

SUMMARY
CRISPR genome engineering has become a powerful tool to functionally investigate the complex mechanisms of immune system regulation. While decades of work have aimed to genetically reprogram innate immunity, the utility of current approaches is restricted by poor knockout efficiencies or limited specificity for mature cell lineages in vivo. Here, we describe an optimized strategy for non-viral CRISPR-Cas9 ribonucleoprotein (cRNP) genomic editing of mature primary mouse innate lymphocyte cells (ILCs) and myeloid lineage cells that results in an almost complete loss of single or double target gene expression from a single electroporation. Furthermore, we describe in vivo adoptive transfer mouse models that can be utilized to screen for gene function during viral infection using cRNP-edited naive natural killer (NK) cells and bone-marrow-derived conventional dendritic cell precursors (cDCPs). This resource will enhance target gene discovery and offer a specific and simplified approach to gene editing in the mouse innate immune system.
Graphical Abstract
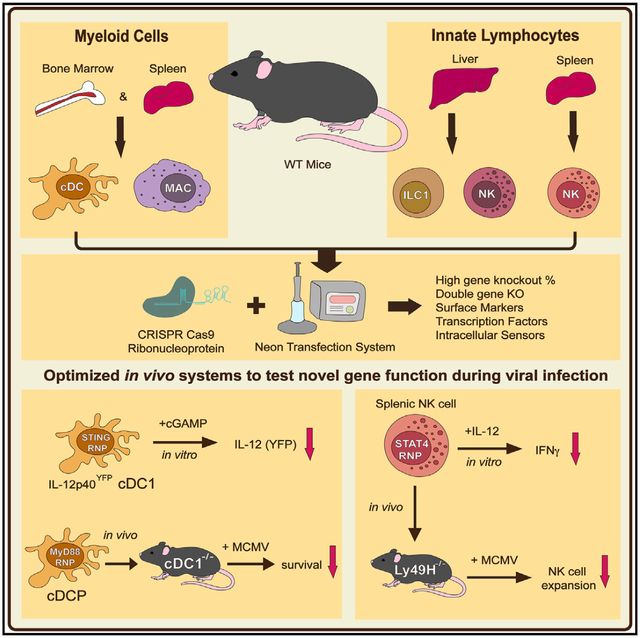
In Brief
Riggan et al. optimize electroporation conditions for high-efficiency cRNP-mediated gene deletion in primary mature mouse innate immune cells and utilize this approach to elucidate gene function of NK cells and cDC1s using two in vivo adoptive transfer models during MCMV infection.
INTRODUCTION
The mammalian immune system consists of both tissue-resident and circulating immune cells. Tissue-resident innate immune cells, such as dendritic cells (DCs), can produce a wide-variety of effector molecules that can directly or indirectly limit pathogen spread and tumor growth in tissue microenvironments (Hildner et al., 2008; Wculek et al., 2020; Weizman et al., 2017). Innate lymphoid cells (ILCs) are tissue-resident cells that produce both proinflammatory and regulatory cytokines in response to local injury, inflammation, pathogen infection, or commensal microbiota perturbation (Vivier et al., 2018). However, persistent inflammatory signals can also lead to unrestrained activation of innate immunity that is associated with inflammatory pathologies such as Crohn’s disease (CD), chronic obstructive pulmonary disease (COPD), type II diabetes mellitus (T2D), and systemic lupus erythematosus (SLE) (Riggan et al., 2019; Vivier et al., 2018). Although understanding and harnessing the cellular and molecular mechanisms that regulate the innate immune system hold promise for the treatment of several inflammatory disorders, a mechanistic understanding of the mammalian innate immune system has been limited by suboptimal cell lineage gene targeting strategies.
Current models to specifically manipulate gene expression in the mouse innate immune system have been confounded by non-lineage-specific Cre mouse transgenic lines. For example, LysMCre, Csf1rCre, and Cx3cr1Cre mice (commonly used to target macrophage populations) contain overlapping expression profiles in several other myeloid lineage cells based on previous fate-mapping studies (Abram et al., 2014), making a Cre-transgenic line specific for macrophages currently impossible. Furthermore, mouse transgenic Cre lines used to selectively target DCs in vivo, such as ItgaxCre and Zbtb46Cre, express the transgene in DCs, macrophages, and natural killer (NK) cells (Itgax) (Abram et al., 2014) or conventional type 1 DCs (cDC1s) and type 2 DCs (cDC2s), as well as endothelial cells (Zbtb46) (Loschko et al., 2016b; Satpathy et al., 2012). While Xcr1Cre mice can be used to selectively target and ablate cDC1s (Hemmi et al., 2016), there are no current tools to specifically target gene expression in cDC2s in vivo. In the ILC lineage, Ncr1Cre mice induce gene deletion in NK cells, type 1 ILCs (ILC1s), and a subset of ILC3s (Narni-Mancinelli et al., 2011). While genetic strategies exist for ablation of each individual ILC lineage in vivo (Oliphant et al., 2014; Rankin et al., 2016; Weizman et al., 2017), there are currently no tools available for specific genetic manipulation in primary mature ILCs without off-target effects in other cell types or potential cell-extrinsic effects derived from whole-body knockout (KO) mice. Thus, the prevalent issue of nonspecific gene targeting of innate immune cells significantly limits the precise mechanistic understanding of the innate immune system in models of host defense and disease in vivo.
Recent advances in CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 genome editing provide and alternative gene manipulation tool that can be used on enriched primary immune cells (Simeonov and Marson, 2019). Cas9 is a DNA endonuclease that can bind to a complementary region of the genome through its associated guide RNA (gRNA) to generate double-strand breaks in the DNA that result in insertions and/or deletions (indels) in coding regions of proteins to permanently suppress their expression (Pickar-Oliver and Gers-bach, 2019). Specifically, viral- and nonviral-based strategies have been used to manipulate gene expression in purified primary human and mouse adaptive lymphocytes by lentiviral or retroviral overexpression of Cas9 and gRNA sequences or electroporation of a Cas9:gRNA ribonucleoprotein complex (Farboud et al., 2018; Seki and Rutz, 2018; Shifrut et al., 2018; Simeonov and Marson, 2019; Wu et al., 2018). While these previous approaches have been used to elucidate novel gene function in mature mammalian adaptive lymphocytes, a method to implement high efficiency CRISPR-Cas9 gene editing in primary mature innate immune cells in vivo has not been described.
Here, we describe an optimized strategy for nonviral cRNP genomic editing of mature primary mouse innate immune cells. Optimal voltage parameters were determined for maximal Cas9 protein electroporation efficiency and viability of primary mature and bone-marrow-derived innate leukocytes. Using these optimized conditions, we were able to achieve high KO efficiency of cell-surface proteins, intracellular signaling proteins, and transcription factors in innate immune cells using cRNP complexes. Furthermore, we describe two in vivo adoptive transfer models using cRNP-edited naive NK cells and conventional DC precursors (cDCPs) to reveal mechanistic details of antiviral gene function in these cell types during mouse cytomegalovirus (MCMV) infection. This general gene editing strategy may be further adapted to other primary immune cell types and in vivo transfer models to investigate protective or pathologic biological processes in the mammalian innate immune system.
RESULTS
Optimized cRNP Electroporation of Primary Splenic Innate Immune Cells
To determine the optimized electroporation efficiencies for Cas9 in primary leukocytes (Figures S1A–S1C), mouse splenocytes were electroporated using the Neon transfection system. Because we determined that primary leukocytes display maximal viability at an electroporation pulse width of 1 × 20 ms (data not shown), we first tested a range of voltages to optimize the maximal frequency of intracellular Cas9+ leukocytes following electroporation. While freshly isolated splenic NK and T cells had lower electroporation efficiencies of Cas9 with increasing voltage, overnight activation with interleukin-15 (IL-15) in vitro increased the frequency of intracellular Cas9+ cells to ~80% in both NK and T cells at all voltages tested (Figures 1A, 1B, S2A, and S2B). In contrast, freshly isolated splenic macrophages, cDC1s, and cDC2s displayed similar frequencies of intracellular Cas9+ cells following electroporation when compared to splenocytes stimulated with macrophage colony-stimulating factor (M-CSF) or FLT3-L overnight at all voltages tested (Figures 1C and S2C). Furthermore, increased concentrations of Cas9 present in the electroporation buffer decreased the frequency of intracellular Cas9+ lymphocytes, with a much more severe reduction in NK cells (~40%) than CD4+ and CD8+ T cells (~20%) at a constant voltage (Figure 1D). However, splenic macrophages, cDC1s, and cDC2s did not display a similar dose-dependent decrease in the frequency of intracellular Cas9+ cells, suggesting that splenic myeloid lineages have an increased capacity of intracellular Cas9 delivery following electroporation at a constant voltage (Figure 1E). Although our results suggested that splenocytes could be reliably used to measure intracellular Cas9 following electroporation in all leukocyte populations tested, it remained possible that these results may not precisely reflect the response of purified leukocytes. However, purified NK cells displayed a similar frequency of intracellular Cas9+ cells as NK cells from splenocytes, suggesting that the presence of other leukocytes in a splenocyte single-cell suspension does not inhibit intracellular Cas9 uptake following electroporation (Figure S2D). These results reveal cell-intrinsic differences in resting and cytokine-stimulated leukocyte electroporation efficiencies of Cas9 that need to be optimized for each cell type and condition of interest.
Figure 1. Optimized Electroporation of Cas9 in Mouse Splenic Leukocytes.
