

Abstract
Multitarget ligands are interesting candidates for drug discovery and development due to improved safety and efficacy. However, rational design and optimization of multitarget ligands is tedious because affinity optimization for two or more targets has to be performed simultaneously. In this study, we demonstrate that, given a molecular fragment, which binds to two targets of interest, computer-aided fragment growing can be applied to optimize compound potency, relying on either ligand- or structure-derived information. This methodology is applied to the design of dual inhibitors of soluble epoxide hydrolase and leukotriene A4 hydrolase.
Keywords: Fragment-based drug design, multitarget drugs, polypharmacology, computer-aided drug design, machine learning
Designed multitarget ligands (DMLs) are the focus of modern drug discovery and offer the advantage of higher efficacy compared to selective ligands.1,2 Diverse strategies exist to generate a lead structure which affects two (or even more) targets of interest.3,4 However, classical approaches such as pharmacophore linking often yield DMLs with unfavorable pharmacokinetic properties due to high molecular weight.5 Fragment-based approaches are very successful to generate high-quality leads with acceptable ligand efficiency, and several studies demonstrated the feasibility of fragment-based discovery of DMLs.6,7 The initial step of fragment identification is often successful and delivers a starting point for further optimization.8 However, established strategies such as fragment growing or merging are much more demanding for two or even more targets. The study on the discovery of indeglitazar, a pan-peroxisome proliferator-activated receptor agonist,9 as well as the study on PLX647, a dual FMS and KIT kinase inhibitor,10 demonstrated fragment growing for simultaneous optimization of potency. The aforementioned studies were performed on related targets in the presence of experimental structural information. However, in many cases, the binding modes of a fragment in complex with all targets of interest are not available. In this case, screening of available derivatives can lead to success,8 while computational approaches offer a rational method for fragment growing.11 Shang et al. implemented an iterative fragment growing strategy, which led to the design of moderately potent dual cyclooxygenase-2 (COX-2)/leukotriene A4 hydrolase (LTA4H) inhibitors.12
In this study we present that fragment growing for DMLs is possible by using ligand-based or structure-based information. We developed two different in silico strategies to identify a DML affecting soluble epoxide hydrolase (sEH) and LTA4H. Both enzymes hydrolyze epoxides of the arachidonic acid. sEH converts the epoxyeicosatrienoic acids toward their corresponding vicinal diol,13 while LTA4H hydrolyzes the instable leukotriene A4 toward the 5,12-dihydroxy derivative leukotriene B4.14 The simultaneous inhibition of both enzymes might lead to synergistic anti-inflammatory effects, which have already been demonstrated for simultaneous inhibition of sEH and 5-lipoxygenase activating protein (FLAP).15 Recently, we demonstrated the feasibility of dual sEH/LTA4H inhibitors which bear the potential as novel anti-inflammatory agents.16
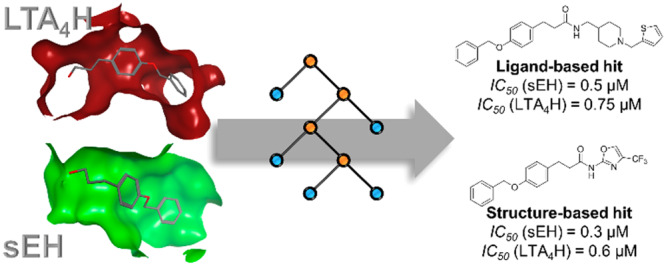
As a first step, a fragment, which can act as a starting point for optimization, was identified. In a previous study by Achenbach et al.8 we demonstrated that self-organizing maps17 (SOMs) offer an opportunity to identify fragments binding to both targets. Therefore, we extracted reported sEH and LTA4H inhibitors from ChEMBL DB18 v24 and trained a SOM using OSIRIS DataWarrior (Idorsia Pharmaceuticals). The analysis of the SOM revealed that LTA4H (blue circles) and sEH (red circles) ligands build distinct clusters (Figure 1). The few compounds which were assigned to the opposite cluster were manually examined. One of these compounds was fragment 1, which was initially identified by Amano et al. as a fragment that binds to sEH and exhibits moderate potency and ligand efficacy.19 The published cocrystal structure of 1 in complex with sEH shows that the highly lipophilic benzyloxy phenyl moiety occupies a lipophilic tunnel in the active site (PDB code 4Y2T; Figure 2A). The hydroxyl group exhibits directed hydrogen bonds toward Asp335, Tyr383, and Tyr466, three residues important for the catalytic activity of sEH. The lipophilic pocket, which is located behind the three aforementioned residues (Figure 2A, gray dashed circle), offers space for fragment growing. We evaluated the inhibition of sEH by 1 in a fluorescence-based enzyme activity assay20 and could measure an IC50 of 79 ± 16 μM. Given the MW = 242 and heavy atom count (HAC) of 18, the ligand efficiency results in LE = 1.4pIC50/(HAC) = 0.31,21 which qualifies it as an acceptable starting point for fragment growing.21 However, the ligand-lipophilicity efficiency LLE = pIC50 – clogP = 0.34 is very low and needs to be improved during optimization.22
Figure 1.
