis. J Natl Cancer Inst 1978;61:1451–9. [PubMed] [Google Scholar]
- [109].Russo J, Wilgus G, Russo IH. Susceptibility of the mammary gland to carcinogenesis. I. Differentiation of the mammary gland as determinant of tumor incidence and type of lesion. Am J Pathol 1979;96:721–36. [PMC free article] [PubMed] [Google Scholar]
- [110].Tay LK, Russo J. 7,12-dimethylbenz[a]anthracene-induced DNA binding and repair synthesis in susceptible and nonsusceptible mammary epithelial cells in culture. J Natl Cancer Inst 1981;67:155–61. [PubMed] [Google Scholar]
- [111].Tay LK, Russo J. Formation and removal of 7,12-dimethylbenz[a]anthracene– nucleic acid adducts in rat mammary epithelial cells with different susceptibility to carcinogenesis. Carcinogenesis 1982;2:1327–33. [DOI] [PubMed] [Google Scholar]
- [112].Russo IH, Russo J. Mammary gland neoplasia in long-term rodent studies. Environ Health Perspect 1996;104:938–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].D’Cruz CM, Moody SE, Master SR, Hartman JL, Keiper EA, Imielinski MB, et al. Persistent parity-induced changes in growth factors, TGF-βeta3, and differentiation in the rodent mammary gland. Mol Endocrinol 2002;16: 2034–51. [DOI] [PubMed] [Google Scholar]
- [114].Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res 1999;59:3379–86. [PubMed] [Google Scholar]
- [115].Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998;94:703–14. [DOI] [PubMed] [Google Scholar]
- [116].Bellone G, Carbone A, Tibaudi D, Mauri F, Ferrero I, Smirne C, et al. Differential expression of transforming growth factors-beta1, -beta2 and -beta3 in human colon carcinoma. Eur J Cancer 2001;37:224–33. [DOI] [PubMed] [Google Scholar]
- [117].Vagenas K, Spyropoulos C, Gavala V, Tsamandas AC. TGFbeta1, TGFbeta2, and TGFbeta3 protein expression in gastric carcinomas: correlation with prognostics factors and patient survival. J Surg Res 2007;139: 182–8. [DOI] [PubMed] [Google Scholar]
- [118].Booth D, Potten CS. Protection against mucosal injury by growth factors and cytokines. J Natl Cancer Inst Monogr 2001;29:16–20. [DOI] [PubMed] [Google Scholar]
- [119].Schrementi ME, Ferreira AM, Zender C, Dipietro LA. Site-specific production of TGF-βeta in oral mucosal and cutaneous wounds. Wound Repair Regen 2008;16:80–6. [DOI] [PubMed] [Google Scholar]
- [120].Bristow RE, Baldwin RL, Yamada SD, Korc M, Karlan BY. Altered expression of transforming growth factor-beta ligands and receptors in primary and recurrent ovarian carcinoma. Cancer 1999;85:658–68. [DOI] [PubMed] [Google Scholar]
- [121].Inan S, Vatansever S, Celik-Ozenci C, Sanci M, Dicle N, Demir R. Immunolo-calizations of VEGF, its receptors flt-1, KDR and TGF-βeta’s in epithelial ovarian tumors. Histol Histopathol 2006;21:1055–64. [DOI] [PubMed] [Google Scholar]
- [122].Rosenberg L, Zhang Y, Coogan PF, Strom BL, Palmer JR. A case-control study of oral contraceptive use and invasive epithelial ovarian cancer. Am J Epidemiol 1994;139:654–61. [DOI] [PubMed] [Google Scholar]
- [123].Rodriguez GC, Nagarsheth NP, Lee KL, Bentley RC, Walmer DK, Cline M, et al. Progestin-induced apoptosis in the Macaque ovarian epithelium: differential regulation of transforming growth factor-beta. J Natl Cancer Inst 2002;94:50–60. [DOI] [PubMed] [Google Scholar]
- [124].Parada D, Arciniegas E, Moreira O, Trujillo E. Transforming growth factor-beta2 and beta3 expression in carcinoma of the prostate. Arch Esp Urol 2004;57:93–9. [PubMed] [Google Scholar]
- [125].Djonov V, Ball RK, Graf S, Mottaz AE, Arnold AM, Flanders K, et al. Transforming growth factor-beta 3 is expressed in nondividing basal epithelial cells in normal human prostate and benign prostatic hyperplasia, and is no longer detectable in prostate carcinoma. Prostate 1997;31:103–9. [DOI] [PubMed] [Google Scholar]
- [126].Luo J, Dunn T, Ewing C, Sauvageot J, Chen Y, Trent J, et al. Gene expression signature of benign prostatic hyperplasia revealed by cDNA microarray analysis. Prostate 2002;51:189–200. [DOI] [PubMed] [Google Scholar]
