

Abstract

The histone acetyltransferases, CREB binding protein (CBP) and EP300, are master transcriptional co-regulators that have been implicated in numerous diseases, such as cancer, inflammatory disorders, and neurodegeneration. A novel, highly potent, orally bioavailable EP300/CBP histone acetyltransferase (HAT) inhibitor, CPI-1612 or 17, was developed from the lead compound 3. Replacement of the indole scaffold of 3 with the aminopyridine scaffold of 17 led to improvements in potency, solubility, and bioavailability. These characteristics resulted in a 20-fold lower efficacious dose for 17 relative to lead 3 in a JEKO-1 tumor mouse xenograft study.
Keywords: p300, CBP, histone acetyltransferase, HAT, KAT
EP300 and CREB binding protein (CBP), also known as KAT3A/3B, are two highly homologous, multidomain, epigenetic regulatory enzymes that affect transcription. Although their transcriptional effects are mediated through multiple mechanisms utilizing the different functional domains of these enzymes, the acetylation of histone lysine residues with the cofactor acetyl coenzyme A (AcCoA) by their histone acetyltransferase (HAT) domains plays a central role in regulating transcription. The HAT domains of EP300 and CBP also modulate the activity of non-histone proteins through post-translational acetylation.1 Both enzymes have been implicated in a wide variety of human diseases, such as cancer, inflammation, fibrosis, and neurodegeneration.2−7 While loss of function in EP300 and/or CBP has been shown to be deleterious, double knockout is lethal in mammals; it has also been shown that overexpression or activating mutations can lead to disease states, particularly in cancer.8,9 As such, small molecule inhibitors of EP300/CBP HAT activity may provide a novel opportunity for therapeutic intervention in a wide range of human diseases.10
We previously described our early drug-discovery efforts toward the identification of leads for the development of novel EP300/CBP HAT inhibitors.11 With an emphasis on improving the potency of the high throughput screen (HTS) hit 1, we independently explored the structure–activity relationship (SAR) of the indole, central benzene, and phenethylamine side chain. To do this, each hydrogen atom of compound 2, the minimum pharmacophore of lead compound 1, was replaced with a chlorine atom or methyl group to establish which vectors may allow for introduction of moieties to optimize the potency and physicochemical properties of the scaffold.12 These efforts culminated in the identification of 3, a tool compound suitable to validate our biochemical assays, interrogate CBP/EP300 HAT biology in vitro, and perform initial in vivo experiments. While compound 3 and its analogues were suitable for early proof of concept studies, we sought to improve the potency, oral bioavailability, solubility, and off-target profile of the series with the goal of advancing an EP300/CBP HAT inhibitor into clinical studies. Herein, we describe our medicinal chemistry lead optimization efforts.
The thorough investigation of the SAR outlined in Figure 1 gave us confidence that we had identified the substitution pattern for optimal potency on the phenethylamine side chain, central benzene, and indole core. Subsequently, we focused our attention on the identification of replacements for the indole system, a portion of the molecule that we had not modified up to this stage of the program, that would retain the potency of 3 while improving upon its suboptimal solubility, bioavailability, and human ether-à-go-go-related gene or Kv 11.1 (hERG) inhibition. Our initial efforts in this vein were centered on adding an additional nitrogen atom to the indole core, either in the form of aza-indole 4 or indazole 5 or by substitution with an indole isostere, such as imidazopyridines 6 and 7 or pyrazolylpyridine 8. Only the pyrazolylpyridine retained the potency of 3 but with decreased stability in mouse liver microsomes (Table 1). Furthermore, we noted that the potency of compounds 3–8 converged with their respective enantiomers in JEKO-1 cell viability experiments (data not shown). We hypothesized that this convergence in potency was due to compound racemization during the viability assay which was of substantially longer duration relative to the scintillation proximity assays (SPAs).13 We also sought to address this later issue with future analogues.
Figure 1.

Development of lead compound 3 from HTS hit 1.
Table 1. Alternative Scaffolds to the Indole in Compound 3.


