

Abstract

Carbamoyl phosphate synthetase 1 (CPS1) is a potential synthetic lethal target in LKB1-deficient nonsmall cell lung cancer, where its overexpression supports the production of pyrimidine synthesis. In other cancer types, CPS1 overexpression and activity may prevent the accumulation of toxic levels of intratumoral ammonia to support tumor growth. Herein we report the discovery of a novel series of potent and selective small-molecule inhibitors of CPS1. Piperazine 2 was initially identified as a promising CPS1 inhibitor through a high-throughput screening effort. Subsequent structure–activity relationship optimization and structure-based drug design led to the discovery of piperazine H3B-616 (25), a potent allosteric inhibitor of CPS1 (IC50 = 66 nM).
Keywords: Carbamoyl-phosphate synthetase 1, CPS1, allosteric inhibitors, high-throughput screening, cancer
Carbamoyl phosphate synthetase 1 (CPS1) catalyzes the first step in ammonia detoxification through the urea cycle, generating carbamoyl phosphate from ammonia, bicarbonate, and two molecules of ATP.1,2 In cancer, CPS1 can be overexpressed in several tumor lineages, where its activity is responsible for preventing the accumulation of toxic intratumoral ammonia in some cancer types,3 while in others carbamoyl phosphate generated by CPS1 feeds into the pyrimidine biosynthetic pathway to support rapid tumor growth by maintaining a constant supply of pyrimidine nucleotides.4−6 Overexpression of CPS1 is associated with poor prognosis in nonsmall cell lung cancer (NSCLC),4,7 colon cancer,8−10 cholangiocarcinoma,11 and a subset of glioblastoma cancers,12 indicating that multiple cancer types may exploit CSP1 overexpression to support aggressive tumor growth through these mechanisms. These observations suggest that small-molecule inhibitors of CPS1 may provide a therapeutic benefit for cancer patients in these indications.
We recently reported the identification of the first small-molecule inhibitor of CPS1, H3B-120 (1), through a high-throughput screening (HTS) campaign (Figure 1).13 Kinetic studies and a cocrystal structure of inhibitor 1 bound to CPS1 demonstrated that 1 achieves selective CPS1 inhibition by binding to an allosteric pocket in the integrating domain. Binding to the allosteric pocket leads to a conformational change in the carbamate synthetase domain, blocking ATP binding and subsequent catalysis. Furthermore, HTS hit compound 1 demonstrated CPS1 cellular target engagement by blocking CPS1 activity in both the urea cycle and support of the pyrimidine biosynthetic pathway.13 However, the use of compound 1 as a biological tool to probe CPS1 cancer biology was limited by both its low potency (1.5 μM) and less desirable CYP-mediated metabolic stability and physicochemical properties.
Figure 1.

Structure of CPS1 inhibitors identified via HTS.
Herein we discuss the structure–activity relationship (SAR) of HTS hit 2, a symmetrical piperazine with moderate activity (IC50 = 7.8 μM) (Figure 1).14 Initial SAR investigation focused on the exploration of the piperazine core, evaluating ring size, substituents, and stereochemistry (3–12, Table 1). Analogues were synthesized initially in a one-pot approach via bisacylation of various piperazine cores.15−17 Alternatively, a stepwise approach using differentially protected piperazine cores could be employed for the rapid synthesis of unsymmetrical analogues.
Table 1. Piperazine Core SAR Exploration.


IC50 values were determined in a biochemical assay measuring ADP formation from CPS1 in the presence of the inhibitor. See the Supporting Information for methods. The reported IC50 values are averages of at least two experimental replicates (N = 2).
Focusing on the piperazine core of 2, removal of a single nitrogen atom (3) or expansion to the corresponding seven-membered diazepine ring (4) led to modest loss of activity, while modification of the piperazine core through the addition of a methyl group (5) retained the activity. Further derivatization of the piperazine core with methyl substitution was tolerated for both the 2,6-dimethyl (6) and 2,5-dimethyl (8) analogues, while 2,3-disubstitution (7) or bridging of the piperazine (9) led to significant loss of inhibition. Building on the identification of 6, we next investigated the effect of stereochemistry on the piperazine core by synthesizing all of the stereoisomers of 6. While isomers 10 and 11 demonstrated activity comparable to that of the mixture 6, the (2R,6R) isomer H3B-374 (12) exhibited an 18-fold enhancement in activity (IC50 = 360 nM, H = 1.2).
