

Abstract

Lysophosphatidic acid (LPA) is a bioactive lipid mediator that elicits a number of biological functions, including smooth muscle contraction, cell motility, proliferation, and morphological change. LPA is endogenously produced by autotaxin (ATX) from extracellular lysophosphatidylcholine (LPC) in plasma. Herein, we report our medicinal chemistry effort to identify a novel and highly potent ATX inhibitor, ONO-8430506 (20), with good oral availability. To enhance the enzymatic ATX inhibitory activity, we designed several compounds by structurally comparing our hit compound with the endogenous ligand LPC. Further optimization to improve the pharmacokinetic profile and enhance the ATX inhibitory activity in human plasma resulted in the identification of ONO-8430506 (20), which enhanced the antitumor effect of paclitaxel in a breast cancer model.
Keywords: Autotaxin, ENPP2, LPA, β-oxidation, in vivo isomerization
Autotaxin (ATX) is an extracellular enzyme1 that has lysophospholipase D (LysoPLD) activity to produce lysophosphatidic acid (LPA) from lysophosphatidylcholine (LPC) in circulating blood.2,3 The primary structure is identical to the ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2).4 Many reports have suggested the involvement of the LPA-producing enzyme ATX in the formation of blood vessels,5 chronic inflammation,6 and pain generation.7 Additionally, multiple studies have reported on the function of the ATX-LPA system in cancer. It has been suggested that the anticancer effects of taxanes, doxorubicin, and radiation may be controlled by ATX-LPA.8−10 The use of ATX inhibitors is expected to ameliorate the disease for patients whose cancer cells are resistant to the effects of these anticancer treatments.
Owing to its attractiveness as a novel medicinal target, intensive efforts on the discovery of potent ATX inhibitors have been conducted. As reviewed by Kokotos et al., many researchers in the pharmaceutical industry have discovered novel ATX inhibitors in the past several years.11,12 Among them, GLPG1690 is currently in phase 3 clinical trials for idiopathic pulmonary fibrosis (IPF).13 Herein, we report our efforts to find a novel, potent and orally available ATX inhibitor.
To identify novel ATX inhibitors, we performed high-throughput screening (HTS) of our in-house compound library and discovered compound 1 as a hit compound (Figure 1). Although 1 showed very weak enzymatic inhibitory activity toward recombinant human ATX (hATX assay, 51.6% inhibition at 30 μM), its modular structure encouraged us to investigate the structure–activity relationship (SAR) to increase the activity.
Figure 1.

Structures of hit compound (1) and 18:2 LPC (2).
As an initial strategy, we focused on the structures of compound 1 and an endogenous substrate of ATX, lysophosphatidylcholine (2, 18:2 LPC). We considered the possibility of both compounds structurally overlapping each other with the fatty-acid side chain of LPC occupying the same binding pocket as the 9-benzyl tetrahydro-β-carboline moiety of compound 1. Based on this hypothesis, we supposed that compound 1 lacked a polar group such as a phosphate group, which is thought to be important for the activity. Indeed, Nishimasu et al. reported that the phosphate group of LPA interacts with Zn2+ and other highly conserved residues among the ENPP family in the crystal structure of ATX bound with LPAs having different side chain lengths and saturations.14 Therefore, we tried to introduce a polar group with various lengths of methylene linker into the acetyl part of compound 1.
As shown in Table 1, we employed a carboxyl group as the introduced polar group and various carboxylic acid analogues were investigated. Our studies revealed that compounds 3 and 4, which have ethylene (C-2) and propylene (C-3) linkers between the amide moiety and carboxyl group, were less potent in the hATX assay (3, IC50 = 5.4 μM; 4, IC50 = 0.83 μM). Conversely, compounds 5 and 6 with butylene (C-4) and pentylene (C-5) linkers, respectively, demonstrated significantly increased potency (5, IC50 = 0.24 μM; 6, IC50 = 0.25 μM). In particular, 5 showed an approximately 100-fold increase in IC50 relative to 1 and was considered to be a suitable compound for further exploration. This SAR illustrated that the existence of a carboxyl group at the appropriate position plays an important role in providing strong ATX inhibitory activity in this chemical series. Substitutions at the benzene ring were also quickly explored, considering the potential metabolic instability of the unsubstituted benzene ring. This exploration showed that lipophilic groups were well tolerated and gave the 3-fluoro substituted analogue 7 as a more potent compound.