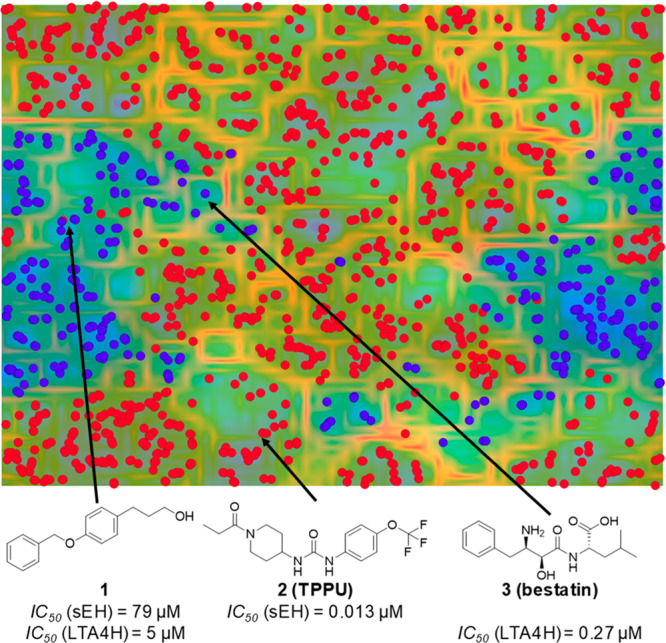
Identification of dual fragments using a self-organizing map. Training a SOM (50 × 50 neurons) with known active sEH (red circles) and LTA4H (blue circles) ligands led to identification of 1, a previously reported sEH inhibitor, which is located within the LTA4H cluster. The reference sEH inhibitor 2 (TPPU) and the LTA4H inhibitor 3 (bestatin) were located within the respective cluster.
Figure 2.

Starting point for fragment growing. (A) Cocrystal structure of 1 with sEH hydrolase domain (PDB code: 4Y2T). (B) Proposed binding mode of 1 in complex with LTA4H, based on cocrystal structure of a similar fragment (PDB code: 3CHO). 1 is shown as orange sticks, the molecular surface of the binding site is colored by lipophilicity (green: lipophilic; magenta: hydrophilic), and a gray dashed circle indicates an unoccupied space in the binding site. (C) Fragment growing strategy toward amides 4a–k. Red arrows indicate H-bond acceptor, and blue arrows indicate H-bond donor capabilities of the hydroxyl moiety, which is bioisosterically replaced by the secondary amide.