The X-ray cocrystal structure of 1 with EP300 led us to hypothesize that the indole of 3 served mainly as a scaffolding element to appropriately position the phenethylamine side chain and the C-6 pyrazole group.11 This reasoning led us to speculate that a bicyclic scaffold was not required and that it could be replaced with scaffolds that would adopt a conformation in which the benzenoid portion of the indole of 3 was coplanar with its carbonyl group.14 Based on this reasoning, we chose to explore anilines and amino-heterocyclic amines as replacements for the indole scaffold. While indoline 9 proved to be a potent inhibitor, the compound had poor microsomal stability. The tetrahydroquinoline 10 was not tolerated as a replacement, likely because the tetrahydroquinoline disrupts the coplanar relationship of the scaffold and carbonyl group. While the aniline amide 11 was approximately 3-fold less potent than 3, it encouraged us to explore additional amino-heterocyclic amides with the hope of identifying a scaffold that would confer improved solubility to this class of inhibitors. This led us to discover the 2-aminopyridine derivative 12, which possessed improved potency and solubility relative to 3. Other aminoheterocyclic scaffolds including the regioisomeric pyridine 13, pyridazine 14, pyrazine 15, and pyrimidine 16 were inferior to the 2-aminopyridine 12.
In addition to its improved potency and solubility, 12 has improved stability to epimerization, permeability, and mouse pharmacokinetics (PK) relative to 3. These findings led us to focus our efforts on the optimization of the aminopyridine scaffold. Based on earlier studies examining the effect of substitution of the phenethylamine side chain, a methyl group was incorporated into the phenethylamine side chain of 12 to provide compound 17 with the expectation that it would improve the potency and lower the in vitro clearance.15 Indeed, this modification provided a compound with a 2- to 3-fold improvement in potency across a range of biochemical (EP300 HAT, full length EP300, and full length CBP) and cell-based assays [H3K18Ac MSD (H3K18 = histone 3 lysine 18, MSD = meso scale discovery) and JEKO-1 proliferation] while at the same time improving the microsomal stability. The stereoisomers of 17 were >40 times less potent than 17.16 Further modifications to 17 were made, including heterocyclic analogues of the pyrazole, methyl-substituted pyridine analogues, and 2- and 3-fluoro-4-cyanophenethylamine analogues.17 While some of these analogues provided modest improvements over 17 for a single parameter, none of them were better than 17 in aggregate and we focused our attention on 17 for further in vivo studies.
The high EP300 potency, promising in vitro profile, and good mouse PK of 17 prompted us to further profile this compound’s pharmacokinetic profile in rats and dogs. While the oral exposure of 17 in dogs (0.5 mg/kg IV; 1.0 mg/kg PO; clearance = 0.42 L/h/kg, Vss = 3.7 L/kg, T1/2 = 5.5 h, F% = 71; AUC/dose = 1691 h·mg/mL) and mice (1 mg/kg IV; 5 mg/kg PO; clearance = 3.8 L/h/kg, Vss = 2.0 L/kg, T1/2 = 0.98 h, F% = 79; AUC/dose = 211 h·mg/mL) is good, the exposure in rats is limited by poor bioavailability (1.0 mg/kg IV; 5.0 mg/kg PO; clearance = 2.6 L/h/kg, Vss = 1.8 L/kg, T1/2 = 1.2 h, F% = 9; AUC/dose = 35.6 h·mg/mL).18 Additionally, because there are many potential indications where a brain-penetrant EP300/CBP HAT inhibitor would be of interest, we performed a mouse PK experiment to monitor for brain exposure.6,7 In this experiment, a single dose of 17 was administered orally to CD-1 mice and brain and plasma exposures of 17 were measured at 0.25, 0.5, 1.0, 2.0, 4.0, and 8.0 h. 17 is highly brain-penetrant, showing a brain-to-plasma ratio of 0.35 after a single oral dose.19
Compound 17 was then characterized for its selectivity against other histone acetyltransferases and a panel of typical antitargets. No inhibitory activity was observed in a panel of seven HAT targets (Tip60, HAT1, PCAF, MYST2, MYST3, MYST4, and GCN5L2) and had minimal activity against the antitargets in the Eurofins Safety44 off-target screening panel. Additionally, 17 showed weak activity in a hERG binding assay (IC50 = 10.4 μM) and displayed moderate inhibition of CYP2C8 (IC50 = 1.9 μM) and CYP2C19 (IC50 = 2.7 μM), but it showed less inhibition of CYP3A4, CYP2D6, CYP1A2, CYP2B6, and CYP2C9 (>50 μM, 34 μM, >50 μM, 8.2 μM, 6.6 μM).
