

Abstract
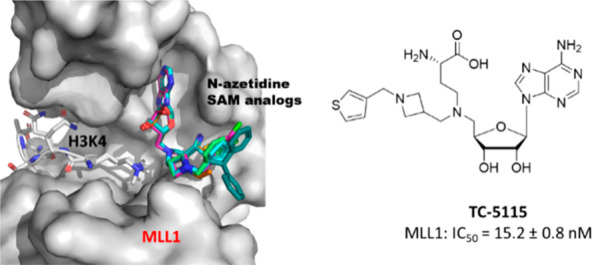
The mixed-lineage leukemia (MLL) protein, also known as MLL1, is a lysine methyltransferase specifically responsible for methylation of histone 3 lysine 4. MLL has been pursued as an attractive therapeutic target for the treatment of acute leukemia carrying the MLL fusion gene or MLL leukemia. Herein, we report the design, synthesis, and evaluation of an S-adenosylmethionine-based focused chemical library which led to the discovery of potent small-molecule inhibitors directly targeting the MLL SET domain. Determination of cocrystal structures for a number of these MLL inhibitors reveals that they adopt a unique binding mode that locks the MLL SET domain in an open, inactive conformation.
Keywords: MLL methyltransferase, H3K4, epigenetics, S-adenosylmethionine, mixed-lineage leukemia
MLL protein is a lysine methyltransferase, responsible for Histone 3 Lysine 4 (H3K4) methylation. Rearrangements of the mixed-lineage leukemia (MLL) gene, also known as MLL1, have been detected in >70% of acute lymphoblastic leukemias (ALL) in infants and 5–10% of acute myelogenous leukemias (AML) in adults.1,2 AML patients carrying the MLL fusion gene, or MLL leukemia, have a very poor prognosis and are unresponsive to current treatments. There is therefore an urgent need to develop new therapeutic strategies for MLL leukemia.
MLL translocations invariably occur on only one MLL allele. Because the MLL fusion proteins contain no SET [Su(var.)3-9, Enhancer-of-zeste and Trithorax] domain, which is the catalytic unit of MLL, MLL translocations lead to a loss of the H3K4 methyltransferase activity, but the remaining wild-type MLL allele retains the H3K4 methyltransferase activity. It has been shown that both the wild-type and fusion MLL proteins are required for MLL-AF9-induced leukemogenesis and maintenance of MLL-AF9-transformed cells. Therefore, inhibition of the H3K4 methyltransferase activity of MLL has been proposed as an attractive therapeutic strategy for the treatment of MLL leukemia.
Although MLL protein contains a catalytic SET domain, the protein itself has very low histone methyltransferase (HMT) activity.3 When MLL protein forms a complex with WDR5, RbBP5, and ASH2L, its HMT activity is dramatically enhanced.3 Therefore, targeting the protein–protein interactions within the MLL complex, in particular the WDR5-MLL protein–protein interaction, has been pursued as a strategy to inhibit the MLL HMT activity. In the past few years, we have reported the discovery of highly potent and cell-permeable peptidomimetics of the WDR5-MLL protein–protein interaction.4 Potent, nonpeptidic, small-molecule inhibitors of the WDR5-MLL interaction have also been reported.5,6
Because MLL is a methyltransferase, another potential therapeutic strategy is to develop small-molecule inhibitors that bind to the active site of MLL and directly block its HMT enzymatic activity. This strategy has been successfully employed for the design of direct inhibitors for a number of histone methyltransferases including MMSET,7 SET7/9,8,9 SMYD3,10 SETD2,11 EZH2,12 DOT1L,13,14 SMYD2,15 PRMT4,16 and PRMT717 but, to date, no direct MLL inhibitor has been reported.
Like other methyltransferases, MLL employs S-adenosylmethionine (SAM or AdoMet) as a major methyl donor which engineers the transfer of a methyl group to histone lysine residues.18,19 In the present study, we describe the discovery of potent, direct MLL inhibitors, through the design, synthesis, and evaluation of a SAM-based focused chemical library.
Our strategy was to synthesize small molecules that mimic the transition state of the methylation chemical reaction of the lysine substrate by incorporation of the cofactor SAM with a portion of the lysine substrate as illustrated in Figure 1(A).
Figure 1.