Table 1. Structure–Activity Relationship of Carboxylic Acid Analogues.
![]()
| compd. | R1 | R2 | n | hATX assaya IC50 (μM) |
|---|---|---|---|---|
| 1 | H | Me | 0 | 51.6% inhibition at 30 μM |
| 3 | H | CO2H | 2 | 5.4 |
| 4 | H | CO2H | 3 | 0.83 |
| 5 | H | CO2H | 4 | 0.24 |
| 6 | H | CO2H | 5 | 0.25 |
| 7 | F | CO2H | 4 | 0.050 |
The inhibitory activity of the compound toward recombinant human ATX was evaluated using 16:0 LPC as a substrate.
With the potent compound (7) in hand, pharmacokinetic profiles of 7 were evaluated to confirm its potential as an orally available drug (Table 2). Single-dose rat pharmacokinetic studies (oral dosing at 3 mg/kg and intravenous dosing at 1 mg/kg) revealed that 7 demonstrated relatively low plasma clearance (CL = 7.5 mL/min/kg) and moderate oral bioavailability (%Frat = 30%), which are thought to be acceptable values as a tool compound for in vivo study. However, more optimization was needed to improve its pharmacokinetic profiles and the ATX inhibitory activity toward a clinical candidate.
Table 2. Introduction of the Dimethyl Group Adjacent to the Carboxylic Group.

| metabolic
stability in hepatocytea (%) |
in
vivo PK (rat) |
|||||||
|---|---|---|---|---|---|---|---|---|
| compd. | R1 | R2 | hATX assay IC50 (μM) | logD7.4 | human | rat | CLb (mL/min/kg) | Fc (%) |
| 7 | H | H | 0.050 | 0.51 | 29 | 10 | 7.5 | 30 |
| 8 | Me | H | 0.024 | 1.54 | 42 | 66 | 3.8 | 75 |
| 9 | H | Me | 0.017 | 1.71 | 92 | 47 | 3.1 | 82 |
The values show the remaining % 2 h after hepatocyte incubation.
Plasma clearance (CL) was calculated based on an intravenous dose of 1 mg/kg (n = 3).
Bioavailability (F) was calculated based on an intravenous dose of 1 mg/kg and an oral dose of 3 mg/kg (n = 3).
When we worked on improving the pharmacokinetic profiles of compound 7, we considered that 7 has no substituents at the α and β carbon positions of the carboxyl group. Therefore, this part of the molecule was susceptible to β-oxidation. In fact, the metabolic stability in the hepatocyte of 7 was very low as shown in Table 2 (remaining % in hepatocyte, human: 29%, rat: 10%). While several approaches have been reported to prevent β-oxidation,15,16 we decided to introduce a dimethyl group adjacent to the carboxyl group in 7 considering its low measured LogD value (LogD7.4 = 0.51). Introduction of dimethyl group at the α and β carbon positions of the carboxyl group in 7 afforded compounds 8 and 9, respectively, which exhibited improved metabolic stability in hepatocytes with an improvement in the hATX inhibitory activity (8, IC50 = 0.024 μM; 9, IC50 = 0.017 μM). Indeed, an evaluation of the pharmacokinetic profiles of compounds 8 and 9 revealed that both compounds displayed decreased plasma clearance (8, CL = 3.8 mL/min/kg; 9, CL = 3.1 mL/min/kg). Furthermore, the oral bioavailability was also improved (8, %Frat = 75%; 9, %Frat = 82%) presumably because of the improvement in the membrane permeability with increased LogD values. As compound 9 showed better pharmacokinetic profiles, especially in the metabolic stability in human hepatocyte, compound 9 was selected for further profiling.
