

Abstract

Novel tricyclic analogues were designed, synthesized, and evaluated as RORγt inverse agonists. Several of these compounds were potent in an IL-17 human whole blood assay and exhibited excellent oral bioavailability in mouse pharmacokinetic studies. This led to the identification of compound 5, which displayed dose-dependent inhibition of IL-17F production in a mouse IL-2/IL-23 stimulated pharmacodynamic model. In addition, compound 5 was studied in mouse acanthosis and imiquimod-induced models of skin inflammation, where it demonstrated robust efficacy comparable to a positive control. As a result of this excellent overall profile, compound 5 (BMS-986251) was selected as a clinically viable developmental candidate.
Keywords: RORγt, RORc, inverse agonist, IL-17, IL-23R, psoriasis
Retinoic acid-related orphan receptor γt (RORγt) is a nuclear hormone receptor (NHR) and a member of the RORγ subfamily.1,2 RORγt is expressed in the thymus and is responsible for the differentiation of CD4+T cells into Th17 cells.3 Hence, RORγt plays a significant role in the production of the pro-inflammatory cytokine IL-17 as well as other cytokines (GM-CSF, IL-21, and IL-22).4 Recently, anti-IL-17 biologics have been shown to be clinically effective against autoimmune diseases such as psoriasis.5−8 As these clinical agents are monoclonal antibodies, there is still a need for small molecule oral therapies modulating IL-17. As a result, there has been much interest in small molecule inhibitors of RORγt, including inverse agonists, as a strategy to suppress IL-17.9−18 Herein, we report our continued optimization of tricyclic19 inverse agonists of RORγt, which culminated in the identification of a viable clinical candidate.
As shown in Figure 1, we recently19 reported the synthesis and evaluation of the potent tricyclic RORγt inverse agonists 1 and 2. In an effort to optimize the potency and overall profile of these RORγt inverse agonists, we examined the X-ray crystal structure of 2(19) in RORγt and observed that the cyclohexane ring of 2 came in close proximity to helix 5 of the receptor (Figure 2). This region of helix 5 contains some lipophilic amino acids, including Ala368. We reasoned that a moiety substituted off the C2 or C3 position of the cyclohexane ring (or cyclic sulfone ring of 1) might bring about a favorable interaction with residues of helix 5 and thereby provide an opportunity to improve affinity for RORγt. From this model, the new C2 or C3 vectors did not appear to disrupt the key carboxylate (or sulfone of compound 1) interactions with Arg367 and Arg364. Likewise, the amide carbonyl of 1 or 2 was still able to engage the backbone NH of Phe377.
Figure 1.

Our previously reported RORγt inverse agonists.
Figure 2.
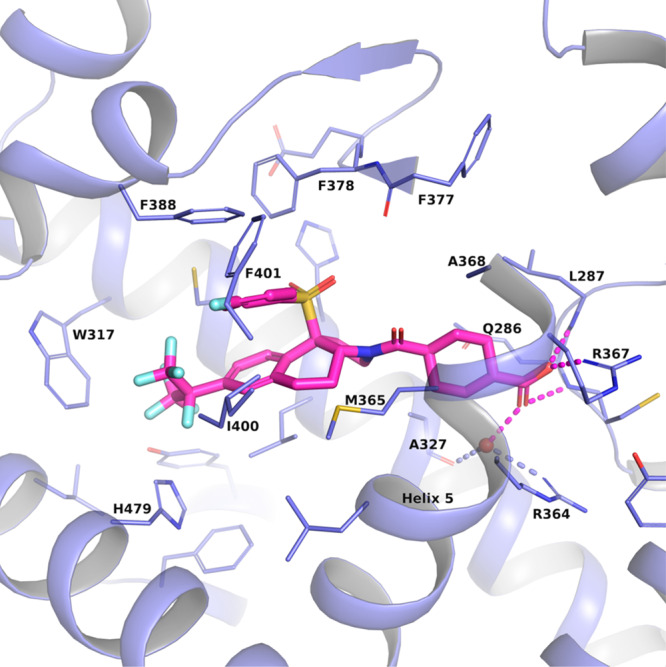
Crystal structure of compound 2 in RORγt (pdb id: 6U25).