Building on the discovery of 12, we investigated the R1 and R2 amide SAR (13–23, Table 2). With regard to R1 (13–18), increasing the size of the methoxy group to ethoxy led to a 2-fold improvement in activity (13), while replacement of the 4-methoxy with 4-methyl (14) or 4-F (15) led to significant loss of activity. Furthermore, switching the methoxy group from the 4-position to either the 3-position (16) or the 5-position (17) led to significant loss of activity. Alternatively, migration of the fluorine group from the 2-position to the 3-position (18) led to only a 2-fold loss of activity. With regard to R2 (19–23), expansion of the 4-methoxy group to ethoxy (19) was tolerated, while a further increase in size to isopropoxy (20) led to significant loss of inhibition relative to 12. Unlike R1, replacement of 4-methoxy with 4-methyl (21) was tolerated, but loss of activity was observed upon substitution with 4-fluoro (22) or 5-methoxy (23). While the SAR generated gave us a better understanding of key structural features required for activity, further increases in potency by modification of R1 and R2 proved unfruitful.
Table 2. Initial Amide SAR for the (2R,6R)-2,6-Dimethylpiperazine Core.
![]()

IC50 values were determined in a biochemical assay measuring ADP formation from CPS1 in the presence of the inhibitor. See the Supporting Information for methods. The reported IC50 values are averages of at least two experimental replicates (N = 2).
To help understand the aforementioned SAR and enable further optimization, we solved a cocrystal structure of 12 bound to CPS1 at 2.62 Å resolution (Figure 2). The crystal structure reveals that 12 binds to an allosteric pocket in the integrating domain of CPS1, immediately adjacent to the ATP binding pocket of the carbamate synthesis domain. Comparison of the structure of 12 with the previously reported structure of 1 (PDB code 6UEL) shows that the two ligands bind in the same allosteric pocket.13 Most of the interactions are hydrophobic, but there are also two key H-bonds formed between backbone amides of the protein (W776 and I851) and the pseudosymmetric amide oxygens on either side of the piperazine (Figure 2). These H-bonds dictate the ligand orientation in the pocket. Moreover, binding of inhibitor 12 to CPS1 stabilizes the flexible K loop (V653–H659) in a conformation that is not compatible with ATP binding and catalysis.13 The conformation of this loop is stabilized by the salt bridge from D654 to R850 and by hydrophobic interactions from M656 to the ligand. This nonproductive conformation explains the inhibitory activity of the compound.
Figure 2.
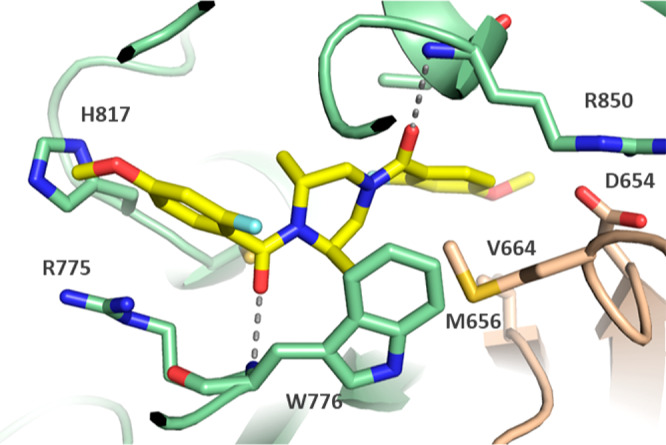
Crystal structure of H3B-374 (12) bound in the allosteric binding pocket of CPS1 (PDB code 6W2J). Green denotes residues from the integrating domain, and wheat color indicates those from the carbamate synthesis domain. The dashed lines indicate H-bonds to backbone amide nitrogens.
