

Abstract
The thermal shift assay is a robust method of discovering protein–ligand interactions by measuring the alterations in protein thermal stability under various conditions. Several thermal shift assays have been developed and their throughput has been advanced greatly by the rapid progress in tandem mass tag-based quantitative proteomics. A recent paper by Gaetani et al. (J. Proteome Res. 2019, 18 (11), 4027–4037) introduced the proteome integral solubility alteration (PISA) assay, further increasing throughput and simplifying the data analysis. Both ΔSm (a proxy of the difference between areas under the melting curves) and fold changes (ratios between integral samples) are readouts of the PISA assay and positively related to ΔTm (shift in melting temperatures). Here, we show that the magnitudes of these readouts are inherently small in PISA assay, which is a challenge for quantitation. Both simulation and experimental results show that the selection of a subset of heating temperatures ameliorates the small difference problem and improves the sensitivity of the PISA assay.
Keywords: protein–ligand interaction, thermal shift assay, shift in melting points, fold change, heating temperature selection, proteomics, mass spectrometry, tandem mass tag
Graphical Abstract
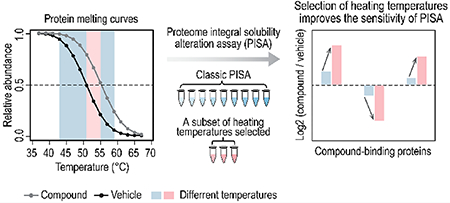
■ INTRODUCTION
Protein–ligand (e.g., metabolites, therapeutic drugs) interactions induce alterations in protein physiochemical properties and regulate protein function and activity in various cellular processes. The thermal shift assay was first employed to measure the change in thermal denaturation temperatures of a purified protein under diverse conditions, thus enabling the detection of ligand-induced changes in thermal stability for target proteins.1,2 Later, the cellular thermal shift assay (CETSA) was developed to investigate protein–ligand binding in cells and tissues.3 Thermal proteome profiling (TPP) further translated CETSA onto a proteome-wide scale using tandem mass tag (TMT)-based quantitative proteomics.4 In a TPP assay, protein abundance across different heating temperatures is measured to generate protein melting curves and calculate the melting points.4 While the TPP assay is effective, analysis of 8–10 temperatures is required to define each melting curve, resulting in long analysis times and relatively low throughput.5 In a recent paper, Gaetani et al. introduced the proteome integral solubility alteration (PISA) assay and greatly increased the throughput of the traditional TPP assay.5 In a PISA assay, samples across an entire temperature gradient are combined. The protein abundance in the pooled sample, which represents the area under the melting curve, is used to measure the effect of a ligand on protein thermal stability.5 The sample pooling step massively increases throughput and reduces both analysis time and sample consumption. Furthermore, the newly introduced TMTpro reagents for proteome-wide quantitation accommodate up to 16 integral samples in one experiment, allowing multiple control and treated samples to be analyzed in the same group without missing values and thus simplifying the data analysis.6
The readout of a PISA assay can be expressed as ΔSm (a proxy of the difference between areas under the melting curves of treated and control samples) or fold changes (integral treated samples versus integral control samples).5 Both ΔSm and fold changes are positively related to ΔTm (shift in melting temperatures) and thus to protein thermal stability. In a PISA assay, the experimental ΔSm is approximated by normalizing the difference between integral treated and integral control samples on integral control samples,5 and fold changes do not require extra normalization steps. Here we characterized the relationship among experimental ΔSm fold changes and ΔTm, the behaviors of experimental ΔSm and fold changes, and showed how selecting a subset of heating temperatures improves the sensitivity of the PISA assay.
■ EXPERIMENTAL PROCEDURES
Ab Initio Simulation
The simulation of sigmoidal curves of 10 000 hypothetical proteins under control and ligand treatment was carried out as described in Gaetani et al.5 in R 3.6.1. Briefly, for a given hypothetical protein, 16 temperatures (T) between 37 and 67 °C with a 2 °C step were chosen; a melting temperature (Tm) under control conditions was selected randomly between 42 and 57 °C; and a shift of the melting temperature (ΔTm) under ligand treatment was selected randomly between −5 °C and +5 °C. Sigmoidal melting curves were simulated by calculating the intensity for a given temperature (T) as
| (1) |
| (2) |
where erf is the error function, and sqrt is the square root function. The simulated intensity(T)control or intensity(T)treated corresponds to a TMT reporter ion intensity normalized to the intensity at the top plateau of the melting curve. The sum of intensity(T)control and the sum of intensity(T)treated values for selected temperatures were used to calculate Smcontrol (i.e., Σ intensity(T)control), Smtreated (i.e., Σ intensity(T)treated), ΔSm (i.e., Sm treated – Sm control), and fold change (i.e., Sm treated/Sm control).