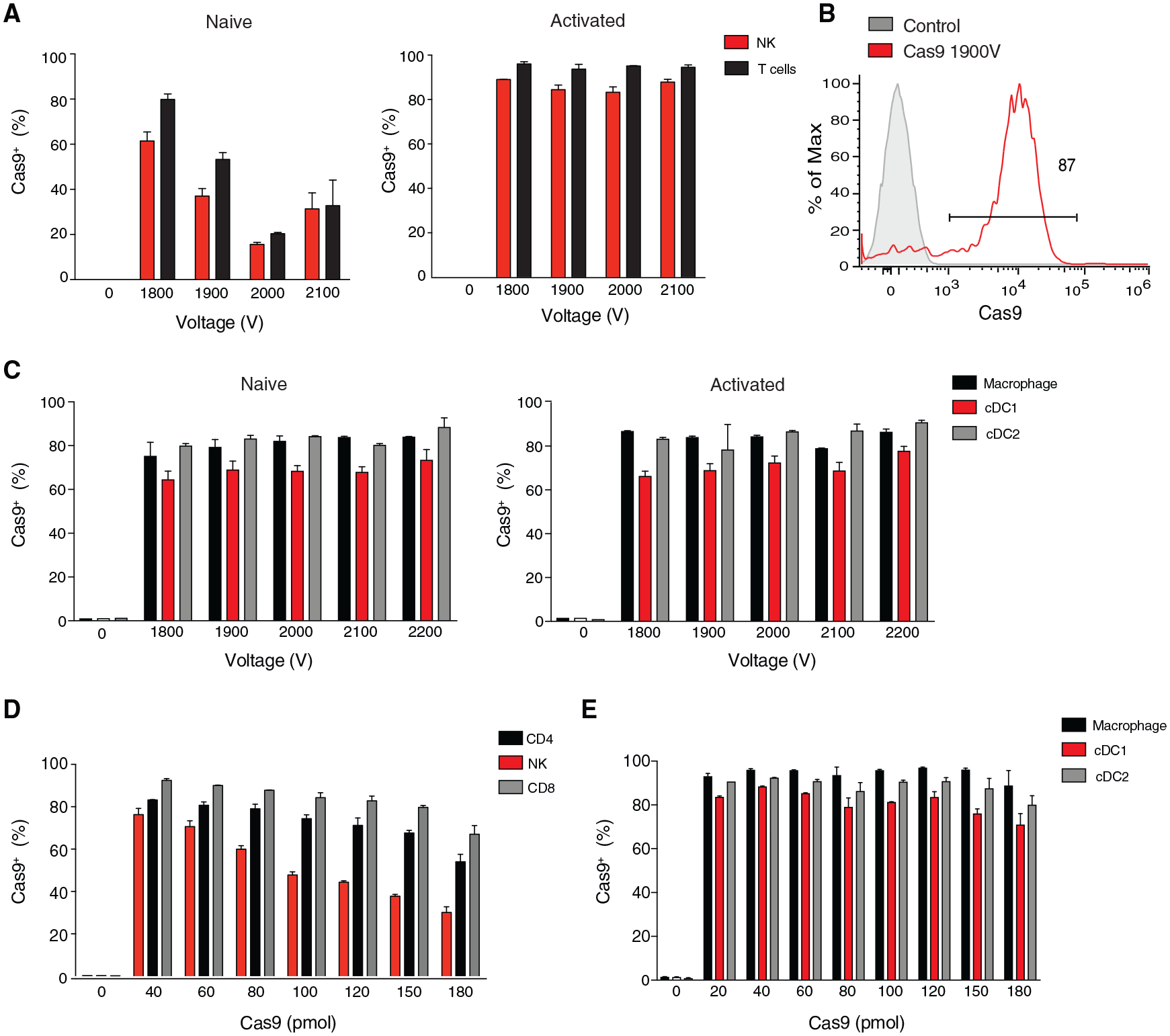
(A–C) Mouse splenocytes were either freshly harvested (naive) or cultured for 16 h with recombinant mouse (rm) IL-15 (activated NK and T cells), FLT3-L (activated cDC1, cDC2), or M-CSF (activated macrophage) in complete media and subsequently electroporated in the presence of 40 pmol Cas9 protein alone. (A) Frequency of intracellular Cas9+ naive and activated NK (TCRβ−CD3ε−NK1.1+) and T cells (TCRβ+CD3ε+NK1.1−) (B) Representative histogram of intracellular Cas9 levels for rmIL-15 stimulated un-electroporated NK cells and NK cells electroporated in the presence of Cas9 protein alone. (C) Frequency of intracellular Cas9+ macrophages (TCRβ−CD3ε−NK1.1−CD64+CD11b+), cDC1s (TCRβ−CD3ε−NK1.1−CD64−CD11β−XCR1+), and cDC2s (TCRβ−CD3ε−NK1.1−CD64−CD11b+XCR1−) following electroporation at the indicated voltages.
(D and E) Splenocytes were either freshly harvested (macrophages, cDC1s, and cDC2s) or cultured for 16 h with rmIL-15 (NK, CD4+, and CD8+ T cells) and subsequently electroporated in the presence of various concentrations of Cas9 protein alone. Frequency of intracellular Cas9+ (D) activated NK, CD4+ T (TCRβ+CD3ε+CD4+NK1.1−), CD8+ T cells (TCRb+CD3ε+CD8+NK1.1−) and (E) naive macrophages, cDC1s, and cDC2s following electroporation at 1,900 V in the presence of the indicated concentrations of Cas9 protein alone is shown. Data are representative of two independent experiments of three mice per group.
Efficient cRNP-Mediated Gene Editing in Primary Circulating and Tissue-Resident Innate Immune Cells Ex Vivo
Group 1 ILCs consist of circulating NK cells and tissue-resident ILC1s that confer host protection during viral infection and liver injury (Nabekura et al., 2020; Riggan et al., 2019; Weizman et al., 2017) while also contributing to tissue pathology during intestinal inflammation and obesity (Bernink et al., 2013; Fuchs et al., 2013; O’Sullivan et al., 2016). However, there are currently no genetic tools available to selectively ablate gene expression in NK cells or ILC1s in vivo (Riggan et al., 2019). In order to achieve specific gene deletion in mature group 1 ILCs, we first purified splenic NK cells and found that electroporation at 1,900 V led to the highest frequency of viable intracellular Cas9+ NK cells (Figure 2A). These results were not influenced by intrinsic differences in NK cell maturation or activation, because all developmental stages of splenic NK cells displayed similar frequencies of intracellular Cas9+ cells, and electroporation at 1,900 V did not induce the expression of markers associated with NK cell activation (Figures S3A and S3B). We next determined that electroporation of purified IL-15-preactivated splenic NK cells in the presence of 40 pmol Cas9:120pmol gRNA Klrb1c (NK1.1) cRNP complex yielded the highest KO efficiency (~90%) (Figures 2B–2E). Purified IL-15-preactivated NK cells and ILC1s from the liver also displayed high frequencies of NK1.1− cells (~80%) using the same conditions, suggesting that cRNP gene editing is not limited to specific group 1 ILCs or lymphocytes from distinct tissues (Figure 2F). However, we observed that gene KO efficiency in primary NK cells is highly dependent on the individual gRNA used and the type of recombinant Cas9. We found that poor efficiency guides targeting similar Klrbc1 exons could be pooled together in separate cRNP complexes within a single electroporation to achieve similar KO efficiencies as the highest efficiency cRNP complex, similar to a previous report in primary T cells (Seki and Rutz, 2018) (Figures S3C and S3D). Furthermore, we observed that different lymphocyte populations could achieve varying levels of KO efficiency using the same Thy1.2 (CD90) cRNP complex (Figure 2G). Together, these results suggest that cell-lineage-intrinsic differences and gRNA editing efficiency contribute to overall KO efficiency following cRNP electroporation in primary lymphocytes.
Figure 2. Efficient cRNP-Complex-Mediated Gene Deletion in Primary Mouse Group 1 ILCs.
