

Abstract

Two novel compounds were identified as Naa50 binders/inhibitors using DNA-encoded technology screening. Biophysical and biochemical data as well as cocrystal structures were obtained for both compounds (3a and 4a) to understand their mechanism of action. These data were also used to rationalize the binding affinity differences observed between the two compounds and a MLGP peptide-containing substrate. Cellular target engagement experiments further confirm the Naa50 binding of 4a and demonstrate its selectivity toward related enzymes (Naa10 and Naa60). Additional analogs of inhibitor 4a were also evaluated to study the binding mode observed in the cocrystal structures.
Keywords: Naa50, N-α-acetyltransferase, acetyl-CoA, DEL
Protein acetylation has received significant attention in the past decade due to its important roles in regulating cell signaling. In particular, N-terminal (Nt) acetylation is known to be a highly abundant cotranslational protein modification that is controlled by a family of acetyltransferases (NAT).1 The Nt-acetylation of a protein can affect its nuclear import and export and can also act as a degradation signal to control the protein’s cellular stability.1 The eukaryotic protein N-terminal acetylation machinery is composed of six NAT protein complexes (NatA-NatF). Knockdown studies have shown that each complex associated with the NAT machinery (i.e., NatA-NatF) is important for cell proliferation and normal cell cycle progression.2 The Nα-terminal acetyltransferase (Naa50) enzyme is a member of the Nα-terminal acetyltransferase NAT protein family. It coexists with Naa10 and Naa15 in the NatE complex and is responsible for the enzymatic function of the complex. Naa50 is also found to be essential for normal sister chromatid cohesion and chromosome condensation.3−5 Therefore, an inhibitor of the Naa50 enzyme might have therapeutic applications in oncology indications.
Recently, a potent inhibitor of Naa50 was described in the literature (compound 1, Figure 1a).6 This molecule combines the acetyl-CoA (AcCoA) cofactor that is known to associate with the enzyme and a tetra-peptide derived from the N-terminal protein sequence common to optimal Naa50 substrates (MLGP).7,8 Construction of inhibitor 1 was also influenced by studies of the Naa50 biochemical mechanism which indicated formation of a ternary complex between the AcCoA cofactor, an appropriate protein substrate, and the enzyme.9 Although compound 1 is a potent Naa50 inhibitor, the molecule is not particularly efficient due to its large molecular weight (ligand efficiency (LE)10 = 0.13). In addition, its high molecular weight (MW = 1223) and high polarity (TPSA = 577 and clogP = −4.1) likely prevents facile permeability across cell membranes and may thus compromise the use of the molecule as a robust in vitro tool compound. In this report, we describe our initial efforts to identify a potent and selective Naa50 inhibitor with improved physiochemical properties relative to compound 1 (i.e., reduced molecular weight and TPSA; increased logD). As part of these studies, we were also interested in finding a more ligand efficient inhibitor that could serve as an appropriate starting point for future medicinal chemistry optimizations.
Figure 1.

(a) Chemical structures of AcCoA, CoA, literature reported bisubstrate inhibitor (1), and its truncated analog: des-A (2). (b) Cocrystal structure of 1 bound to Naa50 (2.3 Å, PDB ID 6WF3). Compound 1 is drawn in orange stick representation and spans the binding pocket, from the cofactor binding site on the left to the peptide binding site on the right. Residues that form hydrogen bonds with the ligand (dashed lines) are drawn as green sticks. Hydrogen bonds are drawn as dashed lines. Protein Cα trace is shown in gray.