Despite the low LLE, 1 bears a benzyloxy phenyl moiety, a typical feature of LTA4H inhibitors, described by Kirkland et al.23 We measured the inhibitory activity of 1 toward LTA4H by using a fluorescence-based enzyme activity assay24 yielding an IC50 of 5 ± 1 μM (LE = 0.40; LLE = 1.54). We used the published X-ray structure of an inhibitor bearing the benzyloxy phenyl moiety (PDB code 3CHP) to predict the binding mode of 1 in complex with LTA4H (Figure 2B).23 The lipophilic tunnel in the binding site of LTA4H, which is important for potent and thermodynamically favorable binding,25 is fully occupied by the lipophilic benzyloxy phenyl residue. The hydroxyl moiety exhibits an H-bond toward the backbone carbonyl of Gly269, which is located near the catalytically important zinc ion. The adjacent pocket is not occupied and can be potentially used for fragment growing (Figure 2B, gray dashed circle).
Given the binding modes of 1 to both enzymes, we decided on bioisosteric replacement of the hydroxyl group by a secondary amide (Figure 2C). This amide exhibits similar H-bond donor and acceptor features and allows the extension of the fragment 1 by coupling of amine building blocks to 3-(4-benzyloxy) phenyl propionic acid 5 (Figure 3C). Therefore, we prepared a virtual combinatorial library of secondary amides extending 5. Commercially available amine building blocks, from six vendors most frequently used in our lab (Acros, Alfa-Aesar, Apollo Scientific, Fluorochem, Sigma-Aldrich, TCI), were extracted from ZINC database26 and duplicates were removed. Filtering for amides and sulfonamides has been performed, in order to remove these epoxide mimetics, which would bias the virtual library toward sEH. The combinatorial library was generated using the Combinatorial Library application in the MOE GUI. The two fragments were combined using a virtual amide condensation reaction. After applying a molecular weight filter (MW ≤ 500 Da) and removing tertiary amides resulting from the condensation procedure, the final combinatorial library contained 20,630 compounds for subsequent computer-aided prioritization (Figure 3A).
Figure 3.

Fragment growing. (A) Compilation of data sets for training various machine learning algorithms. (B) Computational workflow for structure- and ligand-based fragment growing. (C) Reaction conditions for amide coupling. 4a, 4f, 4g, 4j (i) 1.1 equiv of PyBOP, 0.5–1.1 equiv of HOBt·H2O, 1.5–3.0 equiv of DIPEA, THF, rt, 16 h; 4b–e, 4h, 4i (ii) 1.2 equiv of EDC·HCl, 4-DMAP, DCM, 60 °C μw irradiation, 1 h; 4k (iii) (a) 1.5 equiv of 3-(4-(benzyloxy)phenyl)propionic acid, 1.5 equiv of fluoro-N,N,N′,N′-bis(tetramethylen)formamidinium hexafluorophosphate, 4.5 equiv of DIPEA, DCM, 50 °C, 4 h, (b) 1.0 equiv of 4-trifluoromethyl-oxazol-2-ylamine, DCM, 50 °C, 72 h.
In order to demonstrate the applicability of computer-aided design to fragment growing of multitarget ligands, we chose two complementary strategies. The ligand-based strategy relies solely on the information on previously published active ligands. The structure-based design strategy relies on the information contained in the X-ray structures of both enzymes in complex with various inhibitors. Therefore, we compiled data sets (Figure 3A) to train machine learning algorithms to predict the activity toward sEH and LTA4H. First, all cocrystallized ligands for LTA4H and sEH were retrieved from the Protein Data Bank.27 This resulted in 43 unique cocrystallized LTA4H compounds and 92 cocrystallized sEH compounds in complex with the respective targets. Furthermore, active compounds from the ChEMBL database were retrieved. The 1,022 active LTA4H compounds (Target ChEMBL ID: CHEMBL4618) and 2,453 active sEH compounds (Target ChEMBL ID: CHEMBL2409) were further processed. Duplicates and compounds with a binding affinity larger than 1 μM were removed. This results in 382 unique active LTA4H compounds and 1,384 unique active sEH compounds. A data set of random compounds, which were considered to be inactive, was retrieved from ChEMBL. On the 1,727,112 compounds, a molecular weight filter was applied (200–500 Da). A sample of 1,000 randomly selected compounds served as the inactive data set.
