

Abstract

A series of PI3Kβ selective inhibitors derived from a novel 4-(1H-benzo[d]imidazol-1-yl)quinoline chemotype has been rationally designed. Crucial to achieving the desired selectivity over the other class I PI3K isoforms, including the challenging δ-isoform, was the identification of a subset of substituted pyridine hinge binders. This work led to the discovery of (P)-14, a highly selective and orally bioavailable PI3Kβ inhibitor displaying an excellent pharmacokinetic profile in addition to great cellular potency in various PTEN-deficient tumor cell lines. Results from a dog toxicology study revealing structure-related, off-target ocular toxicity are also briefly discussed.
Keywords: PI3K, PTEN, hinge binder, atropisomer, rotational barrier
Class I phosphoinositide 3-kinase (PI3K) is a family of heterodimeric lipid kinases composed of four isoforms of the catalytic subunit (p110α, p110β, p110δ, and p110γ). Class I PI3Ks are responsible for transducing growth, metabolism, and proliferation signals via the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to produce the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3).1 This event triggers the recruitment and activation of proteins containing the PIP3-binding pleckstrin homology (PH) domain to the plasma membrane. Aberrations in this signaling network result in disease states, including many cancers.2,3
The tumor suppressor phosphatase and tensin homologue (PTEN) functions as a negative regulator of the lipid kinase activity, converting PIP3 back to PIP2. Deletion, or, less frequently, mutation of PTEN leads to higher levels of PIP3 and increased activation of downstream signaling pathways. Preclinical studies have shown that PI3Kβ activity is required in PTEN-deficient cancer cells.4,5 Therefore, the development of PI3Kβ-selective inhibitors is an attractive approach for the treatment of PTEN-deficient tumors.
TGX-2216 inspired multiple drug discovery programs searching for selective PI3Kβ inhibitors, including the β-weighted PI3Kβ/δ inhibitors GSK26367717,8 and AZD81869 (Figure 1).10 Docking in a PI3Kβ homology model suggests that TGX-221-R, the most potent of the two enantiomers,11 binds to the ATP binding site of the kinase domain with the morpholine serving as a one-point hinge binder, a common feature of PI3K inhibitors.12 The carbonyl group is believed to interact with the affinity pocket residue Tyr833, presumably via a network of hydrogen bonds involving one bridging water molecule.11 The aniline occupies an induced specificity pocket between Met773 and Trp781. This induced-fit pocket appears to be a central contributor to the β- and δ-isoform selectivity over other isoforms for numerous PI3K inhibitors.10,13
Figure 1.

(Top) Examples of reported β-weighted PI3K inhibitors and docked pose of TGX-221-R in a PI3Kβ homology model built from the cocrystal structure of (P)-19 bound to PI3Kδ (pdb: 6DGT).19 (Bottom) Biochemical potency and selectivity of a novel PI3Kβ chemotype compared to other reported inhibitors.
In an effort to develop β-selective PI3K inhibitors, we designed a new chemotype informed by the aforementioned three compounds while taking advantage of our past success using bicyclic aromatic groups oriented in the specificity pocket.14,15 We also envisaged directing a triazole group toward the polar residues of the affinity pocket (vide infra). Accordingly, we synthesized compound 1 and determined its potency in our class I PI3K biochemical assays (Figure 1). This novel molecule resulted in a β-weighted PI3Kβ/δ inhibitor with potencies of 3 nM and 12 nM for the respective isoforms. From this result, we initiated a medicinal chemistry effort to further improve the selectivity over the α- and δ-isoforms.
Typically, PI3K isoform selectivity can be modulated by varying the substitution patterns around the selectivity group—a quinoline in this case—since the specificity pocket is where most of the nonconserved residues are located.16 Nevertheless, we first decided to explore the hinge binding moiety with the hope of finding a more δ-sparing starting point. As illustrated by compound 2, a single-point pyridine hinge binder can also yield a highly potent PI3Kβ inhibitor (IC50 = 5 nM). Gratifyingly, superior selectivity over the δ-isoform (20×), as well as over the α- and γ-isoforms, was observed.
The presumed binding conformation of 2 favors an orthogonal orientation of the two bicyclic systems (vide infra). Accordingly, we envisioned improving the potency (and potentially other parameters) of this series by restricting the free rotation around the C–N bond, thus generating an axis of atropisomerism.17,18 Torsional scan studies using molecular mechanics (MMFFs force field) revealed that monosubstitution at C5 of the quinoline (4–5) or C2 of the benzimidazole (6) is not enough to hinder rotation effectively and results in class 1 or class 2 atropisomers (Table 1).19 Disubstitution is required to generate stable class 3 atropisomers (7).