One-Dimensional Proteome Integral Solubility Alteration (PISA) Assay
HCT116 cells were purchased from ATCC. Cells were maintained in DMEM with high glucose/pyruvate (Invitrogen) supplemented with 10% FBS (Hyclone) in a 5% CO2 incubator at 37 °C. Cells in a 15 cm dish were harvested at approximately 80% confluency using trypsin, washed once with PBS, and frozen in liquid nitrogen for future use.
Cell pellets were thawed on ice and resuspended in lysis buffer (1× PBS, 1 mM MgCl2, 0.5% NP-40, and protease inhibitor cocktail, pH 7.4). Cells were incubated for 15 min at 4 °C with gentle agitation. Lysates were cleared by centrifugation at 20 000g for 15 min. The soluble fraction was collected and protein concentration was determined by BCA assays (Pierce). Each lysate was diluted to 2 mg/mL in lysis buffer and divided into two aliquots. Each aliquot was mixed in a 1:1 ratio with lysis buffer (control) or lysis buffer with 20 mM NAD+ to yield eight reactions with a final protein concentration of 1 mg/mL and a final NAD+ concentration of 0 (control; N = 4) or 10 mM (N = 4), respectively. Reactions were incubated at room temperature for 10 min with gentle shaking. An equal volume from each reaction was distributed into 10 PCR tubes. Samples were heated in a thermocycler for 3 min across a temperature gradient from 43 to 59 °C. Following heating, samples were equilibrated at room temperature for 5 min. An equal volume was removed from PCR tubes corresponding to selected temperatures and pooled (43 to 59 °C (10 temperatures in total), 52 to 54 °C (3 temperatures in total)). These samples were centrifuged at maximum speed (~21 000g) for 2 h to remove aggregated proteins. An equal volume of the resulting soluble fraction (~50 μg protein) was collected and mixed with an equal volume of buffer comprised of 400 mM EPPS, 2% SDS, and pH 8.5 to yield a final concentration of 200 mM EPPS, 1% SDS, and pH 8.5.
LC–MS Sample Preparation
Samples were reduced with 5 mM TCEP for 30 min, alkylated with 10 mM iodoacetamide for 30 min, and then quenched with 10 mM DTT for 15 min. SP3 protocol7 was used as a basis for sample preparation. Briefly, the reduced and alkylated proteins were bound to SP3 beads, washed three times with 80% ethanol, and subjected to on-bead digestion overnight at 37 °C in 200 mM EPPS, pH 8.5 while shaking with Lys-C protease at a 100:1 protein-to-protease ratio. Trypsin was added to a 100:1 protein-to-protease ratio and the samples were incubated for 6 h at 37 °C while shaking. The beads were removed from the samples and anhydrous acetonitrile was added to a final concentration of ~30%. The ~50 μg of peptides were labeled with ~100 μg of TMT at room temperature for 60 min. The labeled peptides were then quenched with hydroxylamine, pooled, and desalted by Sep-Pak (Waters). Samples were dried, resuspended in 5% acetonitrile and 10 mM ammonium bicarbonate, pH 8 and subjected to high-pH reversed-phase fractionation. Fractions were collected in a 96-well plate and combined for a total of24 fractions prior to desalting and subsequent LC–MS/MS analysis of 12 nonadjacent fractions.
Mass Spectrometry Data Acquisition
Data were collected on Orbitrap Fusion mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Peptides were separated using a 90 min gradient at 525 nL/min on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed inhouse. High-field asymmetric-waveform ion mobility spectroscopy (FAIMS) was enabled during data acquisition with compensation voltages (CVs) set as −40 V, −60 V, and −80 V.8 MS1 data were collected using the Orbitrap (120 000 resolution; maximum injection time 50 ms; AGC 4 × 105). Determined charge states between 2 and 7 were required for sequencing, and a 60 s dynamic exclusion window was used. Data dependent mode was set as cycle time (1 s). MS2 scans were performed in the ion trap with CID fragmentation (isolation window 0.7 Da; Turbo; NCE 35%; maximum injection time 35 ms; AGC 2 × 104). A standard MultiNotch SPS-MS3 was used,9 and MS3 scans were collected in the Orbitrap using a resolution of 50 000, NCE of 65%, maximum injection time of 120 ms, AGC of 1.5 × 105, and number of SPS ions set to 10. Data were collected in positive ion mode and were centroided online.
