

Abstract
Over the past decade, monoclonal antibodies have dramatically impacted the treatment of haematological malignancies, as evidenced by the effect of rituximab on the response rate and survival of patients with follicular and diffuse large B cell non-Hodgkin’s lymphoma. Currently, only two monoclonal antibodies – the anti-CD33 immunotoxin gemtuzumab ozogamicin and the CD52-directed antibody alemtuzumab – are approved for treatment of relapsed acute myeloid leukaemia in older patients and B cell chronic lymphocytic leukaemia, respectively. Although not approved for such treatment, alemtuzumab is also active against T cell prolymphocytic leukaemia, cutaneous T cell lymphoma and Sézary syndrome, and adult T cell leukaemia and lymphoma. In addition, rituximab has demonstrated activity against B cell chronic lymphocytic and hairy cell leukaemia. Monoclonal antibodies targeting CD4, CD19, CD20, CD22, CD23, CD25, CD45, CD66 and CD122 are now being studied in the clinic for the treatment of leukaemia. Here, we discuss how these new antibodies have been engineered to reduce immunogenicity and improve antibody targeting and binding. Improved interactions with Fc receptors on immune effector cells can enhance destruction of target cells through antibody-dependent cellular cytotoxicity and complement-mediated cell lysis. The antibodies can also be armed withcellulartoxinsorradionuclidestoenhancethedestructionofleukaemiacells.
In his Croonian Lecture in 1900, the renowned immunologist Paul Ehrlich proposed that ‘immunisations such as these which are of great theoretic interest may come to be available for clinical application attacking epithelium new formations, particularly carcinoma by means of specific anti-epithelial sera’ (Ref. 1). Unfortunately, Erlich’s dream of the ‘magic bullet’ of antibodies as a cancer treatment remained elusive until the groundbreaking work of Köhler and Milestein in the mid-1970s in which they developed techniques for generating specific monoclonal antibodies (Ref. 2). Despite the enthusiasm for antibodies as an anticancer therapy, early clinical results were discouraging. A number of improvements in understanding and biotechnology were required before the promise could be kept. Only now at the beginning of the twenty-first century have monoclonal antibodies come into their own as a treatment for cancer.
Currently, the US Food and Drug Administration (FDA) has approved nine monoclonal antibodies for the treatment of cancer. Of these, only two, alemtuzumab and gemtuzumab ozogamicin, have indications in leukaemia. Rituximab approved for the treatment of B cell non-Hodgkin’s lymphoma (NHL), has demonstrated activity in B cell chronic lymphocytic leukaemia (CLL) and hairy cell leukaemia (HCL) (Refs 3, 4). In addition, a number of antibodies directed against novel antigens, or that have been engineered to improve effector function, or armed with toxins or radioisotopes to increase killing ability are currently being studied in various leukaemias.
The vast majority of early therapeutic monoclonal antibodies were of rodent origin, and therefore exhibited a number of unfavorable characteristics. Most notably, the nonhuman framework was immunogenic and frequently induced human antirodent antibody responses after very few treatments. This resulted in the loss of therapeutic effect, and increased the risk of infusional reactions and toxicity. In addition, nonhuman antibodies exhibit unfavorable pharmacokinetics with shorter half-lives and reduced Fc receptor binding that is required for antibody-dependent cellular cytotoxicity (ADCC) (Refs 5, 6). To overcome these problems, most approved therapeutic monoclonal antibodies have been modified using recombinant DNA technology to express human framework sequences to reduce immunogenicity and improve pharmacokinetics (Ref. 7). ‘Chimeric’ antibodies have undergone substitution of up to 70% of the nonhuman framework sequences. In ‘humanised’ antibodies, up to 90% of nonhuman sequences have been replaced, leaving only the original nonhuman complementary determining regions, further reducing immunogenicity. Advancements in transgenic technology have allowed the generation of monoclonal antibodies with fully ‘human’ sequences, high antigen affinities and little or no immunogenicity (Ref. 8). Further efforts have focused on engineering the effector function of antibodies by Fc subtype switching to improve the antibody’s ability to activate complement to lyse target cells, and to enhance antibody–Fc-receptor binding on macrophages and other effector cells to increase ADCC (Ref. 9).
The ideal leukaemia antigen for antibody therapy should exhibit certain characteristics: (1) its expression should be restricted to the leukaemic cells. If the antigen is expressed on normal cells, the loss of these cells should not result in serious complications such as life-threatening cytopaenias or prolonged immunosuppression; (2) the target should be expressed at high density on the leukaemic cells to provide an adequate number of antibody binding sites. Studies suggest that tumour responses correlate with target density. The lower responsiveness of CD20-expressing CLL to rituximab compared with follicular B cell NHL appears to be due to the lower level of CD20 expressed in CLL (Ref. 10). Escape mutants that lose antigen expression are unaffected because there is no target for the antibody to bind; (3) for unmodified or unarmed monoclonal antibodies, target antigens should not undergo internalisation (downmodulation). Internalisation of the antigen–antibody complex reduces the number of targets available for binding; however, antigen–antibody internalisation can be an advantage with immunotoxins. In addition, enhancement of antitumour activity is often seen when monoclonal antibodies are used in combination with cytotoxic chemotherapy (Refs 11, 12, 13).
Monoclonal antibodies targeting myeloid leukaemia
Acute myeloid leukaemia (AML) is the most common acute leukaemia of adults and it accounts for about 15% of childhood leukaemias (Ref. 14). At diagnosis, 55% of AML patients are age 65 or older. Between 50% and 80% of previously untreated patients achieve a complete remission with combinations of cytarabine and an anthracycline. For patients achieving a remission, up to 40% will survive for 5 years; however, these outcomes are worse for older patients. Therapeutic options are limited in relapsed patients over the age of 60 because of lower response rates, increased toxicity of treatment, and the frequent comorbidities present in this population. For older patients who failed to respond or relapsed after first-line therapy, the prognosis is often limited. The introduction of the CD33-targeted immunotoxin, gemtuzumab ozogamicin, as well as a number of novel monoclonal antibodies directed against other myeloid antigens currently in clinical development, offer new promise to these patients (Table 1).
Table 1.
Monoclonal antibodies for the treatment of myeloid leukaemia
| Monoclonal antibody | Target antigen | Isotype | Approval and indication | Refs |
|---|---|---|---|---|
| Gemtuzumab ozogamicin (Myelotarg™, hP67.6) | CD33 | Humanised IgG4- γ1-calicheamicin immunotoxin | FDA approved; AML | 18, 24 |
| Lintuzumab (HuM195, SGN-33) | CD33 | Humanised IgG1 | Phase III; AML | 35 |
| Anti-CD44 (A3D8) | Adhesion glycoprotein | Murine IgG1 | Preclinical development; AML | 46 |
| Anti-CD45 (BC8, YTH 24.5, YAMEL568) | Leukocyte protein tyrosine phosphatase | Radiolabelled 131I murine IgG1; 99mTc murine IgG2b; 111In rat IgG2a-imaging/biodistribution | Phase I-II; AML, allogeneic stem cell transplantation, imaging | 39, 40 |
| Anti-CD66 (BW250/183) | CEA family granulocyte antigen | Radiolabelled 188Re 90Y murine IgG1 | Phase I-II; AML, allogeneic stem cell transplantation | 42, 43, 44 |
| Anti-CD123 (26292(Fv)-PE38-KDEL) | CD123 (IL-3Rα) | Murine anti-CD123-Fv-Pseudomonas exotoxin A immunotoxin | Preclinical development; AML | 49 |
Abbreviations: AML, acute myeloid leukaemia; CEA, carcinoembryonic antigen; FDA, Food and Drug Administration; I, iodine; IL-3Rα, interleukin-3 receptor-α; In, indium; Re, rhenium; Tc, technetium; Y, yttrium.
CD33-targeted antibodies
CD33 is a 67 kDa glycoprotein sialoadhesin receptor (Ref. 15). Its intracytoplasmic domain expresses two immunoreceptor tyrosine-based inhibitory motifs (ITIMs). CD33 is believed to have an important role in modulating leukocyte function, inhibiting cellular activation and proliferation, inducing apoptosis, and modulating cytokine secretion (Ref. 16). It is expressed on normal and maturing myeloid progenitor cells, but not on early normal haematopoietic stem cells. Over 90% of acute myeloid leukaemia blast cells express CD33, but it is not found on nonhaematopoietic tissues (Ref. 17).
Gemtuzumab ozogamicin
Gemtuzumab ozogamicin (Mylotarg™, Wyeth, Madison, NJ) is an immunotoxin composed of a humanised IgG4κ monoclonal antibody (hP67.6) that recognises CD33 conjugated to the antitumour antibiotic γ1-calicheamicin, which is active against a broad spectrum of leukaemic and solid tumour cell lines. Gemtuzumab is derived from the murine anti-CD33 antibody p67.6 (Ref. 18) and it is linked to N-acetyl-γ1-calicheamicin (Ref. 19). It is approved for the treatment of CD33-positive AML in patients 60 years of age or older in first relapse.
Upon binding to CD33, the antibody–receptor complex is internalised and γ1-calicheamicin is released through hydrolysis of the N-acetyl linker (Ref. 20). Calicheamicin binds to the minor groove of DNA and results in site-specific double-stranded DNA breaks. Given that CD33 stimulation is normally inhibitory, gentuzumab may also exert effects through receptor binding. In vitro, gemtuzumab ozogamicin is almost 105 times more toxic to CD33-expressing cells than to cells lacking CD33 (Ref. 21).
At least 85% saturation of CD33 on leukaemic blasts is achieved with an initial dose of 9 mg/m2. Saturating levels of the antibody are believed to enhance efficacy. After a 9 mg/m2 dose, gentuzumab achieves a mean peak plasma concentration (Cmax) of 2.86 ± 1.35 μg/ml and has a plasma half-life of 72.4 ± 42.0 hours (Ref. 22). Higher concentrations are observed with second doses. This is thought to be due to reduced tumour burden and lower availability of CD33 to bind gemtuzumab following the initial dose. The area under the curve (AUC) for γ1-calicheamicin tracks with that of gemtuzumab, indicating that γ1-calicheamicin remains conjugated to the antibody in the plasma.
A Phase I trial defined the maximum tolerated dose of gemtuzumab ozogamicin administered as a 2 hour infusion as 9 mg2/m (Ref. 23). recommended treatment course is a total of two doses with at least 14 days between doses. A combined analysis of three open-label Phase II studies in 277 adults with recurrent CD33-positive AML reported an overall response rate of 26% (Ref. 24). The response rate for patients ≥60 years of age was 24% compared with 28% for those less than 60 years old. Gemtuzumab ozogamicin has also demonstrated activity as first-line single-agent therapy, as well as when used as consolidation therapy after remission-induction with chemotherapy, or used in combination with chemotherapy in CD33-postive AML (Refs 25, 26, 27, 28). Improved results were achieved when it was combined with chemotherapy at relapse over treatment with salvage chemotherapy alone (Refs 29, 30). Gemtuzumab ozogamicin combined with arsenic trioxide and all-trans-retinoic acid has also shown promise in recurrent acute promyelocytic leukaemia (Ref. 31).
Toxicity includes chills, fever, nausea, vomiting, tachycardia, headaches, labile blood pressure, myelosuppression, stomatitis, elevation of liver transaminases and bilirubin, and rarely, hepatic veno-occlusive disease. Tumour lysis syndrome and acute adult respiratory distress syndrome have also been reported. Resistance to gemtuzumab ozogamicin has been correlated with expression of the multidrug resistance (MDR-1) p-glycoprotein and increased serum soluble CD33 antigen levels (Refs 32, 33, 34).
Lintuzumab
Lintuzumab (SGN-33, HuM195, Seattle Genetics, Bothell, WA) is a humanised IgG1 anti-CD33 monoclonal antibody derived from the murine M195 antibody that exhibits a high affinity for CD33. Since it is humanised, lintuzumab is less immunogenic than M195. Unlike the IgG4-based gentuzumab, lintuzumab exhibits ADCC and CMC. Similarly to gentuzumab, lintuzumab undergoes internalisation. In a Phase III trial in 191 adults with refractory AML or AML at first relapse, lintuzumab in combination with mitoxantrone, etoposide and cytosine arabinoside was compared with chemotherapy alone (Ref. 35). A response rate of 36% was observed in the combined antibody and chemotherapy group compared with 28% in those undergoing chemotherapy alone, with no difference in overall survival noted between the The two groups. Both M195 and lintuzumab conjugated to α- or β-particle-emitting isotopes have been used to purge bone marrow of leukaemic blasts in patients undergoing stem cell transplantation (Refs 36, 37).