Based on this rationale, we synthesized and evaluated analogues of compound 1 and 2 as outlined in Table 1. These compounds were assessed in our RORγt inverse agonist assay (RORγt GAL4 EC50, GAL-4 reporter assay in Jurkat cell line),19 in an IL-17 human whole blood assay (IL-17 hWB EC50),19 and in liver microsomes (LM t1/2) to assess stability. As shown previously,19 compound 1 was active in both the GAL-4 assay and the whole blood assay with excellent liver microsome stability. Starting with compounds substituted at the C2 of the cyclic sulfone, the first diastereomer 3 was almost 3-fold more active in the GAL-4 assay than compound 1, indicating that a new interaction with the receptor was possible. The second diastereomer 4 also had excellent GAL-4 activity similar to that of compound 3, but neither compound offered a real advantage over compound 1 as far as whole blood activity or liver microsome stability. For this reason, we shifted focus to our previously described carboxylate-based inverse agonists. Carboxylate 5 was 2-fold more active in both the GAL-4 assay and the whole blood assay than compound 1 and equipotent with compound 2. In addition, carboxylate 5 showed excellent microsomal stability. Switching to the 3-fluoro-phenylsulfone of compound 6 did not provide any additional advantage over compound 5. The methyl diastereomer 7 was at least 2-fold less potent in the GAL4/hWB assays as compared to compound 5. Moving the methyl to the C2 position was advantageous, as compound 8 retained the same excellent GAL-4 and whole blood activity as compound 5. As anticipated, based on previous structure–activity relationships (SAR),19 the 1,4-cis-cyclohexane analogues 9 and 11 were less active. Diastereomer 10 was one of the more active GAL-4 compounds with reasonable human whole blood activity.
Table 1. Evaluation of Methyl Substituted Analoguesa.

| # | R | RORγt GAL4 EC50 (nM) | IL-17 hWBb EC50 (nM) | LM t1/2: h, m, rc (min) |
|---|---|---|---|---|
| 1 | Figure 1 | 24 ± 12 | 43 ± 17 | >120, >120, >120 |
| 2 | Figure 1 | 7 ± 6 | 19 ± 8 | >120, >120, >120 |
| 3 | 4-F-Ph | 9 ± 6 (2) | 37 ± 19 (6) | >120, >120, >120 |
| 4 | 4-F-Ph | 9 ± 3 (2) | 44 ± 17 (4) | >120, 98, 33 |
| 5 | 4-F-Ph | 12 ± 6 (3) | 24 ± 6 (8) | >120, >120, >120 |
| 6 | 3-F-Ph | 11 ± 3 (2) | 38 ± 9 (6) | >120, >120, 107 |
| 7 | 4-F-Ph | 39 (1) | 44 ± 25 (2) | >120, >120, >120 |
| 8 | 3-F-Ph | 7 (1) | 27 ± 9 (4) | >120, >120, >120 |
| 9 | 3-F-Ph | 85 (1) | NDd | ND |
| 10 | 4-F-Ph | 7 ± 1 (2) | 40 ± 15 (3) | >120, >120, 73 |
| 11 | 4-F-Ph | 106 (1) | ND | ND |
EC50 values (n) are displayed as ± standard deviation.
Human whole blood assay (hWB).
Liver microsomes (LM) incubation: human (h), mouse (m), and rat (r).
ND = not determined.
With this activity information in hand, we selected three compounds for oral pharmacokinetic (PK) studies in mice. As shown in Table 2, all three compounds showed good oral exposure. Compound 10 had the best 24-h AUC, whereas compounds 5 and 8 were very similar and still promising. Even though compound 10 displayed the best mouse PK of the three compounds tested, it was about 2-fold less active in our human whole blood assay as compared to compound 5. Therefore, based on its overall profile, compound 5 was selected for further evaluation.