Mass Spectrometry Data Analysis
Raw files were first converted to mzXML. Database searching included all human entries from Uniprot (downloaded on July 12, 2019). The database was concatenated with one composed of all protein sequences in the reversed order. Sequences of common contaminant proteins (e.g., trypsin, keratins, etc.) were appended as well. Searches were performed using a 50 ppm precursor ion tolerance and 0.9 Da product ion tolerance. TMT on lysine residues and peptide N termini (+229.1629) and carbamidomethylation of cysteine residues (+57.0215 Da) were set as static modifications, while oxidation of methionine residues (+15.9949 Da) was set as a variable modification.
Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR).10 PSM filtering was performed using linear discriminant analysis (LDA) as described previously,11 while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. Each run was filtered separately. Protein-level FDR was subsequently estimated at a data set level. For each protein across all samples, the posterior probabilities reported by the LDA model for each peptide were multiplied to give a protein-level probability estimate. Using the Picked FDR method,12 proteins were filtered to the target 1% FDR level.
For reporter ion quantification, a 0.003 Da window around the theoretical m/z of each reporter ion was scanned, and the most intense m/z was used. Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMT reagents according to manufacturer specifications. Peptides were filtered to include only those with a summed signal-to-noise (SN) of 200 or greater across all channels. An isolation purity of at least 0.7 (70%) in the MS1 isolation window was used. For each protein, the filtered peptide TMT SN values were summed to generate protein quantification. To control for different total protein loading within a TMT experiment, the summed protein quantities of each channel were adjusted to be equal within the experiment.
Statistical and Bioinformatics Analyses
One-tailed Welch’s t test was employed to test for difference between control and NAD+-treated samples. Proteins showing p-values less than 0.01 and absolute log2 fold change greater than 0.5 were defined as significant changes. A permutation test was performed through randomizing 8 values (4 from control and 4 from treated samples) for each protein. The same cutoffs as described above were applied to the randomized data and 1 000 permutation tests estimated that there are 5% false positives. The STITCH database was used to map known NAD+-binding proteins (combined score ≥700).13 GO analysis was performed with DAVID (6.8)14 using all overlapping quantified proteins as the background, and results were filtered at Benjamini–Hochberg adjusted p-values ≤ 0.05.
Data Availability
The mass spectrometry data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD017346.
■ RESULTS
Relationship among ΔSm, Fold Changes, and ΔTm in Simulated PISA
The fundamental motivation of TPP assays is to detect changes in thermal stability, as reflected by changes in melting temperatures upon ligand binding or other perturbation.4 While most TPP studies have aimed to directly measure ATm (melting temperature shift between treated and control samples) from full melting curves, PISA instead attempts to detect changes in thermal stability from protein abundances in samples pooled across the temperature range.5 While the PISA approach has the potential to greatly increase TPP assay throughput, its utility depends on the sensitivity with which it can detect changes in protein stability. Gaetani et al. performed simulations to show that PISA data relate to ΔTm through the ΔSm (difference between the integral for the treated sample and the integral for the control sample) statistic.5 Following their example, we have performed similar simulations to investigate the relationship between fold changes (integral treated sample versus integral control sample) and ATm in a simulated PISA assay (Figure 1). In line with Gaetani et al., we observed a good linear correlation between simulated ASm and ATm (Figure 1b). The log2 fold changes are positively correlated with ΔTm (Figure 1c), yet the linearity is not as good as between simulated ΔSm and ΔTm in Figure 1b. The magnitude of the fold change is greater for a destabilizer than stabilizer for a given ΔTm and Tm (Figure 1c), which is decided by the attributes of sigmoidal curves (Figure S1a,b). Overall, high Tm proteins tend to have smaller difference, but the magnitudes are also affected by ΔTm (Figure 1c). We next examined the log2 fold change distribution of hypothetical proteins of absolute ΔTm equal or greater than 2 and noticed that the majority of the fold changes is inherently small (Figure 1d). More than 70% and 95% of these proteins showed absolute log2 fold changes smaller than 0.5 and 1.0, respectively.
