

Abstract
The 3-dimensional (3D) structure of therapeutics and other bioactive molecules is an important factor in determining the strength and selectivity of their protein–ligand interactions. Previous efforts have considered the strain introduced and tolerated through conformational changes induced upon protein binding. Herein, we present an analysis of 3-dimentionality for energy-minimized structures from the DrugBank and ligands bound to proteins identified in the Protein Data Bank (PDB). This analysis reveals that the majority of molecules found in both the DrugBank and the PDB tend toward linearity and planarity, with few molecules having highly 3D conformations. Decidedly 3D geometries have been historically difficult to achieve, likely due to the synthetic challenge of making 3D organic molecules, and other considerations, such as adherence to the ‘rule-of-five’. This has resulted in the dominance of planar and/or linear topologies of the molecules described here. Strategies to address the generally flat nature of these data sets are explored, including the use of 3D organic fragments and inorganic scaffolds as a means of accessing privileged 3D space. This work highlights the potential utility of libraries with greater 3D topological diversity so that the importance of molecular shape to biological behavior can be more fully understood in drug discovery campaigns.
Keywords: Drug discovery, conformations, protein−ligand interactions, 3-dimensionality
The recognition of novel therapeutics by their targets is dictated by ligand–protein interactions. Noncovalent interactions, including hydrogen and halogen bonding, salt bridges, and pi–pi stacking, are typically explored through the development of structure–activity relationships (SAR) to produce potent and selective drugs.1−3 In addition to many protein–ligand interactions that can be optimized, the conformation of inhibitors in the solvated vs bound states can also contribute to the energetic favorability of inhibitor binding.4,5 The diversity of druggable protein targets necessitates structural and conformational variability in ligands to generate effective pharmaceuticals.6 The amount of molecular strain and associated energy costs tolerated by drugs upon protein binding has been explored, where it was found that molecules binding to proteins could readily incur 5–9 kcal/mol of strain energy.5 While extensive rearrangements are possible, increased conformational shape diversity has been related to broad biological activity7 and successful clinical outcomes.8 The accessibility and large number of structures in the Protein Data Bank (PDB) has enabled the modeling and docking of 3-dimensional (3D) structures into protein active sites.9−12 These observations and tools have motivated the preparation of libraries with greater topological diversity for drug-discovery campaigns with the aim of improved lead identification and therapeutic success.13−15
The 3D shape of a molecule can be described by the number of sp3 carbons,14 the plane of best fit calculation,16 or normalized principal moment of inertia (PMI) values,7 among other parameters.9,17 These descriptors have facilitated the topological analysis of large fragment18 and high-throughput screening (HTS) libraries;11 to the best of our knowledge, only limited analyses of approved and late-stage drugs have been undertaken.10,12,19 The normalization that is common practice for PMI analysis allows for the comparison of compounds with diverse structural features and molecular weights (MWs) and is a straightforward calculation that can be readily carried out by many commercial software packages. The PMI parameter was demonstrated to be a robust method for evaluating 3D topology when compared to the other methods.16
Using PMI analyses, this work aims to evaluate the 3D conformations of structures reported in the DrugBank, including approved and experimental therapeutics. Of particular interest is the relationship between the energy minimized/optimized conformations of approved therapeutics and the conformation of those molecules when bound to proteins in the PDB. Previous efforts have focused on the exploration of flexible conformations upon protein binding as they relate to in silico screening and predicting binding conformations rather than the inherent change in 3D topology as described herein.5,6,20 Lastly, we consider the role that inorganic chemistry could play in increasing the prevalence of more 3D molecules in the drug discovery pipeline.
3D Diversity in the DrugBank
The DrugBank is an online cheminformatics and bioinformatics database that provides extensive information on both drugs and drug targets.21 The DrugBank repository contains over 13,000 chemical entities, with over 4000 unique targets. Of the 13,000+ chemical entities, 8696 can be downloaded as 3D structures (version 5.1.4), with 8532 having MW > 100 Da. These structures include approved, experimental, investigational, illicit, and withdrawn drugs, as well as nutraceuticals. Molecular Operating Environment (MOE) is a drug discovery software platform that can be used for the curation and evaluation of these structures. MOE was used to determine the moments of inertia for each compound (I1 ≤ I2 ≤ I3), which describe the torque needed for a given angular acceleration. These values were used to determine the normalized PMI values, I1/I3 and I2/I3, for structures with MW > 100 Da. The most common presentation of these data is graphically (Figure 1a), where the [0 1] apex is a linear molecule (dimethylbutadiyne), the [0.5 0.5] corner is a planar molecule (benzene), and the [1 1] apex is a spherical structure (adamantane). Where a conformation falls within these parameters provides an indication of its topology, with molecules having larger scores (I1/I3+ I2/I3) being considered more 3D in character.
Figure 1.