CD45-targeted antibodies
CD45, also known as common leukocyte antigen, is a 200 kDa protein tyrosine phosphatase expressed at high levels on leukocytes and leukocyte precursors, including AML blasts (Ref. 38). CD45 functions as a regulator of T-and B-cell-receptor signalling. CD45 does not undergo internalisation. Anti-CD45 labeled with 131I has been studied in Phase I and Phase II trials in AML patients as part of an allogeneic bone marrow transplant conditioning regimen in combination with cyclophosphamide and total body irradiation, or with busulfan and cyclophosphamide (Refs 39, 40). The 3 year disease-free survival rate was 61% in 46 treated patients. After adjusting for differences in risk, the hazard of mortality of the treatment group was 0.65 (95% CI, 0.39–1.08) compared with 509 similar patients from the International Bone Marrow Transplant Registry, suggesting benefit from this approach (Ref. 40).
CD66-targeted antibodies
CD66 is a heavily glycosylated carcinoembryonic antigen (CEA)-related glycoprotein that is involved in adhesion to E-selectin surface glycoprotein. CD66 is expressed at high levels on granulocytes and it does not appear to be shed or internalised (Ref. 41). Radiolabeled anti-CD66 with stem cell transplantation has been studied in AML (Refs 42, 43, 44). One trial compared the biodistribution of 99mTc-anti-CD66 versus 99mTc-anti-CD45 monoclonal antibodies in 12 AML patients undergoing stem cell transplantation and found that the anti-CD66 antibody (BW250/183) showed a favorable biodistribution and pharmacokinetics compared with the anti-CD45 antibody (Ref. 45). Zenz and colleagues treated 20 adults with allogeneic stem cell transplantation and Rhenium-188 (188Re)-labeled anti-CD66 for Philadelphia-chromosome-positive acute lymphoblastic leukaemia or advanced chronic myeloid leukaemia with acceptable toxicity; however, no reduction in the incidence of leukaemic relapse was observed (Ref. 44).
Antibodies targeted against myeloid leukaemia stem cell antigen (CD44 and CD123)
CD44 (phagocytic glycoprotein I) and CD123 (interleukin-3 receptor a) represent potential targets for antibody-based therapy of myeloid leukaemia (Refs 46, 47). Cancer stem cells are increasingly recognised as a specialised population of selfrenewing tumour cells (Ref. 48). Ultimate success might lie in our ability to effectively target leukaemia stem cells. CD123, the a-subunit of the interleukin-3 receptor (IL-3Ra) is highly expressed on various blasts and leukaemic stem cells. Du and coworkers studied the activity of a series of single-chain Fv antibody fragments fused to a 38 kDa fragment of Pseudomonas exotoxin A (Ref. 49). One of these, 26292(Fv)-PE38-KDEL was cytotoxic to several CD123-expressing cell lines in vitro.
Monoclonal antibodies targeting lymphoid leukaemia: B cell leukaemia antigens
A list of antibodies currently in development or clinical trial for the treatment of B cell leukaemia is provided in Table 2. The various antibodies are considered in detail within their respective groups below.
Table 2.
Monoclonal antibodies for the treatment of B cell leukaemia
| Monoclonal antibody | Target antigen | Isotype | Approval and indication | Refs |
|---|---|---|---|---|
| XmAb 5574 | CD19 | Humanised and Fc enhanced mouse IgG1 | Preclinical development | 92 |
| Rituximab (Rituxan™, MabThera™, C2B8) | CD20 | Humanised mouse IgG1 | FDA approved; follicular and diffuse large B cell NHL | 3, 63 |
| Ofatumumab (HuMax-CD20™) | CD20 | Fully human IgG1 | Phase I-II; follicular B cell NHL | 79, 80 |
| Ocrelizumab | CD20 | Humanised IgG1 | Phase II; follicular B cell NHL | 83, 84 |
| Veltuzumab (IMMUNO-106, hA20) | CD20 | Humanised and enhanced IgG2a | Phase I; follicular B cell NHL and CLL | 85, 86 |
| Epratuzumab (hLL2) | CD22 | Humanised IgG1 | Phase I-II; follicular B cell NHL | 99, 103 |
| Inotuzumab ozogamicin (CMC-544, G5/44) | CD22 | Humanised IgG4-N-acetyl-γ1-calicheamicin | Preclinical development | 107 |
| BL22, HA22 | CD22 | Murine IgG1 Fv(RFB4)-Pseudomonas exotoxin A immunotoxin | Phase I-II; HCL and B cell CLL | 104, 106 |
| Lumiliximab | CD23 | Cynomolgus macaque-human IgG1 | Phase I-II-III; CLL | 111, 113 |
| Alemtuzumab (Campath™, MabCampath™, Campath-IH) | CD52 | Humanised rat IgG2b | FDA approved; B cell CLL | 115, 119 |
Abbreviations: CLL, chronic lymphocytic leukaemia; FDA, Food and Drug Administration; HCL, hairy cell leukaemia; NHL, non-Hodgkin’s lymphoma.
CD20-targeted antibodies
CD20 is a 31–37 kDa antigen expressed on almost all normal B cells and most B cell malignancies (Ref. 50). It is a nonglycosylated phosphoprotein member of the membrane-spanning 4A gene family. Its function is unclear, but CD20 is believed to have a role in B cell activation and regulation of B cell growth, as well as acting as a cell membrane calcium channel (Ref. 51). CD20 is expressed at almost all stages of B cell development, except in pro-B cells and terminally differentiated plasma cells. CD20 is expressed on the malignant B cells of NHL, CLL and HCL. The number of targets varies with the underlying disease; it is greatest in follicular and diffuse large B cell lymphoma and lower in CLL and HCL. CD20 is nonmodulating and it is not shed, making it an ideal target for antibody therapy (Ref. 52).
Rituximab
Rituximab (Rituxan™, MabThera™, Genentech, South San Francisco, CA) is a chimeric anti-CD20 monoclonal antibody. It was generated from the murine anti-human CD20 antibody 2B8 by cloning the light- and heavy-chain complementary determining regions (CDR) on to a human IgG1 heavy-chain and kappa light-chain constant region (Ref. 53). In 1997, Rituximab became the first monoclonal antibody to be licensed for the treatment of cancer. Since then, it has revolutionised the treatment of B cell malignancies. Rituximab is approved by the FDA for: (1) the treatment of relapsed or refractory, low-grade or follicular, CD20-positive, B cell NHL as a single agent; (2) previously untreated follicular, CD20-positive, B cell NHL in combination with chemotherapy; (3) nonprogressing low-grade, CD20-positive B cell NHL, as a single agent, after first-line chemotherapy, and (4) previously untreated diffuse large B cell, CD20-positive NHL in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) or other anthracycline-based chemotherapy regimens.
Rituximab exhibits powerful B-cell-depleting properties. The mechanism of action is complex and may vary by disease (Ref. 54). Its mechanism includes induction of apoptosis, inhibition of cell growth, complement-mediated cytotoxicity (CMC), sensitisation to both chemotherapy and radiation, and ADCC. Rituximab enhances sensitivity to chemotherapy by downregulating expression of Bcl-2 (Ref. 55). Some preclinical work suggested that full cytotoxic activity could only be achieved in mice expressing the stimulatory g-chain of the FcgRIII receptor (Ref. 56). Activation of allelic variants of the FcgRIIIa gene correlates with an improved response to rituximab in patients with follicular NHL, but not in patients with CLL (Refs 57, 58). Rituximab enhances caspase-9, caspase-3 and poly(ADP-ribose) polymerase (PARP) cleavage in CLL cells, suggesting that induction of apoptosis is the dominant mechanism of action in CLL, whereas CMC and ADCC appear to be the dominant effects in NHL (Ref. 59). Mechanisms of resistance to rituximab are unclear, but might involve alterations in CD20 expression, reduced antibody penetration of tumour masses, increased expression of complement inhibitors, enhanced prosurvival pathways and inhibition of ADCC.
Phase I studies failed to identify a maximum tolerated dose and established rituximab 375 mg/m2/week for 4 weeks as standard (Refs 60, 61). Pharmacokinetic studies showed a plasma half-life of 76.3 hours achieved with the first infusion that increased to 205.8 hours after the fourth infusion (Ref. 62). The efficacy of rituximab was established in a pivotal Phase II trial in 166 patients with relapsed NHL (Ref. 63). In low-grade and follicular B cell NHL patients who relapsed or failed primary therapy, extending treatment to eight weekly doses appeared to increase the number of responses, improve complete response rate, time to progression and the duration of response (Ref. 64). The 8 week course is often used for the treatment of bulky or resistant disease, or for patients undergoing retreatment.
Rituximab is not approved for the treatment of CLL or other B cell leukaemias; however, a substantial number of studies demonstrated its activity in previously treated and untreated CLL (Refs 3, 65, 66, 67). One early trial treated a variety of malignant B cell disorders including 15 patients with small lymphocytic lymphoma or chronic lymphocytic leukaemia (SLL/CLL) and mantle cell lymphoma (MCL) in leukaemic phase with rituximab 375 mg/m2/week for 4 weeks, and concluded that the efficacy of rituximab against SLL/CLL and MCL was limited (Ref. 68). O’Brien and colleagues examined the efficacy of higher doses rituximab in CLL (Ref. 69). Forty patients with CLL and ten with other mature B cell leukaemias were treated with four weekly infusions of rituximab. The initial dose was 375 mg/m2 in all patients, with the subsequent doses escalated in groups of patients from 500 to 2250 mg/m2. Toxicity with the first dose was significantly more common in patients with non-CLL B cell leukaemias, occurring in 5 out of 10 patients, versus 1 out of 40 patients with CLL. The overall response rate was 36% in CLL and 60% in the other B cell leukaemias. The lower responsiveness of CLL to rituximab was attributed to the lower density of CD20 targets on CLL cells compared with other B cell malignancies (Ref. 70). Similarly to follicular and diffuse large B cell NHL, combinations of rituximab and chemotherapy demonstrated improved responses and progression-free survival compared with treatment with either chemotherapy or rituximab alone in CLL. The CALGB 9712 clinical trial randomised concurrent treatment with fludarabine and rituximab versus sequential treatment in 104 symptomatic CLL patients (Ref. 71). Patients receiving concurrent treatment experienced greater haematological toxicity and infusional reactions compared with the sequential treatment group; however, the response rate of the concurrent group was 90% compared with 77% in the sequential treatment group. In a retrospective analysis, these data were combined with that of a second multicenter clinical trial of fludarabine in CLL (CALGB 9011) (Ref. 72). In multivariate analyses controlling for pretreatment characteristics, patients receiving fludarabine and rituximab had a significantly better progression-free survival (P < 0.0001) and overall survival (P = 0.0006) than patients receiving fludarabine alone. The number of infectious events was similar between the two treatments. A number of clinical trials in CLL of rituximab in combination with fludarabine and cyclophosphamide at varying drug doses, or sequential or concurrent treatment, demonstrated molecular responses in excess of 85% (Refs 73, 74). Rituximab has also demonstrated activity in a number of other B cell leukaemias including hairy cell leukaemia (HCL), where response rates of 25–80% have been reported (Refs 75, 76).
Rituximab toxicities include acute infusional reactions, fever, chills, nausea, vomiting, bronchospasm and hypotension, most often with the initial infusion. Other serious adverse events include the potentially fatal cytokine release syndrome (Ref. 77). B cell counts nadir 24–72 hours after the initial rituximab infusion and begin to return to normal values at an average of 9 months after therapy. The risk of serious infection is marginally increased in the months following rituximab treatment.
Ofatumumab
Ofatumumab (Arzzera™, GlaxoSmithKline, Philadelphia, PA; HuMax-CD20™, Genmab, Copenhagen, Denmark) is a second-generation anti-CD20 monoclonal antibody (Ref. 78). The lower levels of CD20 expression in CLL and other B cell malignancies suggest that an antibody designed with enhanced Fc effector function may be required. Based on this, a number of CD20-targeted monoclonal antibodies have been generated with enhanced effector function using the IgG1κ immunoglobulin framework. These enhanced antibodies strongly bind to CD20-positive cells, efficiently recruit mononuclear cells (ADCC) and lyse even rituximab-resistant targets (Ref. 79). Ofatumumab exhibits a higher affinity for CD20 than rituximab; it attaches to a different epitope closer to the cell surface, which makes it easier for effector cells to kill the target cell and activate complement.
Ofatumumab has shown activity in low-grade follicular B cell NHL and is currently in clinical trials for CLL (Refs 80, 81). One hundred and thirty-eight patients with fludarabine and alemtuzumab refractory (double refractory, DR) or bulky (>.5 cm lymph nodes) fludarabine-refractory (BFR) CLL received eight weekly infusions of ofatumumab followed by four monthly infusions (Ref. 82). The overall response rate was 51% for the DR group and 44% for the BFR group, with one patient achieving a complete response. Serious toxicities included infections, granulocytopaenia and anaemia. An FDA Oncologic Drugs Advisory Committee has recommended accelerated approval of ofatumumab for the treatment of fludarabine and alemtuzumab-refractory CLL.