Table 1. Rotational Barriers of Related 4-(1H-Benzo[d]imidazol-1-yl)quinolines.

| Cpd | A | B | ΔErot (TSin) (kcal/mol)a | ΔErot (TSout) (kcal/mol)a | Atropisomer classb |
|---|---|---|---|---|---|
| 3 | H | H | 29.6 | 14.1 | 1 |
| 4 | H | F | 33.7 | 15.5 | 1 |
| 5 | H | CH3 | >40 | 22.5 | 2 |
| 6 | CH3 | H | 32.2 | 28.3 | 2 |
| 7 | CH3 | F | 37.7 | 33.9 | 3 |
Using MMFFs force field, the best estimates for the rotational barriers ΔErot via TSin and TSout were obtained from the lowest energy pathway in either direction (clockwise or counterclockwise).19
Class 1: ΔErot < 20 kcal/mol; Class 2: 20 kcal/mol < ΔErot < 30 kcal/mol; Class 3: ΔErot > 30 kcal/mol.
According to these calculations, the rotational barrier of compound 2 is approximately 14 kcal/mol, well below the ideal >30 kcal/mol for stable atropisomer. In theory, the combination of a methyl group at the C2-position of the benzimidazole with a 5,8-difluoroquinoline, such as in (rac)-8, should restrict the free rotation around the C–N bond.20 The rotational barrier of 8 is estimated to be greater than 33 kcal/mol, suggesting a pair of noninterconverting, class 3 atropisomers.21 Upon evaluation in our PI3K biochemical assays, (rac)-8 was shown to exhibit a PI3Kβ IC50 of 9 nM with comparable isoform selectivity to compound 2 (Table 2).
Table 2. Optimization of the Pyridine Hinge Binder.

The activity against each class I PI3K was evaluated in in vitro kinase assays containing 2Km steady state concentrations of ATP (average of ≥2 determinations).
Numbers in parentheses represent fold selectivity (PI3Kα/δ/γ IC50 divided by PI3Kβ IC50).
We then evaluated a subset of pyridine-based analogs in our biochemical assays to determine their potency and selectivity. The 2-chloro- and the 2-methylpyridine hinge binders, (rac)-9 and (rac)-10, are well tolerated in terms of PI3Kβ potency, but only the latter affords a similar selectivity profile to (rac)-8. The 2-methylpyrimidine (rac)-11 analog leads to a loss in PI3Kα and PI3Kδ selectivity, whereas 6-methylpyrimidine (rac)-12 and 2-(difluoromethyl)pyridine (rac)-13 lose both potency and selectivity. Conversely, the 2-cyanopyridine hinge binder found in (rac)-14 leads to a compound with great potency (IC50 = 4 nM), as well as the highest selectivity over the α- and δ-isoforms (120× and 55×, respectively).
Two-point hinge binders, such as 2-aminopyridine (rac)-15, tend to lead to a slight reduction in isoform selectivity when compared to single-point pyridine hinge binders (Table 3). Substitution around the 2-aminopyridine such as a 6- or 3-methyl ((rac)-16-17) results in significant loss in potency, whereas a 2-amino-3-chloropyridine hinge binder ((rac)-18) is reasonably active with a PI3Kβ IC50 of 19 nM. Replacement of the 3-chloro by a 3-fluoro ((rac)-19) is accompanied by a desirable 6-fold increase in potency with acceptable isoform selectivity. The 2-amino-5-fluoropyridine, the 2-amino-3,5-difluoropyridine, and the 2-(N-methylamino)-3-fluoropyridine analogs all result in a significant loss in potency ((rac)-20-22).
Table 3. Optimization of the 2-Aminopyridine Hinge Binder.

We then proceeded to the separation of the most promising analogs ((rac)-8, 10, 14, 15, and 19) by chiral preparative supercritical fluid chromatography (SFC), providing the corresponding single atropisomers for further profiling (Table 4). The absolute configurations of the single atropisomers were unambiguously determined by X-ray crystallography of (P)-19 bound in the PI3Kδ ATP binding site (pdb: 6DGT).19 All five (P)-atropisomers exhibit PI3Kβ potency of less than 10 nM with moderate to good isoform selectivity. (P)-8 was found to be a potent inhibitor of CYP3A4 (IC50 = 1.0 μM), a member of the cytochrome P450 family of metabolic enzymes, invoking the potential for drug–drug interactions. Introduction of a 2-substituent on the pyridine alleviates or eliminates the propensity for CYP3A4 inhibition.