- [127].Chetcuti A, Margan S, Mann S, Russell P, Handelsman D, Rogers J, et al. Identification of differentially expressed genes in organ-confined prostate cancer by gene expression array. Prostate 2001;47:132–40. [DOI] [PubMed] [Google Scholar]
- [128].Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature 2001;412:822–6. [DOI] [PubMed] [Google Scholar]
- [129].Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res 2003;63:3877–82. [PubMed] [Google Scholar]
- [130].Liu P, Ramachandran S, Ali Seyed M, Scharer CD, Laycock N, Dalton WB, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res 2006;66:4011–9. [DOI] [PubMed] [Google Scholar]
- [131].Tomlins SA, Rubin MA, Chinnaiyan AM. Integrative molecular concept modeling of prostate cancer progression. Nat Genet 2007;39:41–51. [DOI] [PubMed] [Google Scholar]
- [132].Kloen P, Gebhardt MC, Perez-Atayde A, Rosenberg AE, Springfield DS, Gold LI, et al. Expression of transforming growth factor-beta (TGF-βeta) isoforms in osteosarcomas: TGF-βeta3 is related to disease progression. Cancer 1997;80:2230–9. [PubMed] [Google Scholar]
- [133].Yang RS, Wu CT, Lin KH, Hong RL, Liu TK, Lin KS. Relation between histological intensity of transforming growth factor-beta isoforms in human osteosarcoma and the rate of lung metastasis. Tohoku J Exp Med 1998;184:133–42. [DOI] [PubMed] [Google Scholar]
- [134].Grainger DJ, Percival J, Chiano M, Spector TD. The role of serum TGF-βeta isoforms as potential markers of osteoporosis. Osteoporos Int 1999;9: 398–404. [DOI] [PubMed] [Google Scholar]
- [135].Chien HH, Lin WL, Cho MI. Expression of TGF-βeta isoforms and their receptors during mineralized nodule formation by rat periodontal ligament cells in vitro. J Periodontal Res 1999;34:301–9. [DOI] [PubMed] [Google Scholar]
- [136].Teare JA, Ramoshebi LN, Ripamonti U. Periodontal tissue regeneration by recombinant human transforming growth factor-beta3 in Papio ursinus. J Periodontal Res 2008;43:1–8. [DOI] [PubMed] [Google Scholar]
- [137].Lee BS, Nowak RA. Human leiomyoma smooth muscle cells show increased expression of transforming growth factor-beta 3 (TGF beta 3) and altered responses to the antiproliferative effects of TGF beta. J Clin Endocrinol Metab 2001;86:913–20. [DOI] [PubMed] [Google Scholar]
- [138].Gaide Chevronnay HP, Cornet PB, Delvaux D, Lemoine P, Courtoy PJ, Henriet P, et al. Opposite regulation of transforming growth factors-{beta}2 and - {beta}3 expression in the human endometrium. Endocrinology 2008;149: 1015–25. [DOI] [PubMed] [Google Scholar]
- [139].Al-Mowallad A, Carr T, Al-Qouzi A, Li C, Byers R, Kumar S. Plasma CD105, TGFbeta-1, TGFbeta-3 and the ligand/receptor complexes in children with acute lymphoblastic leukaemia. Anticancer Res 2006;26:543–7. [PubMed] [Google Scholar]
- [140].Yamamoto T, Akisue T, Marui T, Fujita I, Matsumoto K, Hitora T, et al. Expression of transforming growth factor beta isoforms and their receptors in malignant fibrous histiocytoma of soft tissues. Clin Cancer Res 2004;10:5804–7. [DOI] [PubMed] [Google Scholar]
- [141].Zhang PJ, Goldblum JR, Pawel BR, Pasha TL, Fisher C, Barr FG. PDGF-A, PDGF-Rbeta, TGFbeta3 and bone morphogenic protein-4 in desmoplastic small round cell tumors with EWS-WT1 gene fusion product and their role in stromal desmoplasia: an immunohistochemical study. Mod Pathol 2005;18:382–7. [DOI] [PubMed] [Google Scholar]
- [142].Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986;315:1650–9. [DOI] [PubMed] [Google Scholar]
- [143].Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer 2001;1:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Pedersen TX, Leethanakul C, Patel V, Mitola D, Lund LR, Danø K, et al. Laser capture microdissection-based in vivo genomic profiling of wound keratinocytes identifies similarities and differences to squamous cell carcinoma. Oncogene 2003;22:3964–76. [DOI] [PubMed] [Google Scholar]
- [145].Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2004;2:E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Daopin S, Piez KA, Ogawa Y, Davies DR. Crystal structure of transforming growth factor-beta 2: an unusual fold for the superfamily. Science 1992;257:369–73. [DOI] [PubMed] [Google Scholar]