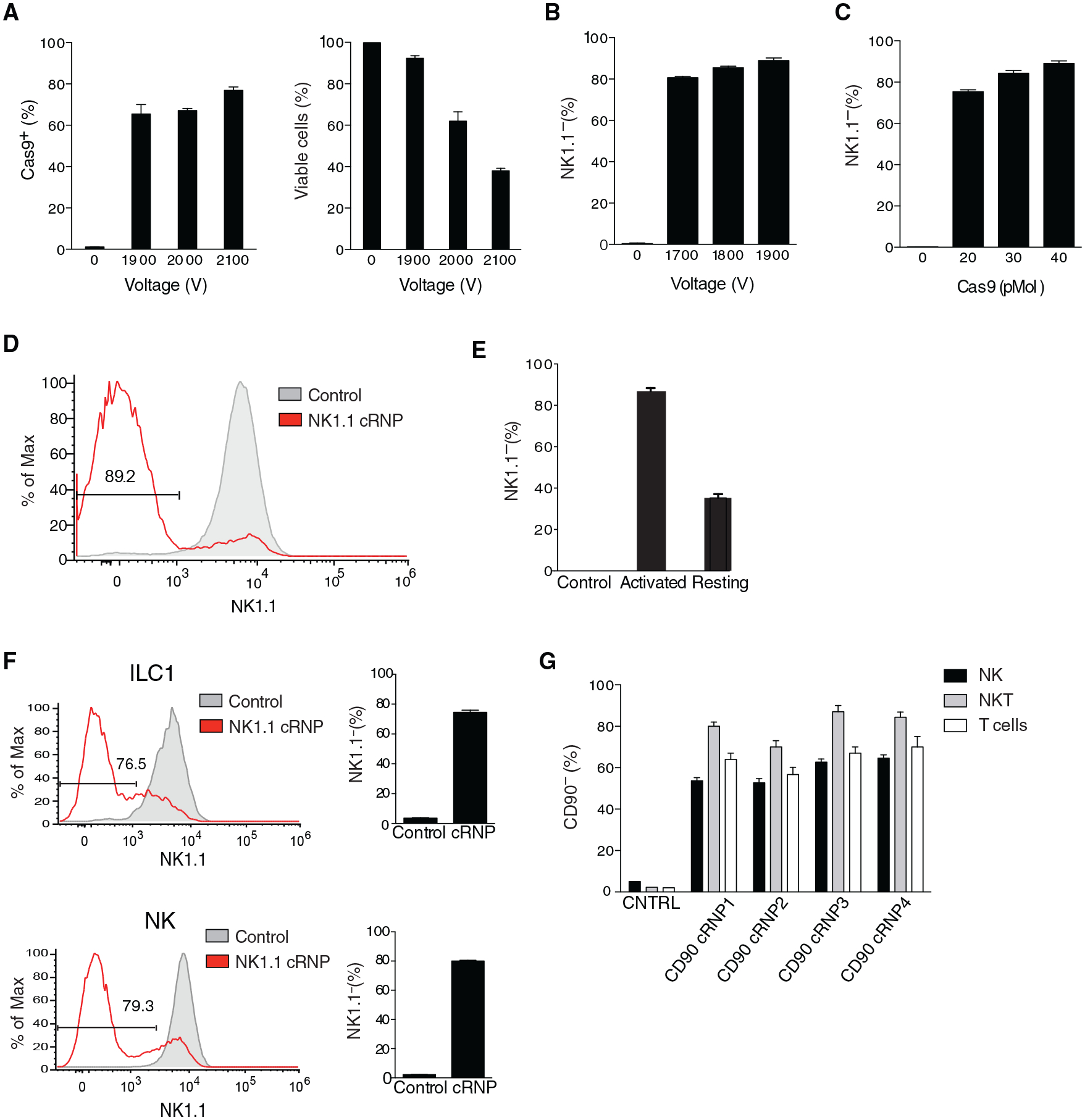
(A–E) Purified splenic NK cells were cultured for 16 h with 50 ng/mL rmIL-15 in complete media or freshly isolated before electroporation. (A) Frequency of intracellular Cas9+ NK cells (left) and percentage of viable NK cells (right) 2 h following electroporation with 40 pmol Cas9 of 5 × 105 rmIL-15-preactivated NK cells at the indicated voltages compared to un-electroporated controls. (B and C) Percentage of NK1.1− rmIL-15-preactivated NK cells 3 days after electroporation with cRNP complex consisting of Cas9 and Klrb1c gRNA compared to un-electroporated controls with varying (B) voltages or (C) concentrations of Cas9 in the Klrb1c cRNP complex electroporated at 1,900 V. Representative histogram of NK1.1 expression (D) and percentage of NK1.1− NK cells (E) 3 days after electroporation at 1,900 V of 5 × 105 rmIL-15-preactivated activated or freshly isolated naive cells compared to controls electroporated in the presence of Cas9 protein alone.
(F) Representative histograms and frequency of NK1.1 expression 3 days after electroporation of 2.5 × 105 rmIL-15-preactivated purified liver NK cells (TCRβ−CD3ε−NK1.1+CD49b+CD200r−) or ILC1s (TCRβ−CD3ε−NK1.1+CD49b−CD200r+) with Klrb1c cRNP complex at 1,900 V compared to controls electroporated in the presence of Cas9 protein alone.
(G) Percentage of indicated CD90− cells 3 days after electroporation of 2.5 × 105 rmIL-15-preactivated liver NK, NKT (TCRβ+CD3ε+NK1.1+) and T cells with various Thy1.2 cRNP complexes at 1,900 V compared to controls electroporated in the presence of Cas9 protein alone. Data are representative of three independent experiments of three mice per group.
Given our results with group 1 ILCs, we next tested a range of voltages and Cas9 concentrations that would lead to optimized cRNP-mediated gene editing in myeloid lineage cells. Bone-marrow-derived macrophages (BMDMs) and cDC1s (BM-cDC1s) did not differ in the frequency of intracellular Cas9+ cells following electroporation at all voltages tested, similar to splenic macrophages and DCs (Figures 3A and 1C). However, higher voltages sequentially decreased the viability of both BMDMs and BM-cDC1s, suggesting that electroporation at 1,900 V was optimal (Figure 3B). Indeed, electroporation of CD11b+ BMDMs or CD11c+ BM-cDC1s with 40 pmol Cas9:120 pmol gRNA Itgam cRNP complex or Itgax cRNP complex lead to ~90% CD11b− BMDMs and ~95% CD11c− BM-cDC1s, respectively (Figures 3C and 4D). Although slightly higher KO efficiencies were achieved with a higher concentration of Cas9 and gRNA cRNP complex, this reduced cell viability in both BMDMs and BM-cDC1s, suggesting that a 40 pmol Cas9:120 pmol gRNA cRNP complex and electroporation at 1,900 V was ideal for maximal gene deletion efficiency and viability for both cell types (Figures 3D and 3E). These results were corroborated by high-efficiency gene KOs of CD11c and CD11b in mature primary splenic DCs and macrophages respectively (Figure 3F). To test whether multiple cRNP complexes could be complexed together in a single electroporation to generate double-KO innate immune cells, we electroporated IL-15-preactivated purified splenic NK cells in the presence of Klrb1c and Thy1.2 cRNP complexes. 3 days after activation with IL-15 in vitro, we observed that ~55% of NK cells were NK1.1−CD90− and retained high viability (Figures 4A–4C). Similarly, ~77% of BMDMs electroporated in the presence of CD11b and β2M cRNP complexes and stimulated with interferon-γ (IFN-γ) were CD11b− major histocompatibility complex class I (MHC class I)− and displayed similar viability to single-cRNP-edited macrophages (Figures 4D–4F and 3E). Together, these results suggest that our cRNP gene editing method can be used to efficiently create single- and double-KO primary innate leukocytes.
Figure 3. High-Efficiency Gene Deletion of Primary and Bone-Marrow-Derived Myeloid Cells Using cRNP Complexes.
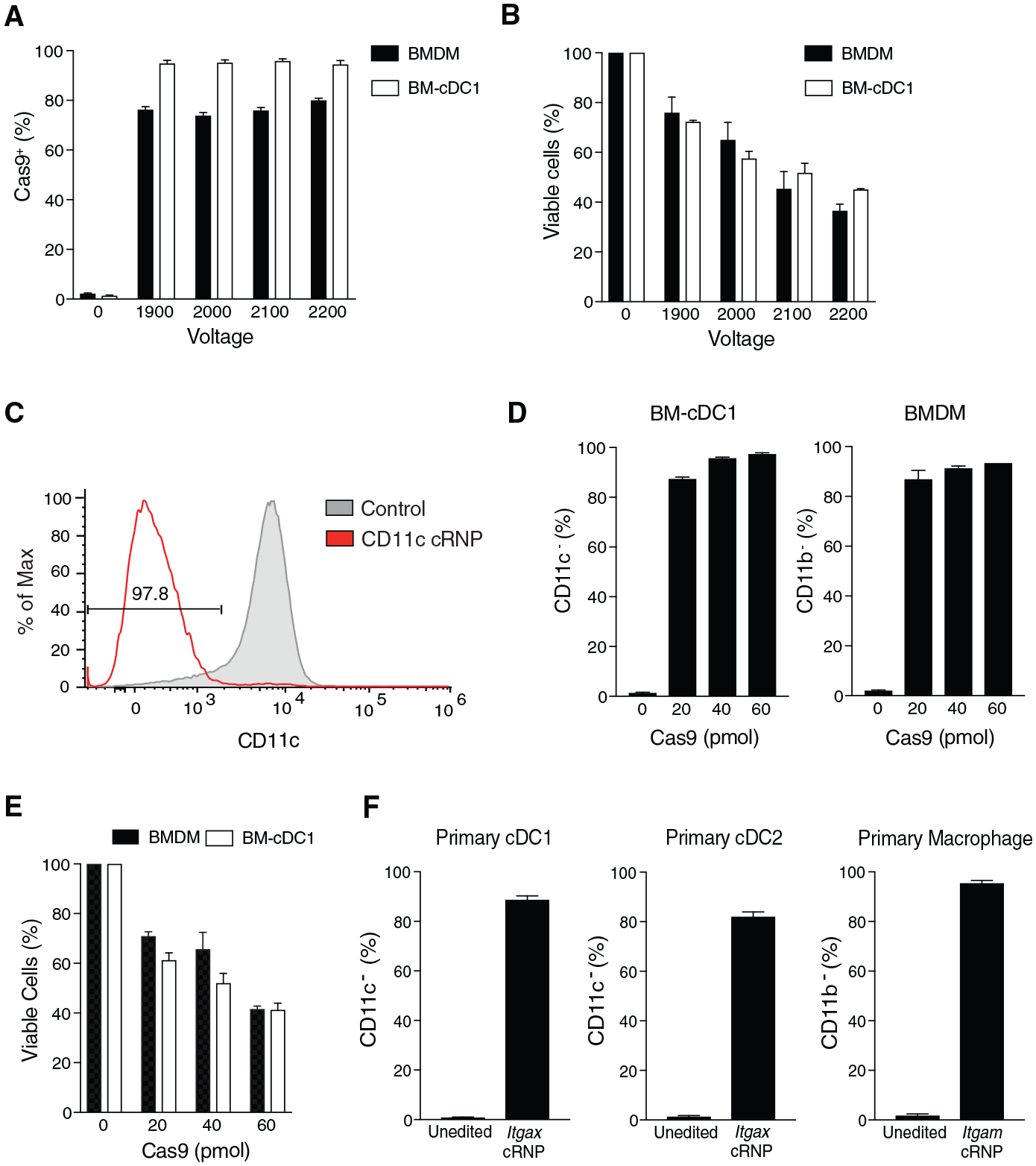
(A) Frequency of intracellular Cas9+ bone-marrow-derived macrophage (BMDM) or bone marrow-derived cDC1s (BM-cDC1s) 2 h post-electroporation with 40 pmol Cas9 compared to un-electroporated controls.
(B) Percent of viable BMDMs or BM-cDC1s 4 or 6 days, respectively, after electroporation of 1 × 106 cells at indicated voltages compared to un-electroporated controls.