Figure 1.

A simulated PISA assay combining samples incubated at temperatures ranging from 37 to 67 °C. (a) Examples of simulated melting curves. (b) Relationship between simulated ΔSm and ΔTm. Simulated ΔSm and ΔTm are linearly correlated when samples heated at 37 to 67 °C are combined. (c) Relationship between simulated log2 fold changes and ΔTm. Log2 fold changes and ΔTm are positively related, but they do not show good linearity as simulated ΔSm and ΔTm. Dashed boxes highlight hypothetical proteins showing |ΔTm| ≥ 2. (d) Distribution of log2 fold changes of hypothetical proteins showing |ΔTm| ≥ 2. The majority of the log2 fold changes are within a narrow range. (e) Equations describing the relationship among simulated ΔSm, fold change (FC), and ΔTm. Intensity(treated) means the TMT reporter ion intensity of integral treated samples (combining from multiple samples covering the full melting curve). Intensity(control) indicates the TMT reporter ion intensity of integral control samples (combining from multiple samples covering the full melting curve). Intensity(treated or control at top plateau) indicates the TMT reporter ion intensity of the individual sample incubated at the top plateau of a melting curve. ΔSm is the difference between integral treated and integral control samples in simulated data. ΔTm is the melting temperature shift between treated and control samples. Fold change (FC) is the ratio of integral treated sample versus integral control sample. (f) Equations describing the relationship between experimental ΔSm and fold change (FC). In a PISA experiment, Intensity(treated or control at top plateau) is not available and experimental ΔSm is approximated by normalizing the difference between integral treated and integral control samples on integral control samples. Experimental fold change (FC) remains the same as simulated fold change. There is a linear relationship between experimental ΔSm and FC (ASm = FC – 1).
From the equations for simulated ΔSm and ΔTm (Figure 1b), we derived the equation describing the relationship between ΔTm and fold changes (Figure 1e). The relationship between ΔTm and fold changes relies on the intensity of the integral control sample and the intensity of the individual sample incubated at the top plateau of the melting curve. In a PISA experiment, the individual sample intensity at the top plateau is not available (see Figure 1e for the equation for simulated ΔSm), and the experimental ΔSm is approximated by normalizing the difference between integral treated and integral control samples on integral control samples5 (Figure 1f). Experimental fold changes remain the same as simulated ones, thus the experimental ΔSm and fold changes are also linearly correlated with each other in a PISA experiment (Figure 1f).
Tm was selected randomly between 42 and 57 °C in the simulation, and intensities were simulated between 37 and 67 °C. The simulated results included proteins of low melting temperatures (see an example in Figure S1c). The top plateau of the melting curve of a low Tm protein may be lower than 37 °C, which is typically the lowest temperature in a TPP experiment. The intensities of low temperature (37 °C) control and treated samples are different in this case (Figure S1c), a scenario which is also observed in a TPP experiment. Since only part of the area under the curve is used to calculate simulated ΔSm (Figure S1c), ΔSm is underestimated and the linearity between simulated ΔSm and ΔTm is compromised for low Tm proteins (Figure 1b, proteins of low Tm and negative ΔTm). This situation is the same for high Tm proteins (Figure S1d). However, high Tm proteins are not affected as much as low Tm proteins in the simulation (Figure 1b, proteins of high Tm and positive ΔTm), as the highest Tm (57 °C) is farther away from the highest simulated temperature (67 °C) than the lowest Tm (42 °C versus 37 °C). Fold change and experimental ΔSm remain unaffected since they are unrelated with the intensity at the top or bottom plateau.
Selection of Heating Temperatures Increases the Magnitudes of Fold Changes in Simulated PISA
The simulation result in Figure 1d shows that the majority of the log2 fold changes is inherently small and lies within a narrow range around zero. This scenario can suffer from even small systematic or technical errors and is extremely challenging for quantitation. The large proportion of small fold changes in Figure 1d remains a difficult problem to tackle. Since there is a linear relationship between experimental ΔSm and fold changes (Figure 1f), using ΔSm faces the same issue (experimental ΔSm = FC — 1). We just focused on fold changes here, as it is more straightforward and easy to evaluate the data from a quantitative point of view.