Ocrelizumab
Ocrelizumab (rhumAb 2H7v.16, Biogen-IDEC, Cambridge, MA) is a recombinant humanised anti-human CD20 antibody (Ref. 83). Ocrelizumab can induce apoptosis in several human lymphoma cell lines in vitro, and has been studied in a multicenter, open-label, dose-escalation Phase I–II trial in relapsed and refractory follicular NHL with an overall response rate of 36%, with 13% in complete remission (Ref. 84). Ocrelizumab is now in a Phase II trial in CLL.
Veltuzumab
Veltuzumab (IMMU-106, Immunomedics, Morris Plains, NJ) is a humanised anti-CD20 (hA20) monoclonal antibody with identical complementary determining regions (CDR) to rituximab except for a single residue in the variable heavy chain, and contains the framework regions of a humanised anti-CD22 antibody, epratuzumab (see CD22-targeted antibodies) (Ref. 85). Compared with rituximab, veltuzumab demonstrated significantly lower off-rates in three human lymphoma cell lines studied, as well as increased CMC. A multicentre open-label, dose-escalation study demonstrated safety and efficacy in patients with NHL. At 80 mg/m2 veltuzumab depleted peripheral blood B cells after a single infusion and five out of eight patients had objective responses, including two complete remissions (Ref. 86). Veltuzumab is currently in a phase II trial in CLL.
CD19-targeted antibodies
CD19 is a pan-B-cell antigen expressed at the pro-B-cell stage up to the level of mature B cells. CD19 is downregulated during the terminal differentiation of plasma cells. It is also expressed in a subset of acute myeloid leukaemias (Refs 87, 88, 89). CD19 functions as a co-receptor in a multimeric complex that includes CD21 and CD81, which activates a Src-family protein tyrosine kinase system that regulates thresholds and intensifies signalling following B cell receptor (BCR) activation (Refs 90, 91). CD19 is expressed in the majority of NHL and B cell CLL and it undergoes internalisation when bound to antibody.
XmAb 5574
XmAb 5574 (Xencor, Monrovia, CA) is a humanised anti-CD19 monoclonal antibody that has an engineered Fc domain to increase binding to Fcg receptors and enhance Fc-mediated effector function. In vitro, XmAb 5574 enhanced ADCC ~1000-fold relative to an anti-CD19 IgG1 analogue against a broad range of B cell lymphoma and leukaemia cell lines including primary ALL and mantle cell lymphoma (Ref. 92). XmAb 5574 increased effector cell phagocytosis and target cell apoptosis. In vivo, XmAb 5574 inhibited tumour growth in established lymphoma mouse xenograft models, and demonstrated more potent antitumour activity than its IgG1 analogue. Comparison with a variant antibody incapable of Fcγ-receptor binding showed that engagement of these receptors was critical for efficacy. In a nonhuman primate model, XmAb 5574 depleted B cells from the peripheral blood, lymph nodes and spleen (Ref. 93). XmAb 5574 has not yet entered clinical trials.
Anti-CD19 antibodies and immunomagnetic separation have been used to deplete B cells from the bone marrow in CLL patients undergoing autologous transplants (Refs 94, 95, 96). In one study, 20 patients with poor-risk CLL underwent stem-cell mobilisation with chemotherapy and granulocyte-colony stimulating factor (Ref. 96). B cell depletion was performed using selection for CD34+ cells and selection against B cells with anti-CD19, -CD20, -CD23 and -CD37 immuno- magnetic beads. With a median follow-up of 20 months, 17 patients were in complete remission.
Anti-CD19 immunotoxins
The internalisation of CD19 after antibody binding suggests that an immunotoxin might be an effective strategy for treatment of CD19-expressing leukaemias. One study examined the use of an anti-CD19-based immunotoxin in acute lymphocytic leukaemia (ALL) for eradication of residual disease after treatment (Ref. 97). After initial chemotherapy, 46 adults with previously untreated CD19-positive ALL received an immunotoxin composed of the murine B4 anti-CD19 antibody linked to modified ricin. The most frequent toxicities were transient elevation of liver transaminases and lymphopaenia. Molecular studies, however, failed to show a consistent decrease in the residual number of leukaemic cells with the immunotoxin treatment.
CD22-targeted antibodies
CD22 is a 140 kDa membrane protein associated with the BCR that possesses seven extracellular immunoglobulin-like domains and is responsible for binding ligands with α(2,6) sialic acid residues (Ref. 98). CD22 has three intracellular immunoreceptor tyrosine inhibitory motifs (ITIMs) and functions as an inhibitory coreceptor. Upon antigen crosslinking of the BCR, the ITIMs are phosphorylated, which in turn activates protein tyrosine phosphatases, including SHP-1, that dephosphorylate BCR components attenuating signalling. CD22 is highly expressed on mature B cells, but is expressed at low levels on immature B cells and on plasma cells. CD22 is expressed in more than 90% of childhood B cell precursor ALL, as well as adult HCL and CLL.
Epratuzumab
Epratuzumab is a humanised IgG1 monoclonal antibody that binds to the extracellular domain of CD22. It was generated from the mouse IgG2a antibody LL2 (HPB-2) (Ref. 99). Epratuzumab was developed in an effort to reduce antibody immunogenicity, prolong half-life, and to increase effector function. After binding, the CD22–epratuzumab complex is rapidly internalised. In vitro studies suggest a role for ADCC, activation of CD22 and inhibition of proliferation as possible mechanisms of action. Unlike rituximab, which appears to be directly cytotoxic to B cells, epratuzumab seems to predominantly modulate B cell activation and signalling (Ref. 100).
Epratuzumab has demonstrated antitumour activity as a single agent and in combination with chemotherapy in patients with relapsed and refractory indolent and high-grade NHL, including rituximab-refractory disease (Refs 101, 102). Epratuzumab was assessed in a Children’s Oncology Group study alone and in combination with chemotherapy in 15 children with relapsed B cell ALL (Ref. 103). Patients received intravenous epratuzumab twice weekly for 2 weeks followed by four weekly doses in combination with reinduction chemotherapy. At the doses used (350 mg/m2), epratuzumab was able to achieve saturation of surface CD22 on the leukaemic cells in all but one patient. Nine patients achieved a complete remission. Epratuzumab treatment with reinduction chemotherapy was feasible and tolerated in relapsed ALL; however, this trial could not assess whether the addition of epratuzumab enhanced the response rate over that achieved with reinduction chemotherapy alone.
BL22 and HA22 immunotoxins
Antibody-bound CD22 undergoes internalisation, suggesting that anti-CD22-based immunotoxins might be more therapeutically effective than unmodified antibodies in CD22-expressing leukaemias (Fig. 1). BL22, a single chain anti-CD22 variable region antibody fragment (scFv) coexpressed as a fusion protein with truncated Pseudomonas exotoxin A has been studied in 46 patients with CD22-expressing B cell malignancies including 31 patients with HCL and 11 CLL patients (Ref. 104). Serious toxicities included haemolytic uraemic syndrome in four patients and cytokine release syndrome in one patient. Vascular leak syndrome and increases in serum transaminases were frequent, but were not considered dose limiting. Responses were seen in 25 HCL patients, with complete remissions observed in 61% of these. Responses were rapid: 99% of circulating hairy cells were cleared within one week. Development of neutralising serum antibodies against the immunotoxin was demonstrated in 11 patients and appeared to have a variable impact on tumour response. A follow-up trial showed that cladribine-resistant HCL responded to BL22 (Ref. 105). HA22 is a similar immunotoxin that has had its CDR sequence modified to increase its affinity for the CD22 antigen. It is currently being evaluated in a Phase I trial (Ref. 106).
Figure 1. Immunotoxin targeting in leukaemia.
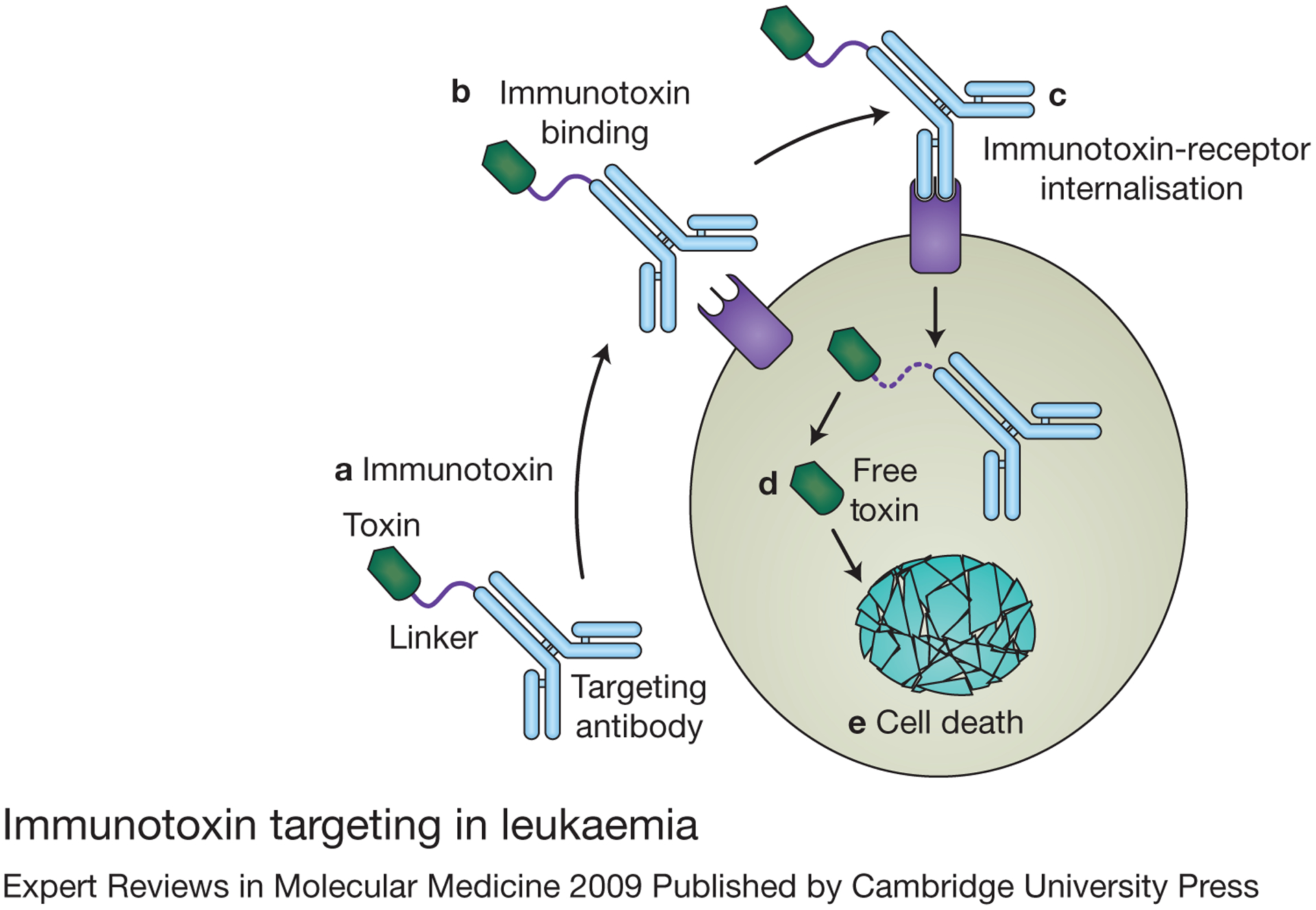
(a) Immunotoxins are therapeutic molecules composed of a monoclonal antibody or antibody fragment linked to a cellular toxin. (b) The antibody directs the toxin to a cell expressing a specific receptor for that antibody. Once bound to the target cell, the immunotoxin–receptor complex is internalised (downmodulated) (c), and the toxin is released intracellularly (d) to kill the targeted leukaemia cell (e).
Inotuzumab ozogamicin
Inotuzumab ozogamicin (CMC-544, Wyeth BioPharma, Andover, MA) is an immunotoxin composed of an IgG4 humanised anti-CD22 monoclonal antibody (G5/44) and the antitumour antibiotic γ1-calicheamicin (Ref. 107). The G5/44 antibody binds to CD22 with nanomolar affinity and is rapidly internalised. After internalisation, γ1-calicheamicin is released and causes double-stranded DNA breaks that result in apoptosis. Inotuzumab ozogamicin inhibited the growth of a panel of human ALL cell lines in vitro and reduced the leukaemic cell load, reduced hind limb paralysis and prolonged the survival of nude mice injected with REH ALL cells (Ref. 108). Inotuzumab ozogamicin is currently in Phase I clinical trials.