(C–F) BM-cDC1s, BMDMs, splenic cDC1s, cDC2s, and macrophages were electroporated at 1,900 V in the presence of Itgax or Itgam cRNP complexes. (C) Representative histogram of CD11c expression in BM-cDC1 6 days after electroporation compared to controls electroporated in the presence of Cas9 protein. Percentage of CD11c− BM-cDC1s and CD11b− BMDMs (D) and viable cells (E) 4 or 6 days, respectively, after electroporation. (F) Frequencies of primary splenic CD11c− cDC1, cDC2, and CD11b− macrophages 4 days after electroporation with indicated cRNP complex compared to controls electroporated in the presence of Cas9 protein alone. Data are representative of three independent experiments or three mice per group.
Figure 4. cRNP Complex-Mediated Double Gene Ablation in ILCs and Myeloid Cells.
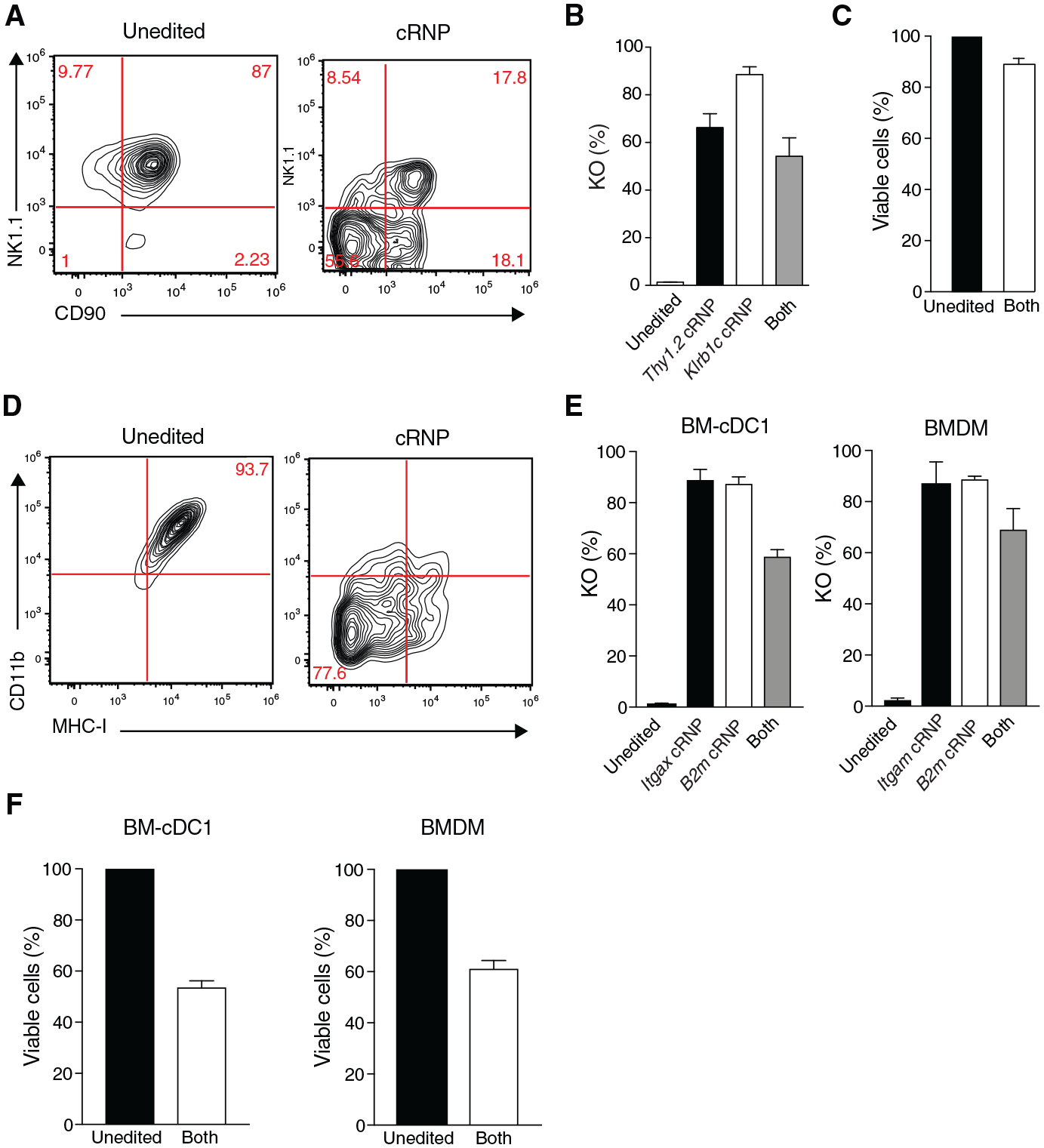
(A–C) NK cells (preactivated 50 ng rmIL-15), BM-cDC1s, and BMDMs were electroporated at 1,900 V in the presence of indicated single cRNP complexes or pooled cRNP1 and cRNP2 complexes targeting Klrb1c and Thy1.2 for NK cells, Itgax and B2m for BM-cDC1s, and Itgam and B2m for BMDMs. (A) Representative flow plots of NK cell NK1.1 and CD90 expression 3 days after electroporation of 5 × 105 cells with (Right panel) pooled cRNP complexes or (Left panel) control Cas9 protein electroporated cells (B) Percent of NK1.1−, CD90− or NK1.1− and CD90− (“Both”) NK cells and (C) percentage of viable NK cells after electroporation in the presence of indicated cRNP complexes relative to un-electroporated controls (unedited).
(D–F) BMDMs and BM-cDC1s were electroporated in the presence of various cRNP complexes and stimulated with 20 ng/mL IFN-γ for 48 h in vitro. (D) Representative flow plots of BMDM CD11b and MHC-I expression 4 days after electroporation of 1 × 106 cells with pooled cRNP complexes (right) or controls (left) electroporated in the presence of Cas9 protein alone. (E) Percentage of CD11c−, MHC-I−, or CD11c− and MHC-I− (“Both”) (left) and percentage of CD11b−, MHC-I−, or CD11b− and MHC-I− (“Both”) BMDM cells (right) 4–6 days after electroporation with indicated cRNP complexes compared to un-electroporated controls (unedited). (F) Percentage of viable BM-cDC1s and BMDMs from pooled cRNP-edited cells normalized to un-electroporated controls (unedited). Data are representative of two independent experiments of three mice per group.
cRNP Editing in NK Cells and cDC1s for In Vitro Functional Assays
To demonstrate the utility of this gene editing approach in vitro, we chose to target the transcription factor Stat4, which is essential for IL-12-mediated IFN-γ production in primary NK cells (Sun et al., 2012; Weizman et al., 2017). Similar to Stat4−/− NK cells, Stat4 cRNP-edited NK cells displayed a significant decrease in IFN-γ production following stimulation with IL-12 ex vivo when compared to a control Rosa26 cRNP (wild-type [WT], non-targeting control[NTC]) NK cells (Figures 5A–5C). Previous studies have suggested that tumor-intrinsic production of the cyclic dinucleotide 2′3′-cGAMP can activate DCs to mediate anti-tumor immunity in a type I IFN-dependent manner (Liu et al., 2019; Marcus et al., 2018). However, whether cGAMP can also stimulate the production of other anti-tumor cytokines in cDC1 is not well understood. Using BM-cDC1s from Il12p40YFP reporter mice, we observed that stimulation with the cyclic dinucleotide 2′3′-cGAMP significantly increased the frequency of YFP+ BM-cDC1s (Figures 5D–5F). IL-12 production by BM-cDC1s required Tmem173 (STING) signaling, because Tmem173 cRNP-edited BM-cDC1s displayed similar frequencies of YFP+ BM-cDC1s when compared to unstimulated and Il12p40YFP × Tmem173−/− controls (Figures 5D–5F). These results were unlikely due to cRNP-extrinsic effects on BM-cDC1 activation or maturation, because resting Rosa26 cRNP-edited (NTC) BM-cDC1s displayed a similar phenotype and background YFP+ frequency as un-electroporated controls (Figures S4A–S4C). Thus, our gene editing system revealed that BM-cDC1s robustly produced IL-12 in response to extracellular cGAMP in a STING-dependent manner in vitro.
Figure 5. cRNP-Edited Innate Immune Cells Are Functionally Deficient In Vitro.