In a proof-of-concept experiment in Gaetani et al., the low-and high-solubility plateaus were excluded and a temperature range of 43 to 59 °C was used.5 We simulated the results with integral samples heated at 43–59 °C (Figure 2a,b). The magnitudes of log2 fold changes increased to some extent, yet there are still a great portion of hypothetical proteins of absolute ΔTm equal or greater than 2 showing small fold changes (40% between −0.5 and 0.5; 79% between −1.0 and 1.0) (Figure 2b).
Figure 2.

Selection of heating temperatures increases the magnitudes of fold changes in a simulated PISA assay. Two temperature ranges (a,b, 43–59 °C; c d, 51–55 °C) are presented. (a) Relationship between simulated log2 fold changes and ΔTm when samples incubated at 43– 59 °C are combined. (b) Distribution of log2 fold changes of hypothetical proteins showing |ΔTm| ≥ 2 when samples are incubated at 43–59 °C are combined. (c) Relationship between simulated log2 fold changes and ΔTm when samples incubated at 51–55 °C are combined. (d) Distribution of log2 fold changes of hypothetical proteins showing |ΔTm| ≥ 2 when samples incubated at 51–55 °C are combined. Compared with results in Figure 1c,d where samples incubated at 37–67 °C are combined, results from a narrower temperature range (43–59 °C) show greater magnitudes of fold changes (a,b). A much narrower temperature range (51–55 °C) further increases the magnitudes of fold changes (c,d). ΔTm is the melting temperature shift between treated and control samples. Fold change (FC) is the ratio of integral treated sample versus integral control sample. Dashed boxes highlight hypothetical proteins showing |ΔTm| ≥ 2.
Gaetani et al. mentioned that the dynamic range of the PISA readout could be increased by narrowing the temperature range to the region of the most significant solubility changes.5 The majority of the human protein Tm lies between 45 and 55 °C, and the proteome-wide denaturation catastrophe happens around 50 °c.15,16 We then simulated the results with integral samples heated from 51 to 55 °C, anticipating much greater magnitudes of fold changes. Encouragingly, the fraction of small fold changes is dramatically reduced (14% between −0.5 and 0.5; 40% between −1.0 and 1.0) (Figure 3c,d). A few small fold changes remain, but overall they are much more manageable, especially when coupled with multiNotch SPS-MS3 quantification9 plus FAIMS8 or real-time search,17,18 which have higher quantitation accuracy. We noted that the fold changes and ΔTm are still positively related with each other, but a certain fold change could indicate varying ΔTm and Tm (Figure 2a,c).
Figure 3.
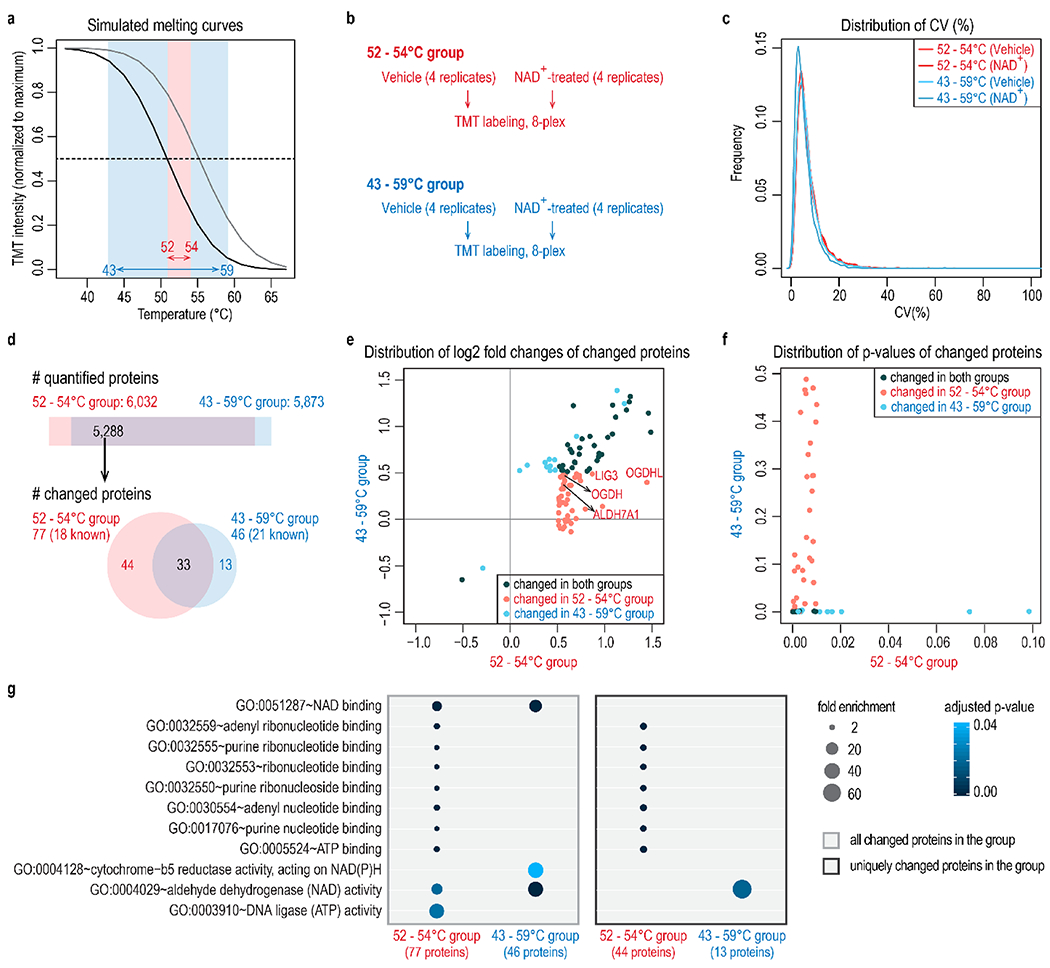