We first explored whether the pyrophosphate adenosine moiety present in compound 1 was required for potent Naa50 inhibition. As shown in Figure 1a, a truncated analog of 1 that lacked this functionality displayed significantly weaker anti-Naa50 activity (compound 2). Separate cocrystal structures obtained with compounds 1 and 2 in complex with Naa50 demonstrated that, in spite of its weaker potency, the latter molecule occupied the same region of the Naa50 active site as the MLGP portion of 1 along with part of the cofactor binding site (Figures 1b and S1, more details regarding the specific interactions formed between compounds 1 and 2 and the Naa50 protein are provided below). This observation afforded confidence that molecules which lacked a pyrophosphate adenosine moiety could appropriately recognize the Naa50 active site and further encouraged our search for new inhibitors that improved upon compounds 1 and 2.
As one approach to the identification of new Naa50 inhibitors, we screened a 2.2 billion-member DNA encoded library (DEL) to find novel ligands with potent affinity to the protein.11,12 Affinity selection was performed using the library with a selectively polyhistidine-tagged human Naa50 protein construct (250 pmol enzyme concentration) in solution incubation for 60 min. The polyhistidine-tagged Naa50 protein was then immobilized to magnetic beads and was washed extensively to remove nonbinding molecules. Bound library members were subsequently eluted from the beads by heat denaturation of the protein at 75 °C. The eluent was then incubated with fresh protein in solution to initiate a new round of affinity selection. A parallel selection against buffer lacking the target protein was also performed to allow for the identification of molecules that were enriched via nontarget affinity mechanisms. After three rounds of such selection, the DNA tags of the eluted molecules were PCR amplified and sequenced, and the sequences were translated to identify the chemical structures of putative Naa50 binders. Importantly, we also screened the library against Naa50 in the presence of either the AcCoA cofactor or inhibitor 1. Inclusion of AcCoA was envisioned to bias screening results toward molecules which mimicked the MLGP tetrapeptide substrate. In contrast, screening in the presence of 1 was anticipated to help identify compounds which bound to Naa50 outside of the catalytic site. To further increase confidence that the identified hits were genuine Naa50 binders, we repeated the above selections using a 5-fold lower enzyme concentration (50 pmol). It was expected that this alternative protein concentration would help us identify the most potent binders to the Naa50 enzyme.13,14
As shown in Figure 2, the described affinity selections afforded a large number of enrichments associated with one specific portion of the 2.2 B-member DEL. This sublibrary (DEL951) was constructed in 3 synthesis cycles and involved derivatization of diamine-containing core scaffolds using a variety of carboxylic acid, aldehyde, sulfonyl chloride, and isocyanate-containing building blocks (Figure 3a, see Supporting Information for more details). The affinity selection results obtained in the presence of the AcCoA cofactor indicated that three R1 building blocks produced multiple library members that were significantly enriched via the selection process (4 vertical rows highlighted in Figure 2, structures 2a–2c). The observed enrichments associated with these R1 moieties were dramatically attenuated when the affinity selections were conducted in the absence of AcCoA (Supporting Information, Figure S2). This outcome suggested that the library members derived from the 2a–2c R1 building blocks cooperatively bound to Naa50 with AcCoA in the vicinity of the cofactor binding site and might possibly mimic the MLGP tetra-peptide substrate. Further support for the hypothesis that the compounds bound to the MLGP site was obtained from affinity selections performed with DEL951 in the presence of compound 1. As shown in Figure S2, these assessments generated no significant enrichments associated with the DEL951 library members consistent with the blocking of the MLGP binding site by 1 during the enrichment process.
Figure 2.