For both strategies, ligand-based and structure-based design, we used the active ligands, the random data set of inactive compounds from ChemblDB, and the cocrystallized compounds as active ligands to train four different widely used classifiers (XGBoost,28 Random Forest,29 AdaBoost,30 and Support Vector Classification31). For the ligand-based strategy, the compounds were encoded using four molecular fingerprints: AtomPair,32 FeatMorgan,33 Morgan,34 and MACCS.35 For the structure-based strategy, Protein Ligand Interaction Fingerprints (PLIFs)36 were generated. Nine potential contacts are integrated in the current PLIF version (side chain hydrogen bonds (donor or acceptor), backbone hydrogen bonds (donor or acceptor), solvent hydrogen bonds (donor or acceptor), ionic interactions, metal binding interactions, and π interactions). Each amino acid residue is classified into these categories describing the binding of small molecules (ligand) in the binding site. For each interaction between a ligand and a residue, the interaction strength is calculated. All models were trained using scikit-learn37 and evaluated by 10-fold cross validation. Accuracy was used as the primary measure of model performance. To calculate the accuracy of the fraction of correct predictions, the accuracy_score function from scikit-learn was used:
where ŷi is the predicted value of the i-th sample, yi is the corresponding true value, and nsamples is the overall sample size (Figure 3B).37
First, the optimal partitioning scheme for splitting training and test set was identified. The accuracies of the models were tested with a partitioning scheme between 75% and 95% training set size. The results can be found in the Supporting Information (SI Table S2). Second, for each machine learning algorithm different parameters were optimized to achieve the most accurate prediction (SI Table S3). In the ligand-based approach, the optimized Random Forest model in combination with the AtomPair fingerprint (SI Table S4) was used to predict compounds for synthesis. The number of predicted compounds was reduced by limiting the fingerprint similarity to a minimum of 0.5 compared to the cocrystallized compounds. This restriction led to the prediction of 116 compounds, from which 6 compounds were cherry picked for synthesis. In the structure-based approach, the optimized Random Forest model in combination with the PLIF fingerprint (SI Table S5) was used to predict compounds for synthesis. The fingerprint similarity was limited to a minimum of 0.5 compared to the cocrystallized compounds and the prediction confidence of the model to a minimum of 0.7. These restrictions led to the prediction of 115 compounds from which 5 compounds were cherry picked for synthesis. Synthetic accessibility, costs of the educts, and uniqueness of the compounds were used as guidelines for cherry picking in both strategies. In more detail, we discarded all compounds bearing a additional primary or a secondary amine or a carboxylate moiety because the synthesis would require additional protection and deprotection steps. sEH pharmacophore requires a NH hydrogen bond donor; therefore, we removed all compounds with a tertiary amide. Furthermore, sEH does not tolerate polar functional groups which are adjacent to the amide, which were also discarded. Compounds which do not exhibit polar groups at all were also not considered for synthesis due to potentially poor solubility. After careful inspection, very similar compounds which differ e.g. only in the phenyl substituents were considered only once. Finally, we removed all compounds for which the building blocks were unavailable or too expensive.
The synthesis was accomplished by typical amide coupling systems (Figure 3C). Either EDC·HCl with 4-DMAP or HOBt·H2O and PyBOP were used. The carbon acid derivative 5 is a weak electrophile, while most amines of the structure-based fragment growing series are weak nucleophiles. This combination is challenging, which is reflected by the moderate yields. Particularly, the coupling with 4-trifluoromethyl-oxazol-2-ylamine needed harsh conditions with fluoro-N,N,N′,N′-bis(tetramethylen)formamidinium hexafluorophosphate and a prolonged reaction time.
Using the ligand-based strategy, the prediction algorithm prioritized compounds exhibiting an N-substituted piperidine or pyrrolidine moiety. These saturated heterocycles are common elements in diverse series of LTA4H38 and sEH39 inhibitors. The 6 selected compounds were subsequently tested in the fluorescence-based enzyme activity assays (Table 1). LTA4H tolerated different variations of the ring, substitution pattern, and N-coupled lipophilic moiety, as long as it contained an ionizable tertiary amine. In contrast, sEH was more restrictive; only compound 4c exhibited submicromolar activity toward both enzymes.