Temperature selection improves the identification of NAD+-binding proteins. (a) Two temperature ranges are compared in this experiment: a broad range of 43 to 59 °C, and a narrow range of 52 to 54 °C. (b) Four replicates of vehicle or NAD+-treated samples were included for each temperature range. Samples were accommodated in two TMT 8-plex experiments. (c) Distribution of coefficient of variation (CV) of four replicates under different conditions shows equivalent data quality. (d) The number of quantified proteins and proteins with altered abundance at different temperature ranges. Both the temperature ranges captured known NAD+-binding proteins. The narrow temperature range of 52–54 °C revealed more targets (77) than the broad temperature range (46). The STITCH database was used to map known NAD+-binding proteins. (e,f) Distribution of log2 fold changes (e) and p-values (f) of changed proteins at either temperature range. The narrow temperature range (52–54 °C) uniquely captured 44 significant target proteins that show small fold changes at a broad temperature range (43–59 °C). Known NAD+-binding proteins uniquely recovered in the 52–54 °C group were highlighted in part e. (g) Gene ontology analysis of proteins with altered abundance in each group. All overlapping quantified proteins were used as the background. p-values were adjusted using the Benjamini-Hochberg method and then filtered at 0.05.
Selection of Heating Temperatures Improves the Identification of NAD+-Binding Proteins
Next, we carried out a PISA experiment to identify NAD+-binding proteins using the two temperature ranges in Figure 2. The temperature ranges were adjusted slightly to fit our thermocycler (Figure 3a). Four replicates were included for each treatment and temperature range, and two TMT 8-plex groups were adopted to accommodate all samples (Figure 3b). Replicates for each treatment and temperature range showed an equivalent coefficient of variance (CV) distribution (Figure 3c). Among the overlapping quantified proteins, the 52–54 °C group revealed 77 target proteins (18 known NAD+-binding proteins), and the 43–59 °C group captured 46 (21 known NAD+-binding proteins) (Figure 3d). In accordance with the simulation results, the 44 target proteins unique in the 52–54 °C group showed greater fold changes compared with the 43–59 °C group (Figure 3e) and, accordingly, smaller p-values (Figure 3f). Gene ontology analysis of proteins with altered abundance in each group showed that, in both cases, the treated samples were significantly enriched for NAD+-binding proteins (Figure 3g).
Because one narrow temperature range is unable to cover the ideal detection region for all proteins, shrinking the heating temperature range may miss some target proteins. As seen in Figure 3d–f, 13 target proteins were identified uniquely in the 43–59 °C group (Figure 3d–f). Overall, the gain in the 52–54 °C group outnumbered the loss with respect to proteins showing significant changes.