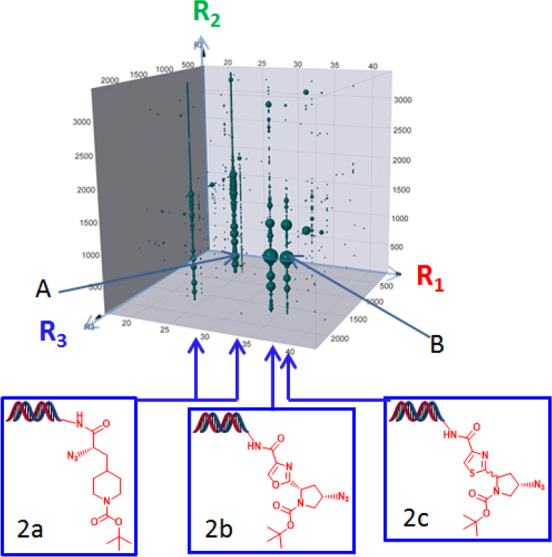
Cube view of DEL951 analysis. Points represent enrichment of library member after 3 rounds of affinity selection using biotinylated Naa50 protein (larger size = greater enrichment). Affinity selection was performed using 250 pmol Naa50 in the presence of the AcCoA cofactor. The cube axes depict the R1, R2, and R3 building blocks employed during construction of the library. R1 building block structures which afforded enrichment of multiple library members are also shown (2a–2c). Note that the 2a building block afforded two vertical rows of enriched library members.
Figure 3.

(a) General construction of DEL951 and (b) specific library members (A and B) initially selected for off-DNA synthesis.
Encouraged by these findings, we initially selected two structurally diverse library members for off-DNA synthesis to confirm affinity of their non-nucleic acid portions with the Naa50 protein (structures A and B, Figure 3b). Both of these members afforded high copy number outcomes from the affinity-selection process using either high (250 pmol) or low (50 pmol) Naa50 concentrations (Figure 3b). The 50 pmol results provided us with increased confidence that the molecules were genuine and relatively potent Naa50 binders. Given the structural similarity between R1 building blocks 2b and 2c (oxazole and thiazole moieties) and the corresponding similar affinity selection data, we chose to only conduct the off-DNA synthesis of the thiazole-containing compound during this initial exploration phase. The off-DNA syntheses of library members A and B and the corresponding diastereomers from Figure 3 are described in Schemes S2–S5 (Supporting Information).
As shown in Figure 4, the synthesized compound (compound 3, Supporting Information) corresponding to DEL library member A increased the thermal stability of the Naa50 protein in the presence of AcCoA using a protein thermal-shift assessment.15 This result provided evidence of direct-target engagement for the compound (Figure S3). As predicted from the affinity selection outcomes performed with DEL951, the thermal shift outcomes were attenuated when the assay was performed in the absence of the AcCoA (Figure 4). This observation suggested that the compound did not bind to the Naa50 AcCoA pocket and instead bound cooperatively with the cofactor. Biochemical testing further demonstrated the functional activity of 3 (IC50 = 2.7 μM). Therefore, both diastereomers of the compound were synthesized (3a and 3b; 3a:Scheme S2, Supporting Information; 3b, Supporting Information) and were further evaluated in surface plasmon resonance (SPR, Table 1, Figure S6) and biochemical assessments (Figure 4). SPR methodology subsequently confirmed specific association of 3a with the Naa50 protein (Kd = 1.0 μM) in the presence of AcCoA, and the corresponding Naa50 biochemical inhibition activity closely matched the SPR binding result (BC IC50 = 2.0 μM, Figure 4). In addition, a cellular thermal shift assay (CETSA)16 conducted using A549 cell lysates demonstrated that compound 3a could stabilize the Naa50 protein against thermal denaturation without impacting the thermal stabilities of the related Naa10 and Naa60 enzymes (Figure S4a). Collectively, these data demonstrated that 3a was a genuine, selective, and relatively potent Naa50 inhibitor. Similar profiling of the 3b diastereomer demonstrated that it inhibited Naa50 in a less potent manner relative to 3a (Kd = 10 μM; BC IC50 = 12 μM, Figure 4). Because of this weaker activity, additional profiling of 3b was not performed.
Figure 4.

Resynthesis hits from DEL951 displayed binding affinity and functional activities.