Table 1. Synthesized Compounds from Ligand-Based Fragment Growing.

All values were measured at least thrice as triplicates (n ≥ 3), mean ± SD is displayed.
The compounds suggested to be active using the structure-based strategy were structurally more heterogeneous than the compounds proposed by the ligand-based approach and exhibited diverse substitution patterns (Table 2). However, all compounds share an aromatic ring directly attached to the amide, a feature that seems to be recognized as important by machine learning. Compound 4j containing a phosphonate ester, which has been described as a tolerated moiety of sEH inhibitors,40 showed moderate dual target activity. Most interestingly, the oxazole-based compound 4k was identified as the most potent compound with unprecedented chemotype for both sEH and LTA4H. The identification of a novel scaffold speaks in favor of using structure-based in silico approaches, which are not biased by previously identified chemotypes.
Table 2. Synthesized Compounds from Structure-Based Fragment Growing.

All values were measured at least thrice as triplicates (n ≥ 3), mean ± SD is displayed.
This study, although successful in yielding dual active structures, has some limitations, which should be kept in mind when transferring the strategy to other target combinations. First, both targets, sEH and LTA4H, convert similar ligands–arachidonic acid epoxides–that leads to similar binding sites, at least concerning the hydrophobicity patterns. It is unclear whether the aforementioned strategy is applicable to completely dissimilar targets. Furthermore, the machine learning algorithm profits from the large number of available active ligands for both targets. Given a novel target without numerous published actives, machine learning will possibly fail to predict activity. Finally, the computational approach just delivers ideas for synthesis which have to be carefully selected by an experienced medicinal chemist able to assess the synthetic accessibility, familiar with the structure–activity relationships of the respective targets, and estimate the potential physicochemical properties of the suggested ligands. Incorporation of more advanced in silico filters could simplify the crucial step of cherry picking.
In this study, we developed a computer-aided fragment-growing strategy for multitarget ligands. We applied it to the design of dual inhibitors of LTA4H and sEH, epoxide hydrolase enzymes located in the arachidonic acid cascade. Starting from fragment 1, a lipophilic dual inhibitor of both proteins with acceptable ligand efficacy, a large combinatorial library of possible expanded ligands was prepared. Machine learning technique, Random Forest, was applied to classify active and inactive compounds based on either structure- or ligand-derived fingerprints. Both, structure- and ligand-based prediction models yielded dual-target ligands, which were confirmed by synthesis in subsequent in vitro evaluation. Thus, this study demonstrates that computer-aided fragment growing is applicable to multitarget ligand design in the presence or absence of structural information.
Glossary
ABBREVIATIONS
- 4-DMAP
4-(dimethylamino)pyridine
- COX-2
cyclooxygenase-2
- DCM
dichloromethane
- DML
designed multitarget ligands
- DIPEA
diisopropylethylamine
- EDC
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide
- FLAP
5-lipoxygenase activating protein
- FMS
Feline McDonough Sarcoma
- GUI
graphical user interface
- HAC
heavy atom count
- HOBt
N-hydroxybenzotriazole
- KIT
tyrosine kinase KIT
- LE
ligand efficiency
- LLE
ligand-lipophilicity efficiency
- LTA4H
leukotriene A4 hydrolase
- MOE
Molecular Operating Environment
- MW
molecular weight
- PDB
Protein Data Bank
- PLIF
Protein Ligand Interaction Fingerprint
- acc
accuracy
- PyBOP
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- sEH
soluble epoxide hydrolase.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00102.