Table 4. Profiles of the Single Atropisomers.

Analytical methods indicate that both purified atropisomers are free of the other by the limit of detection of our technology (>98% ee). The (M)-atropisomer may be completely devoid of PI3Kβ activity, but a 1% impurity of (P)-atropisomer would result in IC50 values between 200 and 1,000 nM.
Determined from human hepatocytes.
Units: 106 cm/s.
In terms of metabolic stability, incubations in the presence of human hepatocytes revealed lower intrinsic and predicted clearances for the potent (P)-atropisomers, compared with higher values for the (M)-atropisomers (except for (P)-14 and (M)-14). In the case of (P)-19 and (M)-19, we previously reported that (M)-19 is primarily metabolized by aldehyde oxidase, whereas (P)-19 is not a substrate for this cytosolic enzyme, resulting in superior metabolic stability.19,22,23
Promising compound (P)-14, bearing a 2-cyanopyridine hinge binder, delivers high forward permeability and low efflux as determined by a Caco-2 cell monolayer assay. To achieve a similar profile in the metabolically more stable 2-aminopyridine series, a fluorine atom at the 3-position was proven effective to restore the forward permeability. This improvement is presumably attributed to a reduced basicity of the pyridine nitrogen; (P)-19 has a measured conjugate acid pKa of 4.8 as opposed to 6.1 for (P)-15. In addition to reducing the basicity of the pyridine nitrogen, the fluorine atom can improve passive permeability by electrostatically shielding the proximal H-bond donor and reduce efflux by mitigating P-gp recognition.24
Among these five compounds, (P)-14 and (P)-19 were identified as the most attractive inhibitors based on their combination of potency, isoform selectivity, and permeability. At a compound concentration of 10 μM, (P)-14 and (P)-19 do not significantly interact with any other kinases, according to the DiscoverX KINOMEscan platform, validating them as potent and selective inhibitors of PI3Kβ (see SI).25 We also evaluated a subset of substituted quinolines and benzimidazoles in combination with both the 2-cyanopyridine and the 2-amino-3-fluoropyridine hinge binders, but none of them exhibited a potency or selectivity advantage (see SI).
With two promising inhibitors in hand, we sought to further profile (P)-14 and (P)-19 by evaluating their cellular potency in comparison to clinical compound GSK2636771 (Table 5). We determined the ability of each compound to inhibit the phosphorylation of AKT, a downstream measure of PI3K activity.9,26 In this study, we assessed potency in five different PTEN-deficient tumor cell lines that show dependence on PI3Kβ for viability: the prostate carcinomas PC3, LNCaP C4-2, and LNCaP, and the breast adenocarcinomas MDA-MB-415 and ZR-75-1. Compounds (P)-14 and (P)-19 were found to have superior potency profiles in these PI3Kβ-dependent cell lines with EC50 values ranging from 5 nM to 53 nM.
Table 5. Activity against AKT1 Ser473 Phosphorylation in PTEN-Deficient Tumor Cell Lines.
| Cell line | GSK2636771 | (P)-14 | (P)-19 |
|---|---|---|---|
| PC3 EC50 (nM)a | 23 | 12 | 9 |
| LNCaP C4-2 EC50 (nM)a | 42 | 24 | 15 |
| LNCaP EC50 (nM)a | 178 | 31 | 53 |
| MDA-MB-415 EC50 (nM)a | 40 | 15 | 12 |
| ZR-75-1 EC50 (nM)a | 28 | 5 | 6 |
Data determined from the ratio of Units pAKT1 Ser473 per nanogram total AKT1.
The synthesis of (P)-19 via intermediate 20 was described in our previous manuscript.19 The sequence to (P)-14 involves a Suzuki reaction between 20 and 4-bromo-2-cyanopyridine to install the hinge binder (21) (Scheme 1). Saponification of the ester, followed by coupling with ammonium chloride, leads to the primary amide 22. The amide is then converted to the corresponding triazole using a one-pot, two-step treatment with dimethylformamide dimethyl acetal and hydrazine, sequentially ((rac)-14). The single atropisomers (P)-14 and (M)-14 are obtained via chiral preparative SFC separation.