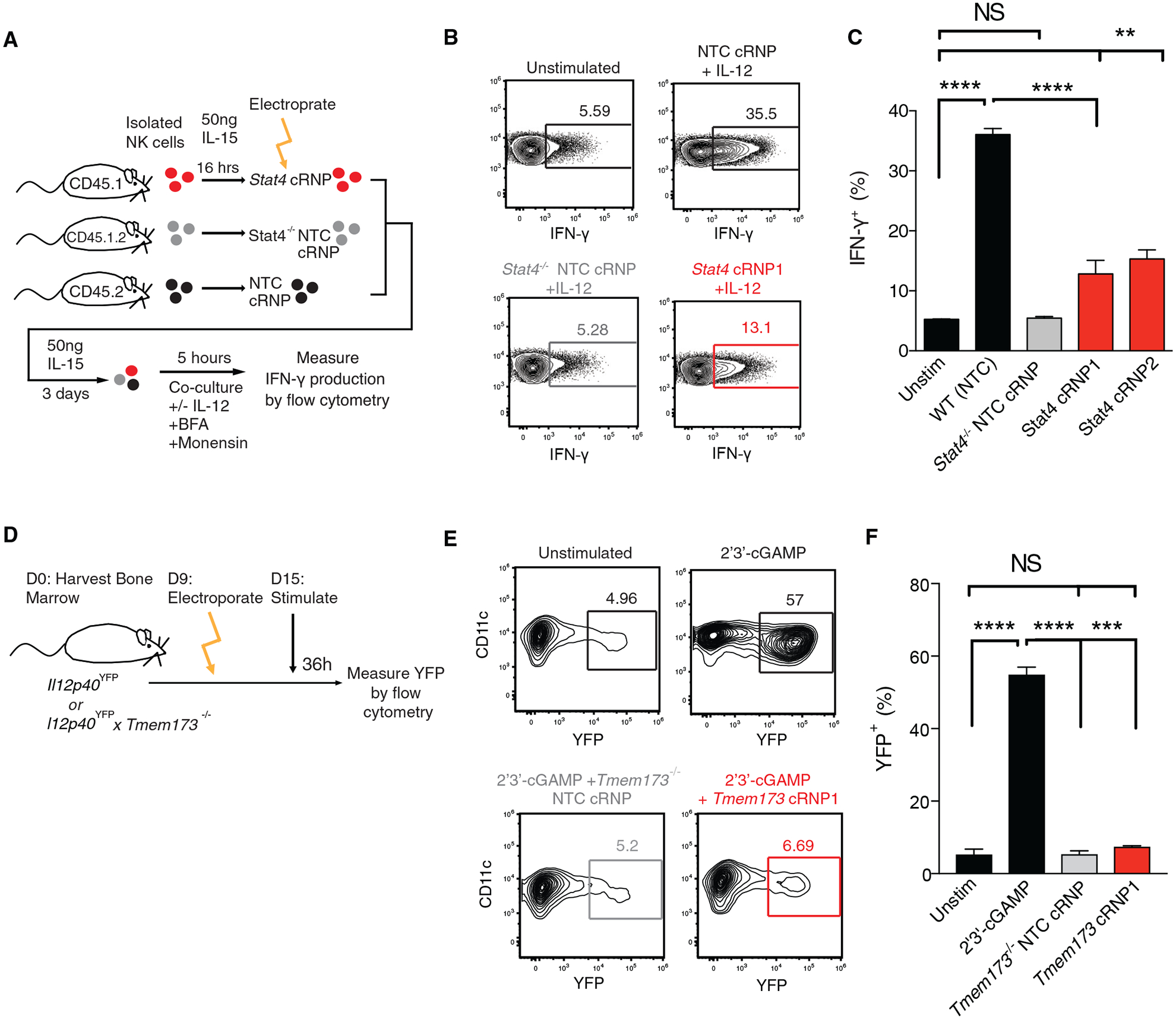
(A–C) Briefly, congenically distinct purified NK cells from WT (CD45.1, CD45.1 × CD45.2) or Stat4−/− (CD45.2) mice were stimulated overnight with 50 ng rmIL-15. NK cells were then electroporated at 1,900 V in the presence of either Rosa26 cRNP (NTC) or Stat4 cRNP complexes, mixed at equal ratios, and cultured in vitro with rmIL-15 for 3 days before stimulation with our without IL-12 in the presence of brefeldin A and monensin. (A) Schematic of experiment. (B) Representative flow plots of intracellular IFN-γ expression in indicated groups 5 h following stimulation. (C) Percentage of IFN-γ+ cells for indicated experimental groups (D–F) Briefly, Il12p40YFP or Il12p40YFP × Tmem173−/− cDCPs were electroporated in the presence either Rosa26 cRNP (NTC) or Tmem173 cRNP complexes and cultured for an additional 6 days in DC medium before stimulation with DC media (Unstim) or DC media containing 2′3′-cGAMP for 36 h. (D) Schematic of experiment. Representative flow plots (E) and percentages of YFP+ BM-cDC1s from each indicated group (F).
Data are representative of two independent experiments of three mice per group. Samples were compared using an unpaired, two-tailed Student’s t test with Welch’s correction, and data are presented as the mean ± SEM (**p < 0.01,***p < 0.001, ****p < 0.0001).
Adoptively Transferred cRNP-Edited Primary Innate Immune Cells Can Be Used to Elucidate Gene Function In Vivo
While previous studies have used cRNP gene editing in adaptive immune cells to elucidate gene function during in vitro functional assays (Seki and Rutz, 2018; Shifrut et al., 2018), there are currently no models to assay the function of cRNP-edited genes in specific innate immune cell lineages in vivo. To address this issue, we modified an adoptive transfer system whereby congenically distinct Ly49H+ NK cells can be transferred to Ly49H−/− hosts to generate effector and memory NK cells following MCMV infection (Sun et al., 2009) (Figure 6A). Specifically, IL-15-preactivated splenic Ly49H+ NK cells were electroporated in the presence of either Rosa26 cRNP (NTC) or Stat4 cRNP complexes and adoptively transferred into Ly49H−/− mice at a 1:1 ratio. Following MCMV infection of recipient mice, we observed that Stat4 cRNP-edited NK cells failed to expand on day 7 post-infection (P.I.) and generated significantly fewer memory NK cells on day 28 P.I., similar to a previous study (Sun et al., 2012) (Figures 6B and 6C). The expansion phenotype of Stat4 cRNP-edited NK cells was not due to off-target effects or influence from congenic mouse strains, as Stat4 cRNP-edited NK cells failed to expand to a similar extent as co-transferred congenically distinct Stat4−/− NK cells (Figures 6D and S5A–S5C). These results suggest that adoptively transferred cRNP-edited primary NK cells can reliably be used to assay gene function during NK cell effector and memory responses to MCMV in vivo.
Figure 6. cRNP-Edited Innate Immune Cells Can Be Used to Elucidate Antiviral Gene Function In Viv.
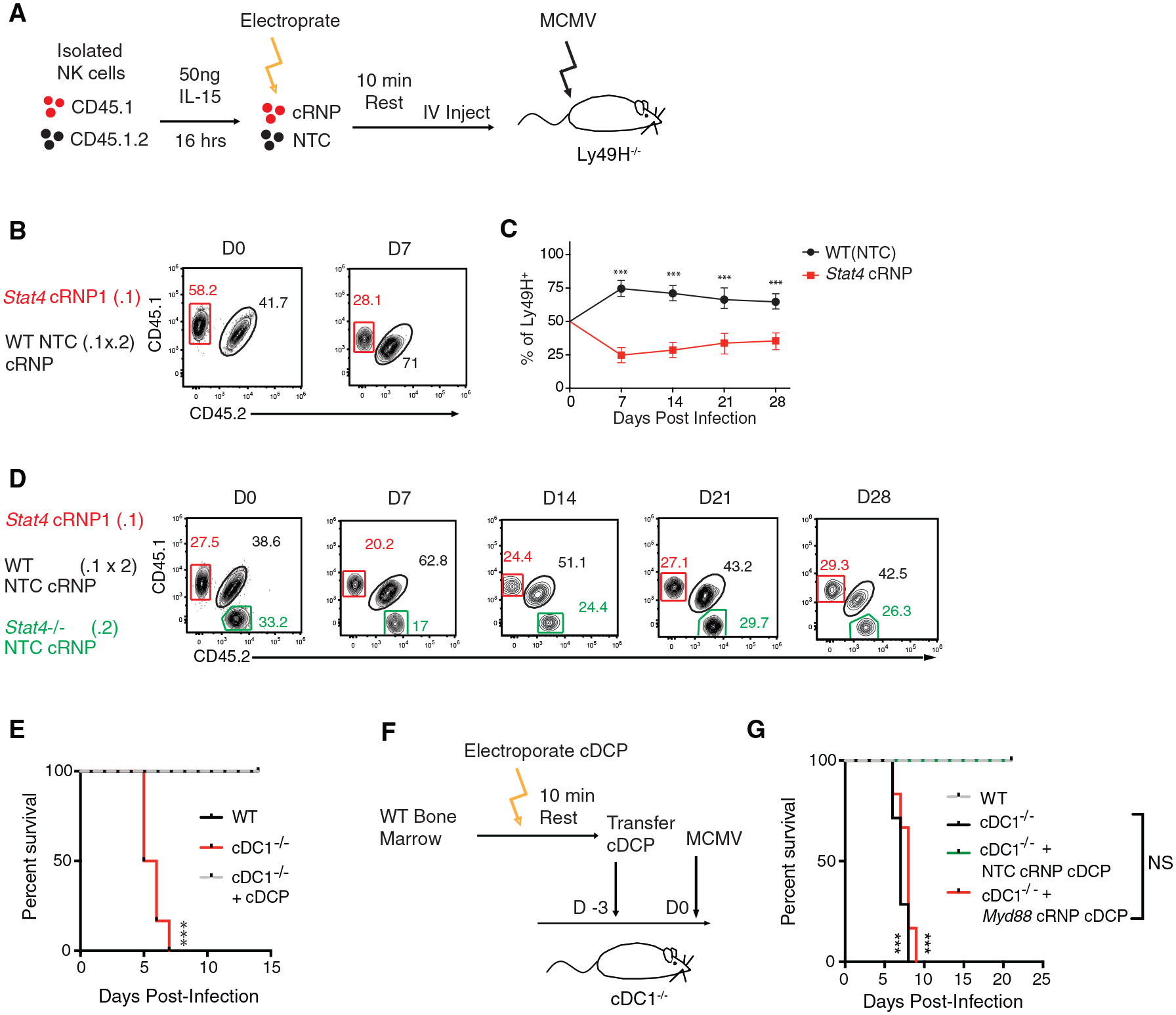
o (A–C) rmIL-15-preactivated splenic WT Ly49H+ NK cells were electroporated in the presence of either Rosa26 cRNP (NTC) (CD45.1 × CD45.2) or Stat4 cRNP complexes (CD45.1) and adoptively transferred i.v. into Ly49H-deficient recipients at a 1:1 ratio. Recipient mice were infected intraperitoneally (i.p.) with MCMV 16 h after adoptive transfer. (A) Schematic of experiment. (B) Representative flow plots of indicated Ly49H+ NK cells before injection (left) and at day 7 P.I. (right). (C) Quantification of adoptively transferred Ly49H+ Rosa26 cRNP (NTC) or Stat4 cRNP-edited NK cells in the blood of recipient mice at various time points P.I. (D) rmIL-15-preactivated splenic WT Ly49H+ NK cells or Stat4−/− (CD45.2) cells were electroporated in the presence of either Rosa26 cRNP (NTC) (CD45.1 × CD45.2) or Stat4 cRNP (CD45.1) complexes and adoptively transferred i.v. into Ly49H-deficient recipients at a 1:1:1 ratio and subsequently infected with MCMV. Representative flow plots of blood Ly49H+ NK cells before injection and at indicated days P.I.