Synthetic procedures, assay descriptions, detailed description of in silico methods, detailed summary of parameter optimization for the machine learning algorithms, and docking modes of compounds 4g–4k (PDF)
Full list of predicted sEH/LTA4H inhibitors from ligand-based approach (PDF)
Full list of predicted sEH/LTA4H inhibitors from structure-based approach (PDF)
Author Contributions
‡ L.H. and K.H. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by the German Research Foundation (DFG; PR1405/2-2, PR1405/4-1, SFB1039, Teilprojekt A07). K.H. was supported by the Else-Kroener-Fresenius-Foundation funding the graduate school ‘Translational Research Innovation Pharma’ (TRIP).
The authors declare no competing financial interest.
Supplementary Material
References
- Morphy R.; Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48 (21), 6523–6543. 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- Ramsay R. R.; Popovic-Nikolic M. R.; Nikolic K.; Uliassi E.; Bolognesi M. L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7 (1), 3. 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morphy R.; Kay C.; Rankovic Z. From Magic Bullets to Designed Multiple Ligands. Drug Discovery Today 2004, 9 (15), 641–651. 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- Proschak E.; Stark H.; Merk D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62 (2), 420–444. 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. The Physicochemical Challenges of Designing Multiple Ligands. J. Med. Chem. 2006, 49 (16), 4961–4970. 10.1021/jm0603015. [DOI] [PubMed] [Google Scholar]
- Morphy R.; Rankovic Z. Fragments, Network Biology and Designing Multiple Ligands. Drug Discovery Today 2007, 12 (3), 156–160. 10.1016/j.drudis.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Mason J. S.; Overington J. P. Can We Rationally Design Promiscuous Drugs?. Curr. Opin. Struct. Biol. 2006, 16 (1), 127–136. 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Achenbach J.; Klingler F.-M.; Blöcher R.; Moser D.; Häfner A.-K.; Rödl C. B.; Kretschmer S.; Krüger B.; Löhr F.; Stark H.; Hofmann B.; Steinhilber D.; Proschak E. Exploring the Chemical Space of Multitarget Ligands Using Aligned Self-Organizing Maps. ACS Med. Chem. Lett. 2013, 4 (12), 1169–1172. 10.1021/ml4002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D. R.; Lin J. J.; Zhang C.; Wang W.; Mehra U.; Perreault M.; Erbe D.; Krupka H. I.; England B. P.; Arnold J.; Plotnikov A. N.; Marimuthu A.; Nguyen H.; Will S.; Signaevsky M.; Kral J.; Cantwell J.; Settachatgull C.; Yan D. S.; Fong D.; Oh A.; Shi S.; Womack P.; Powell B.; Habets G.; West B. L.; Zhang K. Y. J.; Milburn M. V.; Vlasuk G. P.; Hirth K. P.; Nolop K.; Bollag G.; Ibrahim P. N.; Tobin J. F. Scaffold-Based Discovery of Indeglitazar, a PPAR Pan-Active Anti-Diabetic Agent. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (1), 262–267. 10.1073/pnas.0811325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Ibrahim P. N.; Zhang J.; Burton E. A.; Habets G.; Zhang Y.; Powell B.; West B. L.; Matusow B.; Tsang G.; Shellooe R.; Carias H.; Nguyen H.; Marimuthu A.; Zhang K. Y. J.; Oh A.; Bremer R.; Hurt C. R.; Artis D. R.; Wu G.; Nespi M.; Spevak W.; Lin P.; Nolop K.; Hirth P.; Tesch G. H.; Bollag G. Design and Pharmacology of a Highly Specific Dual FMS and KIT Kinase Inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (14), 5689–5694. 10.1073/pnas.1219457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X. H.; Shi Z.; Tan C.; Jiang Y.; Go M. L.; Low B. C.; Chen Y. Z. In-Silico Approaches to Multi-Target Drug Discovery. Pharm. Res. 2010, 27 (5), 739–749. 10.1007/s11095-010-0065-2. [DOI] [PubMed] [Google Scholar]