X-ray cocrystal structures, obtained utilizing the methodology described by Abbvie, clearly show that compounds 12 and 17 bind to the acetylcoenzyme A binding site.11Figure 2 shows an overlay of AcCoA and 12 that highlights the major ligand–protein contacts of 12 to the EP300 HAT domain. Several of the binding interactions are common for indole 1 and aminopyridine 12. These include the SER1400 H-bonding interaction to the phenethylamine N–H and the π-stacking interactions of TRP1466 to the phenethylamine aromatics and of HIS1451 to the indole of 1 or the aminopyridine of 12. A key difference between the crystal structures of 1 and 12 that may account for the improved potency of 12 relative to 3 is a H-bonding network that is established between the aminopyridine of 12 with ASP 1399 and a water bound to the backone N–H of TYR1446. This water is not found in the cocrystal structure of 1 presumably because it has been displaced by the indole N–H.
Figure 2.

Overlay of X-ray cocrystal structures of 12 (cyan) and acetylcoenzyme A (pink) bound to the HAT domain of EP300 with key hydrogen bonds (red) and π-stacking interactions (yellow) of 12 with EP300 highlighted.
While both our series of inhibitors and Abbvie’s occupy the AcCoA pocket of EP300, the contacts the two classes make with the protein are somewhat distinct. While both compounds make hydrogen bonds with SER1400, 17 does so through an N–H bond and A-485 through a carbonyl group from the oxazolidinedione.20,21 The only other interaction common to both inhibitors is a π-stacking interaction with HIS1451. The other interactions the compounds make with the protein are distinct. A-485 makes two hydrogen bonds from a urea moiety with the backbone carbonyl of GLN1455, while 17 makes no direct contact with this residue. In contrast, 17 makes hydrogen bonds with the backbone N–H of TYR1446 and to ASP1399 through the aminopyridine scaffold, while A-485 makes no direct contact with this residue. Another difference in the binding modes are the interactions of the aromatic side chains. The 4-cyanophenethylamine side chain of 17 interacts with TRP1466 through a π-stacking interaction, while the 4-fluorobenzylamide of A-485 fills a pocket formed by TYR1414 and LEU1418. This later pocket is filled by the methyl group on the phenethylamine side chain of 17.
Figure 3.
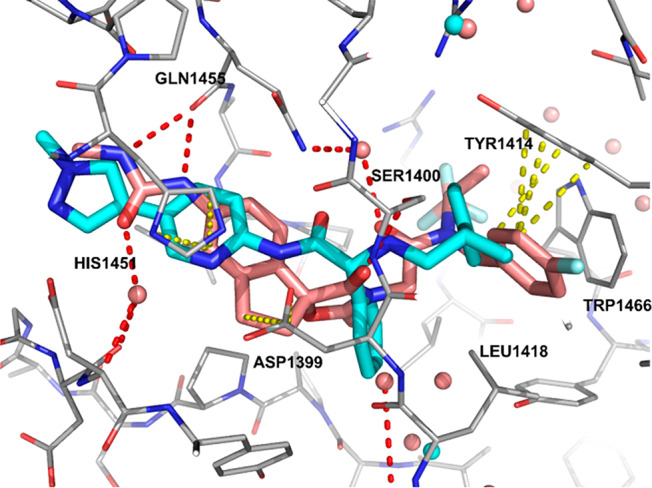
Overlay of X-ray cocrystal structures of 17 (cyan) and A-485 (pink) bound to the HAT domain of EP300 with key hydrogen bonds (red) and π-stacking interactions (yellow) of A-485 highlighted.
The in vivo activity of 17 was assessed by conducting a PK/PD study in C57B6 mice, measuring H3K27Ac (H3K27 = histone 3 lysine 27) reduction in PBMCs and H3K18Ac reduction in spleen tissue (data not shown) after oral administration of 17. As expected, based on its high potency and good pharmacokinetic profile, 17 reduces the histone acetylation in vivo at the primary chromatin acetylation targets of EP300/CBP in a dose- and time-dependent manner (Figure 4).