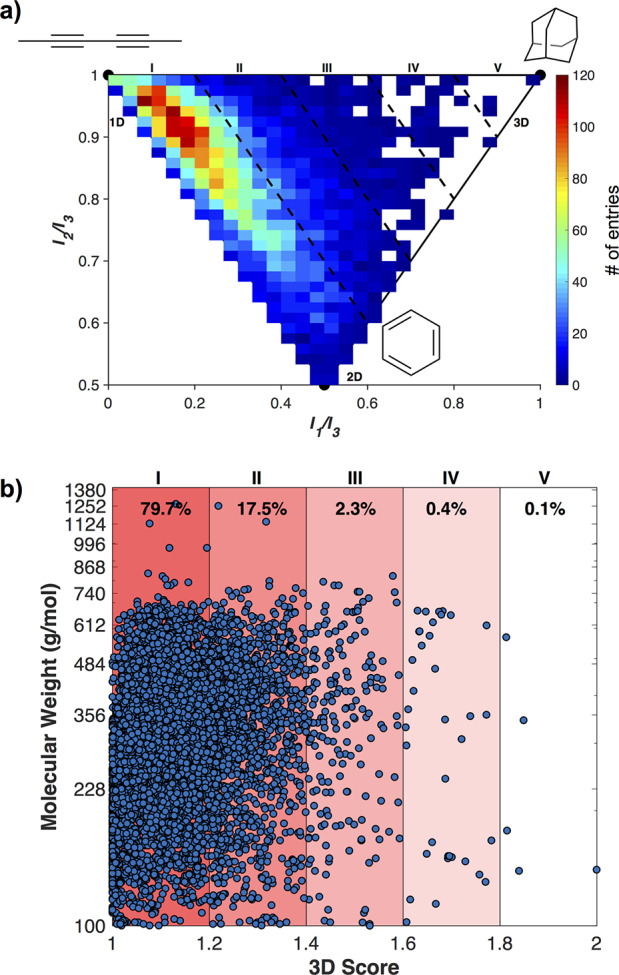
PMI analysis of 8532 entries from the DrugBank, plotted (a) on a typical PMI triangle plot and (b) as 3D Score (I1/I3 + I2/I3) vs MW.
The DrugBank structures, when analyzed by normalized PMIs, are found to have largely linear and planar topologies, with just under 80% having a 3D score < 1.2 (Figure 1). Structures having 3D scores > 1.6 (Figure 1b, IV and V) can be considered “highly” 3D, and only 0.5% of the DrugBank falls in these regions. This is in agreement with a previous report that found molecules in the ChEMBL Database (a database of bioactive compounds with druglike properties) tend toward planarity.22 The topological distribution of the DrugBank structures is also typical of organic libraries used in drug development, such as the ZINC database of virtual fragments, where ∼75% of the fragments have a 3D score of <1.2.18 This is not to imply that contemporary drug discovery does not include concerted design and synthetic efforts at achieving the best 3D molecular shape for a target, only that when libraries of currently and previously used compounds and fragments are retroactively analyzed they are often observed to lack decidedly 3D topologies. The traditional chemical feedstocks used in drug discovery campaigns are dominated by sp2-rich molecules and retrosynthetic analyses of large drug-like libraries have confirmed this, demonstrating the bias toward using planar building blocks in medicinal chemistry.22 Though it has been demonstrated that relatively planar scaffolds may be combined in a way that provides access to a reasonable sampling of chemical and topological space, it has also been shown that constraints in synthetic accessibility limit a more unbiased and diverse sampling of topological space.18,22 Additionally, adherence to the rule of five23 (Ro5) in drug design, which helps select for oral bioavailability, may predispose molecules to flatter geometries, for example, by limiting the number of rotatable bonds.
The 3D scores of the DrugBank structures were evaluated with respect to their MWs, ranging from 100 to 1268 Da. There is no obvious relationship between the MW of a compound and its 3D shape (Figure 1b). Interestingly, in each 3D score range the MWs are broadly distributed between 100 and ∼700 Da. The molecules with the greatest 3D scores (IV) range from 140 to 568 Da. This demonstrates that what might perhaps be the more intuitive notion, that larger molecules have greater 3-dimensionality, is not the case, as has been discussed recently.24
3D Diversity in the PDB
The PDB is the premier repository for biological macromolecular structures, containing over 150,000 total entries and over 11,000 depositions in 2019 alone.25 Within the PDB, ligands bound to protein structures having DrugBank IDs have been identified, and their corresponding ligand coordinates can be downloaded. Limiting these structures to compounds with a MW > 100 Da yields 7411 structures that comprise ∼500 unique chemical entities. The 3D analysis of all the protein-bound structures generates a PMI plot whose general features are similar to those of the DrugBank molecules, with the highest concentration of compounds in the upper left corner (Figure S2). However, this data set is biased by molecules that appear with greater frequency. For example, sapropterin, a dietary supplement administered to reduce blood phenylalanine levels appears in this list 1036 times. To better illustrate the conformational diversity of the PDB, the PMI values for each duplicate molecular entry were averaged over all conformations to yield 502 unique PMI values and 3D scores (Figure 2). As was the case for the DrugBank and individual PBD entries, the averaged PMI values from the different PDB conformations localize primarily in the upper left linear region. Despite the large differences in protein targets and MW values of the structures analyzed here, both the energy minimized conformations (DrugBank) and protein bound conformations remain largely linear and/or planar. There have been in-depth discussions regarding the extensive molecular rearrangements drug molecules are capable of undergoing in order to facilitate protein binding.5,26,27 It is interesting to note that these rearrangements do not translate to an overall change in 3D topology, at least for the molecular structures explored here.