We evaluated the ATX inhibitory activity under physiological conditions in plasma as the LysoPLD activity. As shown in Table 3, the LysoPLD inhibitory activity in human plasma of compound 9 was weakened by 15-fold compared with that evaluated using recombinant human ATX (human plasma LysoPLD assay; IC50 = 0.26 μM vs hATX assay; IC50 = 0.017 μM). One plausible reason for this result may be derived from its high plasma protein binding (>99.9% in human). Therefore, our design strategy was focused toward reducing plasma protein binding.
Table 3. Rigidification around Carboxyl Group.
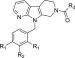

The LysoPLD inhibitory activity of the compound in human plasma was evaluated using 16:0 LPC as a substrate.
Rigidified molecules are often used to increase activity, reduce lipophilicity, and improve metabolic stability.17−19 We considered the possibility that rigidification around the polar carboxyl group would reduce lipophilicity and plasma protein binding. The result is shown in Table 3. First, we synthesized benzoic acid analogues 10 (meta) and 11 (para) to determine the preferred position of the carboxyl group, and demonstrated that putting the carboxyl group at the para position was preferred (hATX assay; 11, IC50 = 0.028 μM vs 10, IC50 = 0.10 μM). Next, the replacement of benzoic acid with cyclohexanecarboxylic acid was attempted to give 12 (cis isomer) and 13 (trans isomer). Fortunately, the trans isomer 13 showed strong enzymatic ATX inhibitory activity (IC50 = 0.017 μM) and reduced plasma protein binding (99.3% in human). Our preliminary explorations revealed that lipophilic groups were well tolerated as substitutions at the benzene ring, and minor modifications on the substituents could be used to modulate the overall properties (e.g., ATX inhibitory activity and DMPK profiles). With these observations in mind, we examined several substitutions for compound 13 and found the difluoro-substituted analogue 15 as a more advanced compound. As we expected, compound 15 demonstrated improved activity in the human plasma LysoPLD assay (human plasma LysoPLD assay; IC50 = 0.0073 μM vs hATX assay; IC50 = 0.014 μM). Notably, the effect of rigidification on increasing the plasma LysoPLD inhibitory activity was supported by the reduced plasma protein binding and measured logD value.
However, as shown in Figure 2, a detailed evaluation of the pharmacokinetic profile revealed that compound 15 was isomerized in vivo to the corresponding cis isomer 14 upon oral administration in a rat (34% of the trans isomer 15 was converted to the cis isomer 14 on an area under the curve (AUC) basis). We also evaluated the metabolic stability of the compounds using hepatocytes from several species to confirm whether there was any possibility of causing this isomerization in other species (Table 4). As a result, the isomerization was observed in all evaluated species (human, rat, dog and monkey), indicating the possibility of making clinical studies complicated. He et al. also reported on a similar in vivo isomerization.20 Therefore, further modification to prevent this isomerization was needed.
Figure 2.

Pharmacokinetic profiles of compound 15.
Table 4. Hepatocyte Incubation Study of Compound 15.
| conversion
ratio in hepatocyte incubation studya (% of
concentration, 14/14 + 15) | |||
|---|---|---|---|
| human | rat | dog | monkey |
| 29.8 | 14.4 | 9.0 | 21.4 |
Conversion ratio was defined as the ratio of concentration 2 h after hepatocyte incubation.
Our first approach to prevent the isomerization was to introduce a methyl group at the α position of the carboxyl group to produce compound 16. Although this compound exhibited slightly decreased activity (human plasma LysoPLD assay; IC50 = 0.025 μM), its good pharmacokinetic profiles prompted us to pursue further modification. Our next approach involved the use of bicyclic systems as in compounds 17 and 18. Bicyclic systems contribute toward keeping the molecules in the biologically active conformation and prevent isomerization. As expected, compounds 17 and 18 displayed single-digit nanomolar potency in the human plasma LysoPLD assay (17, IC50 = 0.0081 μM; 18, IC50 = 0.0079 μM). Thus, a minor modification of the substituents at the benzene ring was conducted again, while retaining the bicyclic systems as in compounds 17 and 18. Ultimately, compound 20 (ONO-8430506) was selected as a preclinical candidate based on its strong potency, good pharmacokinetic profiles, and other overall profiles (Table 5).
Table 5. Attempts to Prevent in vivo Isomerization.