Table 1. Kinetics of Compound Binding to Naa50_apo and Naa50·AcCoA by SPRa.
| Protein | Compound | kon (M–1·s–1) | koff (s–1) | t1/2 (min) | SPR KD (nM) |
|---|---|---|---|---|---|
| Naa50_apo | AcCoA | 1.8 × 105 | 3.1 × 10–3 | 4 | 17.6 ± 2.8 |
| Naa50_apo | CoA | 2.0 × 105 | 3.2 × 10–2 | 0.4 | 156 ± 27 |
| Naa50_apo | 4a | 4.26 × 104 | 4.5 × 10–3 | 2.6 | 105 ± 4.8 |
| Naa50·AcCoA | 4a | 5.5 × 104 | 1.4 × 10–3 | 8.4 | 26.7 ± 7.3 |
| Naa50·AcCoA | 4b | 3.6 × 103 | 1.7 × 10–3 | 8.6 | 470 ± 25 |
| Naa50·AcCoA | 3a | 4.3 × 103 | 6.0 × 10–3 | 3.1 | 1010 ± 963 |
| Naa50·AcCoA | 3b | ND | ND | ND | >10,000 |
Measured at 10 °C in 25 mM HEPES, 150 mM NaCl, 0.5 mM TCEP, 5% glycerol, 0.02% Tween 20, 2% DMSO, pH 7.5.
Since the thiazole-containing library members were prepared and screened as 1:1 diastereomeric mixtures (see Supporting Information), we synthesized a related mixture (4, Supporting Information) as well as both single diastereomers (compounds 4a (Schemes S4 and S5) and 4b (Scheme S3)) corresponding to DEL library member B for off-DNA characterization. As shown in Figure 4, mixture 4 displayed significant thermal stabilization of Naa50 in the thermal shift assay that was stronger than that exhibited by 3 and which was similarly weakened in the absence of AcCoA (Figure S3). The biochemical Naa50 inhibition activity determined for 4 was also correspondingly more potent than that associated with 3 (Figure 4). Both single diastereomers 4a and 4b displayed measurable Naa50 binding affinity and biochemical inhibitory activity with 4a having the better binding affinity and better biochemical potency relative to both 4b and 3a (Figure 4 and Table 1). Compound 4a was also a considerably stronger binder than 3a in CETSA testing (Figure S4a, compare results obtained at 55 and 61 °C for 3a and 4a, respectively, relative to DMSO control at 52 °C). Compound 4a maintained good selectivity for Naa50 over Naa10 and Naa60 just like compound 3a.
To demonstrate the ability of 4a to engage Naa50 in a complex proteome we performed competitive chemoproteomic profiling experiments using Lys-CoA Sepharose.17 This affinity reagent captures Naa50 through a bidentate interaction involving the substrate-binding site,18 which should be completed in the presence of 4a. Consistent with our in vitro inhibition studies, preincubation of cell lysates with 4a led to potent inhibition of Naa50 capture by Lys-CoA Sepharose, with substantial competition observed at concentrations as low as 50 nM (Figure S4b). This closely resembles the binding constant of 4a for recombinant holo (acetyl-CoA bound) Naa50 determined by SPR. These results provide additional evidence that 4a can engage the Naa50 active site even in cellular contexts where it may exist in multiprotein complexes and demonstrates the utility of chemoproteomic capture probes to provide insight into the biochemical mechanism of acetyltransferase inhibitors in complex proteomes.