Figure 2.
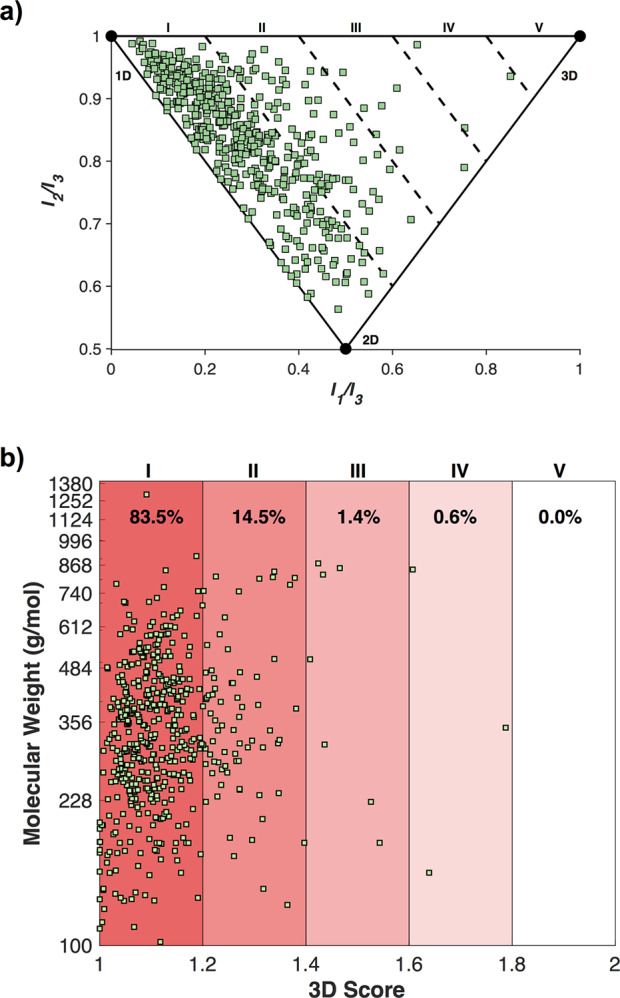
Averaged PMI analysis of 502 entries from the PDB, plotted (a) on a typical PMI triangle plot and (b) as 3D Score (I1/I3 + I2/I3) vs MW.
The 3D score vs MW of the averaged PDB ligand conformations was plotted (Figure 2b) and found to be similar to the outcome from the minimized DrugBank 3D structures (Figure 1b). The 3D scores represent molecules of broad MW distributions, and there is no obvious relationship between MW and 3D topology. As demonstrated in the PMI plot (Figure 2a), the majority (83.5%) of compounds fall into bin I (Figure 2b), with decreasing prevalence in each bin of increasing 3-dimensionality. This follows the same general trend as the geometry minimized DrugBank structures and further highlights the absence of highly 3D topologies in currently used and proposed drug molecules. As described earlier, it is possible that these linear/planar features are the result of the feedstocks and libraries used for drug discovery campaigns.
The energy of protein bound ligand structures in the context of conformational change tolerance has been explored elsewhere.26−29 However, the 3D topologies of these different conformations have not been evaluated through the PMI analysis. Many of the DrugBank structures appear in the PDB with high frequency, though many do not undergo significant changes (<0.1) in 3D score. Diclofenac (Figure 3), an orally administered nonsteroidal anti-inflammatory drug (NSAID), was chosen as a case study to explore both energy and topology. Diclofenac has 51 protein-bound structures in the PDB with 3D scores ranging from 1.03 (linear, I) to 1.52 (moderately 3D, III).
Figure 3.