CD23-targeted antibodies
CD23, the low-affinity receptor for IgE (Fc1 RII), is the only Fc receptor that does not belong to the immunoglobulin gene superfamily (Ref. 109). CD23 functions as a B cell activation antigen and as an adhesion molecule (Ref. 110). CD23 is expressed on the majority of CLL cells. Lumiliximab (Biogen-IDEC, Cambridge, MA), a macaque–human primatised monoclonal antibody, targets CD23 (Ref. 111). Preclinical studies demonstrated that lumiliximab induced the apoptosis of CD23-expressing lymphoid cell lines in vitro and prolonged the survival of immunodeficient mice inoculated with CD23-positive lymphoblastic cell lines (Ref. 112). In a Phase I trial, 46 patients with refractory CLL were treated with doses of lumiliximab up to 500 mg/m2 three times a week for 4 weeks (Ref. 113). No partial or complete responses were observed and toxicity was limited. A randomised registration trial comparing treatment with lumiliximab in combination with fludarabine, cyclophosphamide and rituximab (FCR) to FCR alone has been initiated in patients with CLL.
CD52-targeted antibodies
CD52 is a highly glycosylated 12 amino acid glycospingoinositol (GPI)-anchored surface antigen expressed on B and T cells, monocytes, macrophages and certain epithelial cells of the male genital tract (Ref. 114). CD52 is expressed at higher levels on T cells than on B cells, and is thought to have a role in lymphocyte activation. It is a noninternalising antigen, which makes it a potentially ideal target for antibody therapy.
Alemtuzumab
Alemtuzumab (Campath-1H, Campath™, Mab Campath™, Bayer Healthcare Pharmaceuticals, Pittsburgh, PA) is a humanised antibody derived from the rat monoclonal antibody that defined the CD52 antigen. The Campath series of antibodies were originally developed for T cell depletion of bone marrow to prevent graft-versus-host disease (GvHD). The original antibody, Campath-1M (YTH66.9), which is a rat IgM, proved difficult to manufacture and was replaced by Campath-1G, a rat IgG2b that also exhibited ADCC and CMC (Ref. 115). Alemtuzumab (Campath-1H), the humanised form, recognises the C-terminal region and GPI-anchor of CD52.
Alemtuzumab exhibits a powerful lymphodepleting activity and was approved as a single agent for treatment of alkylating agent and fludarabine-resistant B cell CLL. As such, it is currently the only monoclonal antibody approved for the treatment of a lymphoid leukaemia. Its indication was recently extended to first-line use in CLL. Alemtuzumab exerts its antileukaemic activity through CMC, ADCC and induction of apoptosis (Ref. 116).
Early clinical studies of alemtuzumab showed modest single-agent activity in NHL, with few complete responses (Ref. 117). Subsequent trials demonstrated more meaningful single-agent activity in previously treated, relapsed or chemotherapy-refractory CLL, with responses reported in 33–50% of patients (Refs 118, 119, 120, 121, 122). In 93 CLL patients who were refractory to alkylating agents and fludarabine and received intravenous alemtuzumab 30 mg three times per week, an overall response rate of 33% with two complete remissions was observed. An additional 59% of patients achieved disease stabilisation. Median survival for the entire group was 16 months, which compared very favorably with other salvage treatments. Complete resolution of lymphadenopathy was seen in 64% of patients with lymph nodes less than 2 cm in diameter; however, there was little effect in patients with nodes larger than 5 cm. It is hypothesised that the lower activity of alemtuzumab in bulky disease was the result of the lower concentrations of antibody achieved in large tumours, or decreased ADCC as a result of inhibition or exclusion of effector cells from the tumour environment.
Alemtuzumab response rates in untreated patients with CLL are reported to be as high as 87%, with complete remission seen in up to one third of patients (Ref. 123). In a randomised Phase III trial, previously untreated CLL patients receiving 30 mg alemtuzumab three times a week for up to 12 weeks were compared with patients treated with chlorambucil 40 mg/m2 administered every 28 days for a maximum of 12 months (Ref. 124). The overall response rate was 83% for alemtuzumab compared with 55% for chlorambucil (P < 0.0001), with a complete remission rate of 24% for alemtuzumab versus 2% for chlorambucil (P < 0.0001).
The activity of alemtuzumab is enhanced when it is combined with chemotherapy (Ref. 125). One study investigated the combination of fludarabine and alemtuzumab in refractory CLL patients previously treated with fludarabine, and reported an 83% response rate (Ref. 126). Alemtuzumab in combination with cyclophosphamide, fludarabine and rituximab (CFAR) achieved an overall complete remission rate of 69% in 48 high-risk CLL patients (Ref. 127). Alemtuzumab has also been shown to eliminate minimal residual disease (MRD) from the bone marrow after chemotherapy (Ref. 128). CLL patients who cleared their MRD had longer survival rates than MRD-positive responders (Ref. 129). This was true even for those patients with high-risk CLL with del(17p13) or p53 mutations (Ref. 130). Alemtuzumab can also be subcutaneously administered with a reduced incidence of acute systemic reactions.
In addition, alemtuzumab demonstrated antitumour activity in a number of T cell leukaemias. These are a heterogeneous group of leukaemias that have historically been treated with similar chemotherapy regimens to that used to treat aggressive B cell leukaemias (Ref. 131). With few exceptions, the outcomes have been poor compared with the B cell leukaemias.
Alemtuzumab has been studied in a number of T cell leukaemias, both as a single agent and in combination. Single-agent activity has been shown in T cell prolymphocytic leukaemia (TPLL), with response rates of 51–76% depending on prior treatment (Refs 132, 133, 134). Response rates of 55–100% have also been reported in CTCL and Sézary syndrome (Refs 135, 136, 137). In a preclinical study and two case reports, alemtuzumab demonstrated antitumour activity in HTLV-1-associated adult T cell leukaemia/lymphoma (ATL) (Refs 138, 139, 140). In a Phase II trial, 24 ATL patients were treated with 30 mg alemtuzumab three times per week for up to 12 weeks (Ref. 141). The overall response rate in the 23 evaluable patients was 43.5%. The antibody produced responses in patients with the leukaemic subtype of ATL, but with a short progression-free survival. An ongoing trial is examining alemtuzumab in combination with dose-adjusted EPOCH (etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin) chemotherapy in patients with a variety of T cell neoplasms (Ref. 142).
Alemtuzumab is associated with infusional reactions, fevers, chills, rash, myelosuppression, lymphopaenia and increased risk of opportunistic infection, especially cytomegalovirus (CMV) reactivation. Patients receiving this antibody should also receive prophylaxis against Pneumocystis and CMV.
Monoclonal antibodies targeting lymphoid leukaemia: T cell leukaemia antigens
When combined, T cell leukaemias and lymphomas account for less than 15% of malignant lymphoproliferative diseases (Refs 143, 144, 145). Although the expression of certain antigens is associated with a particular disorder, this association is usually not disease specific. The same antigen may be expressed acros several different T cell leukaemias or lymphomas. Historically, T cell leukaemias were treated with the same chemotherapy regimens that were used to treat high-grade B cell malignancies. With few exceptions, the outcomes were poorer, with lower response rates, shorter time to progression and shorter median survivals than that seen with B cell leukaemias (Ref. 146). To date, no monoclonal antibody has received approval for the treatment of T cell leukaemia; however, a number of novel antibodies are under investigation for the treatment of these diseases (Table 3).
Table 3.
Monoclonal antibodies for treatment of T cell lymphoid leukaemia
| Monoclonal antibody | Target antigen | Isotype | Approval and indication | Refs |
|---|---|---|---|---|
| Siplizumab (MEDI-507) | CD2 | Human IgG1 | Phase I-II; CD2+ T cell leukaemia or lymphoma | 150, 153 |
| Muromonab-CD3 (Orthoclone™, OKT3) | CD3 | Murine IgG2a | FDA approved; renal allograft rejection | 159 |
| Zanolimumab (HuMax-CD4™) | CD4 | Human IgG1 | Phase II-III; CTCL/Sézary syndrome, T cell lymphoma | 163 |
| Daclizumab (Zenapax™) | CD25 (IL-2Rα) | Humanised IgG2a | Phase I-II; HTLV-1-associated ATL | 172, 174, 182 |
| LMB-2 | CD25 (IL-2Rα) | Murine anti-Tac(Fv)-Pseudomonas exotoxin A immunotoxin | Phase I-II; CD25+ lymphoproliferative malignancy | 183, 184 |
| Mik-β1 | CD122 (IL-2R/IL-2Rβ) | Humanised IgG1 | Phase I;T-LGL leukaemia | 193 |
Abbreviations: ATL, adult T cell leukaemia; CTCL, cutaneous T cell lymphoma; FDA, Food and Drug Administration; HTLV-1, human T cell lymphotrophic virus type-1; T-LGL, T cell large granular lymphocyte leukaemia.
CD2-targeted antibodies
CD2 (LFA-2, T11) is a 50 kDa glycoprotein whose extracellular region consists of two immunoglobulin-like domains (Refs 147, 148). It is highly expressed on thymocytes, and on T and NK cells. CD2, whose ligand is LFA-3, is involved in T cell activation and cell adhesion. CD2 enhances the physical interaction between T cells and antigen-presenting cells, as well as between T cells and NK cells, and their targets. CD2 associates with the T cell receptor (TCR) and enhances CD3 signalling during low-affinity interactions with the major histocompatibility complex (MHC) molecules, thereby enhancing MHC class I- and class II-restricted antigen recognition (Ref. 149). CD2 is highly expressed on T cells throughout most of their development and thus represents a pan-T-cell antigen.
Siplizumab
Siplizumab (MEDI-507; MedImmune, Gaithersburg, MD) is a humanised rat monoclonal antibody that recognises human CD2 (Ref. 150). It has been studied as a treatment for psoriasis and for prophylaxis of GvHD (Ref. 151). It demonstrated antitumour activity in a preclinical T cell leukaemia model (Ref. 152). In a Phase I trial, 29 patients with various T cell leukaemias and lymphomas were treated with this antibody (Ref. 153). In the original trial design, 23 patients received siplizumab on two or three consecutive days every 2 weeks. Flow cytometry demonstrated saturation and downmodulation of CD2 on the surface of peripheral blood T cells. In an attempt to increase antibody delivery, additional cohorts of patients received a single dose of siplizumab on days 0 and 14, and once a week thereafter. All except one patient experienced a decline in circulating CD4+ and CD8+ cells, as well as decreases in the numbers of NK cells. Two complete and nine partial responses were reported. CMV reactivation was common, but was not associated with disease; however, four patients developed Epstein–Barr-virus-related B cell lymphoproliferative disease (EBV-LPD) (Ref. 154). EBV-LPD primarily occurred in patients treated on the weekly schedule, which also produced a marked downmodulation in CD2 on residual T and NK cells.
CD3-, CD4- and CD5-targeted antibodies
CD3 represents a series of intermediate molecular weight polypeptides (CD3γ, CD3δ, CD3ε and CDζ) associated with the α- and β-subunits of the TCR (Ref. 155). The intracellular regions of the CD3 subunits are the signalling domains of the TCR complex that mediate T cell activation. CD3 is expressed on most T cells throughout their development and represents a pan-T-cell antigen.
Muromonab-CD3 (Orthoclone™, OKT3; Ortho Biotech Products, Bidgewater, NJ), a murine IgG2a monoclonal antibody directed against the 20 kDa CD3ζ-subunit, is approved for the treatment of acute renal allograft rejection (Ref. 156). It has also been used to deplete T cells from bone marrow allografts to treat or reduce the risk of GvHD (Ref. 157). Muromonab-CD3 results in the rapid clearing of T cells from the peripheral blood and lymphoid tissue through CMC, ADCC, induced apoptosis and the redirection of T cells into other compartments (Ref. 158). Anti-CD3 has been used to treat acute T cell lymphoblastic leukaemia with limited benefit (Ref. 159). Muromonab-CD3 treatment is associated with cytokine release syndrome, suppression of cell-mediated immunity and an increased risk of opportunistic infections and secondary malignancies.
CD4 is a 55 kDa glycoprotein with four immunoglobulin-like domains, a hydrophobic transmembrane domain and a long cytoplasmic tail (Ref. 160). CD4 acts as a coreceptor for the TCR complex and it is expressed on helper and regulatory T cells, where it recognises antigens presented by MHC class II molecules in association with the TCR. Several trials studied the chimeric anti-CD4 antibodies anti-Leu3a (Becton-Dickinson, San Jose, CA) and cM-T412 (Centocor, Horsham, PA) in patients with CTCL or Sézary syndrome and found that these antibodies demonstrated in vivo binding and suppressed peripheral blood CD4+ cell counts (Refs 161, 162). Seven of eight patients receiving cM-T412 responded with a median duration of 25 weeks. Toxicities included infusional reactions, myalgias and rash.
Zanolimumab (HuMax-CD4™; Genmab, Copenhagen, Denmark), a fully human IgG1κ anti-CD4 monoclonal antibody was evaluated in two separate Phase II trials in patients with CTCL or Sézary syndrome (Ref. 163). This antibody inhibits CD4+ T cells through blockade of CD4–TCR signalling and Fc-dependent ADCC (Ref. 164). Patients received zanolimumab weekly for 17 weeks and 13 of 38 (34.2%) previously treated CTCL patients and 2 of 9 (22.2%) Sézary syndrome patients responded. Zanolimumab produces a dose-dependent CD4+ lymphocytopaenia and adverse events were mainly infectious complications. A pivotal study of zanolimumab in CTCL and Sézary syndrome is currently underway.
