

Abstract
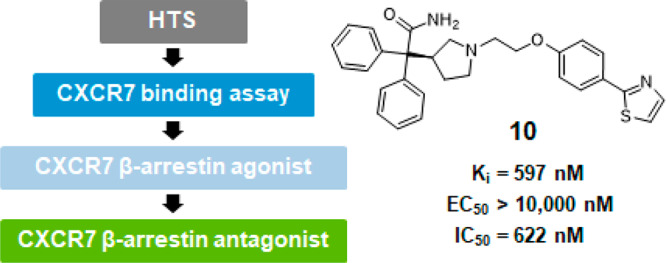
The atypical chemokine receptor CXCR7 has been studied in various disease settings including immunological diseases and heart disease. Efforts to elucidate the role of CXCR7 have been limited by the lack of suitable chemical tools with a range of pharmacological profiles. A high-throughput screen was conducted to discover novel chemical matter with the potential to modulate CXCR7 receptor activity. This led to the identification of a series of diphenylacetamides confirmed in a CXCL12 competition assay indicating receptor binding. Further evaluation of this series revealed a lack of activity in the functional assay measuring β-arrestin recruitment. The most potent representative, compound 10 (Ki = 597 nM), was determined to be an antagonist in the β-arrestin assay (IC50 = 622 nM). To our knowledge, this is the first reported small molecule β-arrestin antagonist for CXCR7, useful as an in vitro chemical tool to elucidate the effects of CXCL12 displacement with β-arrestin antagonism in models for diseases such as cardiac injury and suitable as starting point for hit optimization directed toward an in vivo tool compound for studying CXCR7 receptor pharmacology.
Keywords: CXCR7, ACKR3, HTS, β-arrestin, antagonist, chemical tool
The chemokine receptor CXCR7, later renamed ACKR3 (atypical chemokine receptor 3), is a nonclassical GPCR (G-protein coupled receptor) protein that has been implicated in a variety of disease pathologies,1 including oncology,2,3 immunology,4,5 and cardiovascular diseases.6−8 Two chemokine ligands are the known binding partners for CXCR7: CXCL12 or SDF-1 (stromal cell-derived factor 1) and CXCL11 or I-TAC (interferon-inducible T-cell α chemoattractant).9 In response to ligand binding, β-arrestin is recruited to CXCR7, without noticeable activation of any G-protein coupled pathways, rendering it an atypical member of the GPCR family. The receptor is proposed to act as a decoy receptor or “ligand sink” for CXCL12.10 The binding of CXCL12 to CXCR7 results in β-arrestin recruitment, followed by receptor internalization and degradation.11,12 CXCL12 is also the natural ligand for CXCR4, a classical GPCR with G-protein coupled signaling pathways, relevant for cardiomyocyte survival and angiogenesis. The scavenging activity of CXCR7 is thought to control local plasma levels of CXCL12, thus modulating the activity of CXCR4. Recent reports suggest that in addition to regulating levels of CXCL12, β-arrestin recruitment to CXCR7 may also activate signaling pathways, including EGFR/ERK,13 AKT,14 MAPK,15 and mTOR.16
Several small molecule and peptidic compounds that bind to CXCR7 are reported in peer-reviewed publications and patents (Figure 1).17−20 Compounds 1,212,22 and 3(23) have been shown to be potent CXCR7 modulators. The high-affinity CXCR7 binder 4 has been used to elucidate the role of pharmacological CXCR7 intervention in cardiac repair.24 The macrocyclic peptide-peptoid hybrid 5 was identified as a potent CXCR7 modulator with high selectivity and oral bioavailability.25 The work of others as well as our efforts24 have demonstrated that the above-described literature compounds, while competitively binding with respect to CXCL12, all induce the recruitment of β-arrestin in cell systems and thus can be referred to as agonists of the β-arrestin pathway. At the time we initiated an HTS (high-throughput screening) campaign to find novel chemical matter for CXCR7, no suitable tool compound with CXCR7 receptor binding affinity was available that functionally acted as antagonist of the β-arrestin pathway. Hence, as part of our screening strategy we were open to the possibility of identifying small molecules with novel, hitherto unknown CXCR7 receptor pharmacology that could enable the exploration of both CXCL12 displacement and β-arrestin signaling activities downstream of CXCR7 in relevant disease model systems.
Figure 1.