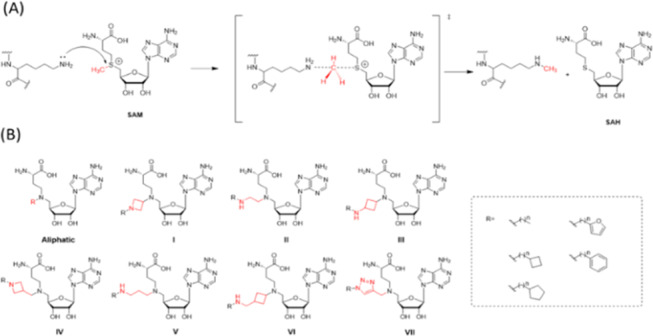
Design of a focused SAM-based library. (A) Proposed mechanism of lysine methylation catalyzed by SAM-dependent lysine methyltransferases. (B) Designed SAM analog library based on the proposed transition state structure. The library contains 8 groups: aliphatic, I (azetidine), II (ethylamine), III (cyclobutane amine), IV (methylene azetidine), V (propyl amine), VI (cyclobutane methylene amine), and VII (triazole). The R functional groups include either an aliphatic or an aromatic hydrophobic fragment.
In our design, the adenosine and the 2-aminobutanoic acid moieties were retained because they are structural moieties necessary for binding to the cofactor binding site. The positively charged sulfur atom in the transition state structure was replaced with a nitrogen atom. To identify optimal moieties that mimic the transition state structure, we systematically explored various primary, secondary, or tertiary amines, as well the linker length between the two nitrogen atoms in the designed inhibitors. We further attached either an aliphatic or an aromatic hydrophobic fragment to the nitrogen to explore the chemical space around the lysine tunnel that is known to be responsible for the selectivity between different methyltransferases. This focused chemical library was categorized, based upon the nature of the group attached to the nitrogen atom mimicking the charged sulfur atom, into the following eight groups: pure aliphatic, cyclobutene amine, cyclobutene methylene amine, triazole, azetidine, ethylamine, propylamine, and methylene azetidine (Figure 1(B)).
We first screened this focused compound library for the ability to inhibit MLL methyltransferase at a single concentration of 10 μM and obtained the results shown in Figure 2. This screening led to identification of 5 compounds, each of which achieved >90% inhibition of the MLL HMT activity. Additionally, 5 other compounds were found to achieve 70–80% inhibition of the MLL HMT activity.
Figure 2.
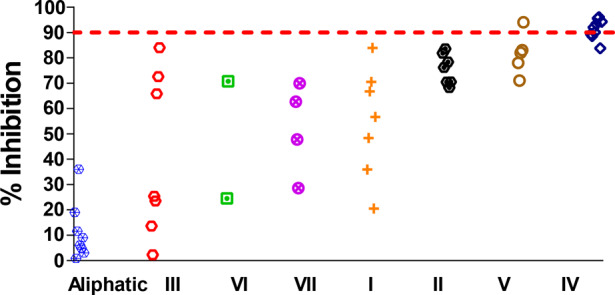
Categorical scatter plot representation of the results of screening a focused library against MLL methyltransferase using an HMT assay at a single concentration of 10 μM.
Among the compounds tested, 4 out of the 5 compounds containing a methylene azetidine (Group IV) inhibit the MLL activity by >90%. In contrast, all the compounds in the aliphatic subset exhibit less than 40% inhibition.
To validate the screening hits, we determined the IC50 values for all compounds which achieved >85% inhibition in the screening using the AlphaLISA assay we developed for MLL.20 For Groups VI and VII, in which no single compound inhibited the MLL activity by >85%, we selected a representative compound with the highest inhibition and evaluated it in the MLL AlphaLISA assay. We included SAH (S-adenosyl-L-homocysteine), a known pan-HMT inhibitor, as a positive control.21 The results of these dose-dependent experiments are summarized in Table 1.
Table 1. Determination of the IC50 Values of Possible Inhibitors Using the AlphaLISA Assay for MLL Methyltransferase.

In the MLL AlphaLISA assay, SAH has an IC50 value of 724 nM. The most potent inhibitors in Groups I–II are 2–3 times more potent than SAH. The most potent inhibitor in Group III has an IC50 of 127 nM and is thus 5-times more potent than SAH. Four compounds in Group IV, including compound 5, achieve the highest potencies with IC50 values of 21–72 nM, and are >10-times more potent than SAH. The most potent compound in Group V has an IC50 value of 201 nM and is 3-times more potent than SAH. The most potent compound in Group VI has an IC50 value of 415 nM, similar to that of SAH. The most potent compound in Group VII has an IC50 value of 132 nM and is thus 5-times more potent than SAH.