We obtained cocrystal structures of compounds 3a and 4a with the CoA-bound Naa50 to further understand their binding modes. CoA was utilized in these experiments to facilitate crystallization. As demonstrated later in this work, the orientation of the bound ligand did not vary significantly when a representative compound (4a) was cocrystallized in the presence of either the CoA or the cofactor, AcCoA. As shown in Figure 5a and 5b, both compounds (3a and 4a) bound to the region of the Naa50 active site adjacent to the CoA-binding pocket. Both 3a and 4a formed H-bond interactions with the side chains of the Tyr31 and Tyr139 residues. They also formed similar common interactions with the backbone carbonyl moieties of Met75 and His112 as well as the backbone NH of Gln114 (Figures 5a and 5b, residues depicted in green sticks). Other H-bond interactions unique to compounds 3a and 4a include the backbone NH moieties of Arg62 (compound 3a) and of Arg141 and Lys140 (compound 4a) (Figures 5a and 5b, residues depicted in orange sticks). The methyl amide moiety, R1 thiazole moiety and the R2 carboxyl amide carbonyl moiety of 4a do not form hydrogen bonds with the protein. The R2 phenyl moiety forms hydrophobic interactions with a hydrophobic pocket created by three tyrosine residues (Y138, Y139, and V29). The R2 tetrazole substituent of 4a forms an H-bond with the backbone NHs of Arg141 and Lys140. It may also form a charge–charge interaction with the side chain of Arg141. However, the precise nature of this association is uncertain as the side chain of Arg141 is disordered in the crystal structures.
Figure 5.

Crystal structures of the complex of Naa50 with CoA and 3a (2.16 Å, PDB ID 6WFG) (a) or 4a (1.87 Å, PDB ID 6WFK) (b). CoA is drawn in yellow stick representation, 3a in blue stick representation, and 4a in magenta stick representation. Residues that form hydrogen bond interactions (dashed lines) that are common to both 3a and 4a are drawn as green sticks, while those that are unique to each ligand are drawn as orange sticks.
To further investigate the binding mechanism, we performed in-depth kinetic analyses of the interactions between 4a and Naa50 in apo as well as AcCoA and CoA complexed forms using SPR (Tables 1, Figure S6). These kinetic measurements suggested that 4a bound to the Naa50·AcCoA complex with ∼4-fold greater affinity than the apo form of enzyme. Notably, the gain in affinity for the Naa50·AcCoA complex was associated with a decrease in the dissociation rate (koff) or increase in the dissociative half-life (t1/2) of the complex (Table 1). To this end, significant evidence exists linking longer t1/2 to binding-induced conformational changes leading to greater steric and electronic complementarity in protein–ligand interactions.19 Interestingly, previous NMR-based biophysical studies determined that a peptide substrate (MLGP-RRR) displayed very weak affinity for the Naa50 protein. Our own SPR assessments confirmed this weak binding affinity (Kd > 100 μM). These results suggest that the 4a inhibitor recognizes a pre-existing Naa50 binding pocket more efficiently than the peptide substrate.
We also obtained a cocrystal structure of Naa50 bound to AcCoA and compound 4a (Figure 6). This structure enabled investigation of the differences in 4a binding modes in the presence of either CoA or AcCoA. In the Naa50·AcCoA crystal structure, the acetyl group of AcCoA formed two hydrogen bonds with Naa50 including one to the backbone NH of Gln114 and one to the side chain of Asn117 (Figure S7). In contrast, in the Naa50·AcCoA·4a complex, 4a formed a hydrogen bond with the NH of Gln114 instead of AcCoA. The acetyl group of AcCoA repositioned to make only a long (3.1 Å) hydrogen bond with the backbone NH of Leu77 of Naa50. Interestingly, this rearrangement was not observed in cocrystal structures obtained with CoA and Naa50 in the presence and absence of 4a. Consistent with the similar Kd values observed for 4a·Naa50 binding with either CoA or AcCoA, the bound inhibitor conformation and protein·ligand interactions were identical in the 4a·CoA·Naa50 and 4a·AcCoA·Naa50 crystal structures. These observations suggested that conclusions drawn via analysis of Naa50·CoA·inhibitor complexes (e.g., Figures 5a and 5b) could also be applied to Naa50·AcCoA·inhibitor structure.
Figure 6.

Crystal structure of the complex of 4a with Naa50 in the presence of AcCoA (1.07 Å, PDB ID 6WFN). View from the top of the binding pocket. AcCoA is drawn in pale blue stick representation while 4a is in magenta stick representation. The binding mode and hydrogen bonds (dashed lines) between 4a and Naa50 are identical to those observed in the presence of CoA.