■ DISCUSSION
Gaetani et al. introduced the proteome integral solubility alteration (PISA) assay and greatly increased the throughput of thermal stability-based identification of ligand-binding proteins.5 We show that the difference (both fold changes and experimental ΔSm) are inherently small and thus challenging to detect in a PISA assay. Detection of thermal stability shifts depends on Tm, ΔTm, heating temperature range, and protein abundance (Figure 2). Combining samples incubated at high heating temperatures is an effective way of getting rid of individual samples of small difference at top plateaus, thus increasing the fold changes of integral samples in the simulation (Figure 2). However, protein abundance decreases dramatically at relatively high heating temperatures in an experimental scenario, especially abundance of low Tm proteins, which potentially makes shift detection difficult. In the simulation and experiment, we picked a narrow temperature range right after the region of the most significant solubility changes to compromise between magnitudes of fold changes and protein abundance, and results showed increased fold change differences and improved sensitivity of detecting the protein thermal stability changes.
We note that one narrow temperature range may miss some target proteins, as it cannot cover the ideal temperature range for all proteins in consideration of Tm, ΔTm and abundance. In the experiment in Figure 3, overall more NAD+-binding proteins were identified in the narrow temperature range group. However, a few target proteins are also missed. The full melting curve can be separated into several segments to achieve a complete recovery of target proteins. Besides, the narrow temperature range employed in this work can be optimized further depending on the specific ligand of interest, to achieve a list of target proteins of both satisfying coverage and high confidence (greater fold changes).
A recently introduced approach named “one-pot analysis” conveyed a similar idea to Gaetani et al.5 and this work but from a different perspective.19 In the “one-pot analysis” strategy, Tm equals to Intensity(average) × (T2 – T1)/Intensity0, where Intensity(average) = Intensity(treated)/16 (16 is the number of temperatures) or Intensity(control)/16, Intensity0 = Intensity(treated or control at top plateau) T2 = 67, and T1 = 37. Then, it can be derived that in one-pot analysis ΔTm equals to 1.875 × (Intensity(treated) – Intensity(control))/Intensity(treated or control at top plateau) (i.e., ΔTm = 1.875 × ΔSm), which matches the equation in Figure 1b (ΔTm = 2.046 × ΔSm – 0.020). In the simulation, the temperature range 37–67 °C does not cover the full melting curve for a small fraction of proteins, which distorts the coefficient of equation in Figure 1b slightly. Theoretically, TMT-based quantitative proteomics can translate the low-throughput Western blotting-based “one-pot analysis” to high-throughput analysis given that Intensity(treated or control at top plateau) is measured in each analysis. Optimization in sample preparation, data acquisition, and normalization will be needed for this purpose. Importantly, to accurately measure Tm, the ratio between Intensity(treated) and Intensity(treated or control at top plateau) and the ratio between Intensity(control) and Intensity(treated or control at top plateau) must be as accurate as possible. Due to the well-known problematic issue of ratio compression in high-resolution MS2 (hrMS2) TMT strategy, multiNotch SPS-MS3 coupled with FAIMS,8 or real-time search17,18 may improve the robust and confident measurement of Tm.
Both PISA assay and one-pot analysis assume that melting curves are sigmoidal and measurable for all proteins. However, a small subset of proteins that do not yield good melting curves exists,20 which potentially generates false positive hits. Selection of a subset of heating temperatures in the PISA assay retains this caveat. Another recent paper introduced the isothermal shift assay (iTSA) using one heating temperature and no effect size cutoff to detect thermal stability shifts.21 It further simplifies the sampling handling process as no sample pooling step is required. However, the iTSA method may suffer more from ill-behaved proteins and technical errors, since only one heating temperature is picked and there is a higher chance that the results are affected. Overall, PISA assay is still a robust tool of screening drug-binding proteins, especially large-scale screenings as the sample pooling step massively enhances the throughput and decreases analysis time. Moreover, the data from a large screening should also enable the detection of some false positive targets given that they may show up more frequently and less uniquely to specific ligand.