Scheme 1. Synthesis of (P)-14.

Reagents and conditions: (a) 4-bromo-2-cyanopyridine, Pd(PPh3)4, K3PO4, dioxane, 90 °C (85%); (b) 1 M LiOH, THF, rt; (c) NH4Cl, EDC, HOBt, DIEA, 50 °C (77% over 2 steps); (d) dimethylformamide dimethyl acetal, 100 °C, then hydrazine, AcOH, 45 °C (68%); (e) CHIRALPAK OJ-H SFC 5 μM 21 × 250 mm column in 30% MeOH/CO2 at 60 mL/min.
We previously reported a crystal structure of PI3Kδ cocrystallized with (P)-19 (pdb: 6DGT).19 A PI3Kβ homology model based on this crystal structure was created to better understand the binding conformation of (P)-14 and related analogs. The 2-cyanopyridine moiety interacts with the hinge residue Val848 by accepting one hydrogen bond, and the orthogonal quinoline (dihedral angle of 96°) occupies the induced specificity pocket between Met773 and Trp781 (Figure 2). The triazole moiety is directed toward the affinity pocket, making hydrogen bond contacts with Lys779, Asp807, and Tyr833. (P)-19 is expected to have a similar binding mode with the 2-aminopyridine serving as a two-point hinge binder to Val848, as observed in its PI3Kδ cocrystallized structure (pdb: 6DGT).
Figure 2.
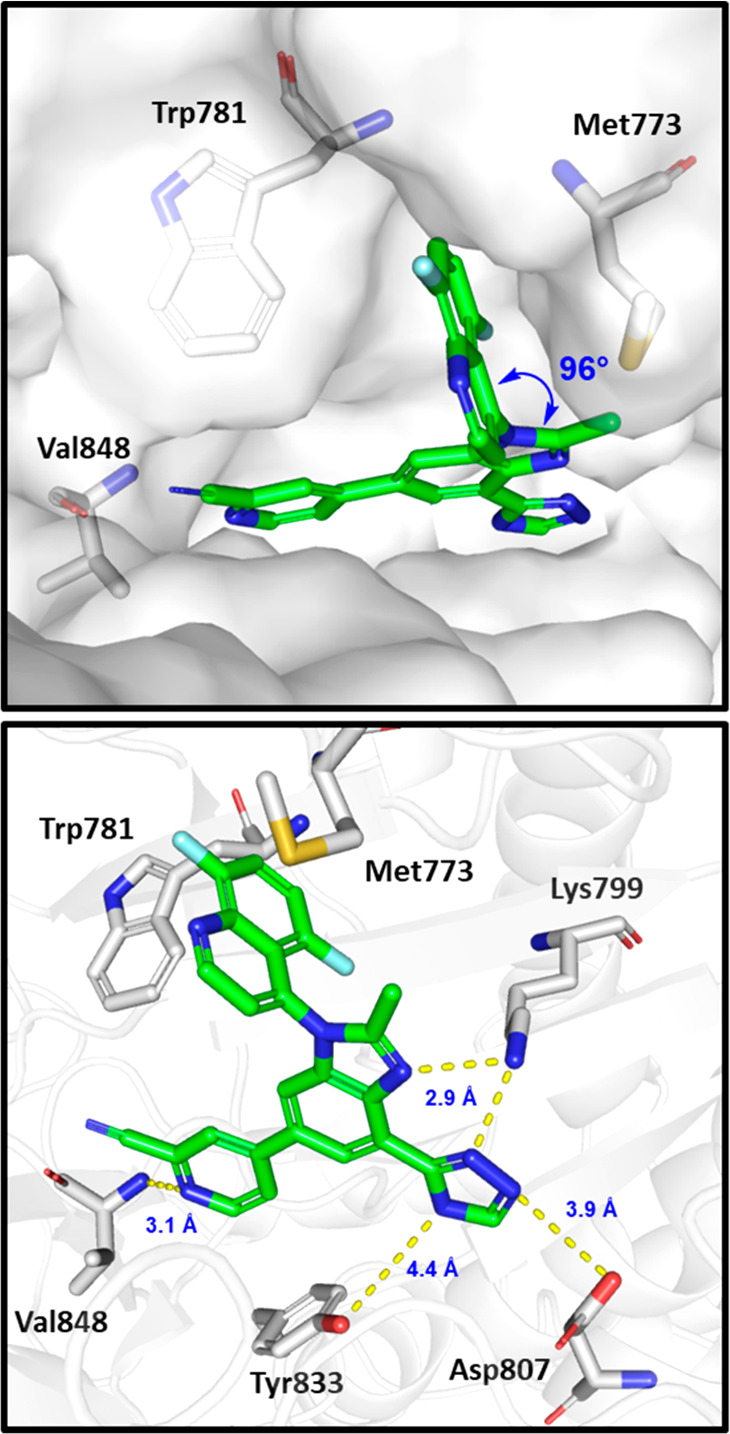
Docked pose of (P)-14 in the homology model of PI3Kβ built from the cocrystal structure of (P)-19 bound to PI3Kδ (pdb: 6DGT).19 Some residues have been removed for clarity, and yellow dashed lines show hydrogen bond contacts between the inhibitor and the protein.