Since compound 4a displayed significantly improved Naa50 binding and inhibition activity compared to 3a (with an improved LE value), we chose to further explore other hits related to 4a from the on-DNA affinity selections as part of our follow-up activities. Accordingly, we expanded the structure–activity relationships associated with 4a by synthesizing several closely related DEL951 members in off-DNA form. As shown in Table 2, these molecules all afforded robust copy numbers at both 250 pmol and 50 pmol Naa50 protein concentrations. The syntheses of compounds 5–8 in Table 2 are described in Schemes S5–S7 (Supporting Information). Replacement of the thiazole moiety in 4a with an oxazole provided a nearly equipotent Naa50 inhibitor (compound 5). This result was consistent with the prior analysis of the DEL951 affinity selection data which suggested that thiazole and oxazole-containing R1 building blocks produced library members with similar Naa50 affinities (2b and 2c in Figure 2). Removal of the R2 substituent from 4a drastically weakened Naa50 biochemical inhibition activity (6, Table 2). Deletion of the R2 amide carbonyl group (7) or repositioning of the tetrazole present in the R2 substituent (8) also weakened anti-Naa50 potency but to a lesser extent than was observed for compound 6 (compound 6 is a truncated analog, not actually present in the original DEL). Interestingly, the biochemical potencies displayed by compounds 4a and 5–8 loosely paralleled the corresponding copy numbers (compounds with the highest copy numbers at low protein concentrations displayed the most potent Naa50 biochemical inhibition). This observation suggested that meaningful SAR information could be extracted from the affinity selection data generated for DEL951 prior to off-DNA synthesis.
Table 2. Biochemical and TPSA Data of Analogs from DEL 951.


In order to investigate whether the cocrystal structure of 4a could be used for further compound optimization, several analogs of 4a were subsequently designed based on the compound’s observed Naa50 interactions and binding mode. The syntheses of compounds 9–14 in Table 3 are described in Schemes S8–S9 (Supporting Information). As shown in Table 3, replacement of the 4a carboxyl amide thiazole substituent with a methyl group afforded similar anti-Naa50 biochemical activity (Table 3, compound 9; compare to 4a). In contrast, replacement of the tetrazole moiety of 4a with hydrogen (10), triazole (11), N-linked triazole (12), and N-linked tetrazole (13) all resulted in significant (>100-fold) loss of biochemical potency (Table 3). Interestingly, the only compound from this subseries which retained reasonable biochemical potency relative to 4a contained a carboxylic acid moiety in the place of the tetrazole (14, Table 3). These results suggested the importance of maintaining a negative charge on the inhibitor even if no specific charge–charge interaction is observed in the 4a·Naa50 cocrystal structure.
Table 3. Biochemical and TPSA Data of Analogs of 4a.

The cocrystal structure of 4b (the less active diastereomer of 4a) in complex with Naa50 was also solved. As shown in Figure S10, 4b formed the same H-bonds with the protein residues as was observed in the 4a·Naa50 cocrystal structure (Figure 5b). These interactions include the tetrazole moiety of 4b forming hydrogen bonds with the backbone NH of Arg141 (as shown in Figure 5b). These observations reinforce the importance of maintaining a negatively charged ligand fragment in close proximity to Arg141 and Lys140. Differences between the 4a and 4b bound conformations occurred in areas where both ligands did not form H-bonds with the protein (e.g., the thiazole methyl amide and the carboxylamide moiety connected to the phenyltetrazole). The bound conformation of 4b exhibited conformational strain in the pendant methyl amide (about 5 kcal/mol calculated at the B3LYP-D3/DFT 6-311g**+ level in the gas phase20). This strain was needed to avoid unfavorable contacts between the ligand and the protein. It seems likely that strain could be partly responsible for the loss of SPR and biochemical potency relative to 4a (compare 4a and 4b in Figure 4).