Supplementary Material
Figure S1, examples of simulated melting curves (PDF)
Table S1, results of NAD+-binding proteins by PISA (XLSX)
■ ACKNOWLEDGMENTS
We would like to thank members of the Gygi Lab, particularly David P. Nusinow, Julian Mintseris, and Devin K. Schweppe for insightful discussions. This work was funded in part by NIH Grants 1R01GM132129 (J.A.P.) and GM67945 (S.P.G) and the Mark Foundation for Cancer Research Fellow of the Damon Runyon Cancer Research Foundation Grant DRG 2359-19 (J.G.V.V.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00063.
The authors declare no competing financial interest.
The mass spectrometry data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD017346
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00063
Contributor Information
Jiaming Li, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Jonathan G. Van Vranken, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States
Joao A. Paulo, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States
Edward L. Huttlin, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Steven P. Gygi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
■ REFERENCES
- (1).Ericsson UB; Hallberg BM; Detitta GT; Dekker N; Nordlund P Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem 2006, 357 (2), 289–98. [DOI] [PubMed] [Google Scholar]
- (2).Vedadi M; Niesen FH; Allali-Hassani A; Fedorov OY; Finerty PJ Jr.; Wasney GA; Yeung R; Arrowsmith C; Ball LJ; Berglund H; Hui R; Marsden BD; Nordlund P; Sundstrom M; Weigelt J; Edwards AM Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. U. S.A 2006, 103 (43), 15835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Molina DM; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao Y; Nordlund P Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341 (6141), 84–87. [DOI] [PubMed] [Google Scholar]
- (4).Savitski MM; Reinhard FBM; Franken H; Werner T; Savitski MF; Eberhard D; Molina DM; Jafari R; Dovega RB; Klaeger S; Kuster B; Nordlund P; Bantscheff M; Drewes G Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346 (6205), 1255784. [DOI] [PubMed] [Google Scholar]
- (5).Gaetani M; Sabatier P; Saei AA; Beusch CM; Yang Z; Lundstrom SL; Zubarev RA Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res 2019, 18 (11), 4027–4037. [DOI] [PubMed] [Google Scholar]
- (6).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hughes CS; Moggridge S; Muller T; Sorensen PH; Morin GB; Krijgsveld J Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 2019, 14 (1), 68–85. [DOI] [PubMed] [Google Scholar]
- (8).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach JJ; Paulo JA; Gygi SP Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem 2019, 91 (6), 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 2014, 86 (14), 7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (11).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villen J; Haas W; Sowa ME; Gygi SP A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143 (7), 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Savitski MM; Wilhelm M; Hahne H; Kuster B; Bantscheff M A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteomics 2015, 14 (9), 2394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Szklarczyk D; Santos A; von Mering C; Jensen LJ; Bork P; Kuhn M STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44 (D1), D380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huang DW; Sherman B T; Lempicki, R. A Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4 (1), 44–57. [DOI] [PubMed] [Google Scholar]
- (15).Sridharan S; Kurzawa N; Werner T; Gunthner I; Helm D; Huber W; Bantscheff M; Savitski MM Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun 2019, 10 (1), 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Miettinen TP; Peltier J; Hartlova A; Gierlinski M; Jansen VM; Trost M; Bjorklund M Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK4/6 inhibitor palbociclib. EMBO J. 2018, 37 (10), e98359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. Proteome Res 2020, DOI: 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Erickson BK; Mintseris J; Schweppe DK; Navarrete-Perea J; Erickson AR; Nusinow DP; Paulo JA; Gygi SP Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res 2019, 18 (3), 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu YK; Chen HY; Chueh PJ; Liu PF A one-pot analysis approach to simplify measurements of protein stability and folding kinetics. Biochim. Biophys. Acta, Proteins Proteomics 2019, 1867 (3), 184–193. [DOI] [PubMed] [Google Scholar]
- (20).Dai L; Prabhu N; Yu LY; Bacanu S; Ramos AD; Nordlund P Horizontal Cell Biology: Monitoring Global Changes of Protein Interaction States with the Proteome-Wide Cellular Thermal Shift Assay (CETSA). Annu. Rev. Biochem 2019, 88, 383–408. [DOI] [PubMed] [Google Scholar]
- (21).Ball KA; Webb KJ; Coleman SJ; Cozzolino KA; Jacobsen J; Jones KR; Stowell MHB; Old WM An isothermal shift assay for proteome scale drug-target identification. Commun. Biol 2020, 3 (1), 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, examples of simulated melting curves (PDF)
Table S1, results of NAD+-binding proteins by PISA (XLSX)
Data Availability Statement
The mass spectrometry data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD017346.