(E) WT, cDC1−/−, and cDC1−/− mice reconstituted with bone-marrow-derived cDCPs were infected with MCMV i.p. Kaplan-Meier survival curves of each indicated cohort are shown.
(F and G) Briefly, 1 × 107 cDCPs were electroporated in the presence of either Rosa26 cRNP (NTC) or Myd88 cRNP complexes and transferred into recipient cDC1−/− mice 3 days before MCMV infection. (F) Schematic of experiment. (G) Kaplan-Meier survival curves of each indicated cohort.
Data are representative of two independent experiments of n = 3–6 mice per group. Samples were compared using an unpaired, two-tailed Student’s t test with Welch’s correction, and data are presented as the mean ± SEM (***p < 0.001).
Previous studies have shown that the transcription factor Batf3 is essential for cDC1 development and protection against viral infection and tumor development (Hildner et al., 2008). However, Batf3 is expressed in CCR6+ ILC3, cDC2, and macrophage subsets in vivo (Immgen) and the functional contribution of Batf3 in these subsets is not well understood in vivo. Thus, the precise contribution of cDC1s during host protection during viral infection is not well defined. To determine the role of cDC1s during viral infection, we used Xcr1Cre/+ × Rosa26LsLDTA (hereafter referred to as cDC1−/−) mice that specifically ablate XCR1+ cDC1 (Hemmi et al., 2016; Ohta et al., 2016). While Xcr1+/+ × Rosa26LsLDTA (hereafter referred to as WT) littermates were protected against MCMV challenge, all cDC1−/− mice rapidly succumbed to MCMV infection by day 7 P.I., indicating an early essential contribution of cDC1s to host protection following MCMV infection (Figure 6E). In order to generate an in vivo transfer model for cRNP-edited cDC1s, we tested whether bone-marrow-derived cDCPs could generate mature cDC1s in cDC1−/− mice. Following adoptive transfer of congenically distinct cDCPs, we observed that donor XCR1+CD11b− cDC1s reconstituted cDC1−/− mice in peripheral organs, albeit to a lesser extent than endogenous cDC1s present in WT littermate controls (Figures S6A–S6C). Because CD11c+ cell-intrinsic MyD88 expression has been shown to be essential for host protection during viral infection (Puttur et al., 2016), we then tested whether cDC1-intrinsic MyD88 signaling was sufficient for host protection during MCMV infection (Figure 6F). While adoptively transferred NTC cRNP-edited cDCPs were sufficient to rescue cDC1−/− mice, MyD88 cRNP-edited cDCPs failed to rescue cDC1−/− mice in a similar manner as untransferred control cDC1−/− mice (Figure 6G). These results suggest that cDC1-intrinsic MyD88 signaling is required for host protection to MCMV. Thus, these adoptive transfer models of cRNP-edited primary innate immune cells can be used and modified in future studies to reliably elucidate gene function in the innate immune system in vivo.
DISCUSSION
Using the Neon electroporation system, we define the conditions necessary for electroporation of cRNP complexes in naive and cytokine-activated mouse leukocytes. It is important to note that we detected minimal nuclear localized Cas9 in leukocytes 2 h post-electroporation (data not shown), suggesting that our optimized conditions are specific to cytoplasmic accumulation of Cas9 in leukocytes at this time point. Irrespective of these findings, we demonstrate that cytoplasmic Cas9 can be used to optimize cRNP gene KO efficiencies and cell viability by varying electroporation voltage conditions. This degree of customization may prove to be more beneficial to optimize cRNP editing in new immune cell types rather than by using proprietary pulse codes from other electroporation platforms. We observed cell-type-specific differences in Cas9 electroporation efficiencies and KO frequencies using set electroporation voltages and concentrations of cRNP complexes. These results may be due to stochastic variations in the accessible chromatin state of each cell lineage that may impact editing efficiency at a given genomic loci or cell-intrinsic differences in cell-surface protein turnover following in vitro culture with specific activating cytokines. Future studies will be necessary to determine the mechanisms behind cell-type-specific electroporation and cRNP-editing efficiencies. Furthermore, utilization of gRNA sequences derived from recent whole-genome-based CRISPR-Cas9 KO libraries (Wang et al., 2017) suggests that many gRNAs have low KO efficiencies for certain genes tested (e.g., Klrb1c) when used as a cRNP complex to edit electroporated primary lymphocytes. Thus, whole-genome-based CRISPR screens in primary T cells may not accurately reflect KO efficiencies in primary innate immune cells and need to be verified independently with validated gRNA sequences in a cell-type-specific manner. Together, our results reveal important cell-type-specific differences that will need to be considered when performing cRNP-based gene editing in mature primary immune cells.
Although previous viral-based CRISPR gene editing strategies have been used to achieve high-efficiency gene deletion in mouse bone-marrow-derived DCs in vitro and hematopoietic stem cell (HSC)-derived macrophages and DCs in vivo (LaFleur et al., 2019; Parnas et al., 2015; Theisen et al., 2018), these approaches require the use of transgenic Cas9 mouse strains, lentiviral or retroviral transduction, or irradiation followed by stem cell transplantation. Lentiviral-based CRISPR gene editing strategies in bone-marrow-derived HSCs cannot achieve gene deletion in specific innate immune lineages and cannot account for the impact of target gene expression on the development of the innate immune system in vivo. While lentiviral transduction of mature innate immune cells or inducible Cas9 expression could overcome these caveats, a previous study has shown that lentivirus does not efficiently transduce primary mature NK cells (~10%) (Boissel et al., 2012), suggesting that viral-based CRISPR strategies may not be optimal for all mature innate immune lineages. Our optimized cRNP genome editing approach in mature innate immune cells offers a robust alternative to previous viral based strategies by eliminating potential developmental defects and nonspecific immune lineage targeting associated with CRISPR gene editing in hematopoietic precursors. cRNP complexes may also minimize Cas9 off-target activity due to the rapid intracellular degradation of cRNP complexes compared to constitutive Cas9 overexpression (Kim et al., 2014; Ramakrishna et al., 2014), although future studies in the innate immune cells will be required to test this hypothesis. Furthermore, our approach leads to high-efficiency gene deletion in several innate immune lineages (ILCs, DCs, and macrophages) and may not be limited by mature cell-type-specific transduction efficiencies observed with viral-based strategies. Therefore, our studies provide a framework for efficient and practical gene editing that eliminates several key caveats from previous viral-based CRISPR gene editing approaches in innate immune cells.
Our study describes the use of two in vivo adoptive transfer models that can be used to elucidate the molecular mechanisms of memory NK cell formation and cDC1 responses to viral infection. However, cRNP-edited primary NK cells and bone-marrow-derived cDCPs could be used in several additional in vivo models. For instance, Ncr1Cre × Eomesfl/fl mice selectively lack circulating NK cells (Pikovskaya et al., 2016) and could be used with adoptively transferred cRNP-edited NK cells to precisely understand the gene regulatory networks induced during anti-tumor responses, obesity, and fetal development (Fu et al., 2017; Pahl and Cerwenka, 2017; Wensveen et al., 2015). In addition, we observed congenically distinct cDC2 in peripheral organs of WT mice following adoptive transfer of bone-marrow-derived cDCPs intravenously (i.v.). Adoptive transfer of cDCPs into cDC2-deficient mice, such as Zbtb46Cre × Irf4fl/fl (Loschko et al., 2016a), could potentially be used to elucidate cDC2-specific gene regulatory networks in vivo. Furthermore, mouse models of inducible monocyte-macrophage deletion, such as Lyz2Cre × Csf1rLsL-DTR (Esterházy et al., 2016), might also be able to be reconstituted with cRNP-edited monocytes to elucidate macrophage-specific gene networks in vivo, although future work will be necessary to confirm these proposed models. Although a wide variety of in vivo models of lineage-specific ablation can be coupled with cRNP-edited primary innate immune cells to overcome previous experimental limitations of gene deletion in specific innate immune lineages in vivo, these models may be limited for other innate immune lineages that cannot be readily adoptively transferred or specifically ablated in vivo.
Our finding that XCR1+ cDC1s are required for host protection during MCMV infection is supported by previous studies demonstrating Batf3 and Irf8-dependent cDC1s are essential for host protection during viral infection (Hildner et al., 2008; Weizman et al., 2017). However, our results expand on these previous findings, because the roles of Batf3 and Irf8 in cDC2s are not well defined and could not be previously excluded during host responses to viral infection. Similarly, recent studies have suggested that TLR7/9 signaling through MyD88 in CD11c+ cells is required for host protection against MCMV (Puttur et al., 2016). Because CD11c is expressed in TLR7/9-expressing macrophages, cDC1s, and cDC2s (Abram et al., 2014; Immgen), our results using cRNP-edited cells suggest that cDC1 expression of MyD88 is required for host protection to MCMV. MyD88 signaling likely acts upstream of IL-12 production in cDC1s, which is essential for group 1 ILC production of IFN-γ to suppress viral replication and confer host protection at initial sites of infection (Weizman et al., 2017). Thus, cRNP-mediated gene editing serves as a useful approach to rapidly elucidate gene function in specific lineages of the innate immune system in vivo. This general gene editing strategy may be further adapted in future studies to other primary immune cell types and in vivo transfer models to investigate protective or pathologic biological processes in the mammalian innate immune system.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources, reagents or materials should be directed to, and will be fulfilled by the Lead Contact, Timothy O’Sullivan (tosullivan@mednet.ucla.edu).
Materials Availability
This study did not create new unique reagents.