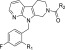

The values show AUC after 0.5 mg/kg oral administration (cassette dosing) for all compounds.
Table 6 shows the pharmacokinetic profiles of compound 20 in several preclinical species. Compound 20 exhibited a relatively low plasma clearance and long half-life. Additionally, an allometric scaling prediction showed that the human clearance would be much lower (predicted human clearance = 2.1 mL/min/kg).
Table 6. Pharmacokinetic Profiles of Compound 20.
| species | route | dose (mg/kg) | Cmaxa (ng/mL) | Vssa (mL/kg) | T1/2a (h) | CLa (mL/min/kg) | Fb (%) |
|---|---|---|---|---|---|---|---|
| mouse | po | 1 | 124 ± 49 | 5.4 ± 2.1 | |||
| rat | iv | 0.3 | 1474 ± 153 | 3.4 ± 0.9 | 8.2 ± 2.3 | ||
| po | 1 | 261 ± 107 | 2.5 ± 0.3 | 51.6 | |||
| dog | iv | 0.3 | 1863 ± 878 | 8.9 ± 5.3 | 4.7 ± 0.7 | ||
| po | 1 | 1670 ± 610 | 5.9 ± 2.0 | 71.1 | |||
| monkey | iv | 0.3 | 2275 ± 235 | 7.9 ± 1.0 | 5.8 ± 0.6 | ||
| po | 1 | 63 ± 38 | NC | 30.8 |
The values are expressed as the mean ± standard deviation (n = 3), NC = not calculated.
F (%) was calculated using the mean of AUC (po) and the mean of AUC (iv).
Previously, we found that compound 20 (ONO-8430506) suppressed LPA formation in plasma and decreased rat intraurethral pressure in a dose-dependent manner during the 20 min after intraduodenal administration in vivo.21 We considered examining the long-term effects of the compound after oral administration. Because it was reported that the antitumor effects of paclitaxel (PTX) were attenuated by LPA stimulation in vitro,8 we confirmed this concept using our compound in vivo.
We evaluated the antitumor effects of compound 20 in combination with PTX in a mouse model of the subcutaneously implanted human breast cancer cell line MDA-MB-231, as shown in Figure 3. Specifically, the enhancing effect of the compound on the antitumor effects of monotherapy PTX at the maximum tolerated dose was evaluated. PTX was administered intraperitoneally at 20 mg/kg (the maximum tolerated dose) three times weekly for 5 weeks.
Figure 3.

Antitumor effect of compound 20 in MDA-MB-231 human breast cancer-bearing mouse model. (a) Change in median tumor volume. (b) Tumor volume at day 32. (b) ***p < 0.001. N.S.: not significant by Student t test. #p < 0.05, ##p < 0.01 by Student t test using a closed testing procedure from the high dose side. (n = 8–9).
Considering the pharmacokinetic profiles of compound 20 in the mouse model, the compound was administered orally at doses of 30 or 100 mg/kg twice daily for 32 days. In the PTX monotherapy group, the tumor volume was significantly lower than that in the vehicle group with TGI (tumor growth inhibition)median at 81.5%. Conversely, there were no significant effects on the tumor volume in the “compound 20 at 100 mg/kg” monotherapy group with TGImedian at 14.7%. In the “PTX and compound 20 combination” group, the tumor volume was significantly lower at all doses in comparison with that in the PTX monotherapy group. This observation was dependent on the dose of compound 20. The TGI medians were 91.2% and 95.5% for the PTX and compound 20 at 30 and 100 mg/kg combination groups, respectively. We also evaluated LysoPLD activity from plasma at 18 h following the final administration of compound 20. Inhibition rates of LysoPLD activity were 97.1% for the “PTX and compound 20 at 30 mg/kg combination” group and 99.3% for the “PTX and compound 20 at 100 mg/kg combination” group. The comparable inhibition of LysoPLD activity was achieved at 30 mg/kg once daily in rat models,21 indicating that a lower dose could be expected in humans.