The pharmacokinetic parameters in preclinical species after intravenous and oral administration of (P)-14 and (P)-19 are listed in Table 6. The total clearance (CL) of (P)-14 was found to be intermediate with respect to hepatic blood flow. The volumes of distribution (Vss) are comparable to or higher than total body water in all species evaluated. On the other hand, the in vivo data indicate that (P)-19 has low to intermediate total clearance (CL). Volumes of distribution (Vss) are close to or lower than total body water, except for rat, in which lower plasma protein binding (PPB) is observed. In agreement with their low to moderate observed clearance, high permeability, and low efflux, both compounds exhibit high oral bioavailability.
Table 6. Summary of Pharmacokinetic Parameters for (P)-14 and (P)-19 in Preclinical Species.
| Parameter | Sprague–Dawley rat | Beagle dog | Cynomolgus monkey | Rhesus monkey |
|---|---|---|---|---|
| SUMMARY FOR (P)-14a | ||||
| PPB (% bound)b | 84 | 86 | 84 | 81 |
| CLpr (L/h/kg)c | 1.65 | 0.27 | 0.25 | 0.81 |
| CL (L/h/kg)d | 1.97 | 0.34 | 0.25 | 0.47 |
| Vss (L/kg)e | 2.5 | 2.5 | 1.3 | 2.6 |
| Terminal t1/2 (h)f | 1.2 | 5.5 | 4.1 | 4.4 |
| MRT (h)g | 1.3 | 7.3 | 5.2 | 5.5 |
| F (%)h | 53 | 76 | 35 | ND |
| SUMMARY FOR (P)-19a | ||||
| PPB (% bound)b | 81 | 92 | 94 | 93 |
| CLpr (L/h/kg)c | 0.60 | 0.47 | 0.22 | 0.41 |
| CL (L/h/kg)d | 0.68 | 0.22 | 0.13 | 0.10 |
| Vss (L/kg)e | 2.7 | 0.9 | 0.5 | 0.6 |
| Terminal t1/2 (h)f | 9.6 | 3.6 | 5.2 | 6.0 |
| MRT (h)g | 3.9 | 4.0 | 4.3 | 6.2 |
| F (%)h | 60 | ND | 64 | ND |
Intravenous doses of 1 mg/kg.
Plasma protein binding.
Predicted clearance from hepatocytes.
In vivo clearance.
Volume of distribution at steady state.
Half-life intravenous.
Mean residence time intravenous.
Bioavailability calculated after oral doses of 5 mg/kg. ND = value not determined.
A significant difference between the two compounds favoring (P)-14 is the human plasma protein binding. The human plasma free fraction of (P)-14 was measured at 11.0%, compared to only 1.4% for (P)-19. With the two compounds having similar potency in serum-free cellular assays (see Table 5), we predicted a lower protein adjusted EC90 for (P)-14 from the average of the measured potency in all cell lines (PA EC90 = 1.0 μM vs 8.0 μM for (P)-19).