- [147].Mittl PR, Priestle JP, Cox DA, McMaster G, Cerletti N, Grütter MG. The crystal structure of TGF-βeta 3 and comparison to TGF-βeta 2: implications for receptor binding. Protein Sci 1996;5:1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wise EL, Akana J, Gerlt JA, Rayment I. Structure of D-ribulose 5-phosphate 3-epimerase from Synechocystis to 1.6 A resolution. Acta Crystallogr D Biol Crystallogr 2004;60:1687–90. [DOI] [PubMed] [Google Scholar]
- [149].Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein–protein and protein–ligand interfaces. Bioinformatics 2006;22:1335–42. [DOI] [PubMed] [Google Scholar]
- [150].Graycar JL, Miller DA, Arrick BA, Lyons RM, Moses HL, Derynck R. Human transforming growth factor-beta 3: recombinant expression, purification, and biological activities in comparison with transforming growth factors-beta 1 and -beta 2. Mol Endocrinol 1989;3:1977–86. [DOI] [PubMed] [Google Scholar]
- [151].Cox DA, et al. Experimental wound healing studies with TGF beta. Wound Repair Regeneration 1994. 2:S208. [Google Scholar]
- [152].Jacobsen SE, Keller JR, Ruscetti FW, Kondaiah P, Roberts AB, Falk LA. Bidirectional effects of transforming growth factor beta (TGF-βeta) on colony-stimulating factor-induced human myelopoiesis in vitro: differential effects of distinct TGF-βeta isoforms. Blood 1991;78:2239–47. [PubMed] [Google Scholar]
- [153].Jacobsen SE, Ruscetti FW, Roberts AB, Keller JR. TGF-βeta is a bidirectional modulator of cytokine receptor expression on murine bone marrow cells. Differential effects of TGF-βeta 1 and TGF-βeta 3. J Immunol 1993;151: 4534–44. [PubMed] [Google Scholar]
- [154].James AW, Xu Y, Lee JK, Wang R, Longaker MT. Differential effects of TGF-beta1 and TGF-βeta3 on chondrogenesis in posterofrontal cranial suture-derived mesenchymal cells in vitro. Plast Reconstr Surg 2009;123:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Potts JD, Dagle JM, Walder JA, Weeks DL, Runyan RB. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta3. Proc Natl Acad Sci USA 1991;88:1516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Nakajima Y, Mironov V, Yamagishi T, Nakamura H, Markwald RR. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor beta3. Dev Dyn 1997;209:296–309. [DOI] [PubMed] [Google Scholar]
- [157].Nguyen AV, Pollard JW. Transforming growth factor beta3 induces cell death during the first stage of mammary gland involution. Development 2000;127:3107–18. [DOI] [PubMed] [Google Scholar]
- [158].ten Dijke P, Iwata KK, Goddard C, Pieler C, Canalis E, McCarthy TL, et al. Recombinant transforming growth factor type beta 3: biological activities and receptor-binding properties in isolated bone cells. Mol Cell Biol 1990;10:4473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Flanders KC, Lüdecke G, Engels S, Cissel DS, Roberts AB, Kondaiah P, et al. Localization and actions of transforming growth factor-beta s in the embryonic nervous system. Development 1991;113:183–91. [DOI] [PubMed] [Google Scholar]
- [160].Lindholm D, Castrén E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol 1992;117:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ren RF, Lah JJ, Diehlmann A, Kim ES, Hawver DB, Levey AI, et al. Differential effects of transforming growth factor-beta(s) and glial cell line-derived neurotrophic factor on gene expression of presenilin-1 in human post-mitotic neurons and astrocytes. Neuroscience 1999;93: 1041–9. [DOI] [PubMed] [Google Scholar]
- [162].Gorvy DA, Herrick SE, Shah M, Ferguson MW. Experimental manipulation of transforming growth factor-beta isoforms significantly affects adhesion formation in a murine surgical model. Am J Pathol 2005;167:1005–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Ask K, Bonniaud P, Maass K, Eickelberg O, Margetts PJ, Warburton D, et al. Progressive pulmonary fibrosis is mediated by TGF-βeta isoform 1 but not TGF-βeta3. Int J Biochem Cell Biol 2008;40:484–95. [DOI] [PMC free article] [PubMed] [Google Scholar]