As shown in Figure S5, compounds 3a and 4a occupied the same region of the Naa50 binding site as the tetrapeptide (MLGP) moiety present in 2. These binding modes were consistent with biochemical experiments which suggested that 3a and 4a bound uncompetitively to Naa50 with respect to the AcCoA cofactor and competitively to the MLGP-containing peptide substrate analog (Figure S8 and Figure S9). In addition to making similar hydrogen bonding interactions with Naa50 observed for the MLGP tetrapeptide, 3a and 4a also formed additional hydrophobic interactions with protein. Therefore, stronger binding affinities were observed for both molecules relative to the MLGP tetrapeptide. Collectively, these results suggest a biochemical inhibition mechanism for compounds 3a and 4a in which AcCoA binding enhances the compound’s binding affinity. Detailed comparisons among the Naa50 binding interactions observed for 3a and 4a, and the MLGP peptide are described in the Supporting Information (Figures S11 and Figure S12).
In conclusion, we utilized DEL technology to identify potent and selective Naa50 inhibitors (compounds 3a and 4a) that bound to the substrate binding pocket. Specifically, compound 4a displayed potent biochemical inhibitory potency (7 nM vs 30 nM), reduced TPSA (224 vs 557), improved LE (0.29 vs 0.13), and increased clogP (0.6 vs −4) relative to compound 1. Further optimization using cocrystal structure information and structure-based design strategy may provide a cell permeable tool compound that could be used to probe the effects of inhibiting Naa50 in cancer cells.
Acknowledgments
The authors thank Simon Bergqvist, Mike Greig, Ben Bolanos, and Simon Planken for their efforts in the early phase of this project. We also thank Sabrina Li for repeating the CETSA experiments and Wei Wang for obtaining the 1H and 13C NMR spectra and HRMS data for compounds 3, 3a, 3b, and 14. We are grateful to Martin Edwards and Martin Wythes for their support and review of the manuscript.
Glossary
Abbreviations
- NAT
N-terminus acetyltransferase
- MLGP
methionine-leucine-glycine-proline
- CoA
coenzyme A
- DEL
DNA-encoded library
- PCR
polymerase chain reaction
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00029.
Details of protein purification, crystallization conditions, compound synthesis, compound characterization, and biochemical and biophysical experiments (PDF)
Accession Codes
PDB numbers: compound 1, 6WF3; compound 2, 6WF5; compound 3a+CoA, 6WFG; compound 4a+CoA, 6WFK; compound 4a+AcCoA, 6WFN; compound 4b+AcCoA, 6WFO.
Author Present Address
⊥ For William K. Sonnenburg: Nurix Therapeutics, 1700 Owens Street, Suite 205, San Francisco, CA 94158, United States.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Aksnes H.; Drazic A.; Marie M.; Arnesen T. First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem. Sci. 2016, 41, 746–760. 10.1016/j.tibs.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Kalvik T. V.; Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene 2013, 32, 269–76. 10.1038/onc.2012.82. [DOI] [PubMed] [Google Scholar]