The piperazine core of 12 adopts a staggered flat confirmation that is enforced by the methyl groups at the 2- and 6-position, allowing R1 and R2 to make optimal interactions within the pocket. Hence, it can be rationalized that the lower IC50 values for 7, 8, and 9 are due to the fact that the core is less amenable to the twisted conformation. Another interesting feature of the protein–ligand interaction is a CH−π engagement between the 2-Me of the piperazine and W776. With regard to R1, the 4-OMe of 12 sits in a pocket created by V664, F890, I773, and I873, a hydrophobic interaction that leads to a 2-fold improvement in potency when R1 is 4-OEt (13). However, the observed decrease in activity for 16 and 17 can be explained by the apparent clashes of the 3-OMe and 5-OMe groups with L813 and D654, respectively. The pocket of R2 is partially solvent-exposed, which is consistent with the relatively tolerant SAR observed for 21 and 22. However, the space around the 5-OMe substituent of 23 is restricted by H817, and this potential steric repulsion could explain the observed 14-fold loss of activity.
Having solved the cocrystal structure of 12 (Figure 2), we focused on optimizing its potency. D654 was identified from the cocrystal structure as a potential binding interaction that could be targeted through R1 optimization. In this regard, analogues of 12 were designed to incorporate functional groups that could form a binding interaction with D654 (Table 3). While addition of the 5-acetamide (24) led to loss of activity, incorporation of 1H-indole (H3B-616, 25; IC50 = 66 nM, H = 1.4) at R1 led to a 5-fold improvement in potency relative to 12. Likewise, the corresponding 2-methyl-1H-indole analogue 26 exhibited an improvement in activity, though the bulkier substituent was slightly less tolerated. To help demonstrate this SAR, N-methylindole 27 was synthesized and, as predicted, was not tolerated (IC50 = 31 μM). This was further demonstrated with the indole regioisomer 28 which negated the gain in activity through Asp654 binding. Furthermore, expansion of the SAR to the corresponding imidazole 29, indoline 30, and 1-methylindoline 31 analogues did not result in any significant gain in activity.
Table 3. SAR Exploration Targeting Asp654.
![]()

IC50 values were determined in a biochemical assay measuring ADP formation from CPS1 in the presence of the inhibitor. See the Supporting Information for methods. The reported IC50 values are averages of at least two experimental replicates (N = 2).
CPS1 is one of two genes with carbamoyl phosphate synthetase activity in the human genome, the second gene being CAD. CAD is a trifunctional enzyme that catalyzes the first three steps of de novo pyrimidine synthesis and exhibits CPS2, aspartate transcarbamoylase, and dihydroorotase activity. Since CAD facilitates de novo pyrimidine synthesis in most cell types, a nonselective CPS inhibitor would be expected to encounter toxicity issues associated with CPS2 inhibition. We therefore assessed the selectivity of compound H3B-616 (25) by measuring inhibition of the carbamoyl phosphate synthetase activity of CPS1 and CPS2 in a biochemical assay.13 While 25 robustly inhibited CPS1 (IC50 = 0.066 μM), it demonstrated no appreciable inhibition of CPS2 (IC50 ≥ 100 μM).
We previously demonstrated targeted cellular inhibition of CPS1 by measuring inhibition of urea production from primary human hepatocytes treated with CPS1 inhibitors.13 In this cellular assay, biochemically active compounds 25 and 26 demonstrated robust cellular inhibition of CPS1 with IC50 values of 0.24 μM and 1.0 μM, respectively, while biochemically inactive compounds 27 and 28 showed no activity (Figure 3).
Figure 3.
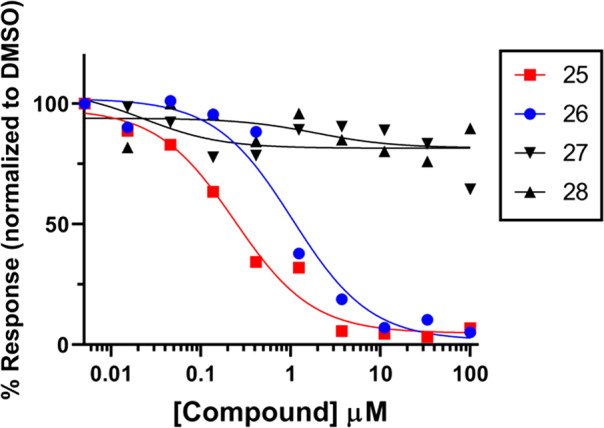
Targeted cellular inhibition of CPS1 activity. Inhibition of urea production from primary human hepatocytes by 25, 26, 27, and 28 is shown. Primary human hepatocytes were incubated with 10 mM NH4Cl in the presence of compound at the indicated concentration. Urea secreted into the medium was measured after 16 h. The data are provided as a representative graph. Replicate experiments yielded similar results.