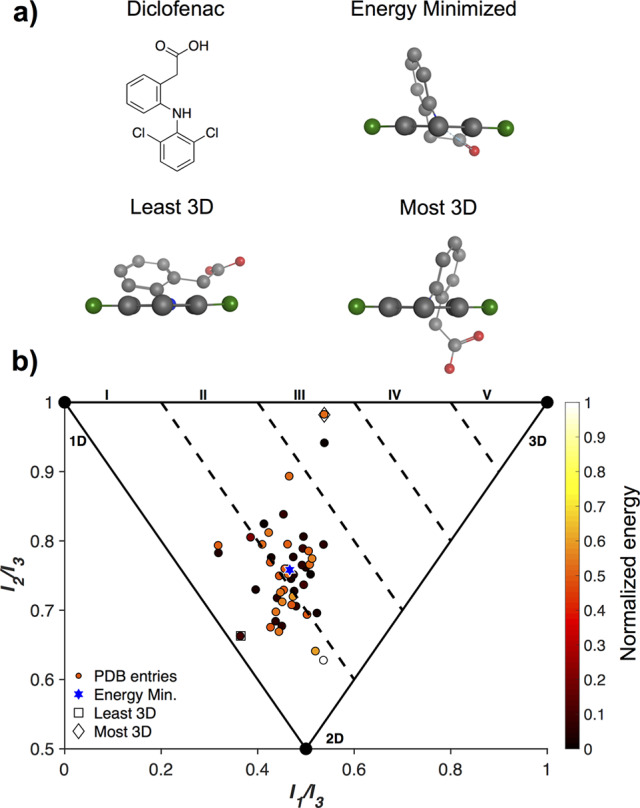
Diclofenac is an NSAID with 51 PDB entries. (a) The most and least 3D conformations of diclofenac, as well as an energy-minimized conformation. (b) An analysis of the relative energies of the various conformations of the diclofenac PDB entries.
Three representative conformations of diclofenac, the least and most 3D, as well as the energy minimized structures, are shown in Figure 3a. These structures have 3D scores of 1.03, 1.52, and 1.22, respectively, and can be visually identified as significant and obvious changes in their 3D conformations. The least 3D structure has the two aromatic rings nearly in plane with each other, while in the most 3D topology the rings are much closer to perpendicular, and the energy minimized conformation falls in between. When the potential energy surfaces of the different conformations are determined, there is no clear relationship to their diverse 3D topologies (Figure 3b). The higher energy conformations (white/orange) include some of the most and least 3D topologies. The lower energy conformations (black) span 3D scores between 1.1 and 1.4, representing significant topological translations. These findings are in agreement with previous reports stating that a wide range of strain energies can be tolerated by therapeutics when bound to target sites.5,6,26,28,29
To more closely examine the effects of a protein active site on a molecule, a PMI analysis of several FDA-approved human immunodeficiency virus (HIV) drugs from two distinct target classes was conducted. Nevirapine and efavirenz are non-nucleoside reverse transcriptase inhibitors (NNRTIs), while nelfinavir, lopinavir, and saquinavir are protease inhibitors.30 Relevant ligand conformations were downloaded from the PDB, and a PMI analysis was performed for each entry (Figure 4).
Figure 4.
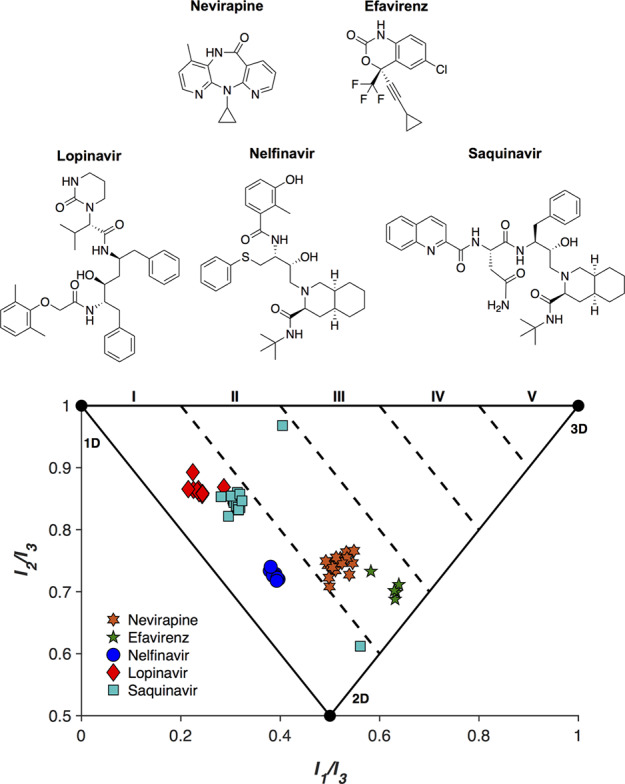
PMI analysis of FDA-approved antiviral therapies, nevirapine, efavirenz (reverse transcriptase inhibitors), nelfinavir, lopinavir, and saquinavir (protease inhibitors).