Figure 4.
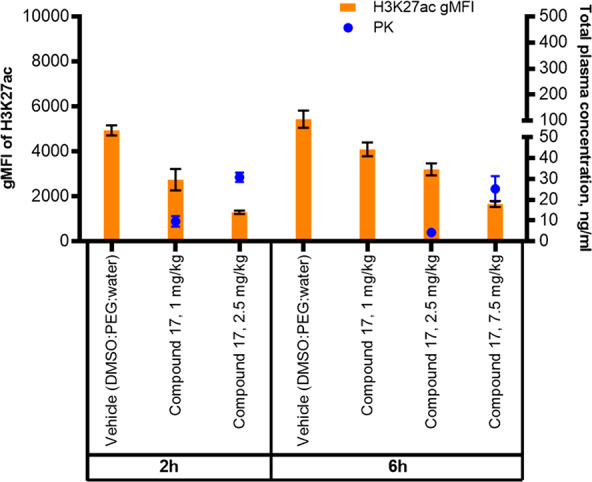
Dose- and time-dependent reduction of H3K27Ac in PBMCs of C57Black6 mice after oral administration of compound 17.
The efficacy of 17 was then assessed in a mouse tumor xenograft model using a B-cell lymphoma line, JEKO-1, which we have shown is sensitive to CBP/EP300 HAT inhibition. 17 showed 67% tumor growth inhibition (TGI) at a dose of 0.5 mg/kg PO BID with concomitant reduction of H3K27Ac in plasma and reduction of H3K18Ac in the tumor. These effects are approximately equivalent to those observed with 3 when dosed at 10 mg/kg BID PO (65% TGI). This represents a 20-fold improvement of in vivo activity for 17 relative to 3, with an improved off-target profile. This improvement in in vivo activity is mainly attributed to the improved potency, permeability, and solubility of 17 relative to 3 (Figure 5).
Figure 5.
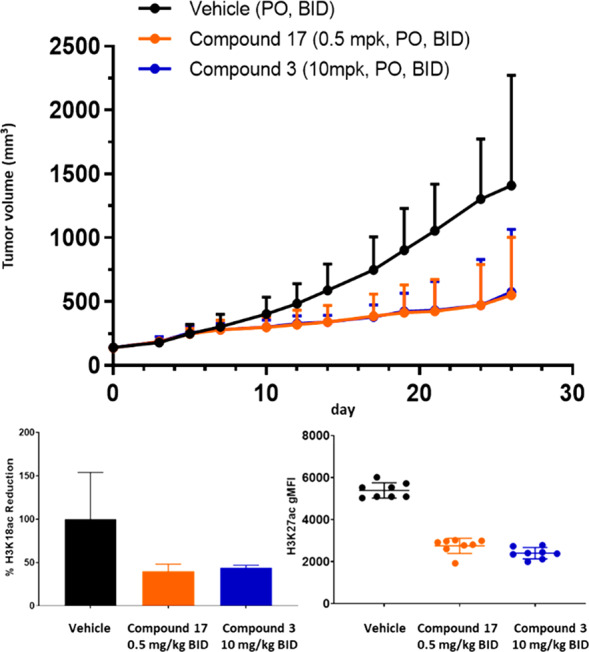
Antitumor activity (top), H3K18Ac reduction in tumor (bottom left), and H3K27Ac reduction in plasma (bottom right) of 17 (orange) and 3 (blue) in a JEKO-1 xenograft model.
In summary, we have detailed the optimization of lead indole compound 3 into the highly potent and selective aminopyridine EP300/CBP HAT inhibitor CPI-1612, 17. A wide variety of scaffold replacements for the indole were surveyed, resulting in the discovery of the aminopyridine core of 12. Further optimization led us to compound CPI-1612 which is potent across a range of biochemical and cellular assays that assess EP300/CBP HAT inhibition, has good pharmacokinetic profiles in mice and dogs, and is devoid of any significant off-target activity. Additionally, CPI-1612 suppresses H3K27 acetylation in vivo and is efficacious in a JEKO-1 mantle cell lymphoma xenograft at a low dose.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00155.