CD5 (Leu-1) is a 67 kDa cysteine-rich scavenger receptor glycoprotein expressed on T cells and the B1a subset of B cells (Ref. 165), where it acts as a coreceptor and regulates the strength of the TCR signal. It has a similar role in B cells where it modulates BCR signalling. CD5 appears to be a key regulator of immune tolerance. Two trials examined murine anti-CD5 monoclonal antibodies in patients with a variety of malignant T cell disorders including Sézary syndrome (Refs 166, 167). Rapid, short-lived decreases in circulating T cells were observed. A radioimmunoconjugate of the anti-CD5 antibody T101 was studied in a Phase I trial in ten patients with advanced or refractory CLL or CTCL (Ref. 168). Imaging demonstrated targeting of skin lesions and involved lymph nodes in the CTCL patients. Toxicity was predominantly bone marrow suppression. All but one of the CTCL patients developed human anti-mouse antibodies (HAMAs) after a single treatment and were not able to be retreated. Partial responses were observed in two patients with CLL and in three with CTCL.
CD25-targeted antibodies
Less than 5% of unstimulated peripheral blood T cells express the 55 kDa a-subunit of the IL-2 receptor (CD25, IL-2Rα, Tac). CD25, however, is highly expressed on activated T and B cells, as well as on many B and T cell neoplasms (Ref. 169). Three different IL-2 receptors are recognised, including a high-affinity heterotrimer composed of a 55 kDa α-subunit, a 75 kDa β-subunit (CD122) that is shared with the IL-15 receptor, and a 64 kDa common γ-chain (CD132) shared with the IL-4, IL-7, IL-9, IL-15 and IL-21 receptors. There is an intermediate-affinity heterodimeric IL-2 receptor composed of the 75 kDa β-subunit and the 64 kDa common γ-chain, and a nonsignalling low-affinity receptor composed solely of the 55 kDa IL-2Rα (Ref. 170). The a-subunit confers the specific binding of IL-2 and upregulates receptor sensitivity; the β-subunit also has a role in IL-2 binding, and the β-subunit and common γ-subunits mediate signal transduction. Overexpression of IL-2Rα has been reported in CLL, ATL, CTCL and Sézary syndrome, HCL, some acute and chronic myeloid leukaemias, and T cell acute lymphocytic leukaemia (Ref. 171).
Daclizumab
In 1981, the murine anti-Tac monoclonal antibody that defined the human IL-2Rα was generated (Ref. 172). Anti-Tac binds IL-2Rα and inhibits its interaction with IL-2 and blocks IL-2 stimulation of T cells (Ref. 173). Murine antibodies are clinically limited because of their immunogenicity and induction of neutralising HAMAs. They also exhibit reduced effector function as a result of limited binding to human Fc receptors. In daclizumab (Zenapax™; Hoffmann-La Roche, Nutley, NJ), approximately 90% of the murine IgG2a has been replaced with a human IgG1κ sequence (Refs 174, 175). It is approved for the prophylaxis of renal allograft rejection in combination with other immunosuppressive agents (Ref. 176).
ATL is an aggressive lymphoproliferative disorder resulting from chronic infection with the human retrovirus HTLV-1 (Ref. 177). Chemotherapy has had little impact on survival (Ref. 178). ATL cells express 10,000–40,000 CD25 molecules per cell, whereas normal resting T cells express fewer than 500. Murine anti-Tac and daclizumab inhibit the ex vivo proliferation of primary ATL cells (Ref. 179).
A series of trials studying these antibodies as therapy for HTLV-1-associated ATL have been carried out. In an early study, 19 patients were treated with murine anti-Tac (Ref. 180). Two patients achieved complete remission and four patients had partial responses. The short serum half-life and the development of HAMAs limited the clinical usefulness of the murine antibody. To enhance the killing ability of the antibody, murine anti-Tac was chelated to the β-emitting radioisotope yttrium-90 (Ref. 181). Eighteen ATL patients were treated with this radiolabeled antibody. Two complete and seven partial responses were observed. Toxicity was primarily granulocytopaenia and thrombocytopaenia; however, a significant fraction of patients still developed HAMAs, which limited the treatment.
In an effort to overcome the immunogenicity, a humanised anti-CD25 monclonal antibody daclizumab was studied. In a Phase I trial, up to 8 mg/kg of daclizumab was administered to 14 ATL patients (Ref. 182) to determine the maximum tolerated dose, the dose required to achieve ≥95% saturation of CD25 on circulating ATL cells and ATL cells in lymph nodes, and to estimate the response rate. Flow cytometry showed that ≥95% saturation of CD25 on circulating ATL cells could be achieved; however, in patients that underwent fine needle aspiration of a lymph node, saturation was documented in only half, suggesting that the impeded access of large antibody molecules into solid tumour is a potential issue. Two partial responses were seen.
Others have studied CD25-targeted immunotoxins in the treatment of T cell leukaemias. An immunotoxin composed of truncated Pseudomonas exotoxin A and a single chain (sc) anti-CD25 Fv-fragment fusion protein (LMB-2) demonstrated activity against CD25- expressing lymphoid malignancies, including HCL, ATL and CTCL (Refs 183, 184). LMB-2 was administered to 35 patients; one HCL patient achieved complete remission and seven others demonstrated a partial response. Toxicity included transient serum transaminase elevations and fever.
CD122-targeted antibodies
CD122 (IL-2R/IL-15Rβ) is the 75 kDa α-subunit shared by the heteromeric IL-2 and IL-15 receptors (Refs 185, 186). CD122 functions during receptor signalling by dimerising with CD132. This leads to activation of JAK1 bound to CD122 and JAK3 bound to CD132. These in turn activate the signal transducer and activator of transcription-3 (STAT3) and STAT5. Phosphorylated STAT proteins translocate to the nucleus where they activate gene expression.
IL-15 induces maturation and enhances the survival of T and NK cells, enhances survival of CD8+ memory T cells, and stimulates expression of TNF-α, IL-1β and other proinflammatory cytokines (Ref. 187). Unlike IL-2, IL-15 is not expressed by lymphocytes, but rather it is produced by macrophages and dendritic cells. IL-2 promotes tolerance through the activation-induced cell death (AICD) of T-lymphocytes. Conversely, IL-15 inhibits self-tolerance (Refs 188, 189). Abnormalities in expression of IL-15 and its receptor have been reported in a number of malignant lymphoproliferative disorders, including ALL, ATL, CTCL and T cell large granular lymphocyte (T-LGL) leukaemia.
Mik-β1
The Mik-β1 antibody directed against the shared IL-2R/IL-15Rβ chain (CD122), inhibits transpresentation of IL-15 by its receptor to NK and T cells (Refs 190, 191, 192). T-LGL leukaemia is a rare lymphoproliferative disorder characterised by elevated numbers of CD3+ CD8+ CD57+ large granular lymphocytes in the peripheral blood, spleen and bone marrow, and is often associated with haemocytopaenias. Both IL-2 and IL-15 are known to induce lymphokine-activated killer (LAK) cell activity in LGLs. Resting NK cells and LGLs lack the IL-2Rα subunit; however, its expression can be induced by interaction of IL-2 with CD122 (Ref. 189). LGLs may also be stimulated by IL-15 through the same receptor. It is hypothesised that the haemocytopaenias in T-LGL leukaemia are a result of enhanced NK and LAK activity.
In a Phase I trial, 12 patients with T-LGL leukaemia received four doses of the murine Mik-b1 antibody 4 days apart (Ref. 193). Surprisingly, none of the patients developed a neutralising antibodies to the murine antibody. Greater than 95% saturation of CD122 was achieved in all patients, and downmodulation of surface CD122 on the LGL cells was observed in some patients. Side effects were minor; however, no responses were seen. The lack of response may be the result of the short half-life of the murine antibody. Humanised Mik-b1 is currently being studied in a Phase I trial in T-LGL.
Summary
The introduction of monoclonal antibodies into clinical practice has changed the treatment paradigm for haematological malignancies, as illustrated by the impact of rituximab on the treatment response and survival of patients with follicular lymphoma and diffuse large B cell NHL. Currently only two monoclonal antibodies are approved for the treatment of leukaemia. These include the CD33-directed immunotoxin gemtuzumab ozogamicin, which is approved for the treatment of older patients with acute myeloid leukaemia in relapse; and alemtuzumab, which is approved for single-agent treatment of CLL. Although not approved for the treatment of leukaemia, rituximab has demonstrated activity in CLL and HCL. Alemtuzumab is also active against T-PLL and CTCL and Sézary syndrome, and to a lesser degree, HTLV-1-associated ATL. To date, no monoclonal antibody has received approval for the treatment of T cell leukaemia. As a general observation, tumour response, duration of response and outcome are often improved when monoclonal antibodies are used in combination with chemotherapy. The optimal timing and doses of these antibody–chemotherapy combinations is an area of ongoing study.
In an effort to improve therapeutic activity and reduce side effects, monoclonal antibodies are being engineered: (1) to bind to their targets with higher affinity through amino acid substitutions made in their antigen-binding regions; (2) to target more effective surface antigens and epitopes; (3) to reduce their immunogenicity using recombinant and transgenic technologies to produce fully human antibodies; and (4) to increase interaction of the antibody Fc region with selected effector cell receptors to enhance ADCC and CMC, and thus target cell killing. One example is the anti-CD20 antibody ofatumumab, which has shown activity in rituximab-resistant disease. A number of monoclonal antibodies and immunotoxins that target novel leukaemia antigens are in various stages of preclinical development and clinical trials. These include antibodies targeting CD2, CD4, CD19, CD20, CD22, CD23, CD25, CD44, CD45, CD66, CD122 and CD123. The future continues to be bright for further improvements in monoclonal antibody therapy of leukaemia.
Acknowledgements and funding
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors wish to thank the reviewers for their thoughtful and helpful comments on this article.