The aforementioned inhibitors were bound to HIV reverse transcriptases and proteases that contained a range of sequence mutations. In this analysis, sequence identity between reverse transcriptases was conserved to at least 98% when compared to a parent sequence (Table S1). Similarly, homology between proteases was maintained to a minimum of 78% in order to ensure comparison of inhibitor structures bound to similar proteins. For the purposes of this analysis, it was assumed that tertiary structure remained largely conserved across the mutations. For each protein-bound inhibitor, the PMI remained mostly static, indicating a high conservation of binding motif, as expected for compounds bound to conserved targets. Nelfinavir and nevirapine showed almost no variation in conformation across 11 and 17 structures, respectively, while saquinavir remained mostly consistent, with two notable exceptions. Both exceptions, at [0.40 0.97] and [0.56 0.61] from PDB entries 3S56 and 3UFN, respectively, are due to a second saquinavir molecule that is bound to an extension of the active site cavity near the protein surface.31,32 These second drug molecules exhibit a more 3D conformation than the saquinavir molecule found in the active site, which in the 3S56 PDB structure has a PMI of [0.31 0.86]. While this more 3D conformation is likely due to both crystal packing and mutational differences between the proteases, it is an interesting example of one molecule displaying significantly different topologies even when interacting with the same protein. In particular, this example demonstrates that an active site can dictate the topology of the bound ligand quite substantially, and any straying from that binding pocket can result in notable changes in conformation. It is interesting to observe that with a single exception across 52 structures, each protease inhibitor adopts a conformation with a 3D score < 1.2. These topologies are likely due to both the shape of the active site of HIV proteases, as well as their chemical structural similarities. Both NNRTIs (efavirenz and nevirapine) also show closely related 3D scores, though slightly higher than those of the protease inhibitors.
These similar PMI values across several drugs designed for the same target highlight the importance of the protein-binding pocket shape in drug discovery. While the need for 3D scaffolds to explore many current and desired target sites exists, it is also true that in some cases a linear/planar molecule will be best suited. It is for this reason that topological shape diversity is desired in library development and is predicated on the ability to generate molecules of high 3-dimensionality, as low 1- and 2-dimensionality is already broadly covered. Attempts to address the size and shape of protein active sites have resulted in a suite of tools that can be used to aid in drug discovery efforts.33−36
Achieving 3-Dimensionality in Medicinal Chemistry
To address the lack of 3-dimensionality in pharmaceuticals, there have been efforts in academic and industry laboratories to design and synthesize libraries of 3D molecules to be used in drug discovery campaigns.13,15,17,18,37,38 These structures include compounds prepared through diversity-oriented syntheses with a higher than typical number of sp3 carbons, as well as chiral and spiro centers. Such strategies are often regarded as possible solutions to the over-representation of flat molecules in fragment libraries and drug discovery feedstocks.7,13,14,18,24,37,38 To evaluate the success of such approaches, a PMI analysis (Figure 5) was carried out on 15 unique fragments from three different reports (Figure S4).18,37,38 These studies each sought to generate organic fragments with high degrees of 3-dimensionality. Of the compounds evaluated, 6/15 (40%) effectively escape the linear/planar region (3D Score > I). These results represent a significant enhancement in 3D character compared to the typical fragment libraries that have 75% of molecules in “flatland.”18 While these results are promising, all organic fragments presented here fail to achieve 3D Scores > II, indicating their relatively limited topological diversity. This is not a comprehensive survey of current efforts in this area, but it does suggest that there is room for further improvement.
Figure 5.
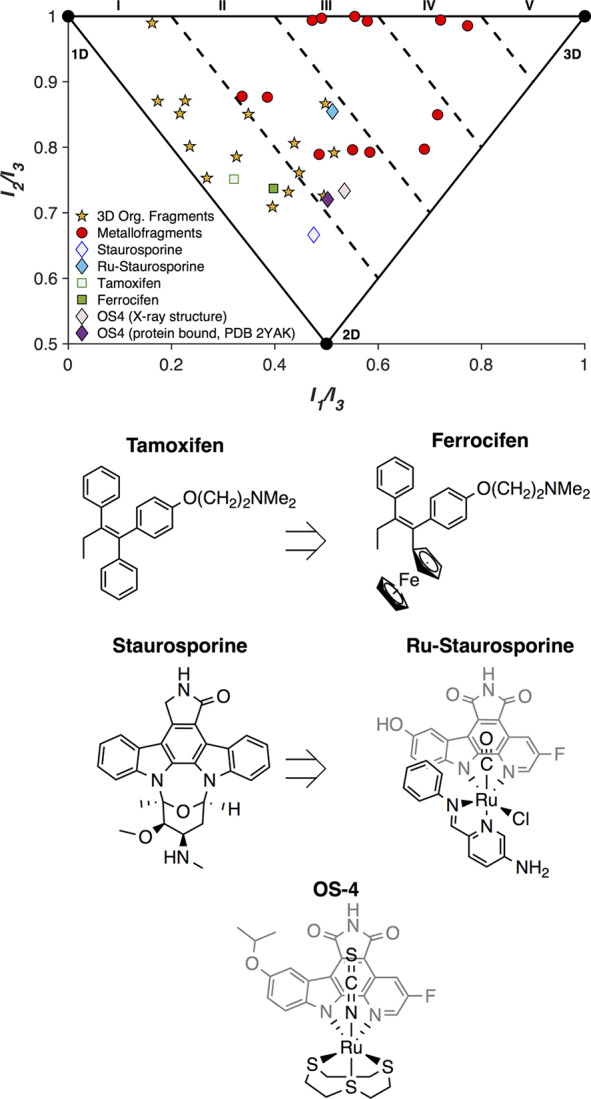
PMI analysis of 3D organic fragments (☆) and metallofragments (○). PMI analysis and structures of tamoxifen and staurosporine, along with their metal containing analogues/derivatives ferrocifen, Ru-staurosporine, and OS-4.