In summary, we developed a novel and highly potent ATX inhibitor that enhances the antitumor effect of PTX in a mouse model of a breast cancer cell line. To increase the enzymatic ATX inhibitory activity, a carboxyl group was successfully incorporated by structurally comparing our hit compound with an endogenous ligand LPC. Further optimization focused on improving the pharmacokinetic profiles and plasma LysoPLD inhibitory activity, leading to compound 20 (ONO-8430506). The detailed pharmacokinetic and pharmacodynamic profiles demonstrated that compound 20 (ONO-8430506) is a suitable candidate for further clinical development.
Acknowledgments
We thank T. Inohara for NMR measurement and N. Nakamura for mass spectrometry analysis.
Glossary
Abbreviations
- ATX
autotaxin
- LysoPLD
lysophospholipase D
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- ENPP2
ectonucleotide pyrophosphatase/phosphodiesterase 2
- PTX
paclitaxel
- TGI
tumor growth inhibition
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00200.
Experimental procedures and characterization data for all compounds. Experimental conditions of the biological and DMPK evaluations. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Stracke M. L.; Krutzsch H. C.; Unsworth E. J.; Arestad A.; Cioce V.; Schiffmann E.; Liotta L. A. Identification, Purification, and Partial Sequence Analysis of Autotaxin, a Novel Motility-stimulating Protein. J. Biol. Chem. 1992, 267, 2524. [PubMed] [Google Scholar]
- Umezu-Goto M.; Kishi Y.; Taira A.; Hama K.; Dohmae N.; Takio K.; Yamori T.; Mills G. B.; Inoue K.; Aoki J.; Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227. 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumura A.; Majima E.; Kariya Y.; Tominaga K.; Kogure K.; Yasuda K.; Fukuzawa K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J. Biol. Chem. 2002, 277, 39436. 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- Murata J.; Lee H. Y.; Clair T.; Krutzsch H. C.; Arestad A. A.; Sobel M. E.; Liotta L. A.; Stracke M. L. cDNA Cloning of the Human Tumor Motility-stimulating Protein, Autotaxin, Reveals a Homology with Phosphodiesterases. J. Biol. Chem. 1994, 269, 30479. [PubMed] [Google Scholar]
- Tanaka M.; Okudaira S.; Ohkawa R.; Iseki S.; Ota M.; Noji S.; Yatomi Y.; Aoki J.; Arai H. Autotaxin Stabilizes Blood Vessels and Is Required for Embryonic Vasculature by Producing Lysophosphatidic Acid. J. Biol. Chem. 2006, 281, 25822. 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- Nikitopoulou I.; Oikonomou N.; Karouzakis E.; Sevastou I.; Nikolaidou-Katsaridou N.; Zhao Z.; Mersinias V.; Armaka M.; Xu Y.; Masu M.; Mills G. B.; Gay S.; Kollias G.; Aidinis V. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925. 10.1084/jem.20112012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai J.; Uchida H.; Matsushita Y.; Yano R.; Ueda M.; Niwa M.; Aoki J.; Chun J.; Ueda H. Autotaxin and lysophosphatidic acid1 receptormediated demyelination of dorsal root fibers by sciatic nerve injury and intrathecal lysophosphatidylcholine. Mol. Pain 2010, 6, 78. 10.1186/1744-8069-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadi N.; Gaetano C.; Goping I. S.; Brindley D. N. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene 2009, 28, 1028. 10.1038/onc.2008.442. [DOI] [PubMed] [Google Scholar]
- Venkatraman G.; Benesch M. G.; Tang X.; Dewald J.; McMullen T. P.; Brindley D. N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: implications for cancer treatment. FASEB J. 2015, 29, 772. 10.1096/fj.14-262659. [DOI] [PubMed] [Google Scholar]
- Brindley D. N.; Lin F. T.; Tigyi G. J. Role of the autotaxin–lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 74. 10.1016/j.bbalip.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbayianni E.; Magrioti V.; Moutevelis-Minakakis P.; Kokotos G. Autotaxin inhibitors: a patent review. Expert Opin. Ther. Pat. 2013, 23, 1123. 10.1517/13543776.2013.796364. [DOI] [PubMed] [Google Scholar]
- Nikolaou A.; Kokotou G. M.; Limnios D.; Psarra A.; Kokotos G. Autotaxin inhibitors: a patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815. 10.1080/13543776.2017.1323331. [DOI] [PubMed] [Google Scholar]
- https://www.glpg.com/IPF.
