

Abstract
Fluctuations of the cytosolic calcium ion (Ca2+) concentration regulate a variety of cellular functions in all eukaryotes. Cells express a sophisticated set of mechanisms to balance the cytosolic Ca2+ levels and the signals that elevate Ca2+ in the cytosol are compensated by mechanisms that reduce it. Alterations in these Ca2+-dependent homeostatic mechanisms are the cause of many prominent diseases in humans, such as heart failure or neuronal death.
The genetic tractability of Toxoplasma gondii and the availability of genetic tools enabled the use of Genetically Encoded Calcium Indicators (GECIs) expressed in the cytoplasm, which started a new era in the studies of Toxoplasma calcium signaling. It was finally possible to see Ca2+ oscillations prior to exit of the parasite from its host cells. This was more than 30 years after Endo [1] showed that ionophores triggered egress and assumed that it was because of a Ca2+ increase. The use of GECIs allowed to visualize specific Ca2+ signals in live intracellular parasites and to distinguish these signals from host cell calcium fluctuations. In this article we present an overview describing “tried and true” methods of our lab who pioneered the first use of GECI’s in Toxoplasma, including GECI choice, methodology for transfection and selection of ideal clones, their characterization, and the use of GECI-expressing parasites for fluorometric and microscopic analysis.
1. Introduction
Ca2+ is a universal signaling molecule that regulates essential cellular functions. Ca2+ is controlled spatiotemporally, and intracellular organelles balance Ca2+ influx and efflux during Ca2+ signaling events [2]. Fluorescence Ca2+ indicators, whose brightness is sensitive to Ca2+ concentration, have been extensively utilized for decades in order to visualize Ca2+ dynamics [3]. Ca2+ indicators designed as organic synthetic dyes have improved over the years and exhibit excellent dynamic range and favorable kinetics. Genetically Encoded Calcium Indicators (GECIs) have become powerful tools for the study of calcium signaling and current efforts in protein engineering have significantly increased their performance to match those of organic dyes [4–8]. GECIs have the advantage of enabling noninvasive imaging of defined cells and compartments, whereas organic dyes are prone to organellar compartmentalization and leakage from the cytosol. Targeting of GECIs to various subcellular compartments, using targeting sequences, together with high-resolution microscopy have opened the possibility of selectively monitoring the dynamics of Ca2+ with unprecedented spatial resolution. State-of-the-art GECIs include the single-wavelength intensiometric (the intensity of the fluorescence increases proportionally to Ca2+ increase) GCaMP’s, which are based on circularly permuted green fluorescent protein (cpGFP), calmodulin (CaM), and the Ca2+/CaM-binding “M13” peptide (M13pep) and FRET based indicators, which are ratiometric and require dual-channel recording [6, 9]. Single-wavelength intensiometric GECI’s have a larger dynamic range; and thus, tend to be preferred over FRET based indicators. In mammalian cells a large number of probes have been produced for a variety of uses, including targeting to the endoplasmic reticulum, nucleus, mitochondria, Golgi, and endosome/peroxisomes [9]. The GCaMP6 series are highly sensitive probes with three variants available that differ in their kinetics [10] (Table 1). GCaMP6s has the slowest kinetics, GCaMP6m is faster while maintaining a high response, and GCaMP6f is the fastest variant [10].
Table 1:
Genetically Encoded Calcium Indicators that have been used in Toxoplasma gondii
| GECIs | Kd for Ca2+ | Dynamic Range (Fmax/Fmin) | References | Comments |
|---|---|---|---|---|
| GCaMP3 | 542 nM | 13.5 | Tian et al., 2009 [6] Borges et al, 2015 [14] |
Improved dynamic range over first generation |
| GCaMP5G | 460 nM | 32.7 | Akerboom et al, 2012 [44] Sidik et al, 2016 [15] |
Improved dynamic range over GCaMP3 |
| R-GECO | 482 nM | 16.0 | Campbell et al., 2011 [7] Borges et al, 2015 [14] |
First generation red GECI |
| B-GECO | 164 nM | 7 | Campbell et al., 2011 [7] Borges et al, 2015 [14] |
First generation blue GECI |
| GCaMP6f | 375 nM | 51.8 | Kim et al., 2013 [10] Borges et al, 2015 [14] Brown et al, 2016 [19] |
Fast on/off kinetics |
| GCaMP6s | 144 nM | 63.2 | Kim et al., 2013 [10] Stewart et al, 2017 [20] Kuchipudi et al, 2016 [45] |
Slow on/off kinetics |
| LAR-GECO1 | 24 μM | 10 | Campbell et al., 2014 [22] | Low affinity, endoplasmic reticulum targeted |
| LAR-GECO1.2 | 12 μM | 8.7 | Campbell et al., 2014 [22] | Low affinity, Mitochondria targeted |
| jRGECO1a | 148 nM | 11.6 | Kim et al., 2016 [12] | Second generation variant of R-GECO |
| jRCaMP1a | 214 nM | 3.2 | Kim et al., 2016 [12] | Second generation red GECI |
| jRCaMP1b | 712 nM | 7.2 | Kim et al., 2016 [12] | Second generation red GECI |
The specific characteristics of these indicators have been recently reviewed [11]. Multicolor GECI’s are also available like the red shifted variants jRGECO1a, jRCaMP1a/b, RCaMP, R-GECO, O-GECO, and CAR-GECO and the blue-shifted variants like B-GECO and GEM-GECO [7, 8]. Red indicators are of particular interest because red fluorescence is absorbed much less than green fluorescence, especially in mammalian cells, and the recently developed jGECO1a has acquired favorable kinetics to match GCaMP6s, giving red fluorophores a competitive advantage for in vivo imaging [12]. GECIs have been applied in a number of studies including neuronal physiology, Drosophila, mice, plants, primates, and parasites [13, 14].