Table 2. Mouse PK Data for Select Compoundsa.
| # | Cmax (μM) | AUC24h (μM*h) | C24h (μM) |
|---|---|---|---|
| 5 | 11 ± 8 | 68 ± 5 | 0.52 ± 0.13 |
| 8 | 12 ± 2 | 72 ± 18 | 0.78 ± 0.65 |
| 10 | 9 ± 1 | 134 ± 10 | 2.2 ± 0.47 |
Balb/c mice dosed at 10 mg/kg PO. Values are means from three mice. Vehicle: 5% NMP; 76% PEG 400; 19% TPGS.
Table 3 outlines the full in vitro profile of compound 5. Compound 5 was selective not only against ROR family members (RORα and RORβ) but also against other nuclear receptors (PXR, LXRα, and LXRβ). Compound 5 was tested in a mouse reporter assay, and this correlated very well with the human values. The Caco-2 data confirms that compound 5 has good permeability with a low efflux ratio, which is consistent with the mouse PK study shown above. Compound 5 did not inhibit any of the CYP’s tested. With this promising profile in hand, compound 5 was selected for additional PK studies in other species (Table 4).
Table 3. Compound 5 Profilea.
| Assay | Result |
|---|---|
| RORγt GAL4 EC50 | 12 ± 6 nM |
| RORα GAL4 EC50 | >10000 nM |
| RORβ GAL4 EC50 | >10000 nM |
| IL-17 hWB EC50 | 24 ± 6 nM |
| mouse Th17 EC50 | 11 ± 2 nM |
| PXR/LXRα/LXRβ EC50b | >5000/>7500/>7500 nM |
| rCYP 1A2/2C8/2C9/2C19 IC50c | >20/16/>20/>20 μM |
| rCYP 2D6/3A4 BFC IC50 | >20/>20 μM |
| Caco-2 A-B (nm/s) | 240 nm/s |
| Caco-2 efflux ratio | 0.5 |
| Protein binding % free h/m/r | 1.2/1.6/2.1 |
Protein binding: human (h), mouse (m), rat (r).
PXR, pregnane X receptor; LXR, liver X receptor.
rCyP, recombinant cytochrome P450.
Table 4. Pharmacokinetic Data for Compound 5 in Preclinical Speciesa.
|
iv |
po |
||||||
|---|---|---|---|---|---|---|---|
| species | dose (mg/kg) iv/po | CL (mL min–1 kg–1) | Vss (L/kg) | t1/2 (h) | Cmax (μM) | AUC24h (μM h) | F (%) |
| mouse | 2/4 | 2.7 | 1.9 | 7.7 | 4.8 ± 0.3 | 37 | ∼100 |
| rat | 2/4 | 1.3 ± 0.3 | 1.2 ± 0.3 | 11 ± 0.8 | 4.7 ± 0.5 | 64 ± 3.4 | 94 |
| dog | 1/1 | 0.18 ± 0.04 | 0.5 ± 0.1 | 36 ± 3 | 6.4 ± 1.0 | 120 ± 21 | ∼100 |
| cyno | 1/1 | 1.1 ± 0.2 | 2.0 ± 0.4 | 33 ± 4 | 3.1 ± 0.3 | 35 ± 3.1 | ∼100 |
Values are means obtained from three or more animals.
As shown in Table 4, compound 5 displayed excellent oral bioavailability across all the four species in addition to exhibiting low clearance. The half-life in rodent was excellent (7.7 h to 11 h); however, the half-life in dog and cyno did trend high (33 h to 36 h). Given these favorable PK results, compound 5 was selected for mouse in vivo efficacy studies.
Compound 5 was first assessed in an IL-2/IL-23 stimulated mouse pharmacodynamic (PD) model.19 In this model, naïve C57BL/6 female mice (7–9 weeks of age from Charles River) were injected intraperitoneally with 5 μg/ms of IL-2 at −24, 0, and 23 h and 10 μg/ms at 7 h. In addition, IL-23 (dose of 1 μg/ms) was injected intraperitoneally at 0, 7, and 23 h. Compound 5 was dosed once a day orally at 0.13, 0.79, and 4.76 mg/kg (vehicle: 1-methyl-2-pyrrolidinone (NMP):PEG300:TPGS, 5:76:19 v/v) 30 min prior to the 0 h time point. Blood was collected for exposure and serum was collected for IL-17F luminex analysis at the 30 h time point. As shown in Figure 3, compound 5 displayed a dose-dependent reduction of the IL-17F produced. Compound 5 also performed better than compound 2 in this PD model (data not shown). These promising PD results prompted us to take compound 5 into the mouse acanthosis model (a preclinical model of psoriasis).