Structures of representative chemotypes of CXCR7 modulators described in the literature acting as agonists of the β-arrestin pathway.
Our primary research effort was aimed at modulating the CXCR7-mediated degradation of circulating CXCL12. We hypothesized that increasing the bioavailability of CXCL12 in blood plasma by blocking CXCL12 from binding to CXCR7 and lowering its subsequent degradation would lead to an increased protection in a disease setting of cardiac injury. However, it was unclear if the intracellular signaling downstream of the β-arrestin recruitment, either activated by β-arrestin agonism or down-regulated by β-arrestin antagonism, will lead to different phenotypes with distinct therapeutic outcomes. While genetic knockout data can be edifying to study the mechanistic role of CXCR7,26−28 the pharmacological antagonism of CXCR7 by a small molecule modulator can have a unique phenotype with therapeutic relevance. In the course of triaging the chemical matter from HTS based on their pharmacological profiles, we identified a novel series of diphenylacetamide compounds that bind competitively with CXCL12 and act as functional antagonists of the β-arrestin pathway.
The overall screening cascade to identify novel chemical matter for modulating CXCR7 receptor pharmacology is shown in Figure 2. The Pfizer screening library of 2.8 million small molecules was evaluated in a CXCR7 Tag-lite binding assay (Cisbio) enabled for high-throughput screening.29 In this HTRF (homogeneous time-resolved fluorescence) assay, CXCR7 is labeled with a fluorophore via a small fusion tag (SNAP-tag) and overexpressed in a cell line. The level of displacement of fluorescent-labeled CXCL12 by nonlabeled screening compounds is detected by changes of the fluorescence signal. Approximately 6,500 primary screening hits with measurable binding affinity (greater than 40% effect at 10,000 nM) were identified and further confirmed in a dose–response format using the same assay format, resulting in ca. 1,000 confirmed hits. These were selected for evaluation in an orthogonal filter binding assay, which measures the level of displacing radiolabeled 125I-CXCL12 from membranes obtained from a cell line overexpressing the CXCR7 receptor.20 Of these, 400 hits were selected for follow-up in a functional CXCR7 β-arrestin assay to measure their ability to induce β-arrestin recruitment in a cell line overexpressing the CXCR7 receptor.30 While the majority of hits confirmed as functionally active in the β-arrestin assay, preliminary results indicated that a few compounds binding to CXCR7 were inactive in the β-arrestin assay. Since this assay was initially run in agonist mode to identify functional β-arrestin agonists, we were interested to determine if these inactive compounds are potentially functional antagonists in the β-arrestin assay. To test them in antagonist mode, CXCL12 at an EC80 concentration (28 nM) was added as a challenge to compete with the screening compounds. Since CXCR4 is a related chemokine receptor to CXCR7 sharing the same ligand CXCL12, we employed a CXCR4 binding assay using a Jurkat cell line to check for selectivity of compound hits against CXCR7.
Figure 2.

Screening cascade to identify selective CXCR7 modulators with different pharmacologies.
Upon resynthesis (Scheme 1) and repeated testing, only a single compound, the diphenylacetamide hit 10, was confirmed to have CXCR7 binding affinity (Ki = 597 nM) using the radiolabeled 125I-filter binding assay, but without functional agonist activity (EC50 > 10,000 nM) in the CXCR7 β-arrestin assay (Table 1). Subsequent hit expansion efforts led to the identification of additional active analogs structurally similar to 10, which suggested the emergence of a novel chemical series with a potentially unique β-arrestin antagonist pharmacology. All diphenylacetamide analogs contain a central ethylene linker flanked by aromatic groups on both sides. One side has an N-linked pyrrolidine ring, substituted with the diphenylacetamide moiety. The other side of the ethylene group is connected to an O-linked phenol with a heterocycle at the para position. This heterocycle constitutes the single point of structural diversity in this analog set. Compound 10 contains a 2-thiazolyl ring and was found to be the most potent analog. Replacing the thiazole group with an oxadiazole in 11 (Ki = 4,087 nM) led to an approximately 6-fold loss in affinity compared to 10. Additional analogs dihydrooxazole 12 and Me-oxadiazole 13 exhibited weaker binding affinities to CXCR7, with Ki = 6,544 nM and Ki = 11,757 nM, respectively. In addition to 10, compounds 11–13 were all inactive (EC50 > 10,000 nM) in the β-arrestin assay.