Compound 5, with an IC50 value of 21.8 nM is the most potent MLL inhibitor identified in the focused chemical library. We performed further modifications of the azetidine in 5 to gain a further understanding of the SAR for this site with the results summarized in Table 2.
Table 2. Structure–Activity Relationship Studies of the Group IV Compounds.
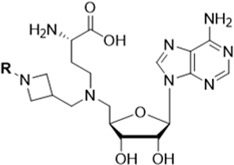

Substitution of the azetidine NH in 5 with a benzyl group resulted in compound 11, which has an IC50 value of 47 nM and is thus 2-times less potent than 5. Substitution of a chlorine atom onto each of three different positions of the phenyl ring in 11 led to a modest decrease in inhibitory potency (2–3 times). Replacing the phenyl group in 11 with 2-thiophenyl or 3-thiophenyl yielded 16 and 15, respectively. While 16 has an IC50 value of 67 nM, which is 3-times less potent than 5, compound 15 is equipotent with 5.
To gain precise structural insights into the potent inhibition by those MLL inhibitors in Group IV, we solved the cocrystal structure of MLL1 SET domain in complex with four methylene azetidine inhibitors (compounds 16 (TC-5109), 14 (TC-5139), 12 (TC-5140), and 18 (TC-5153)) at resolutions ranging from 1.95 to 2.1 Å (Figure 3 and Table S1, PDB ID: 6U9M, 6U9N, 6U9R, 6U9K).
Figure 3.
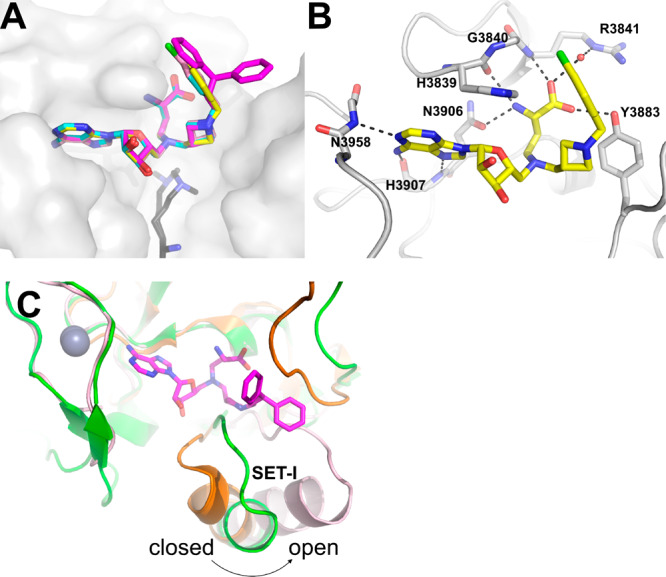
(A) Crystal structures of inhibitors TC-5109 (cyan sticks), TC-5139 (pink sticks), TC-5140 (yellow sticks), and TC-5153 (magenta sticks) bound to the MLL SET domain (gray surface). Phenyl substituents of inhibitors are projected toward bulk solvent opposite to the channel occupied by N-methyllysine (black sticks, based on overlay with PDB ID: 2W5Z). (B) Detailed view of compound TC-5140 (yellow sticks) interactions with MLL cofactor binding site. Dashed lines represent hydrogen bonds. (C) Superposed structure of MLL SET domain (shown in pink) bound to compound 18 (TC-5153, magenta sticks) complex with MMSET/SETD2 (PDB ID: 5LSY, shown in green) and EZH2 (PDB ID: 5WG6, shown in orange).
These cocrystal structures showed that the binding modes of adenosine and the α-amino-acid moieties of these 4 inhibitors occupy the cofactor binding site and form the same contacts with the MLL SET domain as SAH in previous structures.20,22 Unexpectedly, the methylene azetidine moiety of these inhibitors is not directed into the lysine tunnel but instead extends the aromatic substituents of each inhibitor toward bulk solvent on the exterior surface of the protein (Figure 3A). Ligand density (Supporting Information Figure S1) suggests that the azetidine ring and methylene bridge of the phenyl substituent are well-ordered due to hydrophobic packing interactions with the α-amino acid of the inhibitors and with the side chain of Tyr3883 as shown in the MLL/TC-5140 structure (Figure 3B). The chlorophenyl substituent of TC-5140 (Figure 3B) and the thiophene ring of TC-5109 form an edge-to-face π–π interaction with the side chain of His3839. These crystal structures suggest that the azetidine group in our inhibitors induces the “open” conformation of the lysine binding site permitting additional interactions between the tail groups of the inhibitors and the MLL SET domain.