Figure 3.

Oral dosing of compound 5 inhibits IL-17F production in a mouse PD model. *: P < 0.05 (one-way ANOVA) versus vehicle group.
As shown in Figure 4, acanthosis20,21 was induced with recombinant humanized IL-23 injected every other day into the right ear of C57BL/6 female mice until the last injection on day 9. A starting “baseline” measurement of the ear (before first injection) was made on day 0, and thereafter, ear thickness was measured every other day prior to the next ear injection. Compound 5 was dosed orally approximately 2 h before the first IL-23 injection and continued twice daily until day 9 (study ended on day 10). The placebo/control group was dosed with blank vehicle, and a human anti-IL-23 was administered SC as a positive control at doses of 3 mg/kg on days 0, 3, and 7 (human anti-IL-23 dosed at maximal efficacy). As shown in Figure 4, all doses of compound 5 resulted in reduced ear thickness, and the 15 mg/kg dose of 5 was comparable to the human anti-IL-23.
Figure 4.

Oral efficacy of compound 5 in mouse acanthosis model.
Compound 5 was also tested in the well-known imiquimod (IMQ)-induced model22−24 of skin inflammation. In this model, IMQ cream was applied to the backs of C57BL/6 mice from day 0 to day 7. The percent change from baseline for skin thickness was measured daily (except for day 5) over 8 days. Compound 5 was dosed orally at 2, 7, and 20 mg/kg BID each day from day 0 to day 7 (Figure 5). Compound 5 significantly reduced IMQ-induced skin thickening at all dose levels compared to the placebo/control group, which was dosed with blank vehicle. An anti-mouse IL-23 antibody was dosed as a positive control at 10 mg/kg on day −1 and day 3. As shown in Figure 5, compound 5 dosed at 20 mg/kg BID was comparable in efficacy to the anti-mouse IL-23 antibody.
Figure 5.

Efficacy of compound 5 dosed orally in IMQ mouse model.
The synthesis of compound 5 is illustrated in Scheme 1, starting from the previously described tricyclic intermediate 12.19 Compound 12 was coupled to (1R,2S)-2-methyl-4-oxocyclohexane-1-carboxylic acid,25 which was generated via a pig liver esterase (PLE)26 cleavage of the corresponding ester. The resulting ketone 13 was converted to a mixture of regioisomeric enol triflates 14.27 A palladium-catalyzed carbonylation28 of 14 in the presence of methanol gave a regioisomeric mixture of methyl esters 15. This ester mixture 15 was hydrogenated with Crabtree’s catalyst29,30 to give a single diastereomer 16. As a last step, ester 16 was saponified with LiOH to give the carboxylate 5.
Scheme 1. Synthesis of Compound 5.

Reagents and conditions: (a) (1R,2S)-2-methyl-4-oxocyclohexane-1-carboxylic acid, HATU, NMM, DMF, 78%; (b) KN(TMS)2, N-phenyl-bis(trifluoromethanesulfonimide), THF, −78 °C; (c) Pd(PPh3)2Cl2, CO (1 atm), MeOH, DMF, 75%; (d) Crabtree’s catalyst, H2 (1 atm), DCM, 89%; (e) LiOH, MeOH, H2O, THF, 65%.
As shown in Figure 6, we were able to obtain a crystal structure of compound 5 in RORγt (pdb id: 6VQF), and it confirmed the normal interactions of the carboxylate to Arg364/Arg367 and amide carbonyl to Phe377 (Figure 6A). However, the structure of compound 5 was unique, as the cyclohexane of 5 was rotated approximately 75 degrees from that observed for the cyclohexane in the crystal structure of compound 2 (see Figure 6B). It appears that the C3 methyl of compound 5 forces the cyclohexane to rotate back away from helix 5 to make an interaction with Ala368 (distance of C3-methyl of 5 to Ala368 side chain methyl is 3.6 Å). This in turn brings the bottom portion of the cyclohexane of 5 in close proximity to helix 5 as it packs up against the helix. As mentioned, this is a unique orientation of the cyclohexane ring that we have not observed in other crystal structures.