- Shang E.; Yuan Y.; Chen X.; Liu Y.; Pei J.; Lai L. De Novo Design of Multitarget Ligands with an Iterative Fragment-Growing Strategy. J. Chem. Inf. Model. 2014, 54 (4), 1235–1241. 10.1021/ci500021v. [DOI] [PubMed] [Google Scholar]
- Shen H. C.; Hammock B. D. Discovery of Inhibitors of Soluble Epoxide Hydrolase: A Target with Multiple Potential Therapeutic Indications. J. Med. Chem. 2012, 55 (5), 1789–1808. 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeggström J. Z. Leukotriene Biosynthetic Enzymes as Therapeutic Targets. J. Clin. Invest. 2018, 128 (7), 2680–2690. 10.1172/JCI97945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garscha U.; Romp E.; Pace S.; Rossi A.; Temml V.; Schuster D.; König S.; Gerstmeier J.; Liening S.; Werner M.; Atze H.; Wittmann S.; Weinigel C.; Rummler S.; Scriba G. K.; Sautebin L.; Werz O. Pharmacological Profile and Efficiency in Vivo of Diflapolin, the First Dual Inhibitor of 5-Lipoxygenase-Activating Protein and Soluble Epoxide Hydrolase. Sci. Rep. 2017, 7 (1), 9398. 10.1038/s41598-017-09795-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesinger K.; Schott A.; Kramer J. S.; Blöcher R.; Witt F.; Wittmann S. K.; Steinhilber D.; Pogoryelov D.; Gerstmeier J.; Werz O.; Proschak E. Design of Dual Inhibitors of Soluble Epoxide Hydrolase and LTA4 Hydrolase. ACS Med. Chem. Lett. 2020, 11, 298. 10.1021/acsmedchemlett.9b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T. Self-Organized Formation of Topologically Correct Feature Maps. Biol. Cybern. 1982, 43 (1), 59–69. 10.1007/BF00337288. [DOI] [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Krüger F. A.; Light Y.; Mak L.; McGlinchey S.; Nowotka M.; Papadatos G.; Santos R.; Overington J. P. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42 (Database issue), D1083–1090. 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano Y.; Tanabe E.; Yamaguchi T. Identification of N-Ethylmethylamine as a Novel Scaffold for Inhibitors of Soluble Epoxide Hydrolase by Crystallographic Fragment Screening. Bioorg. Med. Chem. 2015, 23 (10), 2310–2317. 10.1016/j.bmc.2015.03.083. [DOI] [PubMed] [Google Scholar]
- Wolf N. M.; Morisseau C.; Jones P. D.; Hock B.; Hammock B. D. Development of a High-Throughput Screen for Soluble Epoxide Hydrolase Inhibition. Anal. Biochem. 2006, 355 (1), 71–80. 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultes S.; de Graaf C.; Haaksma E. E. J.; de Esch I. J. P.; Leurs R.; Krämer O. Ligand Efficiency as a Guide in Fragment Hit Selection and Optimization. Drug Discovery Today: Technol. 2010, 7 (3), e157–e162. 10.1016/j.ddtec.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. The Thermodynamic Basis for the Use of Lipophilic Efficiency (LipE) in Enthalpic Optimizations. Bioorg. Med. Chem. Lett. 2013, 23 (21), 5992–6000. 10.1016/j.bmcl.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Kirkland T. A.; Adler M.; Bauman J. G.; Chen M.; Haeggström J. Z.; King B.; Kochanny M. J.; Liang A. M.; Mendoza L.; Phillips G. B.; Thunnissen M.; Trinh L.; Whitlow M.; Ye B.; Ye H.; Parkinson J.; Guilford W. J. Synthesis of Glutamic Acid Analogs as Potent Inhibitors of Leukotriene A4 Hydrolase. Bioorg. Med. Chem. 2008, 16 (9), 4963–4983. 10.1016/j.bmc.2008.03.042. [DOI] [PubMed] [Google Scholar]