To further differentiate (P)-14 and (P)-19, we assessed their respective human pharmacokinetic projections. Using tritiated material, the human predicted hepatic clearances of (P)-14 and (P)-19 were determined to be 0.11 L/h/kg and 0.02 L/h/kg, respectively. (P)-14 has significantly higher Vss in preclinical species, leading to a predicted human Vss of 2.2 L/kg (average of Vss in all species). In contrast, the low Vss observed in cynomolgus and rhesus monkeys for (P)-19 result in a predicted human Vss of 0.70 L/kg (average of Vss in dog and monkeys), a value lower than the total body water.27 Consequently, the predicted human half-life for each compound was estimated to be adequate for once-daily dosing (18 h for (P)-14 and 40 h for (P)-19). Assuming 30% bioavailability for (P)-14 and 50% for (P)-19, the doses to cover the protein adjusted EC90 at trough were calculated to be approximatively 250 mg for both compounds. Accordingly, the projected human exposure and maximum plasma concentration were considerably lower in the case of (P)-14 (AUC0–24 = 27 μM·h vs 293 μM·h for (P)-19 and Cmax = 1.5 μM vs 14 μM for (P)-19).
Lastly, the toxicologic profile of (P)-14 was evaluated in a 7 day repeat dose dog study. Male beagle dogs (three per group) were dosed daily with (P)-14 at 10, 40, and 100 mg/kg/day. Based on poor tolerability, high dose animals were euthanized after the second dose. Dosing was suspended in the 40 mg/kg/day group on day 4 and resumed on day 5 at 30 mg/kg/day. Margins of exposure at the clinically efficacious dose were estimated to be 1.8- and 8.1-fold at 10 and 40/30 mg/kg/day (see SI).
Clinical observations indicated decreased response to visual stimuli, dilated pupils, red conjunctiva, and ocular discharge in all animals, beginning on day 4 through the end of the study, at the low and mid dose levels. Detailed ocular assessments revealed both structural and functional damage, likely irreversible (see SI). Microscopically, retinal degeneration/necrosis/detachment and choroidal inflammation associated with decreased electroretinography response were noted in all animals.28 These data, combined with the absence of ocular toxicity with another structurally distinct PI3Kβ candidate,14 suggest that the effect of (P)-14 is related to the compound scaffold. Based on these findings, all efforts on this series of PI3Kβ inhibitors were terminated.
In summary, we have discovered a series of potent and selective PI3Kβ inhibitors. The initial morpholine hinge binder was replaced by a pyridine to yield improved isoform selectivity. Further modifications of the hinge binder ultimately led to a 2-cyanopyridine. This novel hinge binder was found to provide great selectivity over the other isoforms, including the elusive δ-isoform. This work culminated in (P)-14, a potent and selective inhibitor of PI3Kβ with good pharmacokinetic properties. Unfortunately, evaluation of (P)-14 in a dog toxicology study revealed structure-related, off-target ocular toxicity.
Glossary
Abbreviations Used
- AKT
protein kinase B
- pAKT
phosphorylated protein kinase B
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- PTEN
phosphatase and tensin homologue
- CLpr
predicted clearance
- CYP
cytochromes P450
- MMFF
molecular mechanics force field
- TS
transition state
- Pgp
P-glycoprotein
- hERG
human Ether-à-go-go-Related Gene EDC, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
hydroxybenzotriazole
- DIEA
N,N-diisopropylethylamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00095.
Synthetic procedures, characterization of final compounds, all protocols for in vitro and in vivo experiments, ocular assessment, and kinase selectivity (PDF)
Author Present Address
§ E.-I.L.: Pliant Therapeutics, 700 Saginaw Drive, Suite 150, Redwood City, California 94063, United States.
Author Present Address
∥ K.S. and N.A.T.: Department of Chemistry, Princeton University, Princeton, New Jersey, 08544, United States.
The authors declare the following competing financial interest(s): The authors are employees of Gilead Sciences except for E.-I.L. and K.S. who were employed and N.A.T. who was an intern during this research. All authors except N.A.T. are shareholders of Gilead Sciences.
Supplementary Material
References
- Thorpe L. M.; Yuzugullu H.; Zhao J. J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F.; Yap T. A.; Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway?. Nat. Rev. Clin. Oncol. 2018, 15, 273–291. 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- Fruman D. A.; Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discovery 2014, 13, 140–156. 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S.; Wiederschain D.; Maira S.-M.; Loo A.; Miller C.; deBeaumont R.; Stegmeier F.; Yao Y.-M.; Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13057–13062. 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S.; Liu Z.; Zhang S.; Liu P.; Zhang L.; Hyun S.; Zhang L. J.; Signoretti S.; Loda M.; Roberts T. M.; Zhao J. J. Essential roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Chiang G. G.; Alaimo P. J.; Kenski D. M.; Ho C. B.; Coan K.; Abraham R. T.; Shokat K. M. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg. Med. Chem. 2004, 12, 4749–4759. 10.1016/j.bmc.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Blackman S. C.; Gainer S. D.; Suttle B. B.; Skordos K. W.; Greshock J. D.; Motwani M.; Roadcap L. T.; Hardwicke M. A. A.; Wooster R. F.. A phase I/IIa, first time in human, open-label dose escalation study of GSK2636771 in subjects with advanced solid tumors with PTEN deficiency. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research (AACR), Chicago, March 31–April 4, 2012, Abstract 1752.