The medicinal inorganic community has considered the inclusion of metal centers into existing drugs as an intriguing method for augmenting existing therapeutics.39 Tamoxifen is a chemotherapeutic agent that has been modified to include a ferrocene group (ferrocifen), and the anticancer activity against estrogen-receptor positive cancers increases upon the inclusion of the iron moiety. An unanticipated consequence of this modification is that the 3-dimensionality of the molecule increases slightly from tamoxifen to ferrocifen (Figure 5). With an understanding of the inherent 3-dimensionality of metal-complexes, Meggers and co-workers set out to incorporate organometallic groups into kinase inhibitors as deliberately 3D moieties.39−41 When a representative compound from Meggers’ work is analyzed using the PMI method, the inclusion of a Ru(II) center increases the 3-dimensionality of staurosporine quite significantly (Figure 5). In one representative example they crystallized a DAPK1 (PDB 2YAK) selective staurosporine derivative OS4 with the protein kinase.42 When the molecular structure is compared to the PDB structure, there is no significant change in the 3D topology, suggesting the shape is relevant to the selectivity of the inhibitor. While these and prior efforts aim to augment existing organic therapeutics, a recent report sought to address the feedstock issue through the creation of a 3D metallofragment library (Figure S5).15 A PMI analysis of 13 compounds from this library, including metallocenes, piano-stool, and octahedral complexes, found that 100% of the compounds fall outside of flatland (Figure 5). This metallofragment library broadly covers 3D chemical space with a relatively small number of molecules and starts to address the need for topologically diverse feedstocks for drug discovery campaigns.
These efforts to prepare both organic and inorganic libraries with high 3-dimensionality will allow for exploration of biological space that has previously been inaccessible with the limited 1D/2D molecular entities.
In both energy minimized (DrugBank) and protein-bound conformations (PDB) small molecule drugs are overwhelmingly linear and planar. This holds true across a wide range of MWs. Less than 1% of any of the molecular conformations surveyed fall in the highest 3D-area (V, 3D Score > 1.8). While the 3D score of any particular molecule is partly due to the intrinsic ability of the molecule to adopt a 3D conformation, the capacity of the target protein to accept a 3D conformation also plays a critical role in determining the final bound shape of the molecule. The features of an active site, such as nearby or reactive residues, are already taken into consideration in drug design and in the modeling of bound inhibitors. This analysis demonstrates the relevance of the overall shape of the binding site to the final protein–drug complex.
The true need for topological diversity in feedstocks and final drug molecules remains unclear given the overwhelming number of linear and planar drugs. The question remains as to whether more 3D compounds represent attractive and untapped therapeutic space or if more linear/planar molecules are indeed the best topologies for bioactive molecules. Similarly, is the linearity and planarity of current ligands because they produce the most bioactive compounds, or are they simply a consequence of the scarcity of highly 3D molecules in the pipeline? Given the range of proteins currently considered undruggable43,44 or even those targets for which no therapy has been successfully designed, it is anticipated that a broader survey of protein target sites will yield greater shape diversity. Furthermore, the development of topologically rich and varied feedstocks will subsequently yield elaborated bioactive molecules with great 3-dimensionality that may allow for the development of molecules with greater potency and specificity than their planar predecessors.
Acknowledgments
We thank Prof. David Wishart (U. Alberta, Canada) for helpful communications about DrugBank during the revision of this manuscript.
Glossary
Abbreviations
- FDA
Food and Drug Administration
- HIV
human immunodeficiency virus
- MOE
molecular operating environment
- MW
molecular weight
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- NSAID
nonsteroidal anti-inflammatory drug
- PDB
Protein Database
- PMI
principal moment of inertia
- Ro5
Rule of 5
- SAR
structure–activity relationship
- 3D
three-dimensional
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00121.
Methods used to curate and analyze the DrugBank and PDB entries. Additional PMI analyses of the DrugBank and PDB entries, 3D fragment library chemical structures, and PDB ID values for HIV drug conformations. (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
This work was supported by grants from the National Institutes of Health (R21 AI138934), a Natural Sciences and Engineering and Research Council of Canada Postdoctoral Fellowship (to K.E.P.), and the National Science Foundation Graduate Research Fellowship Program (DGE-1650112 to R.W.S.).