Figure 6.

(A) Overlay of two crystal structures in RORγt: compound 5 (green; pdb id: 6VQF) and compound 2 (compound in purple and protein excluded). (B) Overlay of the crystal structure ligands of compounds 5 and 2.
In summary, we have investigated the optimization of our tricyclic RORγt inverse agonists via a potential interaction with the receptor’s helix 5. Evaluation of these new analogues led to the identification of compound 5 as a potent and selective RORγt inverse agonist. Compound 5 demonstrated excellent oral bioavailability and metabolic stability across species. In addition, compound 5 was taken into a mouse PD model where it showed a dose-dependent reduction of the pro-inflammatory cytokine IL-17F. Compound 5 was also studied in mouse acanthosis and imiquimod-induced models (preclinical models of psoriasis), where it demonstrated robust efficacy comparable to that of a positive control. As a result of the above profile, compound 5 (BMS-986251) was selected as a viable clinical candidate (clinical trial ID: NCT03329885).
Acknowledgments
We would like to thank our colleagues in the Department of Discovery Synthesis at the Biocon-BMS R&D Center (Bengaluru, India) for the synthesis of intermediate 12.
Glossary
Abbreviations
- Ala
alanine
- Arg
arginine
- AUC
area under the curve
- bid
twice a day
- BMS
Bristol-Myers Squibb
- Boc
butyloxycarbonyl
- C
concentration
- CL
clearance
- DCM
dichloromethane
- DMF
dimethylformamide
- EC
efficacious concentration
- F
bioavailability
- Glu
glutamic acid
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HATU
hexafluorophosphate azabenotriazole tetramethyl uronium
- hWB
human whole blood
- IL
interleukin
- IMQ
imiquimod
- iv
intravenous
- Leu
leucine
- LM
liver microsome
- LXR
liver X receptor
- M
molar
- ND
not determined
- NMP
N-methylpyrrolidinone
- NR
nuclear receptor
- PD
pharmacodynamics
- PEG
polyethylene glycol
- PBS
phosphate-buffered saline
- Phe
phenyl alanine
- PK
pharmacokinetic
- po
per os, oral dose
- PXR
pregnane X receptor
- qd
once a day
- rCyP
recombinant cytochrome P450
- ROR
receptor-related orphan receptor
- SAR
structure activity relationship
- SC
subcutaneous
- SFC
supercritical fluid chromatography
- Th17
T helper 17 cells
- THF
tetrahydrofuran
- TPGS
tocopheryl polyethylene glycol succinate
- Vss
volume of distribution.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00063.
Experimental details and compound characterization data for all target compounds are available. Details of the IMQ-induced model, the liver microsome t1/2 assay, and the cocrystal structure method for compound 5 in RORγt (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jetten A. M. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signaling 2009, 7, 1–32. 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S.; Cua D. J. The emerging landscape of RORγt biology. Immunity 2014, 40, 451–452. 10.1016/j.immuni.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Ivanov I. I.; McKenzie B. S.; Zhou L.; Tadokoro C. E.; Lepelley A.; Lafaille J. J.; Cua D. J.; Littman D. R. The orphan nuclear receptor RORct directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Pappu R.; Ramirez-Carrozzi V.; Ota N.; Ouyang W.; Hu Y. The IL-17 family cytokines in immunity and disease. J. Clin. Immunol. 2010, 30, 185–195. 10.1007/s10875-010-9369-6. [DOI] [PubMed] [Google Scholar]