This overall fold of the SET domain bound to our inhibitors is consistent with previous structural studies, in which the MLL SET domain demonstrates a more open lysine binding site than other histone methyltransferases.22,23 Superposition of our MLL SET domain structures with three closely aligned lysine methyltransferases (MMSET, SETDB2, EZH2) shows that MLL is in a more “open” conformation based on a critical displacement of the SET-I helix with respect to the C-terminal flanking region23 (Figure 3C). The alignment further indicates that the loop preceding SET-I helix in MMSET/SETDB2 and EZH2 would sterically clash with the N-azetidine moiety in our inhibitors. Previous studies also suggest that the SET-I helix must be reoriented to adopt a “closed” conformation in order to exert full catalytic activity on SAH.23,24 Our cocrystal structures therefore suggest that our designed MLL inhibitors may lock the MLL SET domain in the “open” state observed in the cocrystal structures and consequently inhibit the MLL SET domain by stabilizing an inactive conformation.
Taken together, our cocrystal structures provide a structural basis for the inhibitory activity of these SAM-based inhibitors of MLL and guidance for further structure-based optimization.
The synthesis of compounds 11–18 containing a substituted N-azetidine series is illustrated in Scheme 1. Reductive amination of compound 19 with tert-butyl (S)-2-((tert-butoxycarbonyl)-amino)-4-oxobutanoate produced compound 20. Compound 21 was prepared by reductive amination of compound 20 with benzyl 3-formylazetidine-1-carboxylate, NaHB(OAc)3 and DCE followed by removal of the protecting group by hydrogenation. Reductive amination of various aldehydes (RCHO) with compound 21 and the subsequent deprotection provided the final compounds 11–18.
Scheme 1. Synthesis of Compounds 11–18.

tert-Butyl (S)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate, NaHB(OAc)3, DCE, rt, 4 h.
Benzyl 3-formylazetidine-1-carboxylate, NaHB(OAc)3, DCE, rt, 3 h.
H2, Pd/C, MeOH, rt, 1 h.
RCHO, NaHB(OAc)3, DCE, rt, 2 h.
TFA, DCM, rt, 1 h.
In summary, we designed a focused library using a bisubstrate strategy and discovered a number of potent inhibitors of MLL methyltransferase activity. Among them, compound 16 (TC-5115) is the most potent and exhibits an IC50 value of 16 nM. Cocrystal structures of MLL in complex with 4 potent inhibitors were determined, which reveal that these inhibitors may lock the MLL SET domain in an open, inactive conformation. Further optimization of compound 16 may ultimately lead to the development of a new therapy for the treatment of human MLL leukemia.
Glossary
Abbreviations
- H3K4
histone H3 Lysine 4
- MLL
mixed-lineage leukemia
- AML
acute myelogenous leukemias
- ALL
acute lymphoblastic leukemias
- SET
Su(var.), E(z), Trithorax
- SAM
S-adenosyl-l-methionine
- SAH
S-adenosyl-l-homocysteine
- HMT
histone methyltransferase
- MMSET
multiple myeloma SET domain
- SMYD3
SET- and MYND-domain containing 3
- EZH2
Enhancer of Zeste Homologue 2
- DOT1L
Disruptor of telomeric silencing 1-like
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00229.
Detailed in vitro histone methyltransferase assay, detailed synthesis and chemical data of tested compounds, and protein expression and crystallization details. (PDF)
Author Present Address
† (T.-R.C. and D.B.) Atomwise, Inc., 717 Market Street, Suite 800, San Francisco, CA 94103.