- Nishimasu H.; Okudaira S.; Hama K.; Mihara E.; Dohmae N.; Inoue A.; Ishitani R.; Takagi J.; Aoki J.; Nureki O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205. 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- Guilford W. J.; Bauman J. G.; Skuballa W.; Bauer S.; Wei G. P.; Davey D.; Schaefer C.; Mallari C.; Terkelsen J.; Tseng J. L.; Shen J.; Subramanyam B.; Schottelius A. J.; Parkinson J. F. Novel 3-Oxa Lipoxin A4 Analogues with Enhanced Chemical and Metabolic Stability Have Anti-inflammatory Activity in Vivo. J. Med. Chem. 2004, 47, 2157. 10.1021/jm030569l. [DOI] [PubMed] [Google Scholar]
- Ye Q.; Chourey S.; Wang R.; Chintam N. R.; Gravel S.; Powell W. S.; Rokach J. Structure-activity relationship study of β-oxidation resistant indole-based 5-oxo-6,8,11,14-eicosatetraenoic acid (5-oxo-ETE) receptor antagonists. Bioorg. Med. Chem. Lett. 2017, 27, 4770. 10.1016/j.bmcl.2017.08.034. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Song Y.; Zhan P.; Zhang Q.; Liu X. Conformational restriction: an effective tactic in ’follow-on’-based drug discovery. Future Med. Chem. 2014, 6, 885. 10.4155/fmc.14.50. [DOI] [PubMed] [Google Scholar]
- Jecs E.; Miller E. J.; Wilson R. J.; Nguyen H. H.; Tahirovic Y. A.; Katzman B. M.; Truax V. M.; Kim M. B.; Kuo K. M.; Wang T.; Sum C. S.; Cvijic M. E.; Schroeder G. M.; Wilson L. J.; Liotta D. C. Synthesis of Novel Tetrahydroisoquinoline CXCR4 Antagonists with Rigidified Side-Chains. ACS Med. Chem. Lett. 2018, 9, 89. 10.1021/acsmedchemlett.7b00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman P. J.; Schreier J. D.; McGaughey G. B.; Bogusky M. J.; Cox C. D.; Hartman G. D.; Ball R. G.; Harrell C. M.; Reiss D. R.; Prueksaritanont T.; Winrow C. J.; Renger J. J. Design and synthesis of conformationally constrained N,N-disubstituted 1,4-diazepanes as potent orexin receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 2311. 10.1016/j.bmcl.2010.01.138. [DOI] [PubMed] [Google Scholar]
- He S.; Hong Q.; Lai Z.; Yang D. X.; Ting P. C.; Kuethe J. T.; Cernak T. A.; Dykstra K. D.; Sperbeck D. M.; Wu Z.; Yu Y.; Yang G. X.; Jian T.; Liu J.; Guiadeen D.; Krikorian A. D.; Sonatore L. M.; Wiltsie J.; Liu J.; Gorski J. N.; Chung C. C.; Gibson J. T.; Lisnock J.; Xiao J.; Wolff M.; Tong S. X.; Madeira M.; Karanam B. V.; Shen D.-M.; Balkovec J. M.; Pinto S.; Nargund R. P.; DeVita R. J. Discovery of a Potent and Selective DGAT1 Inhibitor with a Piperidinyl-oxy-cyclohexanecarboxylic Acid Moiety. ACS Med. Chem. Lett. 2014, 5, 1082. 10.1021/ml5003426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga H.; Ohhata A.; Hayashi A.; Katoh M.; Maeda T.; Mizuno H.; Takada Y.; Komichi Y.; Ota H.; Matsumura N.; Shibaya M.; Sugiyama T.; Nakade S.; Kishikawa K. A Novel Highly Potent Autotaxin/ENPP2 Inhibitor Produces Prolonged Decreases in Plasma Lysophosphatidic Acid Formation In Vivo and Regulates Urethral Tension. PLoS One 2014, 9, e93230 10.1371/journal.pone.0093230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.