References
- 1.Ehrlich P (1899–1900) Croonian Lecture: On immunity with special reference to cell life Proceedings of the Royal Society of London 66, 424–448 [Google Scholar]
- 2.Köhler G and Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 [DOI] [PubMed] [Google Scholar]
- 3.Byrd JC et al. (2001) Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. Journal of Clinical Oncology 19, 2153–2164 [DOI] [PubMed] [Google Scholar]
- 4.Hagberg H (1999) Chimeric monoclonal anti-CD20 antibody (rituximab) – an effective treatment for a patient with relapsing hairy cell leukaemia. Medical Oncology 16, 221–222 [DOI] [PubMed] [Google Scholar]
- 5.Swann PG et al. (2008) Considerations for the development of therapeutic monoclonal antibodies. Current Opinion in Immunology 20, 493–499 [DOI] [PubMed] [Google Scholar]
- 6.Liu XY, Pop LM and Vitetta ES (2008) Engineering therapeutic monoclonal antibodies. Immunological Reviews 222, 9–27 [DOI] [PubMed] [Google Scholar]
- 7.Nissim A and Chernajovsky Y (2008) Historical development of monoclonal antibody therapeutics In Handbook of Experimental Pharmacology: Therapeutic Antibodies. (Chernajovsky Y and Nissim A, eds.) 181, pp. 3–18, Springer; [DOI] [PubMed] [Google Scholar]
- 8.Lonberg N (2008) Human monoclonal antibodies from transgenic mice In Handbook of Experimental Pharmacology: Therapeutic Antibodies, (Chernajovsky Y and Nissim A, eds.) 181, pp. 69–97, Springer; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veri MC et al. (2007) Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 121, 392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perz J Topaly J, Fruehauf S, Hensel M, and Ho AD (2002) Level of CD 20-expression and efficacy of rituximab treatment in patients with resistant or relapsing B-cell prolymphocytic leukemia and B-cell chronic lymphocytic leukemia. Leukemia and Lymphoma 43, 149–151 [DOI] [PubMed] [Google Scholar]
- 11.Coiffier B et al. (2002) CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. New England Journal of Medicine 346, 235–242 [DOI] [PubMed] [Google Scholar]
- 12.Sehn LH et al. (2005) Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. Journal of Clinical Oncology 23, 5027–5033 [DOI] [PubMed] [Google Scholar]
- 13.Marcus R et al. (2008) Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. Journal of Clinical Oncology 26, 4579–4586 [DOI] [PubMed] [Google Scholar]
- 14.Kantarjian H et al. (2008) Therapeutic advances in leukemia and myelodysplastic syndrome over the past 40 years. Cancer 113 (Suppl.), 1933–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeman SD, Kelm S, Barber EK, and Crocker PR (1995) Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 85, 2005–2012 [PubMed] [Google Scholar]
- 16.Crocker PR and Redelinghuys P (2008) Siglecs as positive and negative regulators of the immune system. Biochemical Society Transactions 36, 1467–1471 [DOI] [PubMed] [Google Scholar]
- 17.Dinndorf PA, Andrews RG, Benjamin D, Ridgway D, Wolff L and Bernstein ID (1986) Expression of normal myeloid-associated antigens by acute leukemia cells. Blood 67, 1048–1053 [PubMed] [Google Scholar]
- 18.Hamann PR et al. (2002) An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chemistry 13, 40–66 [DOI] [PubMed] [Google Scholar]
- 19.Zein N, Sinha AM, McGahren WJ and Ellestad GA (1988) Calicheamicin gamma 1: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 240, 1198–1201 [DOI] [PubMed] [Google Scholar]
- 20.van Der Velden VH et al. (2001) Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 97, 3197–3204 [DOI] [PubMed] [Google Scholar]
- 21.Naito K et al. (2000) Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab ozogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on P-glycoprotein-expressing sublines. Leukemia 14, 1436–1443 [DOI] [PubMed] [Google Scholar]
- 22.Dowell JA, Korth-Bradley J, Liu H, King SP and Berger MS (2001) Pharmacokinetics of gemtuzumab ozogamicin, an antibody-targeted chemotherapy agent for the treatment of patients with acute myeloid leukemia in first relapse. Journal of Clinical Pharmacology 41, 1206–1214 [DOI] [PubMed] [Google Scholar]
- 23.Sievers EL et al. (1999) Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 93, 3678–3684 [PubMed] [Google Scholar]
- 24.Larson RA et al. (2005) Final report of the efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. Cancer 104, 1442–1452 [DOI] [PubMed] [Google Scholar]
- 25.Kell WJ et al. (2003) A feasibility study of simultaneous administration of gemtuzumab ozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood 102, 4277–4283 [DOI] [PubMed] [Google Scholar]
- 26.Tsimberidou AM et al. Mylotarg, fludarabine, cytarabine (ara-C), and cyclosporine (MFAC) regimen as post-remission therapy in acute myelogenous leukemia. Cancer Chemotherapy and Pharmacology 52, 449–452 [DOI] [PubMed] [Google Scholar]
- 27.Amadori S et al. (2004) Sequential administration of gemtuzumab ozogamicin and conventional chemotherapy as first line therapy in elderly patients with acute myeloid leukemia: a phase II study (AML-15) of the EORTC and GIMEMA leukemia groups. Haematologica 89, 950–956 [PubMed] [Google Scholar]
- 28.Nabhan C et al. (2005) Phase II pilot trial of gemtuzumab ozogamicin (GO) as first line therapy in acute myeloid leukemia patients age 65 or older. Leukemia Research 29, 53–57 [DOI] [PubMed] [Google Scholar]
- 29.Chevallier P et al. (2005) Administration of Mylotarg 4 days after beginning of a chemotherapy including intermediate-dose aracytin and mitoxantrone (MIDAM regimen) produces a high rate of complete hematologic remission in patients with CD33þ primary resistant or relapsed acute myeloid leukemia. Leukemia Research 29, 1003–1007 [DOI] [PubMed] [Google Scholar]
- 30.Chevallier P et al. (2008) Long-term disease-free survival after gemtuzumab, intermediate-dose cytarabine, and mitoxantrone in patients with CD33(þ) primary resistant or relapsed acute myeloid leukemia. Journal of Clinical Oncology 26, 5192–5197 [DOI] [PubMed] [Google Scholar]
- 31.Aribi A et al. (2007) Combination therapy with arsenic trioxide, all-trans-retinoic acid, and gemtuzumab ozogamicin in recurrent acute promyelocytic leukemia. Cancer 109, 1355–1359 [DOI] [PubMed] [Google Scholar]
- 32.Linenberger ML et al. (2001) Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 98, 988–994 [DOI] [PubMed] [Google Scholar]
- 33.van Der Velden VH et al. (2004) High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia 18, 983–988 [DOI] [PubMed] [Google Scholar]
- 34.Chevallier P et al. (2008) Persistence of CD33 expression at relapse in CD33(þ) acute myeloid leukaemia patients after receiving Gemtuzumab in the course of the disease. British Journal of Haematology 143, 744–746 [DOI] [PubMed] [Google Scholar]
- 35.Feldman EJ et al. (2005) Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. Journal of Clinical Oncology 23, 4110–4116 [DOI] [PubMed] [Google Scholar]
- 36.Schwartz MA et al. (1993) Dose-escalation trial of M195 labeled with iodine 131 for cytoreduction and marrow ablation in relapsed or refractory myeloid leukemias. Journal of Clinical Oncology 11, 294–303 [DOI] [PubMed] [Google Scholar]
- 37.Sgouros G et al. (1999) Pharmacokinetics and dosimetry of an alpha-particle emitter labeled antibody: 213Bi-HuM195 (anti-CD33) in patients with leukemia. Journal of Nuclear Medicine 40, 1935–1946 [PubMed] [Google Scholar]
- 38.Mustelin T, Vang T and Bottini N (2005) Protein tyrosine phosphatases and the immune response. Nature Reviews Immunology 5, 43–57 [DOI] [PubMed] [Google Scholar]
- 39.Matthews DC et al. (1999) Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood 94, 1237–1247 [PubMed] [Google Scholar]
- 40.Pagel JM et al. (2006) 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood 107, 2184–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stocks SC, Ruchaud-Sparagano MH, Kerr MA, Grunert F, Haslett C and Dransfield I (1996) CD66: role in the regulation of neutrophil effector function. European Journal of Immunology 26, 2924–2932 [DOI] [PubMed] [Google Scholar]
- 42.Bunjes D et al. (2001) Rhenium 188-labeled anti-CD66 (a, b, c, e) monoclonal antibody to intensify the conditioning regimen prior to stem cell transplantation for patients with high-risk acute myeloid leukemia or myelodysplastic syndrome: results of a phase I-II study. Blood 98, 565–572 [DOI] [PubMed] [Google Scholar]
- 43.Ringhoffer M et al. (2005) 188Re or 90Y-labelled anti-CD66 antibody as part of a dose-reduced conditioning regimen for patients with acute leukaemia or myelodysplastic syndrome over the age of 55: results of a phase I-II study. British Journal of Haematology 130, 604–613 [DOI] [PubMed] [Google Scholar]
- 44.Zenz T et al. (2006) Targeted marrow irradiation with radioactively labeled anti-CD66 monoclonal antibody prior to allogeneic stem cell transplantation for patients with leukemia: results of a phase I-II study. Haematologica 91, 285–286 [PubMed] [Google Scholar]
- 45.Buchmann I et al. (2003) A comparison of the biodistribution and biokinetics of (99 m)Tc-anti-CD66 mAb BW 250/183 and (99 m)Tc-anti-CD45 mAb YTH 24.5 with regard to suitability for myeloablative radioimmunotherapy. European Journal of Nuclear Medicine and Molecular Imaging 30, 667–673 [DOI] [PubMed] [Google Scholar]
- 46.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, and Dick JE (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature Medicine 12, 1167–1174 [DOI] [PubMed] [Google Scholar]
- 47.Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K and Schmetzer H (2004) Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. European Journal of Haematology 72, 89–106 [DOI] [PubMed] [Google Scholar]
- 48.Misaghian N et al. (2009) Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia 23, 25–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du X, Ho M and Pastan I (2007) New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. Journal of Immunotherapy 30, 607–613 [DOI] [PubMed] [Google Scholar]
- 50.Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF and Stashenko P (1981) A unique cell surface antigen identifying lymphoid malignancies of B cell origin. The Journal of Clinical Investigation 67, 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riley JK and Sliwkowski MX (2000) CD20: a gene in search of a function. Seminars in Oncology 27 (Suppl. 12), 17–24 [PubMed] [Google Scholar]
- 52.Press OW, Howell-Clark J, Anderson S and Bernstein I (1994) Retention of B-cell-specific monoclonal antibodies by human lymphoma cells. Blood 83, 1390–1397 [PubMed] [Google Scholar]
- 53.Reff ME et al. (1994) Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83, 435–445 [PubMed] [Google Scholar]
- 54.Smith MR (2003) Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene 22, 7359–7368 [DOI] [PubMed] [Google Scholar]
- 55.Stolz C et al. (2008) Targeting Bcl-2 family proteins modulates the sensitivity of B-cell lymphoma to rituximab-induced apoptosis. Blood 112, 3312–3321 [DOI] [PubMed] [Google Scholar]
- 56.Clynes RA, Towers TL, Presta LG and Ravetch JV Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets Nature Mediane 6, 443–446 [DOI] [PubMed] [Google Scholar]
- 57.Cartron G et al. (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor Fcgamma RIIIa gene. Blood 99, 754–758 [DOI] [PubMed] [Google Scholar]
- 58.Farag SS, Flinn IW, Modali R, Lehman TA, Young D and Byrd JC (2004) Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 103, 1472–1474 [DOI] [PubMed] [Google Scholar]
- 59.Byrd JC, et al. (2002) The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood 99, 1038–1043 [DOI] [PubMed] [Google Scholar]
- 60.Maloney DG et al. (1994) Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood 84, 2457–2466 [PubMed] [Google Scholar]
- 61.Maloney DG et al. (1997) IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. Journal of Clinical Oncology 15, 3266–3274 [DOI] [PubMed] [Google Scholar]
- 62.Berinstein NL et al. (1998) Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Annals of Oncology 9, 995–1001 [DOI] [PubMed] [Google Scholar]
- 63.McLaughlin P et al. (1998) Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. Journal of Clinical Oncology 16, 2825–2833 [DOI] [PubMed] [Google Scholar]
- 64.Piro LD et al. (1998) Extended rituximab (anti-CD20 monoclonal antibody) therapy for relapsed or refractory low-grade or follicular non-Hodgkin’s lymphoma. Annals of Oncology 10, 655–661 [DOI] [PubMed] [Google Scholar]
- 65.Nguyen DT, Amess JA, Doughty H, Hendry L and Diamond LW (1999) IDEC-C2B8 anti-CD20 (rituximab) immunotherapy in patients with low-grade non-Hodgkin’s lymphoma and lymphoproliferative disorders: evaluation of response on 48 patients. European Journal Haematology 62, 76–82 [DOI] [PubMed] [Google Scholar]
- 66.Ladetto M, Bergui L, Ricca I, Campana S, Pileri A and Tarella C (2000) Rituximab anti-CD20 monoclonal antibody induces marked but transient reductions of peripheral blood lymphocytes in chronic lymphocytic leukaemia patients. Medical Oncology 17, 203–210 [DOI] [PubMed] [Google Scholar]