Author Contributions
T.-R. Chern designed and synthesized all the compounds and wrote the paper; L. Liu developed the AlphaLISA assay and tested the compounds; J. Stuckey and E. Petrunak expressed the MLL protein and determined the cocrystal structures and wrote the paper; M. Wang tested the compounds in the AlphaLISA assay and wrote the paper; D. Bernard performed computational modeling and wrote the paper; H. Zhou provided advice for the synthesis and wrote the paper; S. Lee developed the HMT assay and tested the compounds in the HMT assay; Y. Dou designed the HMT assay and supervised the testing of compounds in the HMT assay; S. Wang designed the project, supervised the experiments, and wrote the paper. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work is supported in part by the Rogel Cancer Center Core Grant from the National Cancer Institutes, NIH (P30 CA046592). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
The authors declare no competing financial interest.
Supplementary Material
References
- Djabali M.; Selleri L.; Parry P.; Bower M.; Young B. D.; Evans G. A. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992, 2 (2), 113–8. 10.1038/ng1092-113. [DOI] [PubMed] [Google Scholar]
- Tkachuk D. C.; Kohler S.; Cleary M. L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71 (4), 691–700. 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- Dou Y.; Milne T. A.; Ruthenburg A. J.; Lee S.; Lee J. W.; Verdine G. L.; Allis C. D.; Roeder R. G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13 (8), 713–9. 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Cao F.; Townsend E. C.; Karatas H.; Xu J.; Li L.; Lee S.; Liu L.; Chen Y.; Ouillette P.; Zhu J.; Hess J. L.; Atadja P.; Lei M.; Qin Z. S.; Malek S.; Wang S.; Dou Y. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53 (2), 247–61. 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senisterra G.; Wu H.; Allali-Hassani A.; Wasney G. A.; Barsyte-Lovejoy D.; Dombrovski L.; Dong A.; Nguyen K. T.; Smil D.; Bolshan Y.; Hajian T.; He H.; Seitova A.; Chau I.; Li F.; Poda G.; Couture J. F.; Brown P. J.; Al-Awar R.; Schapira M.; Arrowsmith C. H.; Vedadi M. Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 2013, 449 (1), 151–9. 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F.; Vedadi M.; Getlik M.; Giambruno R.; Grover A.; Avellino R.; Skucha A.; Vittori S.; Kuznetsova E.; Smil D.; Barsyte-Lovejoy D.; Li F.; Poda G.; Schapira M.; Wu H.; Dong A.; Senisterra G.; Stukalov A.; Huber K. V. M.; Schonegger A.; Marcellus R.; Bilban M.; Bock C.; Brown P. J.; Zuber J.; Bennett K. L.; Al-Awar R.; Delwel R.; Nerlov C.; Arrowsmith C. H.; Superti-Furga G. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat. Chem. Biol. 2015, 11 (8), 571–578. 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisi D.; Chiarparin E.; Tamanini E.; Pathuri P.; Coyle J. E.; Hold A.; Holding F. P.; Amin N.; Martin A. C. L.; Rich S. J.; Berdini V.; Yon J.; Acklam P.; Burke R.; Drouin L.; Harmer J. E.; Jeganathan F.; van Montfort R. L. M.; Newbatt Y.; Tortorici M.; Westlake M.; Wood A.; Hoelder S.; Heightman T. D. Structure of the Epigenetic Oncogene MMSET and Inhibition by N-Alkyl Sinefungin Derivatives. ACS Chem. Biol. 2016, 11 (11), 3093–3105. 10.1021/acschembio.6b00308. [DOI] [PubMed] [Google Scholar]
- Mori S.; Iwase K.; Iwanami N.; Tanaka Y.; Kagechika H.; Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18 (23), 8158–8166. 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Niwa H.; Handa N.; Tomabechi Y.; Honda K.; Toyama M.; Ohsawa N.; Shirouzu M.; Kagechika H.; Hirano T.; Umehara T.; Yokoyama S. Structures of histone methyltransferase SET7/9 in complexes with adenosylmethionine derivatives. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69 (4), 1–8. (2013), D69, 595–602 10.1107/S0907444912052092. [DOI] [PubMed] [Google Scholar]