- Papp K. A.; Leonardi C.; Menter A.; Ortonne J.-P.; Krueger J. G.; Kricorian G.; Aras G.; Li J.; Russell C. B.; Thompson E. H. Z.; Baumgartner S. Brodalumab, an anti–interleukin-17–receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- Leonardi C.; Matheson R.; Zachariae C.; Cameron G.; Li L.; Edson-Heredia E.; Braun D.; Banerjee S. Anti–interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- Langley R. G.; Elewski B. E.; Lebwohl M.; Reich K.; Griffiths C. E. M.; Papp K.; Puig L.; Nakagawa H.; Spelman L.; Sigurgeirsson B.; Rivas E.; Tsai T.-F.; Wasel N.; Tyring S.; Salko T.; Hampele I.; Notter M.; Karpov A.; Helou S.; Papavassilis C. Secukinumab in plaque psoriasis — results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- Prinz I.; Sandrock I.; Mrowietz U. Interleukin-17 cytokines: effectors and targets in psoriasis — a breakthrough in understanding and treatment. J. Exp. Med. 2020, 217, 1–12. 10.1084/jem.20191397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N.; Guo H.; Wang Y. Retinoic acid receptor-related orphan receptor gamma-t (RORγt) inhibitors in clinical development for the treatment of autoimmune diseases: a patent review (2016-present). Expert Opin. Ther. Pat. 2019, 29, 663–674. 10.1080/13543776.2019.1655541. [DOI] [PubMed] [Google Scholar]
- Pandya V. B.; Kumar S.; Sachchidanand; Sharma R.; Desai R. C. Combating autoimmune diseases with retinoic acid receptor-related orphan receptor-γ (RORγ or RORc) inhibitors: hits and misses. J. Med. Chem. 2018, 61, 10976–10995. 10.1021/acs.jmedchem.8b00588. [DOI] [PubMed] [Google Scholar]
- Jetten A. M.; Cook D. N. (Inverse) Agonists of retinoic acid–related orphan receptor γ: regulation of immune responses, inflammation, and autoimmune disease. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 371–390. 10.1146/annurev-pharmtox-010919-023711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Yang X.; Liang Y.; Xie H.; Dai Z.; Zheng G. Transcription factor retinoid-related orphan receptor γt: a promising target for the treatment of psoriasis. Front. Immunol. 2018, 9, 1–8. 10.3389/fimmu.2018.01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner S. M.; Zbieg J. R.; Crawford J. J. RORc antagonists and inverse agonists: a patent review. Expert Opin. Ther. Pat. 2017, 27, 101–112. 10.1080/13543776.2017.1236918. [DOI] [PubMed] [Google Scholar]
- Cyr P.; Bronner S. M.; Crawford J. J. Recent progress on nuclear receptor RORc modulators. Bioorg. Med. Chem. Lett. 2016, 26, 4387–4393. 10.1016/j.bmcl.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Kojetin D. J.; Burris T. P. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discovery 2014, 13, 197–216. 10.1038/nrd4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauber B. P.; Magnuson S. Modulators of the nuclear receptor retinoic acid receptor-related orphan receptor-c (RORc or RORc). J. Med. Chem. 2014, 57, 5871–5892. 10.1021/jm401901d. [DOI] [PubMed] [Google Scholar]
- Dhar T. G. M.; Zhao Q.; Markby D. W. Targeting the nuclear hormone receptor RORγt for the treatment of autoimmune and inflammatory disorders. Annu. Rep. Med. Chem. 2013, 48, 169–182. 10.1016/B978-0-12-417150-3.00012-0. [DOI] [Google Scholar]