- 67.Hainsworth JD et al. (2003) Single-agent rituximab as first-line and maintenance treatment for patients with chronic lymphocytic leukemia or small lymphocytic lymphoma: a phase II trial of the Minnie Pearl Cancer Research Network. Journal of Clinical Oncology 21, 1746–1751 [DOI] [PubMed] [Google Scholar]
- 68.Foran JM et al. (2000) European phase II study of rituximab (chimeric anti-CD20 monoclonal antibody) for patients with newly diagnosed mantle-cell lymphoma and previously treated mantle-cell lymphoma, immunocytoma, and small B-cell lymphocytic lymphoma. Journal of Clinical Oncology 18, 317–324 [DOI] [PubMed] [Google Scholar]
- 69.O’Brien SM et al. (2001) Rituximab dose-escalation trial in chronic lymphocytic leukemia. Journal of Clinical Oncology 19, 2165–2170 [DOI] [PubMed] [Google Scholar]
- 70.Perz J, Topaly J, Fruehauf S, Hensel M and Ho AD (2002) Level of CD 20-expression and efficacy of rituximab treatment in patients with resistant or relapsing B-cell prolymphocytic leukemia and B-cell chronic lymphocytic leukemia. Leukmia and Lymphoma 43, 149–151 [DOI] [PubMed] [Google Scholar]
- 71.Byrd JC et al. (2003) Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 101, 6–14 [DOI] [PubMed] [Google Scholar]
- 72.Byrd JC et al. (2005) Addition of rituximab to fludarabine may prolong progression-free survival and overall survival in patients with previously untreated chronic lymphocytic leukemia: an updated retrospective comparative analysis of CALGB 9712 and CALGB 9011. Blood 105, 49–53 [DOI] [PubMed] [Google Scholar]
- 73.Lamanna N et al. (2009) Sequential therapy with fludarabine, high-dose cyclophosphamide, and rituximab in previously untreated patients with chronic lymphocytic leukemia produces high-quality responses: molecular remissions predict for durable complete responses. Journal of Clinical Oncology 27, 491–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foon KA et al. (2009) Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. Journal of Clinical Oncology 27, 498–503 [DOI] [PubMed] [Google Scholar]
- 75.Lauria F et al. (2001) Efficacy of anti-CD20 monoclonal antibodies (Mabthera) in patients with progressed hairy cell leukemia. Haematologica 86, 1046–1050 [PubMed] [Google Scholar]
- 76.Nieva J, Bethel K and Saven A (2003) Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood 102, 810–813 [DOI] [PubMed] [Google Scholar]
- 77.Winkler U, Jensen M, Manzke O, Schulz H, Diehl V and Engert A (1999) Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 94, 2217–2224 [PubMed] [Google Scholar]
- 78.Robak T (2008) Ofatumumab, a human monoclonal antibody for lymphoid malignancies and autoimmune disorders. Current Opinion in Molecular Therapeutics 10, 294–309 [PubMed] [Google Scholar]
- 79.Teeling JL et al. (2004) Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 104, 1793–1800 [DOI] [PubMed] [Google Scholar]
- 80.Hagenbeek A et al. (2008) First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: results of a phase 1/2 trial. Blood 111, 5486–5495 [DOI] [PubMed] [Google Scholar]
- 81.Coiffier B et al. (2008) Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1–2 study. Blood 111, 1094–1100 [DOI] [PubMed] [Google Scholar]
- 82.Osterborg A et al. (2008) Ofatumumab (HuMax-CD20), a novel CD20 monoclonal antibody, is an active treatment for patients with CLL refractory to both fludarabine and alemtuzumab or bulky fludarabine-refractory disease: Results from the planned interim analysis of an international pivotal trial. Blood 112, 328 [Abstract] [Google Scholar]
- 83.Liu AY, Robinson RR, Murray ED Jr., Ledbetter JA, Hellström I and Hellström KE (1987) Production of a mouse-human chimeric monoclonal antibody to CD20 with potent Fc-dependent biologic activity. Journal of Immunology 139, 3521–3526 [PubMed] [Google Scholar]
- 84.Morschhauser F et al. (2007) Interim results of a phase I/II study of ocrelizumab, a new humanised anti-CD20 antibody in patients with relapsed/refractory follicular non-Hodgkin’s lymphoma. Blood 110, 645 [Abstract] [Google Scholar]
- 85.Stein R et al. (2004) Characterization of a new humanized anti-CD20 monoclonal antibody, IMMU-106, and Its use in combination with the humanized anti-CD22 antibody, epratuzumab, for the therapy of non-Hodgkin’s lymphoma. Clinical Cancer Research 10, 2868–2878 [DOI] [PubMed] [Google Scholar]
- 86.Morschhauser F et al. (2007) Low doses of humanized anti-CD20 antibody, IMMU-106 (hA20), in refractory or recurrent NHL: Phase I/II results. J Clin Oncol, ASCO Annual Meeting Proceedings 25, 8032 [Abstract] [Google Scholar]
- 87.Dorshkind K and Montecino-Rodriguez E (2007) Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nature Reviews Immunology 7, 213–219 [DOI] [PubMed] [Google Scholar]
- 88.Ball ED et al. (1991) Prognostic value of lymphocyte surface markers in acute myeloid leukemia. Blood 77, 2242–2250 [PubMed] [Google Scholar]
- 89.Bradstock K, Matthews J, Benson E, Page F and Bishop J (1994) Prognostic value of immunophenotyping in acute myeloid leukemia. Australian Leukaemia Study Group. Blood 84, 1220–1225 [PubMed] [Google Scholar]
- 90.Carter RH and Fearon DT (1992) CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science 256, 105–107 [DOI] [PubMed] [Google Scholar]
- 91.Bobbitt KB and Justement LB (2000) Regulation of MHC class II signal transduction by the B cell coreceptors CD19 and CD22. Journal of Immunology 165, 5588–5596 [DOI] [PubMed] [Google Scholar]
- 92.Horton HM et al. (2008) Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Research 68, 8049–8057 [DOI] [PubMed] [Google Scholar]
- 93.Zalevsky J et al. (2009) The impact of Fc engineering on an anti-CD19 antibody: increased Fc-gamma receptor affinity enhances B-cell clearing in nonhuman primates. Blood 113, 3735–3743 [DOI] [PubMed] [Google Scholar]
- 94.Khouri IF et al. (1994) Autologous and allogeneic bone marrow transplantation for chronic lymphocytic leukemia: preliminary results. Journal of Clinical Oncology 12, 748–758 [DOI] [PubMed] [Google Scholar]
- 95.Paulus U, Schmitz N, Viehmann K, von Neuhoff N and Dreger P (1997) Combined positive/negative selection for highly effective purging of PBPC grafts: towards clinical application in patients with B-CLL. Bone Marrow Transplant 20, 415–420 [DOI] [PubMed] [Google Scholar]
- 96.Dreger P et al. (2000) A prospective study of positive/negative ex vivo B-cell depletion in patients with chronic lymphocytic leukemia. Experimental Hematology 28, 1187–1196 [DOI] [PubMed] [Google Scholar]
- 97.Szatrowski TP et al. (2003) Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and Leukemia Group B Study 9311. Cancer 97, 1471–1480 [DOI] [PubMed] [Google Scholar]
- 98.Walker JA and Smith KG (2008) CD22: An inhibitory enigma. Immunology 123, 314–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leung SO et al. (1994) Chimerization of LL2, a rapidly internalizing antibody specific for B cell lymphoma. Hybridoma 13, 469–476 [DOI] [PubMed] [Google Scholar]
- 100.Carnahan J et al. (2007) Epratuzumab, a CD22-targeting recombinant humanized antibody with a different mode of action from rituximab. Molecular Immunology 44, 1331–1341 [DOI] [PubMed] [Google Scholar]
- 101.Leonard JP et al. (2004) Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin’s lymphoma: phase I/II clinical trial results. Clin Cancer Res 10, 5327–5334 [DOI] [PubMed] [Google Scholar]
- 102.Micallef IN et al. (2006) A pilot study of epratuzumab and rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated, diffuse large B-cell lymphoma. Cancer 107, 2826–2832 [DOI] [PubMed] [Google Scholar]
- 103.Raetz EA et al. (2008) Chemoimmunotherapy reinduction with epratuzumab in children with acute lymphoblastic leukemia in marrow relapse: a Children’s Oncology Group Pilot Study. Journal of Clinical Oncology 26, 3756–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kreitman RJ et al. (2005) Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. Journal of Clinical Oncology 23, 6719–6729 [DOI] [PubMed] [Google Scholar]
- 105.Kreitman RJ et al. (2001) Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. New England Journal of Medicine 345, 241–247 [DOI] [PubMed] [Google Scholar]
- 106.Bang S, Nagata S, Onda M, Kreitman RJ and Pastan I (2005) HA22 (R490A) is a recombinant immunotoxin with increased antitumor activity without an increase in animal toxicity. Clinical Cancer Research 11, 1545–1550 [DOI] [PubMed] [Google Scholar]
- 107.DiJoseph JF et al. (2004) Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugateofcalicheamicinforthetreatment of B-lymphoid malignancies. Blood 103, 1807–1814 [DOI] [PubMed] [Google Scholar]
- 108.DiJoseph JF, Dougher MM, Armellino DC, Evans DY and Damle NK (2007) Therapeutic potential of CD22-specific antibody-targeted chemotherapy using inotuzumab ozogamicin (CMC-544) for the treatment of acute lymphoblastic leukemia. Leukemia 21, 2240–2245 [DOI] [PubMed] [Google Scholar]
- 109.Delespesse G et al. (1991) Expression, structure, and function of the CD23 antigen. Advances in Immunology 49, 149–191 [DOI] [PubMed] [Google Scholar]
- 110.Bonnefoy JY et al. (1997) Structure and functions of CD23. International Reviews of Immunology 16, 113–128 [DOI] [PubMed] [Google Scholar]
- 111.Reichert JM (2004) Technology evaluation: lumiliximab, Biogen Idec. Current Opinion in Molecular Therapeutics 6, 675–683 [PubMed] [Google Scholar]
- 112.Pathan NI, Chu P, Hariharan K, Cheney C, Molina A and Byrd J (2008) Mediation of apoptosis by and antitumor activity of lumiliximab in chronic lymphocytic leukemia cells and CD23+ lymphoma cell lines. Blood 111, 1594–1602 [DOI] [PubMed] [Google Scholar]
- 113.Byrd JC et al. (2007) Phase 1 study of lumiliximab with detailed pharmacokinetic and pharmacodynamic measurements in patients with relapsed or refractory chronic lymphocytic leukemia. Clinical Cancer Research 13, 4448–4455 [DOI] [PubMed] [Google Scholar]
- 114.Treumann A, Lifely MR, Schneider P and Ferguson MA (1995) Primary structure of CD52. Journal of Biological Chemistry 270, 6088–6099 [DOI] [PubMed] [Google Scholar]
- 115.Hale G et al. (1983) Removal of T cells from bone marrow for transplantation: a monoclonal antilymphocyte antibody that fixes human complement. Blood 62, 873–882 [PubMed] [Google Scholar]
- 116.Nückel H, Frey UH, Röth A, Dührsen U and Siffert W (2005) Alemtuzumab induces enhanced apoptosis in vitro in B-cells from patients with chronic lymphocytic leukemia by antibody-dependent cellular cytotoxicity. European Journal of Pharmacology 514, 217–224 [DOI] [PubMed] [Google Scholar]
- 117.Hale G et al. (1988) Remission induction in non-Hodgkin lymphoma with reshaped human monoclonal antibody CAMPATH-1H. Lancet 2, 1394–1399 [DOI] [PubMed] [Google Scholar]
- 118.Osterborg A et al. (1997) Phase II multicenter study of human CD52 antibody in previously treated chronic lymphocytic leukemia. European Study Group of CAMPATH-1H Treatment in Chronic Lymphocytic Leukemia. Journal of Clinical Oncology 15, 1567–1574 [DOI] [PubMed] [Google Scholar]
- 119.Keating MJ et al. (2002) Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 99, 3554–3561 [DOI] [PubMed] [Google Scholar]
- 120.Rai KR et al. (2002) Alemtuzumab in previously treated chronic lymphocytic leukemia patients who also had received fludarabine. Journal of Clinical Oncology 20, 3891–3897 [DOI] [PubMed] [Google Scholar]
- 121.Ferrajoli A et al. (2003) Phase II study of alemtuzumab in chronic lymphoproliferative disorders. Cancer 98, 773–778 [DOI] [PubMed] [Google Scholar]
- 122.Cortelezzi A et al. (2005) A pilot study of low-dose subcutaneous alemtuzumab therapy for patients with hemotherapy-refractory chronic lymphocytic leukemia. Haematologica 90, 410–412 [PubMed] [Google Scholar]
- 123.Lundin J et al. (2002) Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 100, 768–773 [DOI] [PubMed] [Google Scholar]
- 124.Hillmen P et al. (2007) Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. Journal of Clinical Oncology 25, 5616–5623 [DOI] [PubMed] [Google Scholar]
- 125.Elter T et al. (2005) Fludarabine in combination with alemtuzumab is effective and feasible in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: results of a phase II trial. Journal of Clinical Oncology 23, 7024–7031 [DOI] [PubMed] [Google Scholar]
- 126.Kennedy B et al. (2002) Campath-1H and fludarabine in combination are highly active in refractory chronic lymphocytic leukemia. Blood 99, 2245–2247 [DOI] [PubMed] [Google Scholar]
- 127.Wierda WG et al. (2008) CFAR, an active frontline regimen for high-risk patients with CLL including those with del 17p. Blood 112, 2095 [Abstract] [Google Scholar]
- 128.O’Brien SM et al. (2003) Alemtuzumab as treatment for residual disease after chemotherapy in patients with chronic lymphocytic leukemia. Cancer 98, 2657–2663 [DOI] [PubMed] [Google Scholar]
- 129.Moreton P et al. (2005) Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. Journal of Clinical Oncology 23, 2971–2979 [DOI] [PubMed] [Google Scholar]
- 130.Lozanski G et al. (2004) Alemtuzumab is an effective therapy forchronic lymphocytic leukemia with p53 mutations and deletions. Blood 103, 3278–3281 [DOI] [PubMed] [Google Scholar]