- Van Aller G. S.; Graves A. P.; Elkins P. A.; Bonnette W. G.; McDevitt P. J.; Zappacosta F.; Annan R. S.; Dean T. W.; Su D.-S.; Carpenter C. L.; Mohammad H. P.; Kruger R. G. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24 (5), 774–781. 10.1016/j.str.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Ibáñez G.; Wu H.; Blum G.; Zeng H.; Dong A.; Li F.; Hajian T.; Allali-Hassani A.; Amaya M. F.; Siarheyeva A.; Yu W.; Brown P. J.; Schapira M.; Vedadi M.; Min J.; Luo M. Sinefungin Derivatives as Inhibitors and Structure Probes of Protein Lysine Methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134 (43), 18004–18014. 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung P. P.; Huang B.; Zehnder L.; Tatlock J.; Bingham P.; Krivacic C.; Gajiwala K.; Diehl W.; Yu X.; Maegley K. A. SAH derived potent and selective EZH2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25 (7), 1532–7. 10.1016/j.bmcl.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122 (6), 1017–25. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Song Y. A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem. 2013, 56 (22), 8972–83. 10.1021/jm4007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. P.; Swewczyk M.; Kennedy S.; Trush V. V.; Wu H.; Zeng H.; Dong A.; Ferreira de Freitas R.; Tatlock J.; Kumpf R. A.; Wythes M.; Casimiro-Garcia A.; Denny R. A.; Parikh M. D.; Li F.; Barsyte-Lovejoy D.; Schapira M.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Owen D. R. Selective, Small-Molecule Co-Factor Binding Site Inhibition of a Su(var)3–9, Enhancer of Zeste, Trithorax Domain Containing Lysine Methyltransferase. J. Med. Chem. 2019, 62 (17), 7669–7683. 10.1021/acs.jmedchem.9b00112. [DOI] [PubMed] [Google Scholar]
- Cai X. C.; Zhang T.; Kim E. J.; Jiang M.; Wang K.; Wang J.; Chen S.; Zhang N.; Wu H.; Li F.; Dela Sena C. C.; Zeng H.; Vivcharuk V.; Niu X.; Zheng W.; Lee J. P.; Chen Y.; Barsyte D.; Szewczyk M.; Hajian T.; Ibanez G.; Dong A.; Dombrovski L.; Zhang Z.; Deng H.; Min J.; Arrowsmith C. H.; Mazutis L.; Shi L.; Vedadi M.; Brown P. J.; Xiang J.; Qin L. X.; Xu W.; Luo M., A chemical probe of CARM1 alters epigenetic plasticity against breast cancer cell invasion. eLife 2019, 8, 10.7554/eLife.47110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk M. M.; Ishikawa Y.; Organ S.; Sakai N.; Li F.; Halabelian L.; Ackloo S.; Couzens A. L.; Eram M.; Dilworth D.; Fukushi H.; Harding R.; dela Seña C. C.; Sugo T.; Hayashi K.; McLeod D.; Zepeda C.; Aman A.; Sánchez-Osuna M.; Bonneil E.; Takagi S.; Al-Awar R.; Tyers M.; Richard S.; Takizawa M.; Gingras A.-C.; Arrowsmith C. H.; Vedadi M.; Brown P. J.; Nara H.; Barsyte-Lovejoy D. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 2020, 11 (1), 2396. 10.1038/s41467-020-16271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecave M.; Atta M.; Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 2004, 29 (5), 243–9. 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Magnusson O. T. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin?. Chem. Rev. 2003, 103 (6), 2129–48. 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- Karatas H.; Li Y.; Liu L.; Ji J.; Lee S.; Chen Y.; Yang J.; Huang L.; Bernard D.; Xu J.; Townsend E. C.; Cao F.; Ran X.; Li X.; Wen B.; Sun D.; Stuckey J. A.; Lei M.; Dou Y.; Wang S. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J. Med. Chem. 2017, 60 (12), 4818–4839. 10.1021/acs.jmedchem.6b01796. [DOI] [PubMed] [Google Scholar]
- Deguchi T.; Barchas J. Inhibition of transmethylations of biogenic amines by S-adenosylhomocysteine. Enhancement of transmethylation by adenosylhomocysteinase. J. Biol. Chem. 1971, 246 (10), 3175–81. [PubMed] [Google Scholar]
- Campagna-Slater V. r.; Mok M. W.; Nguyen K. T.; Feher M.; Najmanovich R.; Schapira M. Structural Chemistry of the Histone Methyltransferases Cofactor Binding Site. J. Chem. Inf. Model. 2011, 51 (3), 612–623. 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- Southall S. M.; Wong P.-S.; Odho Z.; Roe S. M.; Wilson J. R. Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33 (2), 181–191. 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Mittal A.; Reid J.; Reich S.; Gamblin S. J.; Wilson J. R. Evolving Catalytic Properties of the MLL Family SET Domain. Structure 2015, 23 (10), 1921–1933. 10.1016/j.str.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.