Scheme 1. Synthesis of Compound 10.

For description of synthesis of compounds see the Supporting Information. Reagents and conditions: (i) 2-bromoethoxy-tert-butyldimethylsilane, K2CO3, MeCN, 80 °C, 70%; (ii) HCl in dioxane, CH2Cl2, rt, 41%; (iii) SOCl2, CHCl3, 85 °C, 92%; (iv) Cs2CO3, DMF, 100 °C, 40%.
Table 1. Biological Activitiesa of CXCR7 HTS Hit 10 and SAR Analogs.


For detailed description of assay formats and conditions see the Supporting Information.
Values are reported as geometric means of at least three independent experiments with pKi ± SD in parentheses.
As the most potent analog in this series, compound 10 was selected for further in vitro characterization (Table 2). Running the β-arrestin assay in antagonist mode in the presence of CXCL12 at EC80 concentration, the functional antagonist activity was confirmed (IC50 = 622 nM). Since CXCL12 is also a known ligand for the related chemokine receptor CXCR4, we tested compound 10 for binding to CXCR4. No measurable binding affinity against CXCR4 was detected (IC50 > 15,000 nM), indicating selectivity of 10 toward the CXCR7 receptor. Additional profiling showed that 10 is moderately lipophilic (logD = 3.1) with low passive permeability (Papp = 1.1 × 10–6 cm/s) and high metabolic turnover in human and mouse liver microsomes (CLint = 109 μL/min/mg and 737 μL/min/mg, respectively).
Table 2. Characterization of CXCR7 β-Arrestin Antagonist 10.

| 10 | |
|---|---|
| CXCR7 Ki (nM)a | 597 (6.22 ± 0.27) |
| CXCR4 IC50 (nM) | >15,000 |
| CXCR7 β-arrestin EC50 (nM) agonist mode | >10,000 |
| CXCR7 β-arrestin IC50 (nM)aantagonist mode | 622 (6.20 ± 0.38) |
| logDb | 3.1 |
| Solubility (μg/mL)c | 98.8 |
| Papp (10–6 cm/s)d | 1.1 |
| HLM CLint (μL/min/mg)e | 109 |
| MLM CLint (μL/min/mg)f | 737 |
Values are reported as geometric means of at least three independent experiments with pKi or pIC50 ± SD in parentheses.
logD was measured by the shake flask method at pH 7.4.
Kinetic stability was measured at pH 6.5.
Passive permeability was determined using a Madin–Darby canine kidney (MDCK) cell line.
Microsomal stability was determined in human liver microsomes (HLM).
Microsomal stability was determined in mouse liver microsomes (MLM).
While compound 10 demonstrated interesting in vitro pharmacology, it was not a suitable compound for oral administration due to its low passive permeability and high metabolic turnover. Instead, it was dosed subcutaneously in male CD-1 mice (n = 3) to determine its utility as an in vivo tool to interrogate CXCR7 β-arrestin antagonist pharmacology in mice (Table 3, Figure 3). Following an administered dose of 30 mg/kg a Cmax = 1,450 ng/mL or 2,996 nM was observed. However, when corrected for mouse plasma protein binding (fu,p = 0.0075) the unbound maximum plasma exposure was low (Cu,max = 22 nM). In fact, it was determined to be ∼30-fold lower than the measured CXCR7 in vitro binding affinity (Ki = 597 nM). Consequently, compound 10 would have to be further optimized toward improved potency or lowering metabolic clearance, in order to serve as a suitable in vivo tool to interrogate the effect of a CXCR7 binder that is a functional antagonist of the β-arrestin recruitment pathway.
Table 3. Pharmacokinetics of CXCR7 β-Arrestin Antagonist 10 in Mice after Subcutaneous Administrationa.
| Tmax (h) | Cmax (ng/mL) | Cmax (nM) | AUC (ng·h/mL) | T1/2 (h) | fu,p |
|---|---|---|---|---|---|
| 2.0 | 1,450 | 2,996 | 9,960 | 2.7 | 0.0075 |
Pharmacokinetic parameters were calculated from plasma concentration–time data in male CD-1 mice (n = 3) following subcutaneous administration (30 mg/kg) in 10% DMSO, 50% PEG-400, 40% water.
Figure 3.

Pharmacokinetic evaluation of 10.