The preparation of compounds 3–17, experimental details for in vivo experiments and ADME experiments, details for X-ray crystallography of compounds 12 and 17, and PDB deposition validation reports (PDF)
Author Present Address
‡ F.B.: Foghorn Therapeutics, 100 Binney Street, Suite 610, Cambridge, MA, USA 02142.
Author Present Address
§ A.H.: Kymera Therapuetics, 300 Technology Square, Cambridge, MA, USA 02142.
Author Present Address
∥ R.J.S.: Third Rock Ventures, 29 Newbury St #301, Boston, MA, USA 02116.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dancy B. M.; Cole P. A. Protein Lysine Acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar N.; Kurdistani S. K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harbor Perspect. Med. 2017, 7, a026534. 10.1101/cshperspect.a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farria A.; Li W.; Dent S. Y. R. KATs in Cancer: Functions and Therapies. Oncogene 2015, 34, 4901–4913. 10.1038/onc.2014.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Varga J. The Transcriptional Coactivator and Acetyltransferase p300 in Fibroblast Biology and Fibrosis. J. Cell. Physiol. 2007, 213, 663–671. 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- Ghizzoni M.; Haisma H. J.; Maarsingh H.; Dekker F. J. Histone acetyltransferases are crucial regulators in NF-kB mediated inflammation. Drug Discovery Today 2011, 16, 504–511. 10.1016/j.drudis.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor L. M.; Viosca J.; Lopez-Atalaya J. P.; Barco A. Lysine Acetyltransferases CBP and p300 as Therapeutic Targets in Cognitive and Neurodegenerative Disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. 10.2174/13816128113199990382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S.-W.; Chen X.; Tracy T. E.; Li Y.; Zhou Y.; Wang C.; Shirakawa K.; Minami S. S.; Defensor E.; Mok S. A.; Sohn P. D. M.; Schilling B.; Cong X.; Ellerby L.; Gibson B. W.; Johnson J.; Krogan N.; Shamloo M.; Gestwicki J.; Masliah E.; Verdin E.; Gan L. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficit. Nat. Med. 2015, 21, 1154–1162. 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Naruse I.; Maekawa T.; Masuya H.; Shiroishi T.; Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: A partial similarity with Rubinstein–Taybi syndrome. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10215–10220. 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T. P.; Oh S. P.; Fuchs M.; Zhou N. D.; Ch’ng L. E.; Newsome D.; Bronson R. T.; Li E.; Livingston D. M.; Eckner R. Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator p300. Cell 1998, 93, 361–372. 10.1016/S0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Lasko L. M.; Jakob C. S.; Edalji R. P.; Qiu W.; Montgomery D.; Digiammarino E. L.; Hansen T. M.; Risi R. M.; Frey R.; Manaves V.; Shaw B.; Algire M.; Hessler P.; Lam L. T.; Uziel T.; Faivre E.; Ferguson D.; Buchanan F. G.; Martin R. L.; Torrent M.; Chiang G. G.; Karukurichi K.; Langston J. W.; Weinert B. T.; Choudhary C.; DeVries P.; VanDrie J. H.; McElligott D.; Kesicki E.; Marmorstein R.; Sun C.; Cole P. A.; Rosenberg S.; Michaelides M. R.; Lai A.; Bromberg K. D. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumors. Nature 2017, 550, 128–132. 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. E.; Huhn A.; Gardberg A. S.; Poy F.; Brucelle F.; Vivat V.; Patel G.; Patel C.; Cummings R.; Sims R.; Levell J.; Audia J. E.; Bommi-Reddy A.; Cantone N. Early Drug Discovery Efforts Towards the Identification of EP300/CBP Histone Acetyltransferase (HAT) Inhibitors. ChemMedChem 2020, 10.1002/cmdc.202000007. [DOI] [PubMed] [Google Scholar]
- Data for chlorine- and nitrogen-“walk” studies can be found in the associated Supporting Information document for ref (11).
- The EP300 HAT SPA enzymatic assay was run for 1 h, while the JEKO-1 cell viability assay was run for 72 h.
- The X-ray cocrystal structure of 1 clearly shows that the indole scaffold and the 3-carbonyl group are coplanar. The X-ray structure of 1 with an EP300 HAT construct has been deposited in the PDB (6v8b). See ref (11).