- Huh J. R.; Littman D. R. Small molecule inhibitors of RORγt: Targeting Th17 cells and other applications. Eur. J. Immunol. 2012, 42, 2232–2237. 10.1002/eji.201242740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux D.; Duan J. J. W.; Shi Q.; Cherney R. J.; Srivastava A. S.; Cornelius L.; Batt D. G.; Liu Q.; Beaudoin-Bertrand M.; Weigelt C. A.; Khandelwal P.; Vishwakrishnan S.; Selvakumar K.; Karmakar A.; Gupta A. K.; Basha M.; Ramlingam S.; Manjunath N.; Vanteru S.; Karmakar S.; Maddala N.; Vetrichelvan M.; Gupta A.; Rampulla R. A.; Mathur A.; Yip S.; Li P.; Wu D.-R.; Khan J.; Ruzanov M.; Sack J. S.; Wang J.; Yarde M.; Cvijic M. E.; Li S.; Shuster D. J.; Borowski V.; Xie J. H.; McIntyre K. W.; Obermeier M. T.; Fura A.; Stefanski K.; Cornelius G.; Hynes J.; Tino J. A.; Macor J. E.; Salter-Cid L.; Denton R.; Zhao Q.; Carter P. H.; Dhar T. G. M. Rationally designed, conformationally constrained inverse agonists of RORγt — identification of a potent, selective series with biologic-like in vivo efficacy. J. Med. Chem. 2019, 62, 9931–9946. 10.1021/acs.jmedchem.9b01369. [DOI] [PubMed] [Google Scholar]
- Rizzo H. L.; Kagami S.; Phillips K. G.; Kurtz S. E.; Jacques S. L.; Blauvelt A. IL-23–mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J. Immunol. 2011, 186, 1495–1502. 10.4049/jimmunol.1001001. [DOI] [PubMed] [Google Scholar]
- See our protocols:Duan J. J.-W.; Lu Z.; Jiang B.; Stachura S.; Weigelt C. A.; Sack J. S.; Khan J.; Ruzanov M.; Galella M. A.; Wu D.-R.; Yarde M.; Shen D.-R.; Shuster D. J.; Borowski V.; Xie J. H.; Zhang L.; Vanteru S.; Gupta A. K.; Mathur A.; Zhao Q.; Foster W.; Salter-Cid L. M.; Carter P. H.; Dhar T. G. M. Structure-based discovery of phenyl (3-Phenylpyrrolidin-3-yl)sulfones as selective, orally active RORγt inverse agonists. ACS Med. Chem. Lett. 2019, 10, 367–373. 10.1021/acsmedchemlett.9b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L.; Mourits S.; Voerman J. S.; Kant M.; Boon L.; Laman J. D.; Cornelissen F.; Mus A.-M.; Florencia E.; Prens E. P.; Lubberts E. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- Flutter B.; Nestle F. O. TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur. J. Immunol. 2013, 43, 3138–3146. 10.1002/eji.201343801. [DOI] [PubMed] [Google Scholar]
- Hawkes J. E.; Gudjonsson J. E.; Ward N. L. The snowballing literature on imiquimod-induced skin inflammation in mice: a critical appraisal. J. Invest. Dermatol. 2017, 137, 546–549. 10.1016/j.jid.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A.; Benetti S.; Bianchi A.; Casolari A.; Pollini G. P.; Romagnoli R.; Spalluto G.; Zanirato V. Enantioselective synthesis of the hexahydronaphthalene nucleus of (−)-compactin from ethyl (1R,2S)-2-methyl-4-oxocyclohexanecarboxylate and 2-(3-nitropropyl)-1,3-dioxolane as four carbon bifunctional annelating agent. Tetrahedron 1994, 50, 11743–11754. 10.1016/S0040-4020(01)85667-2. [DOI] [Google Scholar]
- Tamm C. Pig liver esterase catalyzed hydrolysis: substrate specificity and stereoselectivity. Pure Appl. Chem. 1992, 64, 1187–1191. 10.1351/pac199264081187. [DOI] [Google Scholar]
- Mc Murry J. E.; Scott W. J. A method for the regiospecific synthesis of enol triflates by enolate trapping. Tetrahedron Lett. 1983, 24, 979–982. 10.1016/S0040-4039(00)81581-6. [DOI] [Google Scholar]
- Cacchi S.; Morera E.; Ortar G. Palladium-catalyzed carbonylation of enol triflates. A novel method for one-carbon homologation of ketones to α,β-unsaturated carboxylic acid derivatives. Tetrahedron Lett. 1985, 26, 1109–1112. 10.1016/S0040-4039(00)98525-3. [DOI] [Google Scholar]
- Crabtree R. H.; Davis M. W. Directing effects in homogeneous hydrogenation with [Ir(cod)(PCy3)(py)]PF6. J. Org. Chem. 1986, 51, 2655–2661. 10.1021/jo00364a007. [DOI] [Google Scholar]
- Verendel J. J.; Pàmies O.; Diéguez M.; Andersson P. G. Asymmetric hydrogenation of olefins using chiral Crabtree-type catalysts: scope and limitations. Chem. Rev. 2014, 114, 2130–2169. 10.1021/cr400037u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.