In summary, we have identified and developed a series of allosteric small-molecule inhibitors of carbamoyl phosphate synthetase 1 (CPS1). Starting from HTS hit 2 (IC50 = 7.8 μM), initial SAR followed by structure-based drug design furnished CPS1 inhibitor H3B-616 (25) with potent biochemical (IC50 = 66 nM) and cellular (IC50 = 240 nM) activity. A cocrystal structure of 12 bound to CPS1 demonstrated an allosteric pocket in the integrating domain. In comparison to our previously reported inhibitor 1, 25 demonstrated improved biochemical potency (66 nM vs 1.5 μM) and a significant improvement in human hepatocyte stability (8.7 vs 35 μL/min/10-6 cells), favorable permeability (Caco2 A-B = 12.3 × 10–6 cm/s), and good kinetic solubility (148 μM, pH 7.4). Further studies are ongoing to investigate the application of this tool compound in the context of liver kinase B1 (LKB1)-deficient NSCLC.
Acknowledgments
We thank all of the employees of H3 Biomedicine Inc. for their helpful discussions related to this project.
Glossary
Abbreviations
- CPS1
carbamoyl-phosphate synthase 1
- HTS
high-throughput screening
- Bn
benzyl
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00145.
Experimental details, spectral data for selected compounds, X-ray crystallographic analysis, and biological assay protocols (PDF)
Accession Codes
The coordinates have been deposited in the PDB with accession code 6W2J.
Author Contributions
The manuscript was written through contributions of all authors.
This work was funded by H3 Biomedicine Inc.
The authors declare no competing financial interest.
Supplementary Material
References
- Adeva M. M.; Souto G.; Blanco N.; Donapetry C. Ammonium Metabolism in Humans. Metab., Clin. Exp. 2012, 61, 1495–1511. 10.1016/j.metabol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- de Cima S.; Polo L. M.; Díez-Fernández C.; Martínez A. I.; Cervera J.; Fita I.; Rubio V. Structure of Human Carbamoyl Phosphate Synthetase: Deciphering the on/off Switch of Human Ureagenesis. Sci. Rep. 2015, 5, 16950. 10.1038/srep16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Mao Y.; Zhao L.; Li L.; Wu J.; Zhao M.; Du W.; Yu L.; Jiang P. P53 Regulation of Ammonia Metabolism through Urea Cycle Controls Polyamine Biosynthesis. Nature 2019, 567 (7747), 253–256. 10.1038/s41586-019-0996-7. [DOI] [PubMed] [Google Scholar]
- Kim J.; Hu Z.; Cai L.; Li K.; Choi E.; Faubert B.; Bezwada D.; Rodriguez-Canales J.; Villalobos P.; Lin Y.-F.; et al. CPS1 Maintains Pyrimidine Pools and DNA Synthesis in KRAS/LKB1-Mutant Lung Cancer Cells. Nature 2017, 546, 168–172. 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çeliktaş M.; Tanaka I.; Chandra Tripathi S.; Fahrmann J. F.; Aguilar-Bonavides C.; Villalobos P.; Delgado O.; Dhillon D.; Dennison J. B.; Ostrin E. J.; et al. Role of CPS1 in Cell Growth, Metabolism, and Prognosis in LKB1-Inactivated Lung Adenocarcinoma. J. Natl. Cancer Inst. 2017, 109, djw231. 10.1093/jnci/djw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S.; Adler L.; Karathia H.; Carmel N.; Rabinovich S.; Auslander N.; Keshet R.; Stettner N.; Silberman A.; Agemy L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570. 10.1016/j.cell.