- Hou F.; Chu C. W.; Kong X.; Yokomori K.; Zou H. The acetyltransferase activity of San stabilizes the mitotic cohesin at the centromeres in a shugoshin-independent manner. J. Cell Biol. 2007, 177 (4), 587–97. 10.1083/jcb.200701043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. C.; Garrett-Engele C. M.; Li Z.; Williams E. V.; Rosenman E. D.; Goldberg M. L. Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr. Biol. 2003, 13, 2025–36. 10.1016/j.cub.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Pimenta-Marques A.; Tostoes R.; Marty T.; Barbosa V.; Lehmann R.; Martinho R. G. Differential requirements of a mitotic acetyltransferase in somatic and germ line cells. Dev. Biol. 2008, 323, 197–206. 10.1016/j.ydbio.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyn H.; Jones J. E.; Lewallen D.; Narawane R.; Varhaug J. E.; Thompson P. R.; Arnesen T. Design, synthesis, and kinetic characterization of protein N-terminal acetyltransferase inhibitors. ACS Chem. Biol. 2013, 8, 1121–7. 10.1021/cb400136s. [DOI] [PubMed] [Google Scholar]
- Evjenth R.; Hole K.; Karlsen O. A.; Ziegler M.; Arnesen T.; Lillehaug J. R. Human Naa50p (Nat5/San) displays both protein Nα- and Nϵ-acetyltransferase activity. J. Biol. Chem. 2009, 284, 31122–31129. 10.1074/jbc.M109.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P.; Evjenth R.; Foyn H.; Demeyer K.; De Bock P. J.; Lillehaug J. R.; Vandekerckhove J.; Arnesen T.; Gevaert K.. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(alpha)-acetyltransferases and point to hNaa10p as the post-translational actin N(alpha)-acetyltransferase. Mol. Cell. Proteomics 2011, 10, M110.004580. 10.1074/mcp.M110.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evjenth R. H.; Brenner A. K.; Thompson P. R.; Arnesen T.; Froystein N. A.; Lillehaug J. R. Human protein N-terminal acetyltransferase hNaa50p (hNAT5/hSAN) follows ordered sequential catalytic mechanism. J. Biol. Chem. 2012, 287, 10081–10088. 10.1074/jbc.M111.326587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C. H.; Tounge B. A.; Bembenek S. D. Ligand binding efficiency: trends, physical basis, and implications. J. Med. Chem. 2008, 51, 2432–2438. 10.1021/jm701255b. [DOI] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Ding Y.; Franklin G. J.; Franzen K. D.; Gefter M. L.; Hale S. P.; Hansen N. J. V.; Israel D. I.; Jiang J.; Kavarana M. J.; Kelley M. S.; Kollmann C. S.; Li F.; Lind K.; Mataruse S.; Medeiros P. F.; Messer J. A.; Myers P.; O’Keefe H.; Oliff M. C.; Rise C. E.; Satz A. L.; Skinner S. R.; Svendsen J. L.; Tang L.; van Vloten K.; Wagner R. W.; Yao G.; Zhao B.; Morgan B. A. Design, synthesis and selection of DNA-encoded small -molecule libraries. Nat. Chem. Biol. 2009, 5, 647–654. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Satz A. L. DNA encoded library selections and insights provided by computational simulations. ACS Chem. Biol. 2015, 10, 2237–2245. 10.1021/acschembio.5b00378. [DOI] [PubMed] [Google Scholar]
- Satz A. L. What do you get from DNA-encoded libraries?. ACS Med. Chem. Lett. 2018, 9, 408–410. 10.1021/acsmedchemlett.8b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y.; Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Montgomery D. C.; Garlick J. M.; Kulkarni R. A.; Kennedy S.; Allali-Hassani A.; Kuo Y. M.; Andrews A. J.; Wu H.; Vedadi M.; Meier J. L. Global Profiling of Acetyltransferase Feedback Regulation. J. Am. Chem. Soc. 2016, 138 (20), 6388–91. 10.1021/jacs.6b03036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S.; Magin R. S.; Wei X.; Pan B.; Petersson E. J.; Marmorstein R. Structure and Mechanism of Acetylation by the N-Terminal Dual Enzyme NatA/Naa50 Complex. Structure 2019, 27 (7), 1057–1070. e4 10.1016/j.str.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15, 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Sellers B. D.; James N. C.; Gobbi A. A Comparison of Quantum and Molecular Mechanical Methods to Estimate Strain Energy in Druglike Fragments. J. Chem. Inf. Model. 2017, 57 (6), 1265–1275. 10.1021/acs.jcim.6b00614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.