- 131.Morris JC, Waldmann TA and Janik JE (2008) Receptor-directed therapy of T-cell leukemias and lymphomas. Journal of Immunotoxicology 5, 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pawson R et al. (1997) Treatment of T-cell prolymphocytic leukemia with human CD52 antibody. Journal of Clinical Oncology 15, 2667–2672 [DOI] [PubMed] [Google Scholar]
- 133.Dearden CE et al. (2001) High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood 98, 1721–1726 [DOI] [PubMed] [Google Scholar]
- 134.Keating MJ et al. (2002) Campath-1H treatment of T-cell prolymphocytic leukemia in patients for whom at least one prior chemotherapy regimen has failed. Journal of Clinical Oncology 20, 205–213 [DOI] [PubMed] [Google Scholar]
- 135.Lundin J et al. (2003) Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome. Blood 101, 4267–4272 [DOI] [PubMed] [Google Scholar]
- 136.Kennedy GA et al. (2003) Treatment of patients with advanced mycosis fungoides and Sézary syndrome with alemtuzumab. European Journal of Haematology 71, 250–256 [DOI] [PubMed] [Google Scholar]
- 137.Alinari L, Geskin L, Grady T, Baiocchi RA, Bechtel MA and Porcu P (2008) Subcutaneous alemtuzumab for Sézary syndrome in the very elderly Leukemia Research 32, 1299–1303 [DOI] [PubMed] [Google Scholar]
- 138.Zhang Z, Zhang M, Goldman CK, Ravetch JV and Waldmann TA (2003) Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD52 monoclonal antibody, Campath-1H. Cancer Research 63, 6453–6457 [PubMed] [Google Scholar]
- 139.Mone A et al. (2005) Durable hematologic complete response and suppression of HTLV-1 viral load following alemtuzumab in zidovudine/IFNalpha-refractory adult T-cell leukemia. Blood 106, 3380–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ravandi F and Faderl S (2006) Complete response in a patient with adult T-cell leukemia (ATL) treated with combination of alemtuzumab and pentostatin. Leukemia Research 30, 103–105 [DOI] [PubMed] [Google Scholar]
- 141.Sharma S et al. (2008) Alemtuzumab (Campath-1H) in patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Blood 112, 2010 [Abstract] [Google Scholar]
- 142.Janik JE et al. (2005) A pilot trial of Campath-1H and dose-adjusted EPOCH in CD52-expressing aggressive T-cell malignancies. Blood 106, 3348 [Abstract]16051743 [Google Scholar]
- 143.Non-Hodgkin’s Lymphoma Classification Project. (1997) A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 89, 3909–3918 [PubMed] [Google Scholar]
- 144.Armitage JO and Weisenburger DD (1998) New approach to classifying non-Hodgkin’s lymphomas: Clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. Journal of Clinical Oncology 16, 2780–2795 [DOI] [PubMed] [Google Scholar]
- 145.Groves FD, Linet MS, Travis LB and Devesa SS (2000) Cancer surveillance series: non-Hodgkin’s lymphoma incidence by histologic subtype in the United States from 1978 through 1995. Journal of the National Cancer Institute 92, 1240–1251 [DOI] [PubMed] [Google Scholar]
- 146.Gisselbrecht C et al. (1998) Prognostic significance of T-cell phenotype in aggressive non-Hodgkin’s lymphomas. Groupe d’Etudes des Lymphomes de l’Adulte (GELA). Blood 92, 76–82 [PubMed] [Google Scholar]
- 147.Moingeon P et al. (1989) The structural biology of CD2. Immunological Reviews 111, 111–144 [DOI] [PubMed] [Google Scholar]
- 148.Moingeon PE et al. (1991) Complementary roles for CD2 and LFA-1 adhesion pathways during T-cell activation. European Journal of Immunology 21, 605–610 [DOI] [PubMed] [Google Scholar]
- 149.van der Merwe PA and Davis SJ (2003) Molecular interactions mediating T cell antigen recognition. Annual Review of Immunology 21, 659–684 [DOI] [PubMed] [Google Scholar]
- 150.Xu Y et al. (2004) The anti-CD2 monoclonal antibody BTI-322 generates unresponsiveness by activation-associated T cell depletion. Clinical and Experimental Immunology 138, 476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shaffer J et al. (2007) Regulatory T-cell recovery in recipients of haploidentical nonmyeloablative hematopoietic cell transplantation with a humanized anti-CD2 mAb, MEDI-507, with or without fludarabine. Experimental Hematology 35, 1140–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang Z, Zhang M, Ravetch JV, Goldman C and Waldmann TA (2003) Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD2 monoclonal antibody, MEDI-507. Blood 102, 284–288 [DOI] [PubMed] [Google Scholar]
- 153.O’Mahony D et al. (2007) A Phase I trial of siplizumab in CD2-positive lymphoproliferative disease. AIDS Research and Human Retroviruses 23, 598 [Abstract] [Google Scholar]
- 154.O’Mahony D et al. (2009) EBV-related lymphoproliferative disease complicating therapy with the anti-CD2 monoclonal antibody, siplizumab, in patients with T-cell malignancies. Clinical Cancer Research 15, 2514–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Garcia KC and Adams EJ (2005) How the T-cell receptor sees antigen- A structural view. Cell 122, 333–336 [DOI] [PubMed] [Google Scholar]
- 156.Kirk AD (2006) Induction immunosuppression. Transplantation 82, 593–602 [DOI] [PubMed] [Google Scholar]
- 157.Knop S et al. (2007) Treatment of steroid-resistant acute GVHD with OKT3 and high-dose steroids results in better disease control and lower incidence of infectious complications when compared to high-dose steroids alone: A randomized multi-center trial by the EBMT Chronic Leukemia Working Party. Leukemia 21, 1830–1833 [DOI] [PubMed] [Google Scholar]
- 158.Chatenoud L (2003) CD3-specific antibody-induced active tolerance: From bench to bedside. Nature Reviews Immunology 3, 123–132 [DOI] [PubMed] [Google Scholar]
- 159.Gramatzki M et al. (1995) Therapy with OKT3 monoclonal antibody in refractory T-cell acute lymphoblastic leukemia induces interleukin-2 responsiveness. Leukemia 9, 382–390 [PubMed] [Google Scholar]
- 160.Leahy DJ (1995) A structural view of CD4 and CD8. FASEB Journal 9, 17–25 [DOI] [PubMed] [Google Scholar]
- 161.Knox SJ et al. (1991) Observations on the effect of chimeric anti-CD4 monoclonal antibody in patients with mycosis fungoides. Blood 77, 20–30 [PubMed] [Google Scholar]
- 162.Knox S et al. (1996) Treatment of cutaneous T-cell lymphoma with chimeric anti-CD4 monoclonal antibody. Blood 87, 893–899 [PubMed] [Google Scholar]
- 163.Kim YH et al. (2007) Clinical efficacy of zanolimumab (HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood 109, 4655–4662 [DOI] [PubMed] [Google Scholar]
- 164.Rider DA et al. (2007) A human CD4 monoclonal antibody for the treatment of T-cell lymphoma combines inhibition of T-cell signaling by a dual mechanism with potent Fc-dependent effector activity. Cancer Research 67, 9945–9953 [DOI] [PubMed] [Google Scholar]
- 165.Lozano F et al. (2000) CD5 signal transduction: Positive or negative modulation of antigen receptor signaling. Critical Reviews in Immunology 20, 347–358 [PubMed] [Google Scholar]
- 166.Miller RA, Oseroff AR, Stratte PT and Levy R (1983) Monoclonal antibody therapeutic trials in seven patients with T-cell lymphoma. Blood 62, 988–995 [PubMed] [Google Scholar]
- 167.Dillman RO, Shawler DL, Dillman JB and Royston I (1984) Therapy of chronic lymphocytic leukemia and cutaneous T-cell lymphoma with T101 monoclonal antibody. Journal of Clinical Oncology 2, 881–891 [DOI] [PubMed] [Google Scholar]
- 168.Foss FM et al. (1998) Phase I study of the pharmacokinetics of a radioimmunoconjugate, 90Y-T101, in patients with CD5-expressing leukemia and lymphoma. Clinical Cancer Research 4, 2691–2700 [PubMed] [Google Scholar]
- 169.Robb RJ, Munck A and Smith KA (1982) T-cell growth factor receptors. Journal of Experimental Medicine 154, 1455–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Morris JC and Waldmann TA (2000) Advances in interleukin 2 receptor targeted treatment. Annals of the Rheumatic Diseases 59 (Suppl. 1), 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Waldmann TA (1986) The structure, function, and expression of interleukin-2 receptors on normal and malignant T-cells. Science 232, 727–732 [DOI] [PubMed] [Google Scholar]
- 172.Uchiyama T, Broder S and Waldmann TA (1981) A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T-cells. I. Production of anti-Tac monoclonal antibody and distribution of Tac (+) cells. Journal of Immunology 126, 1393–1397 [PubMed] [Google Scholar]
- 173.Uchiyama T, Nelson DL, Fleisher TA and Waldmann TA (1981) A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T-cells. II. Expression of Tac antigen on activated cytotoxic killer T-cells, suppressor cells, and on one of two types of helper T-cells. Journal of Immunology 126, 1398–1403 [PubMed] [Google Scholar]
- 174.Queen C et al. (1989) A humanized antibody that binds to the interleukin 2 receptor. Proceedings of the National Academy of Sciences of the United States of America 86, 10029–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang M et al. (2004) Activating Fc receptors are required for anti-tumor efficacy of the antibodies directed toward CD25 in a murine model of adult T-cell leukemia. Cancer Research 64, 5825–5829 [DOI] [PubMed] [Google Scholar]
- 176.Nashan B, Light S, Hardie IR, Lin A and Johnson JR (1999) Reduction of acute renal allograft rejection by daclizumab. Daclizumab Double Therapy Study Group. Transplantation 67, 110–115 [DOI] [PubMed] [Google Scholar]
- 177.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD and Gallo RC (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 77, 7415–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Matutes E (2007) Adult T-cell leukaemia/lymphoma. Journal of Clinical Pathology 60, 1373–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tendler CL et al. (1990) Transactivation of interleukin 2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: Pathogenic implications and a rationale for immunotherapy. Proceedings of the National Academy of Sciences of the United States of America 87, 5218–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Waldmann TA et al. (1993) The interleukin-2 receptor: A target for monoclonal antibody treatment of human T-cell lymphotrophic virus I-induced adult T-cell leukemia. Blood 82, 1701–1712 [PubMed] [Google Scholar]
- 181.Waldmann TA et al. (1995) Radioimmunotherapy of interleukin-2Ra-expressing adult T-cell leukemia with yttrium-90-labeled anti-Tac. Blood 86, 4063–4075 [PubMed] [Google Scholar]
- 182.Morris JC et al. (2003) A Phase I trial of humanized anti-Tac (daclizumab) for the treatment of human T-cell lymphotrophic virus type-1-associated leukemia/lymphoma. Journal of Clinical Oncology, American Society of Clinical Oncology Annual Meeting Proceedings 22, 695 [Abstract] [Google Scholar]
- 183.Kreitman RJ, Bailon P, Chaudhary VK, FitzGerald DJ and Pastan I (1994) Recombinant immunotoxins containing anti-Tac(Fv) and derivatives of Pseudomonas exotoxin produce complete regression in mice of an interleukin-2 receptor-expressing human carcinoma. Blood 83, 426–434 [PubMed] [Google Scholar]
- 184.Kreitman RJ et al. (2000) Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. Journal of Clinical Oncology 18, 1622–1636 [DOI] [PubMed] [Google Scholar]
- 185.Tsudo M, Kozak RW, Goldman CK and Waldmann TA (1986) Demonstration of a non-Tac peptide that binds interleukin 2: a potential participant in a multichain interleukin 2 receptor complex. Proceedings of the National Academy of Sciences of the United States of America 83, 9694–9698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tsudo M, Kitamura F and Miyasaka M (1989) Characterization of the interleukin 2 receptor beta chain using three distinct monoclonal antibodies. Proceedings of the National Academy of Sciences of the United States of America 86, 1982–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shanmugham LN et al. (2006) IL-15 an immunoregulatory and anti-cancer cytokine. Recent advances. Journal of Experimental and Clinical Cancer Research 25, 529–536 [PubMed] [Google Scholar]
- 188.Waldmann TA (2006) The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nature Reviews Immunology 6, 595–601 [DOI] [PubMed] [Google Scholar]
- 189.Kanegane H and Tosato G (1996) Activation of naive and memory T-cells by interleukin-15. Blood 88, 230–235 [PubMed] [Google Scholar]
- 190.Guex-Crosier Y et al. (1997) Humanized antibodies against the alpha-chain of the IL-2 receptor and against the b-chain shared by the IL-2 and IL-15 receptors in a monkey uveitis model of autoimmune diseases. Journal of Immunology 158, 452–458 [PubMed] [Google Scholar]
- 191.Alileche A, Goldman CK and Waldmann TA (2001) Differential effects of IL-2 and IL-15 on expression of IL-2 receptor-a. Biochemical and Biophysical Research Communications 285, 1302–1308 [DOI] [PubMed] [Google Scholar]
- 192.Giri JG, Anderson DM, Kumaki S, Park LS, Grabstein KH and Cosman D (1995) IL-15, a novel T-cell growth factor that shares activities and receptor components with IL-2. Journal of Leukocyte Biology 57, 763–766 [DOI] [PubMed] [Google Scholar]
- 193.Morris JC et al. (2006) Preclinical and phase I clinical trial of blockade of IL-15 using Mik-b-1 monoclonal antibody in T-cell large granular lymphocyte leukemia. Proceedings of the National Academy of Sciences of the United States of America 103, 401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]