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham-Danis C.; Gehrke S.; Danis E.; Rozhok A. I.; Daniels M. W.; Gao D.; Collins C.; Paola J. T. D.; D’Alessandro A.; DeGregori J. Urea Cycle Sustains Cellular Energetics upon EGFR Inhibition in EGFR-Mutant NSCLC. Mol. Cancer Res. 2019, 17, 1351–1364. 10.1158/1541-7786.MCR-18-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May D.; Pan S.; Crispin D. A.; Lai K.; Bronner M. P.; Hogan J.; Hockenbery D. M.; McIntosh M.; Brentnall T. A.; Chen R. Investigating Neoplastic Progression of Ulcerative Colitis with Label-Free Comparative Proteomics. J. Proteome Res. 2011, 10, 200–209. 10.1021/pr100574p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan A.; Ramar K.; Ramalingam S. Computational Identification of Novel Stage-Specific Biomarkers in Colorectal Cancer Progression. PLoS One 2016, 11, e0156665 10.1371/journal.pone.0156665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-Y.; Li C.-F.; Lin C.-Y.; Lee S.-W.; Sheu M.-J.; Lin L.-C.; Chen T.-J.; Wu T.-F.; Hsing C.-H. Overexpression of CPS1 Is an Independent Negative Prognosticator in Rectal Cancers Receiving Concurrent Chemoradiotherapy. Tumor Biol. 2014, 35, 11097–11105. 10.1007/s13277-014-2425-8. [DOI] [PubMed] [Google Scholar]
- Ma S.-L.; Li A.-J.; Hu Z.-Y.; Shang F.-S.; Wu M.-C. Co-Expression of the Carbamoyl-Phosphate Synthase 1 Gene and Its Long Non-Coding RNA Correlates with Poor Prognosis of Patients with Intrahepatic Cholangiocarcinoma. Mol. Med. Rep. 2015, 12, 7915–7926. 10.3892/mmr.2015.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milinkovic V.; Bankovic J.; Rakic M.; Stankovic T.; Skender-Gazibara M.; Ruzdijic S.; Tanic N. Identification of Novel Genetic Alterations in Samples of Malignant Glioma Patients. PLoS One 2013, 8, e82108. 10.1371/journal.pone.0082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S.; Nguyen T.-V.; Rolfe A.; Agrawal A. A.; Ke J.; Peng S.; Colombo F.; Yu S.; Bouchard P.; Wu J.; Huang K.-C.; Bao X.; Omoto K.; Selvaraj A.; Yu L.; Ioannidis S.; Vaillancourt F. H.; Zhu P.; Larsen N. A.; Bolduc D. M. Small molecule inhibition of CPS1 activity through an allosteric pocket. Cell Chem. Biol. 2020, 27, 259–268. 10.1016/j.chembiol.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 2 was identified from a biochemical high-throughput screen of ∼350 000 compounds after a series of counterscreens and biophysical assays to verify direct binding.
- Wang Y.; Zhang Z.; Meanwell A. N. Regioselective Monobenzoylation of Unsymmetrical Piperazines. J. Org. Chem. 2000, 65, 4740–4742. 10.1021/jo000005e. [DOI] [PubMed] [Google Scholar]
- Wang T.; Kadow J. F.; Zhang Z.; Yin Z.; Gao Q.; Wu D.; Parker D. D.; Yang Z.; Zadjura L.; Robinson B. A.; Gong Y.-F.; Blair W. S.; Shi P.-Y.; Yamanaka G.; Lin P.-F.; Meanwell N. A. Inhibitors of HIV-1 attachment. Part 4: A study of the effect of piperazine substitution patterns on antiviral potency in the context of indole-based derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 5140–5145. 10.1016/j.bmcl.2009.07.076. [DOI] [PubMed] [Google Scholar]
- Liu F.; Dawadi S.; Maize K. M.; Dai R.; Park S. W.; Schnappinger D.; Finzel B. C.; Aldrich C. C. Structure-Based Optimization of Pyridoxal 5′-Phosphate-Dependent Transaminase Enzyme (BioA) Inhibitors that Target Biotin Biosynthesis in Mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 5507–5520. 10.1021/acs.jmedchem.7b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.