- Yu H.; Moore M. L.; Erhard K.; Hardwicke M. A.; Lin H.; Luengo J. I.; McSurdy-Freed J.; Plant R.; Gu J.; Raha K.; Rominger C. M.; Schaber M. D.; Spengler M. D.; Rivero R. A. [3a,4] dihydropyrazolo[1,5a]pyrimidines: novel, potent, and selective phosphatidylinositol-3-kinase β -nhibitors. ACS Med. Chem. Lett. 2013, 4, 230–234. 10.1021/ml300330m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlaam B.; Cosulich S.; Degorce S.; Fitzek M.; Green S.; Hancox U.; Lambert-Van der Brempt C.; Lohmann J.-J.; Maudet M.; Morgentin R.; Pasquet M.-J.; Péru A.; Plé P.; Saleh T.; Vautier M.; Walker M.; Ward L.; Warin N. Discovery of (R)-8-(1-(3,5-difluorophenylamino)ethyl)-N, N-dimethyl-2-morpholino-4-oxo-4Hchromene-6-carboxamide (AZD8186): a potent and selective inhibitor of PI3Kf and PI3K for the treatment of PTEN-deficient cancers. J. Med. Chem. 2015, 58, 943–962. 10.1021/jm501629p. [DOI] [PubMed] [Google Scholar]
- Garces A. E.; Stocks M. J. Class 1 PI3K clinical candidates and recent inhibitor design strategies: a medicinal chemistry perspective. J. Med. Chem. 2019, 62, 4815–4850. and references cited therein. 10.1021/acs.jmedchem.8b01492. [DOI] [PubMed] [Google Scholar]
- Lin H.; Erhard K.; Hardwicke M. A.; Luengo J. I.; Mack J. F.; McSurdy-Freed J.; Plant R.; Raha K.; Rominger C. M.; Sanchez R. M.; Schaber M. D.; Schulz M. J.; Spengler M. D.; Tedesco R.; Xie R.; Zeng J. J.; Rivero R. A. Synthesis and structure-activity relationships of imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2230–2234. 10.1016/j.bmcl.2012.01.092. [DOI] [PubMed] [Google Scholar]
- Andrs M.; Korabecny J.; Jun D.; Hodny Z.; Bartek J.; Kuca K. Phosphatidylinositol 3-kinase (PI3K) and phosphatidylinositol 3-kinase-related kinase (PIKK) inhibitors: importance of the morpholine ring. J. Med. Chem. 2015, 58, 41–71. 10.1021/jm501026z. [DOI] [PubMed] [Google Scholar]
- Berndt A.; Miller S.; Williams O.; Le D. D.; Houseman B. T.; Pacold J. I.; Gorrec F.; Hon W.-C.; Liu Y.; Rommel C.; Gaillard P.; Ruckle T.; Schwarz M. K.; Shokat K. M.; Shaw J. P.; Williams R. L. The p110δ structure: mechanisms for selectivity and potency of new PI3K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. and references therein. 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault S.; Chandrasekhar J.; Cui Z.-H.; Evarts J.; Hao J.; Kaplan J. A.; Kashishian A.; Keegan K. S.; Kenney T.; Koditek D.; Lad L.; Lepist E.-I.; McGrath M. E.; Patel L.; Phillips B.; Therrien J.; Treiberg J.; Yahiaoui A.; Phillips G. Discovery of a phosphoinositide 3-kinase (PI3K) i// Inhibitor for the treatment of phosphatase and tensin homolog (PTEN) deficient tumors: building PI3Ku potency in a PI3Ko-selective template by targeting non-conserved Asp856. J. Med. Chem. 2017, 60, 1555–1567. 10.1021/acs.jmedchem.6b01821. [DOI] [PubMed] [Google Scholar]
- Patel L.; Chandrasekhar J.; Evarts J.; Forseth K.; Haran A. C.; Ip C.; Kashishian A.; Kim M.; Koditek D.; Koppenol S.; Lad L.; Lepist E.-I.; McGrath M. E.; Perreault S.; Puri K. D.; Villasenor A. G.; Somoza J. R.; Steiner B. H.; Therrien J.; Treiberg J.; Phillips G. Discovery of orally efficacious phosphoinositide 3-kinase D inhibitors with improved metabolic stability. J. Med. Chem. 2016, 59, 9228–9242. 10.1021/acs.jmedchem.6b01169. [DOI] [PubMed] [Google Scholar]