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou P.; Huang J.; Tian F. Specific noncovalent interactions at protein-ligand interface: implications for rational drug design. Curr. Med. Chem. 2012, 19 (2), 226–38. 10.2174/092986712803414150. [DOI] [PubMed] [Google Scholar]
- Southall N. T.; Dill K. A.; Haymet A. D. J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106 (3), 521–533. 10.1021/jp015514e. [DOI] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56 (4), 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Mobley D. L.; Dill K. A. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get. Structure 2009, 17 (4), 489–498. 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E.; Charifson P. S. Conformational Analysis of Drug-Like Molecules Bound to Proteins: An Extensive Study of Ligand Reorganization upon Binding. J. Med. Chem. 2004, 47 (10), 2499–2510. 10.1021/jm030563w. [DOI] [PubMed] [Google Scholar]
- Friedrich N.-O.; Simsir M.; Kirchmair J. How Diverse Are the Protein-Bound Conformations of Small-Molecule Drugs and Cofactors?. Front. Chem. 2018, 6, 68. 10.3389/fchem.2018.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer W. H. B.; Schwarz M. K. Molecular Shape Diversity of Combinatorial Libraries: A Prerequisite for Broad Bioactivity. J. Chem. Inf. Model. 2003, 43 (3), 987–1003. 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- Kombo D. C.; Tallapragada K.; Jain R.; Chewning J.; Mazurov A. A.; Speake J. D.; Hauser T. A.; Toler S. 3D Molecular Descriptors Important for Clinical Success. J. Chem. Inf. Model. 2013, 53 (2), 327–342. 10.1021/ci300445e. [DOI] [PubMed] [Google Scholar]
- Kortagere S.; Krasowski M. D.; Ekins S. The importance of discerning shape in molecular pharmacology. Trends Pharmacol. Sci. 2009, 30 (3), 138–147. 10.1016/j.tips.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S. R.; Moore J. B.; Schymura Y.; Churchill G. C. Shape-Based Reprofiling of FDA-Approved Drugs for the H1 Histamine Receptor. J. Med. Chem. 2012, 55 (16), 7054–7060. 10.1021/jm300671m. [DOI] [PubMed] [Google Scholar]
- Kalászi A.; Szisz D.; Imre G.; Polgár T. Screen3D: A Novel Fully Flexible High-Throughput Shape-Similarity Search Method. J. Chem. Inf. Model. 2014, 54 (4), 1036–1049. 10.1021/ci400620f. [DOI] [PubMed] [Google Scholar]
- Gadhe C. G.; Lee E.; Kim M.-h. Finding new scaffolds of JAK3 inhibitors in public database: 3D-QSAR models & shape-based screening. Arch. Pharmacal Res. 2015, 38 (11), 2008–2019. 10.1007/s12272-015-0607-6. [DOI] [PubMed] [Google Scholar]
- Morton D.; Leach S.; Cordier C.; Warriner S.; Nelson A. Synthesis of Natural-Product-Like Molecules with Over Eighty Distinct Scaffolds. Angew. Chem., Int. Ed. 2009, 48 (1), 104–109. 10.1002/anie.200804486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52 (21), 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Morrison C. N.; Prosser K. E.; Stokes R. W.; Cordes A. L.; Metzler-Nolte N.; Cohen S. M. Expanding Medicinal Chemistry into 3D Space: Metallofragments as 3D Scaffolds for Fragment-Based Drug Discovery. Chem. Sci. 2020, 11, 1216–1225. 10.1039/C9SC05586J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth N. C.; Brown N.; Blagg J. Plane of Best Fit: A Novel Method to Characterize the Three-Dimensionality of Molecules. J. Chem. Inf. Model. 2012, 52 (10), 2516–2525. 10.1021/ci300293f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. R.; Gillet V. J.; Lewis R. A.; Taylor R. Three-Dimensional Pharmacophore Methods in Drug Discovery. J. Med. Chem. 2010, 53 (2), 539–558. 10.1021/jm900817u. [DOI] [PubMed] [Google Scholar]
- Hung A. W.; Ramek A.; Wang Y.; Kaya T.; Wilson J. A.; Clemons P. A.; Young D. W. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (17), 6799. 10.1073/pnas.1015271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48 (3), 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Friedrich N.-O.; Meyder A.; de Bruyn Kops C.; Sommer K.; Flachsenberg F.; Rarey M.; Kirchmair J. High-Quality Dataset of Protein-Bound Ligand Conformations and Its Application to Benchmarking Conformer Ensemble Generators. J. Chem. Inf. Model. 2017, 57 (3), 529–539. 10.1021/acs.jcim.6b00613. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Guo A. C.; Lo E. J.; Marcu A.; Grant J. R.; Sajed T.; Johnson D.; Li C.; Sayeeda Z.; Assempour N.; Iynkkaran I.; Liu Y.; Maciejewski A.; Gale N.; Wilson A.; Chin L.; Cummings R.; Le D.; Pon A.; Knox C.; Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46 (D1), D1074–d1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J.; Carter M.; Mok N. Y.; Brown N. On the origins of three-dimensionality in drug-like molecules. Future Med. Chem. 2016, 8 (14), 1753–1767. 10.4155/fmc-2016-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46 (1–3), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lipkus A. H.; Watkins S. P.; Gengras K.; McBride M. J.; Wills T. J. Recent Changes in the Scaffold Diversity of Organic Chemistry As Seen in the CAS Registry. J. Org. Chem. 2019, 84 (21), 13948–13956. 10.1021/acs.joc.9b02111. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boström J.; Norrby P.-O.; Liljefors T. Conformational energy penalties of protein-bound ligands. J. Comput.-Aided Mol. Des. 1998, 12 (4), 383–383. 10.1023/A:1008007507641. [DOI] [PubMed] [Google Scholar]