Data and Code Availability
This study did not generate datasets/code. The data that support the findings of this study are available from the corresponding author upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Mice were bred at UCLA in accordance with the guidelines of the institutional Animal Care and Use Committee (IACUC). The following mouse strains were used this study: C57BL/6 (CD45.2) (Jackson Labs, #000664), B6.SJL (CD45.1) (Jackson Labs, #002114), Klra8−/− (Ly49H-deficient), Il12btm1.1Lky/J (Il12p40YFP), Xcr1-Cre (Hemmi et al., 2016), Rosa26LsL-DTA, Tmem173−/−, and Stat4−/−. Experiments were conducted using 6–8 week old age- and gender-matched mice in accordance with approved institutional protocols.
Viruses and In vivo Infection Models
MCMV (Smith) was serially passaged through BALB/c hosts three times, and then salivary gland viral stocks were prepared with a dounce homogenizer for dissociating the salivary glands of infected mice 3 weeks after infection. Experimental mice in studies were infected with MCMV by i.p. injection of 7.5 × 103 plaque-forming units (PFU) in 0.5 mL of PBS.
Primary cell and in-vitro bone marrow-derived macrophage and cDC1 culture
To generate bone marrow derived macrophages and cDC1, bone marrow leukocytes were resuspended at 1.5×107 cells/10mL in DC media (RMPI 1640 + 25mM HEPES + 10% FBS, 1% L-glutamine, 1% 200mM sodium pyruvate, 1% MEM-NEAA, 1% penicillin-streptomycin, 0.5% sodium bicarbonate, 0.01% 55 mM 2-mercaptoethanol supplemented with 200 ng/mL FLT3-L and 50 ng/mL GMCSF) or macrophage media (DMEM + 10%FBS, 1% L-glutamine, 1% penicillin-streptomycin, 0.5% sodium pyruvate, supplemented with 50 ng/ml M-CSF and then plated in 10cm non-TC treated culture dishes (Corning). Bone marrow leukocytes were then cultured for 9 days in DC media with an additional 5 mL of DC media added on D5. cDCP were either harvested on D9 or media was changed and BM-cDC1 were used for experiments on D15 as described previously (Mayer et al., 2014). BMDM are cultured for 7 days in macrophage media, with a media change on D3. Isolated ILC1 and NK cells were cultured in CR-10 (RMPI 1640 + 25 mM HEPES + 10% FBS, 1% L-glutamine, 1% 200 mM sodium pyruvate, 1% MEM-NEAA, 1% penicillin-streptomycin, 0.5% sodium bicarbonate, 0.01% 55 mM 2-mercaptoethanol) supplemented with 50ng rmIL-15 (Peprotech). Purified ILC1’s and NK cells were cultured 16 hours prior to electroporation in TC treated plates at ~5 × 105 cells / cm2.
Method Details
Isolation and ex vivo culture of mouse leukocytes
Mouse spleens, livers, lungs, adipose tissue and bone marrow were harvested and prepared into single cell suspensions as described previously (O’Sullivan et al., 2016; Weizman et al., 2017). Briefly, spleens were dissociated using glass slides and filtered through a 100-mm strainer. To isolate liver lymphocytes, the tissues were physically dissociated using a glass tissue dounce homogenizer and purified using a discontinuous gradient of 40% over 60% Percoll (VWR). To isolate cells from the lung, the tissue was physically dissociated using scissors and incubated for 45 min in digest solution (1mg/ml type D collagenase (Roche) in CR-10). Resulting dissociated tissue was passed through 100-mm strainers, centrifuged, and lymphocytes were removed from the supernatant. To isolate the stromal vascular fraction (SVF) of adipose tissue, visceral adipose tissue was physically dissociated using scissors and incubated for 45 min in adipose digest solution (2mg/ml type II collagenase (Worthington Biochem) in HBSS +MgCl+CaCl, 5% fetal calf serum, 1% L-glutamine, 1% penicillin-streptomycin, and 50ug/mL DNase1 (Sigma)). Resulting dissociated tissue was passed through 100-mm strainers, centrifuged, and adipocytes were removed from the supernatant. To harvest bone marrow, the tibia, fibula, and femur were removed and then crushed using mortar and pestle in CR-10. Resulting mixture was filtered using 100-mm strainers and centrifuged. Red blood cells in spleen, adipose, liver, lung, and bone marrow were lysed using ACK lysis buffer. For purified NK and ILC1 isolation, liver or splenic single cell suspensions were resuspended in EasySep buffer (Stemcell) and processed using the EasySep Mouse NK Cell Isolation Kit according to the manufacturers protocol. Tissues were harvested from both male and female 6–8 week old mice to isolate primary cells.
Guide RNA Design
Synthetic gRNAs were purchased from SYNTHEGO. gRNA sequences were derived from a mouse whole genome CRISPR library described previously (Wang et al., 2017). 10 gRNA sequences from this dataset were ranked according to predicted indel percentage and low off-target score using the inDelphi machine-learning algorithm for each gene target (Shen et al., 2018). The top 3–7 guides were validated for high indel percentages by sanger sequencing before utilization in experiments.
cRNP complex formation
gRNAs (Synthego) were diluted to 100μM (100 pMol/μL) in 1× TE buffer (Synthego). 1.2uL (120 pMol) of gRNA, 0.9uL of 100 μM Alt-R® Cas9 Electroporation Enhancer (IDT) and 3.9 uL water were added to a 1.5mL tube per sample for a total of 6 μL. 2 μL of recombinant Cas9 (40pMol) (Synthego or QB3 Macrolab) was added to 4 μL water in a separate 1.5mL tube. 6 mL of diluted Cas9 was added to 6 μL of gRNA-enhancer mixture for a total of 12 mL cRNP complex at a 1:3 molar ratio. The cRNP complex was allowed to incubate for at least 10 minutes at room temperature (RT). For double KO experiments, 3 μL of diluted Cas9 was added to 3 μL of each gRNA-enhancer mixture (as prepared above) at a 1:3 molar ratio and complexed separately at RT. Each RNP complex was then added together for a total of 12uL per reaction. For other experiments, various concentrations of Cas9 were used to make cRNP complexes as indicated.
Electroporation
12uL of cRNP complex and 5 × 105 − 3 × 106 leukocytes resuspended in 100 μL of Buffer T were mixed (112uL total per reaction) and electroporated using the Neon Transfection system (Thermo-Fisher) at pulse code (20μs × 1 pulse) using 100ul Neon tips (Thermo-Fisher) at various voltages (1800–2200V). In knockout experiments, cRNP complexes consisting of 40pMol Cas9 were electroporated at 1,900 V unless indicated otherwise. Immediately following electroporation, cells were slowly pipetted into 1.5mL Eppendorf tubes containing pre-warmed culture media (CR-10 with 50ng rmIL-15 for NK cells, DC media, or Macrophage media) and inverted several times to dilute the T buffer. Cells were then incubated at 37°C for 90 minutes before centrifugation and resuspension in cell specific media before culturing in vitro. Cells were cultured in vitro for 3–6 days following electroporation prior to reading out gene editing efficiency by flow cytometry or sanger sequencing.
Adoptive transfer experiments
1 × 107 bone marrow-derived cDCP were transferred i.v. into recipient cDC1−/− mice 3 days before MCMV infection. In other experiments, cDCP were electroporated and then rested for 10 minutes and 5 × 106 cells were transferred into cDC1−/− recipient mice 3 days before MCMV infection. Adoptive NK cell co-transfer studies were performed by injecting a total of 1 × 106 NK cells; Rosa26 cRNP-edited Stat4−/−, Rosa26 cRNP-edited WT, and Stat4 cRNP-edited WT NK cells purified from spleens of congenically distinct WT mice (CD45.1, CD45.1.2 or CD45.2) into Klra8–/– mice 16 hours prior to MCMV infection.
Flow Cytometry
Cells were analyzed for cell-surface markers using fluorophore-conjugated antibodies (BioLegend, eBioscience). Cell surface staining was performed in 1X PBS and intracellular staining was performed using the eBioscience Foxp3/Transcription Factor or BD Cytofix/Cytoperm kits. Flow cytometry was performed using the Attune NxT and data were analyzed with FlowJo software (Tree Star). Cell surface and intracellular staining was performed using the following fluorophore-conjugated antibodies: CD45.1 (A20), CD45.2 (104), NK1.1 (PK136), CD49b (DX5), KLRG1 (2F1), TCRβ (H57–597), CD90.2 (30-H12), CD3 (17A2), Ly49H (3D10), CD200r1 (OX-110), CD8α (53–6.7), CD4 (GK1.5), IFN-γ (XMG1.2), I-A/I-E (M5/114.15.2), B220 (RA3–6B2), CD19 (6D5), CD11c (N418), CD11b (M1/70), CD64 (X54–5/7.1), XCR1 (Zet), Ly6G (1A8), and Cas9 (7A9–3A3 Cell Signaling Technology).
Ex vivo stimulation of lymphocytes
~5 × 105 NK cells were stimulated for 5 hours in CR-10 containing 50ng/mL rmIL-15, Brefeldin A (1:1000; BioLegend) and Monensin (2uM; BioLegend) with or without recombinant mouse IL-12 (20 ng/ml; Peprotech). ~1 × 106 BMDC1 were stimulated for 36h in DC media with or without 2′3′-cGAMP (5 μg/mL; InvivoGen). Cells were cultured in media alone as a negative control.
PCR and Sanger Sequencing
DNA from primary leukocytes was isolated using the DNeasy Blood and Tissue kits (QIAGEN). DNA concentration was measured using the NanoDrop OneC Microvolume UV-Vis Spectrophotometer (Thermo Scientific) and then diluted to 50 ng/μl in water before PCR amplification of cRNP-targeted genomic regions of approximately 500–1000 base pairs. PCR samples from WT and cRNP-edited cells were submitted for sanger sequencing (GENEWIZ) and then indel percentage was calculated using ICE analysis (SYNTHEGO) (see Table S1 for sgRNA and PCR primers).