- In all cases we have examined, addition of a methyl group at the carbon atom adjacent to the aromatic group that is part of the phenethylamine side chain provides approximately a 2- to 3-fold improvement in potency in the EP300 HAT domain SPA assay. See the Supporting Information for additional data.
- See the Supporting Information for additional information.
- Wilson J. E.; Brucelle F.; Levell J. R.. Amino amides as P300/CBP HAT inhibitors and their preparation. Pat. Appl. WO2019161162.
- The data in the text are for single compound PK experiments with 17, while those in Table 2 are for experiments with a cassette of at least three compounds each. For comparison, the single compound mouse PK for compound 3 is as follows: 1 mg/kg IV; 10 mg/kg PO; clearance = 3.0 L/h/kg, Vss = 1.9 L/kg, T1/2 = 0.98 h, F% = 26; AUC/dose = 90 h·mg/mL.
- Compound 17 was administered orally to 18 male CD-1 mice. Plasma, brain, and CSF levels of compound 17 were then determined at multiple time points. See the Supporting Information for experimental details and additional data.
- Michaelides M. R.; Kluge A.; Patane M.; Van Drie J. H.; Wang C.; Hansen T. M.; Risi R. M.; Mantei R.; Hertel C.; Karukurichi K.; Nesterov A.; McElligott D.; de Vries P.; Langston J. W.; Cole P. A.; Marmorstein R.; Liu H.; Lasko L.; Bromberg K. D.; Lai A.; Kesicki E. A. Discovery of Spiro Oxazolidinediones as Selective, Orally Bioavailable Inhibitors of p300/CBP Histone Acetyltransferases. ACS Med. Chem. Lett. 2018, 9, 28–33. 10.1021/acsmedchemlett.7b00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The structure of A-485
is shown below.
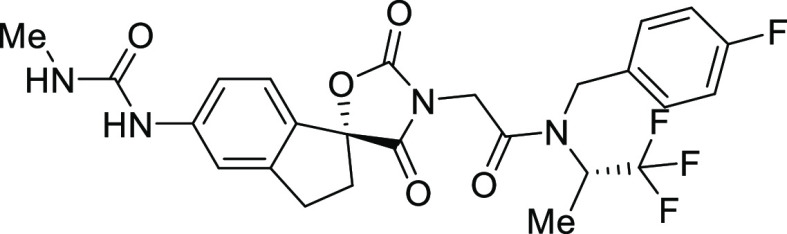

Table 2. Structure of Compound 17 and Profiles of Indole Lead 3 and Aminopyridine Analogues 12 and 17.

| compound | 3 | 12 | 17 |
|---|---|---|---|
| In Vitro Assaysa | |||
| EP300 HAT SPA IC50 (μM) | 0.040 | 0.019 | 0.0081 |
| full length EP300 SPA IC50 (μM) | 0.0025 | <0.0005 | <0.0005 |
| full length CBP SPA IC50 (μM) | 0.0032 | 0.0067 | 0.0029 |
| H3K18Ac MSD EC50 (μM) | 0.037 | 0.025 | 0.014 |
| JEKO-1 GI50 (μM) | 0.044 | 0.017 | <0.0079 |
| hERG (% inh. @ 10 μM) | 97 | n.a. | 9.2 |
| PAMPA pe (nm/s) | <0.066 | 13.8 | 11.9 |
| mouse PPB (%) | 99.1 | 98.3 | 99.1 |
| MLM Cl (μL/min/mg) | 46.6 | 215 | 178 |
| Mouse PKb | |||
| clearance (L/h/kg) | 3.49 | 2.30 | 1.88 |
| Vss obs | 1.92 | 0.88 | 0.99 |
| T1/2 (h) | 1.44 | 1.24 | 1.40 |
| F% | 43 | 88 | 47 |
All biochemical assays are the average of two or more runs. Parallel artificial membrane permeation assay (PAMPA), mouse plasma protein binding (PPB), and mouse liver microsome (MLM) assays are a single run.
Mouse PK data are derived from cassette PK experiments, using 0.5 mg/kg intravenous (IV) and 2.5 mg/kg per os (PO) doses.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.