- Nicklaus M. C.; Wang S.; Driscoll J. S.; Milne G. W. A. Conformational changes of small molecules binding to proteins. Bioorg. Med. Chem. 1995, 3 (4), 411–428. 10.1016/0968-0896(95)00031-B. [DOI] [PubMed] [Google Scholar]
- Günther S.; Senger C.; Michalsky E.; Goede A.; Preissner R. Representation of target-bound drugs by computed conformers: implications for conformational libraries. BMC Bioinf. 2006, 7, 293–293. 10.1186/1471-2105-7-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieth M.; Hirst J. D.; Brooks C. L. Do active site conformations of small ligands correspond to low free-energy solution structures?. J. Comput.-Aided Mol. Des. 1998, 12 (6), 563–572. 10.1023/A:1008055202136. [DOI] [PubMed] [Google Scholar]
- Margolis A. M.; Heverling H.; Pham P. A.; Stolbach A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10 (1), 26–39. 10.1007/s13181-013-0325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Y.; Wang Y. F.; Boross P. I.; Chiu T. Y.; Ghosh A. K.; Tozser J.; Louis J. M.; Harrison R. W.; Weber I. T. Critical differences in HIV-1 and HIV-2 protease specificity for clinical inhibitors. Protein Sci. 2012, 21 (3), 339–350. 10.1002/pro.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agniswamy J.; Shen C.-H.; Aniana A.; Sayer J. M.; Louis J. M.; Weber I. T. HIV-1 Protease with 20 Mutations Exhibits Extreme Resistance to Clinical Inhibitors through Coordinated Structural Rearrangements. Biochemistry 2012, 51 (13), 2819–2828. 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Edelsbrunner H.; Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998, 7 (9), 1884–1897. 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinf. 2009, 10 (1), 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel M.; Proschak E.; Schneider G. Pocket Picker: analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007, 1, 7. 10.1186/1752-153X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L.; Govindaraj R. G.; Lemoine J. M.; Wu H.-C.; Brylinski M. DeepDrug3D: classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019, 15 (2), e1006718 10.1371/journal.pcbi.1006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg D. G.; Kondo N.; Mitchell S. L.; Galloway W. R. J. D.; Sore H. F.; Madin A.; Spring D. R. Partially Saturated Bicyclic Heteroaromatics as an sp3-Enriched Fragment Collection. Angew. Chem., Int. Ed. 2016, 55 (40), 12479–12483. 10.1002/anie.201606496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd S. L.; Osberger T. J.; Mateu N.; Sore H. F.; Spring D. R. Recent Applications of Diversity-Oriented Synthesis Toward Novel, 3-Dimensional Fragment Collections. Front. Chem. 2018, 6, 460. 10.3389/fchem.2018.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggers E. Targeting proteins with metal complexes. Chem. Commun. 2009, (9), 1001–1010. 10.1039/b813568a. [DOI] [PubMed] [Google Scholar]
- Maksimoska J.; Feng L.; Harms K.; Yi C.; Kissil J.; Marmorstein R.; Meggers E. Targeting Large Kinase Active Site with Rigid, Bulky Octahedral Ruthenium Complexes. J. Am. Chem. Soc. 2008, 130 (47), 15764–15765. 10.1021/ja805555a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggers E.; Atilla-Gokcumen G. E.; Bregman H.; Maksimoska J.; Mulcahy S. P.; Pagano N.; Williams D. S. Exploring chemical space with organometallics: ruthenium complexes as protein kinase inhibitors. Synlett 2007, 2007 (08), 1177–1189. 10.1055/s-2007-973893. [DOI] [Google Scholar]
- Feng L.; Geisselbrecht Y.; Blanck S.; Wilbuer A.; Atilla-Gokcumen G. E.; Filippakopoulos P.; Kräling K.; Celik M. A.; Harms K.; Maksimoska J.; Marmorstein R.; Frenking G.; Knapp S.; Essen L.-O.; Meggers E. Structurally Sophisticated Octahedral Metal Complexes as Highly Selective Protein Kinase Inhibitors. J. Am. Chem. Soc. 2011, 133 (15), 5976–5986. 10.1021/ja1112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V.; Reddy E. P.; Shokat K. M.; Soucek L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17 (8), 502–508. 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.; Aiello C.; Grieco P.; Marasco D. Targeting “Undruggable” Proteins: Design of Synthetic Cyclopeptides. Curr. Med. Chem. 2016, 23 (8), 748–62. 10.2174/0929867323666160112122540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.