- Amzel L. M.; Huang C.-H.; Mandelker D.; Lengauer C.; Gabelli S. B.; Vogelstein B. Structural comparisons of class I phosphoinositide 3-kinase. Nat. Rev. Cancer 2008, 8, 665–669. 10.1038/nrc2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante S. R.; Fader L. D.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P. F.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- Smyth J. E.; Butler N. M.; Keller P. A. A twist of nature - the significance of atopisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. 10.1039/C4NP00121D. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar J.; Dick R.; Van Veldhuizen J.; Koditek D.; Lepist E. I.; McGrath M. E.; Patel L.; Phillips G.; Sedillo K.; Somoza J. R.; Therrien J.; Till N. A.; Treiberg J.; Villasenor A. G.; Zherebina Y.; Perreault S. Atropisomerism by design: discovery of a selective and stable phosphoinositide 3-kinase (PI3K) beta inhibitor. J. Med. Chem. 2018, 61, 6858–6868. 10.1021/acs.jmedchem.8b00797. [DOI] [PubMed] [Google Scholar]
- Closely related analogs (rac)-5-fluoro-4-(2-methyl-6-(pyridin-4-yl)-4-(4H-1,2,4-triazol-3-yl)-1H-benzo[d]imidazol-1-yl)quinoline and (rac)-8-chloro-5-fluoro-4-(2-methyl-6-(pyridin-4-yl)-4-(4H-1,2,4-triazol-3-yl)-1H-benzo[d]imidazol-1-yl)quinoline resulted in approximately 3-fold loss in potency against PI3K.
- A rotational barrier of 33 kcal/mol corresponds to a t95 of approximately 750 years at 40 °C. t95 refers to the time required to reach a 95:5 atropisomeric ratio (t95) from enantiopure (P)- or (M)-atropisomer.19
- Manevski N.; King L.; Pitt W. R.; Lecompte F.; Toseli F. Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J. Med. Chem. 2019, 62, 10955–10994. 10.1021/acs.jmedchem.9b00875. [DOI] [PubMed] [Google Scholar]
- Pryde D. C.; Dalvie D.; Hu Q.; Jones P.; Obach R. S.; Tran T.-D. Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. 10.1021/jm100888d. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Advanced characterization of (P)-14 and (P)-19 revealed that they do not inhibit major human CYP450 enzymes (IC50 > 25 uM) or hERG (IC50 > 25 μM) in binding assays. (P)-14 and (P)-19 were nonmutagenic in an Ames test and no substantial activity was noted when tested for off-target activity in a CEREP panel screen at 10 μM.
- Certal V.; Halley F.; Virone-Oddos A.; Delorme C.; Karlsson A.; Rak A.; Thompson F.; Filoche-Rommé B.; El-Ahmad Y.; Carry J.-C.; Abecassis P.-Y.; Lejeune P.; Vincent L.; Bonnevaux H.; Nicolas J.-P.; Bertrand T.; Marquette J.-P.; Michot N.; Benard T.; Below P.; Vade I.; Chatreaux F.; Lebourg G.; Pilorge F.; Angouillant-Boniface O.; Louboutin A.; Lengauer C.; Schio L. Discovery and optimization of pyrimidone indoline amide PI3KP inhibitors for the treatment of phosphatase and tensin homologue (PTEN)-deficient cancers. J. Med. Chem. 2014, 57, 903–920. 10.1021/jm401642q. [DOI] [PubMed] [Google Scholar]
- For these two compounds, the average method of predicting human Vss was shown to be comparable to the allometric method:; Berry L. M.; Li C.; Zhao Z. Species differences in distribution and prediction of human Vss from preclinical data. Drug Metab. Dispos. 2011, 39, 2103–2116. 10.1124/dmd.111.040766. [DOI] [PubMed] [Google Scholar]
- Maggs D. J.; Miller P. E.; Ofri R.. Slatter’s Fundamentals of Veterinary Ophthalmology, 6th ed.; Elsevier Inc.: St Louis, MO, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.