In summary, a high-throughput screening campaign using a CXCR7 Tag-lite binding assay was conducted to identify compounds that bind to the CXCR7 receptor. Screening hits were followed up in a β-arrestin functional assay, wherein a single hit, diphenylacetamide 10, was confirmed to be binding to CXCR7 (Ki = 597 nM) selectively over CXCR4 (IC50 > 15,000 nM), but unlike any other CXCR7 modulators was inactive in the β-arrestin recruitment assay (EC50 > 10,000 nM). Compound 10 was subsequently confirmed as a CXCR7 antagonist of the β-arrestin pathway with moderate activity (IC50 = 622 nM). This compound showed a suboptimal in vitro ADME profile with low passive permeability and high metabolic turnover, which was corroborated by pharmacokinetic data in mice. Nonetheless, as a first reported CXCR7 modulator with novel β-arrestin antagonist pharmacology, compound 10 is a useful in vitro tool to elucidate the effects of CXCL12 displacement with β-arrestin antagonism in models for diseases such as cardiac injury and can serve as an important starting point for physicochemical property and potency optimization toward a suitable tool compound for studying CXCR7 receptor pharmacology in vivo.
Glossary
Abbreviations
- ACKR3
atypical chemokine receptor 3
- CXCR7
C-X-C chemokine receptor 7
- CXCL12
C-X-C chemokine ligand 12
- SDF-1
stromal cell-derived factor 1
- CXCR4
C-X-C chemokine receptor 4
- HTS
high-throughput screening
- Papp
apparent permeability
- MDCK
Madin–Darby canine kidney cell line
- ADME
absorption, distribution, metabolism, and excretion
- Cmax
maximal plasma concentration
- CLint
intrinsic clearance
- AUC
area under the time–concentration curve
- T1/2
half-life
- Tmax
time where maximal plasma concentration occurred
- fu,p
fraction unbound to plasma protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00163.
Description of in vitro assays and PK experiment; detailed synthesis procedures and analytical data for all compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Wang C.; Chen W.; Shen J. CXCR7 Targeting and Its Major Disease Relevance. Front. Pharmacol. 2018, 9, 641. 10.3389/fphar.2018.00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Toub M.; Almohawes M.; Vishnubalaji R.; Alfayez M.; Aldahmash A.; Kassem M.; Alajez N. M. CXCR7 signaling promotes breast cancer survival in response to mesenchymal stromal stem cell-derived factors. Cell Death Discovery 2019, 5, 87. 10.1038/s41420-019-0169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian T.; Liu Y.; Dong Y.; Zhang L.; Dong Y.; Sun Y.; Sun D. CXCR7 regulates breast tumor metastasis and angiogenesis in vivo and in vitro. Mol. Med. Rep. 2017, 17, 3633–3639. 10.3892/mmr.2017.8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngamsri K.-C.; Müller A.; Bösmüller H.; Gamper-Tsigaras J.; Reutershan J.; Konrad F. M. The Pivotal Role of CXCR7 in Stabilization of the Pulmonary Epithelial Barrier in Acute Pulmonary Inflammation. J. Immunol. 2017, 198, 2403–2413. 10.4049/jimmunol.1601682. [DOI] [PubMed] [Google Scholar]
- Chang H. C.; Huang P. H.; Syu F. S.; Hsieh C. H.; Chang S. L.; Lu J.; Chen H. C. Critical involvement of atypical chemokine receptor CXCR7 in allergic airway inflammation. Immunology 2018, 154, 274–284. 10.1111/imm.12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro F.; Biben C.; Martinez-Munoz L.; Mellado M.; Ransohoff R. M.; Li M.; Woehl B.; Leung H.; Groom J.; Batten M.; Harvey R. P.; Martinez A. C.; Mackay C. R.; Mackay F. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14759–14764. 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromage D. I.; Davidson S. M.; Yellon D. M. Stromal derived factor 1α: A chemokine that delivers a two-pronged defence of the myocardium. Pharmacol. Ther. 2014, 143, 305–315. 