- Moser D.; Wittmann S. K.; Kramer J.; Blöcher R.; Achenbach J.; Pogoryelov D.; Proschak E. PENG: A Neural Gas-Based Approach for Pharmacophore Elucidation. Method Design, Validation, and Virtual Screening for Novel Ligands of LTA4H. J. Chem. Inf. Model. 2015, 55 (2), 284–293. 10.1021/ci500618u. [DOI] [PubMed] [Google Scholar]
- Wittmann S. K.; Kalinowsky L.; Kramer J. S.; Bloecher R.; Knapp S.; Steinhilber D.; Pogoryelov D.; Proschak E.; Heering J. Thermodynamic Properties of Leukotriene A4 Hydrolase Inhibitors. Bioorg. Med. Chem. 2016, 24 (21), 5243–5248. 10.1016/j.bmc.2016.08.047. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC--a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45 (1), 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J. H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. 10.1214/aos/1013203451. [DOI] [Google Scholar]
- Breiman L. Random Forests. Machine Learning 2001, 45 (1), 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Freund Y.; Schapire R. A Decision-Theoretic Generalization of on-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. 10.1006/jcss.1997.1504. [DOI] [Google Scholar]
- Vapnik V.; Golowich S. E.; Smola A. J.. Support Vector Method for Function Approximation, Regression Estimation and Signal Processing. In Advances in neural information processing systems; 1997; pp 281–287. [Google Scholar]
- Carhart R. E.; Smith D. H.; Venkataraghavan R. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Model. 1985, 25 (2), 64–73. 10.1021/ci00046a002. [DOI] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Morgan H. L. The Generation of a Unique Machine Description for Chemical Structures-a Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5 (2), 107–113. 10.1021/c160017a018. [DOI] [Google Scholar]
- Cereto-Massagué A.; Ojeda M. J.; Valls C.; Mulero M.; Garcia-Vallvé S.; Pujadas G. Molecular Fingerprint Similarity Search in Virtual Screening. Methods 2015, 71, 58–63. 10.1016/j.ymeth.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein- Ligand Binding Interactions. J. Med. Chem. 2004, 47 (2), 337–344. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12 (Oct), 2825–2830. [Google Scholar]
- Sandanayaka V.; Mamat B.; Mishra R. K.; Winger J.; Krohn M.; Zhou L.-M.; Keyvan M.; Enache L.; Sullins D.; Onua E.; Zhang J.; Halldorsdottir G.; Sigthorsdottir H.; Thorlaksdottir A.; Sigthorsson G.; Thorsteinnsdottir M.; Davies D. R.; Stewart L. J.; Zembower D. E.; Andresson T.; Kiselyov A. S.; Singh J.; Gurney M. E. Discovery of 4-[(2S)-2-{[4-(4-Chlorophenoxy)Phenoxy]Methyl}-1-Pyrrolidinyl]Butanoic Acid (DG-051) as a Novel Leukotriene A4 Hydrolase Inhibitor of Leukotriene B4 Biosynthesis. J. Med. Chem. 2010, 53 (2), 573–585. 10.1021/jm900838g. [DOI] [PubMed] [Google Scholar]
- Shen H. C.; Ding F.-X.; Wang S.; Deng Q.; Zhang X.; Chen Y.; Zhou G.; Xu S.; Chen H.-S.; Tong X.; Tong V.; Mitra K.; Kumar S.; Tsai C.; Stevenson A. S.; Pai L.-Y.; Alonso-Galicia M.; Chen X.; Soisson S. M.; Roy S.; Zhang B.; Tata J. R.; Berger J. P.; Colletti S. L. Discovery of a Highly Potent, Selective, and Bioavailable Soluble Epoxide Hydrolase Inhibitor with Excellent Ex Vivo Target Engagement. J. Med. Chem. 2009, 52 (16), 5009–5012. 10.1021/jm900725r. [DOI] [PubMed] [Google Scholar]
- Kim I.-H.; Park Y.-K.; Nishiwaki H.; Hammock B. D.; Nishi K. Structure-Activity Relationships of Amide-Phosphonate Derivatives as Inhibitors of the Human Soluble Epoxide Hydrolase. Bioorg. Med. Chem. 2015, 23 (22), 7199–7210. 10.1016/j.bmc.2015.10.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.