QUANTIFICATION AND STATISTICAL ANALYSIS
For graphs, data are shown as mean ± SEM, and unless otherwise indicated, statistical differences were evaluated using a Student’s t test with Welch’s correction to a assume a non-normal variance in our data distribution. p < 0.05 was considered significant. Graphs were produced and statistical analyses were performed using GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse NK1.1 | BioLegend | PK136 |
| Anti-Mouse CD3e | BioLegend | 17A2 |
| Anti-Mouse Ly6G | BioLegend | 1A8 |
| Anti-Mouse B220 | BioLegend | RA3-6B2 |
| Anti-Mouse IFN- γ | BioLegend | XMG1.2 |
| Anti-Mouse CD11b | BioLegend | M1/70 |
| Anti- Mouse CD27 | Biolegend | LG.3A10 |
| Anti-Mouse CD19 | BioLegend | 6D5 |
| Anti-Mouse CD49b/DX5 | BioLegend | DX5 |
| Anti-Mouse KLRG1 | BioLegend | 2F1 |
| Anti-Mouse CD45.1 | BioLegend | A20 |
| Anti-Mouse CD45.2 | BioLegend | 104 |
| Anti-Mouse CD8α | BioLegend | 53-6.7 |
| Anti-Mouse CD4 | BioLegend | GK1.5 |
| Anti-Mouse TCRβ | BioLegend | H57-597 |
| Anti-Mouse CD90.2 | BioLegend | 30-H12 |
| Anti-Mouse CD200r1 | BioLegend | OX-110 |
| Anti-Mouse CD11c | BioLegend | N418 |
| Anti-Mouse XCR1 | BioLegend | ZET |
| Anti-Mouse MHCII | BioLegend | M5/114.15.2 |
| Anti-Mouse CD64 | BioLegend | X54-5/7.1 |
| Anti-Mouse Ly49H | BioLegend | 3D10 |
| Anti-Cas9 | Cell Signaling Technology | 7A9-3A3 |
| Bacterial and Virus Strains | ||
| Murine Cytomegalovirus | O’Sullivan (PI) | Smith Strain |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Brefeldin A | BioLegend | Cat# 420601 |
| Monensin (Golgistop) | BioLegend | Cat# 420701 |
| Recombinant Mouse IL-12 | Peprotech | Cat# 200-12 |
| Recombinant Mouse IL-15 | Peprotech | Cat# 210-15 |
| 2′3′-cGAMP | InvivoGen | Cat# tlrl-nacga23 |
| Cas9-NLS | SYNTHEGO | N/A |
| Cas9-NLS | qb3 UC Berkley | N/A |
| Alt-R® Cas9 Electroporation Enhancer, 10 nmol | IDT | Cat# 1075916 |
| Synthetic Guide RNAs | SYNTHEGO | N/A |
| Critical Commercial Assays | ||
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| Cytofix/Cytoperm | BD Biosciences | Cat# 554714 |
| EasySep Mouse NK Cell Isolation Kit | Stem Cell | Cat# 19855 |
| EasySep Buffer | Stem Cell | Cat# 20104 |
| Experimental Models: Organisms/Strain | ||
| Mouse: C57BL/6 (CD45.2) | Jackson Lab | Stock # 000664 |
| Mouse: B6.SJL (CD45.1) | Jackson Lab | Stock #002114 |
| Mouse: Il12p40-YFP | Jackson Lab | Stock # 006412 |
| Mouse: Tmem173−/− | Jackson Lab | Stock # 017537 |
| Mouse: Stat4−/−, Klra8−/− (Ly49H-deficient), Xcr1Cre/+, Rosa26LsLDTA | O’Sullivan (PI) | N/A |
| Oligonucleotides | ||
| sgRNA and sgRNA PCR Primers see Table S1 | Multiple | N/A |
| Software and Algorithms | ||
| FlowJo, Version 9.9.6 | Ashland, OR: Becton, Dickinson and Company | https://www.flowjo.com/solutions/flowjo |
| Prism | GraphPad | http://www.graphpad.com/scientific-software/prism |
| ICE Analysis | SYNTHEGO | https://ice.synthego.com/ |
| Other | ||
| Neon Transfection System | ThermoFisher | Cat# MPK5000 |
| Neon 100 uL transfection kit | ThermoFisher | Cat# MPK10096 |
Highlights.
Optimized electroporation of cRNP complexes in primary mature innate immune cells
High knockout efficiency of signaling adaptors and transcription factors
cRNP-mediated knockout of Stat4 in primary NK cells during MCMV infection
cRNP knockout reveals MyD88 is required for cDC1-dependent control of MCMV infection
ACKNOWLEDGMENTS
We thank members of the O’Sullivan, Lanier, Fregoso, and Sun labs for helpful discussion. A.D.H. was supported by the Ruth L. Kirschstein National Research Service Award (AI007323). T.E.O. was supported by the NIH (P30DK063491 and AI145997).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107651.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, et al. (2013). Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol 14, 221–229. [DOI] [PubMed] [Google Scholar]
- Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, and Klingemann H (2012). Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk. Lymphoma 53, 958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterházy D, Loschko J, London M, Jove V, Oliveira TY, and Mucida D (2016). Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat. Immunol 17, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B, Jarvis E, Roth TL, Shin J, Corn JE, Marson A, Meyer BJ, Patel NH, and Hochstrasser ML (2018). Enhanced genome editing with Cas9 ribonucleoprotein in diverse cells and organisms. J. Vis. Exp 135, 57350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Zhou Y, Ni X, Tong X, Xu X, Dong Z, Sun R, Tian Z, and Wei H (2017). Natural killer cells promote fetal development through the secretion of growth-promoting factors. Immunity 47, 1100–1113.e1106. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Hoshino K, and Kaisho T (2016). In vivo ablation of a dendritic cell subset expressing the chemokine receptor XCR1. Methods Mol. Biol 1423, 247–253. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, and Kim JS (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, Brown FD, Gillis JE, Coxe DJ, Doench JG, et al. (2019). A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat. Commun 10, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Crowe WN, Wang L, Lu Y, Petty WJ, Habib AA, and Zhao D (2019). An inhalable nanoparticulate STING agonist synergizes with radio-therapy to confer long-term control of lung metastases. Nat. Commun 10, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschko J, Rieke GJ, Schreiber HA, Meredith MM, Yao KH, Guermonprez P, and Nussenzweig MC (2016a). Inducible targeting of cDCs and their subsets in vivo. J. Immunol. Methods 434, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loschko J, Schreiber HA, Rieke GJ, Esterházy D, Meredith MM, Pedicord VA, Yao KH, Caballero S, Pamer EG, Mucida D, and Nussenzweig MC (2016b). Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J. Exp. Med 213, 517–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 49, 754–763.e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stüve P, Puttur F, Merad M, and Sparwasser T (2014). Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood 124, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura T, Riggan L, Hildreth AD, O’Sullivan TE, and Shibuya A (2020). Type 1 innate lymphoid cells protect mice from acute liver injury via interferon-gamma secretion for upregulating Bcl-xL expression in hepatocytes. Immunity 52, 96–108.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. (2011). Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. USA 108, 18324–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, and Sun JC (2016). Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity 45, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Sugiyama M, Hemmi H, Yamazaki C, Okura S, Sasaki I, Fukuda Y, Orimo T, Ishii KJ, Hoshino K, et al. (2016). Crucial roles of XCR1-expressing dendritic cells and the XCR1-XCL1 chemokine axis in intestinal immune homeostasis. Sci. Rep 6, 23505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, Englezakis A, Barlow JL, Hams E, Scanlon ST, et al. (2014). MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl J, and Cerwenka A (2017). Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 222, 11–20. [DOI] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015). A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar-Oliver A, and Gersbach CA (2019). The next generation of CRISPRCas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikovskaya O, Chaix J, Rothman NJ, Collins A, Chen YH, Scipioni AM, Vivier E, and Reiner SL (2016). Cutting edge: eomesodermin is sufficient to direct type 1 innate lymphocyte development into the conventional NK lineage. J. Immunol 196, 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttur F, Francozo M, Solmaz G, Bueno C, Lindenberg M, Gohmert M, Swallow M, Tufa D, Jacobs R, Lienenklaus S, et al. (2016). Conventional dendritic cells confer protection against mouse cytomegalovirus infection via TLR9 and MyD88 signaling. Cell Rep 17, 1113–1127. [DOI] [PubMed] [Google Scholar]
- Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, and Kim H (2014). Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 24, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, Girard-Madoux MJ, Seillet C, Mielke LA, Kerdiles Y, Fenis A, Wieduwild E, Putoczki T, Mondot S, Lantz O, et al. (2016). Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol 17, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggan L, Freud AG, and O’Sullivan TE (2019). True detective: unraveling group 1 innate lymphocyte heterogeneity. Trends Immunol. 40, 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med 209, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A, and Rutz S (2018). Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med 215, 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, and Marson A (2019). CRISPR-based tools in immunity. Annu. Rev. Immunol 37, 571–597. [DOI] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, and Lanier LL (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, and Lanier LL (2012). Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J. Exp. Med 209, 947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen DJ, Davidson JT 4th, Briseño CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, et al. (2018). WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate lymphoid cells: 10 years on. Cell 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903.e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, and Sancho D (2020). Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol 20, 7–24. [DOI] [PubMed] [Google Scholar]
- Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE (2017). ). ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Štimac D, Wunderlich FT, et al. (2015). NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol 16, 376–385. [DOI] [PubMed] [Google Scholar]
- Wu CM, Roth TL, Baglaenko Y, Ferri DM, Brauer P, Zuniga-Pflucker JC, Rosbe KW, Wither JE, Marson A, and Allen CDC (2018). Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. J. Immunol. Methods 457, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets/code. The data that support the findings of this study are available from the corresponding author upon request.