10.1016/j.pharmthera.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E. S.; Miller L.; Patel A. N.; Anderson R. D.; Mendelsohn F. O.; Traverse J.; Silver K. H.; Shin J.; Ewald G.; Farr M. J.; Anwaruddin S.; Plat F.; Fisher S. J.; AuWerter A. T.; Pastore J. M.; Aras R.; Penn M. S. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: the STOP-HF randomized Phase II trial. Eur. Heart J. 2015, 36, 2228–2238. 10.1093/eurheartj/ehv254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann U.; Cameroni E.; Pruenster M.; Mahabaleshwar H.; Raz E.; Zerwes H.-G.; Rot A.; Thelen M. CXCR7 Functions as a Scavenger for CXCL12 and CXCL11. PLoS One 2010, 5, e9175 10.1371/journal.pone.0009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S.; Kim J.; Ahn S.; Craig S.; Lam C. M.; Gerard N. P.; Gerard C.; Lefkowitz R. J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 628–632. 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins N. L.; Trakimas D.; Chang S. L.; Ehrlich A.; Ray P.; Luker K. E.; Linderman J. J.; Luker G. D. CXCR7 controls competition for recruitment of beta-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS One 2014, 9, e98328 10.1371/journal.pone.0098328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F.; Muller W.; Schutz D.; Penfold M. E.; Wong Y. H.; Schulz S.; Stumm R. Rapid uptake and degradation of CXCL12 depend on CXCR7 carboxyl-terminal serine/threonine residues. J. Biol. Chem. 2012, 287, 28362–28377. 10.1074/jbc.M111.335679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. K.; Lokeshwar B. L. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 2011, 71, 3268–3277. 10.1158/0008-5472.CAN-10-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M.; Zheng J.; Hou K.; Wang J.; Chen X.; Lu X.; Bo J.; Xu C.; Shen K.; Wang J. Role of chemokine receptor CXCR7 in bladder cancer progression. Biochem. Pharmacol. 2012, 84, 204–214. 10.1016/j.bcp.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Lin L.; Han M. M.; Wang F.; Xu L. L.; Yu H. X.; Yang P. Y. CXCR7 stimulates MAPK signaling to regulate hepatocellular carcinoma progression. Cell Death Dis. 2014, 5, e1488-e1488 10.1038/cddis.2014.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieranò C.; Santagata S.; Napolitano M.; Guardia F.; Grimaldi A.; Antignani E.; Botti G.; Consales C.; Riccio A.; Nanayakkara M.; Barone M. V.; Caraglia M.; Scala S. CXCR4 and CXCR7 transduce through mTOR in human renal cancer cells. Cell Death Dis. 2014, 5, e1310-e1310 10.1038/cddis.2014.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlere I.; Caspar B.; Arimont M.; Dekkers S.; Visser K.; Stuijt J.; de Graaf C.; Stocks M.; Kellam B.; Briddon S.; Wijtmans M.; de Esch I.; Hill S.; Leurs R. Modulators of CXCR4 and CXCR7/ACKR3 function. Mol. Pharmacol. 2019, 96, 737–752. 10.1124/mol.119.117663. [DOI] [PubMed] [Google Scholar]
- Gustavsson M.; Wang L.; van Gils N.; Stephens B. S.; Zhang P.; Schall T. J.; Yang S.; Abagyan R.; Chance M. R.; Kufareva I.; Handel T. M. Structural basis of ligand interaction with atypical chemokine receptor 3. Nat. Commun. 2017, 8, 14135. 10.1038/ncomms14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Maishi N.; Akiyama K.; Towfik Alam M.; Ohga N.; Kawamoto T.; Shindoh M.; Takahashi N.; Kamiyama T.; Hida Y.; Taketomi A.; Hida K. CXCL12–CXCR7 axis is important for tumor endothelial cell angiogenic property. Int. J. Cancer 2015, 137, 2825–2836. 10.1002/ijc.29655. [DOI] [PubMed] [Google Scholar]
- Zabel B. A.; Wang Y.; Lewen S.; Berahovich R. D.; Penfold M. E.; Zhang P.; Powers J.; Summers B. C.; Miao Z.; Zhao B.; Jalili A.; Janowska-Wieczorek A.; Jaen J. C.; Schall T. J. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009, 183, 3204–3211. 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- Yoshikawa Y.; Oishi S.; Kubo T.; Tanahara N.; Fujii N.; Furuya T. Optimized method of G-protein-coupled receptor homology modeling: its application to the discovery of novel CXCR7 ligands. J. Med. Chem. 2013, 56, 4236–4251. 10.1021/jm400307y. [DOI] [PubMed] [Google Scholar]
- Wijtmans M.; Maussang D.; Sirci F.; Scholten D. J.; Canals M.; Mujic-Delic A.; Chong M.; Chatalic K. L.; Custers H.; Janssen E.; de Graaf C.; Smit M. J.; de Esch I. J.; Leurs R. Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur. J. Med. Chem. 2012, 51, 184–192. 10.1016/j.ejmech.2012.02.041. [DOI] [PubMed] [Google Scholar]
- Uto-Konomi A.; McKibben B.; Wirtz J.; Sato Y.; Takano A.; Nanki T.; Suzuki S. CXCR7 agonists inhibit the function of CXCL12 by down-regulation of CXCR4. Biochem. Biophys. Res. Commun. 2013, 431, 772–776. 10.1016/j.bbrc.2013.01.032. [DOI] [PubMed] [Google Scholar]
- Menhaji-Klotz E.; Hesp K. D.; Londregan A. T.; Kalgutkar A. S.; Piotrowski D. W.; Boehm M.; Song K.; Ryder T.; Beaumont K.; Jones R. M.; Atkinson K.; Brown J. A.; Litchfield J.; Xiao J.; Canterbury D. P.; Burford K.; Thuma B. A.; Limberakis C.; Jiao W.; Bagley S. W.; Agarwal S.; Crowell D.; Pazdziorko S.; Ward J.; Price D. A.; Clerin V. Discovery of a Novel Small-Molecule Modulator of C-X-C Chemokine Receptor Type 7 as a Treatment for Cardiac Fibrosis. J. Med. Chem. 2018, 61, 3685–3696. 10.1021/acs.jmedchem.8b00190. [DOI] [PubMed] [Google Scholar]
- Boehm M.; Beaumont K.; Jones R.; Kalgutkar A. S.; Zhang L.; Atkinson K.; Bai G.; Brown J. A.; Eng H.; Goetz G. H.; Holder B. R.; Khunte B.; Lazzaro S.; Limberakis C.; Ryu S.; Shapiro M. J.; Tylaska L.; Yan J.; Turner R.; Leung S. S. F.; Ramaseshan M.; Price D. A.; Liras S.; Jacobson M. P.; Earp D. J.; Lokey R. S.; Mathiowetz A. M.; Menhaji-Klotz E. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide-Peptoid Hybrid CXCR7 Modulators. J. Med. Chem. 2017, 60, 9653–9663. 10.1021/acs.jmedchem.7b01028. [DOI] [PubMed] [Google Scholar]
- Hao H.; Hu S.; Chen H.; Bu D.; Zhu L.; Xu C.; Chu F.; Huo X.; Tang Y.; Sun X.; Ding B. S.; Liu D. P.; Hu S.; Wang M. Loss of Endothelial CXCR7 Impairs Vascular Homeostasis and Cardiac Remodeling After Myocardial Infarction: Implications for Cardiovascular Drug Discovery. Circulation 2017, 135, 1253–1264. 10.1161/CIRCULATIONAHA.116.023027. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin L.; Sanchez-Mateos P.; Cabanas C. CXCR7 impact on CXCL12 biology and disease. Trends Mol. Med. 2013, 19, 12–22. 10.1016/j.molmed.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Yang M.; Zeng C.; Li P.; Qian L.; Ding B.; Huang L.; Li G.; Jiang H.; Gong N.; Wu W. Impact of CXCR4 and CXCR7 knockout by CRISPR/Cas9 on the function of triple-negative breast cancer cells. OncoTargets Ther. 2019, 12, 3849–3858. 10.2147/OTT.S195661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwier J. M.; Roux T.; Cottet M.; Durroux T.; Douzon S.; Bdioui S.; Gregor N.; Bourrier E.; Oueslati N.; Nicolas L.; Tinel N.; Boisseau C.; Yverneau P.; Charrier-Savournin F.; Fink M.; Trinquet E. A Fluorescent Ligand-Binding Alternative Using Tag-lite® Technology. J. Biomol. Screening 2010, 15, 1248–1259. 10.1177/1087057110384611. [DOI] [PubMed] [Google Scholar]
- Bassoni D. L.; Raab W. J.; Achacoso P. L.; Loh C. Y.; Wehrman T. S. Measurements of beta-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol. Biol. 2012, 897, 181–203. 10.1007/978-1-61779-909-9_9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.