The role of Ca2+ regulation and homeostasis in the lytic cycle-egress, extracellular motility, invasion, and replication-of Toxoplasma has been very well documented. In Toxoplasma, Ca2+ signaling results in the stimulation of gliding motility, microneme secretion, conoid extrusion, invasion and egress. Many studies of Ca2+ signaling during the lytic cycle of Toxoplasma gondii were done indirectly by loading extracellular parasites with fluorescent dyes to follow Ca2+ changes during their gliding motility; through using Ca2+ ionophores, and other exogenous agents to elevate Ca2+ in extracellular parasites and stimulate conoid extrusion or microneme secretion; or using intracellular or extracellular Ca2+ chelators to prevent host-cell invasion or egress. The potential use of GECIs across a wide diversity of applications is still beginning to emerge and some pioneer studies have utilized GECIs in high-throughput screens of compounds that disrupt Ca2+ signaling in both mammalian cells and parasites [13, 15]. In this article we describe the methods used for the creation of Toxoplasma tachyzoites expressing GECIs, and their validation and use.
2. Instruments and Materials
Hitachi fluorescence spectrophotometer models F-4500 and F-7000 (Hitachi High Technologies Corporation)
BioTek Synergy H1 hybrid multi-mode reader (BioTeck)
Deltavision Elite or similar system for time-lapse experiments
Hamilton microliter syringes with fixed needles, 5, 10, 25 and 50 μl sizes (Fisher catalog, 80075, 1482453A, 148247, 148245)
Toxoplasma gondii tachyzoites wild type, RH strain
Human fibroblasts (immortalized via overexpression of the Human Telomerase Reverse Transcriptase gene (hTERT)) were originally from BD Biosciences.
Human epithelial HeLa cells (ATCC CCL-2™)
Tissue culture supplies
GECI plasmids:
GCaMP6f (Addgene 40755), GCaMP6m (Addgene 40754), GCaMP6s (Addgene 40753), R-GECO (Addgene 32462), B-GECO (Addgene 32448), jRGECO1a (Addgene 61563), jRCaMP1a (Addgene 61562), jRCaMP1b (Addgene 63136), ER-LAR-GECO1 (Addgene 32444), mito-LAR-GECO1.2 (Addgene 32461)
Toxoplasma plasmids:
pCTH3 [16], pDT7S4H3 (a gift from Boris Striepen) [17], pTH3 (modified variant of pCTH3 in which the chloramphenicol cassette has been removed), [14], and pTUBSAG1-IEα_Dsred_DHFR_sag1CATsag1 [14, 18] and for UPRT locus: pUPRT::DHFR-GCaMP6(f/s) [19, 20].
Extracellular (Ringer) buffer:
155 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 3 mM NaH2PO4, 10 mM Hepes, pH 7.3, and 10 mM glucose
Intracellular buffer:
140 mM potassium gluconate, 10 mM NaCl, 2.7 mM MgSO4, 2 mM ATP (sodium salt), 1 mM glucose, 200 μM EGTA, 65 μM CaCl2 (90 nM free Ca2+), and 10 mM Tris/Hepes, pH 7.3.
Buffer A plus glucose:
116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM D -glucose, and 50 mM Hepes, pH 7.2
PolyJet (SignaGen Laboratories, SL100688)
Fura2-AM (ThermoFisher F1221)
Ionomycin (Santa Cruz Biotechnology sc-3592)
Thapsigargin (Abcam ab120286)
Saponin (Sigma S-7900–25g)
Zaprinast (Sigma 684500–25MG)
35 mm glass bottom cover dishes (MatTek Corp P35G-1.5–20-C)
Tissue Culture Cloning Cylinders (Bel-Art 978470100)
3. Methods
3.1. Plasmid construction
It is ideal and possible to generate T. gondii tachyzoites stably expressing GECIs. This can be done by stable integration of GECI genes into the genome of T. gondii. This has been achieved either through nonhomologous, random integration into the genome [14] or by integration of the GECI genes into a locus of the genome like the UPRT (uracil phosphoribosyltransferase) locus, which is not essential for parasite survival in vitro [20]. GECI genes that are introduced at the genomic locus of the UPRT gene are expressed uniformly because they are under the control of the same cis-element across different clones (Note 1). Plasmids for nonhomologous/random integration include pCTH3, pTH3 and pDT7S4H3, while plasmids for integration into the UPRT locus using homologous recombination include pUPRT::DHFR-GCaMP6(f/s) [19, 20].
Without a targeting sequence, GECI genes are expressed in the cytosol. The addition of the superoxide dismutase (SOD2) targeting sequence at the 5’-end of the GECI gene confers localization to the mitochondria [21]. In our lab we have successfully constructed strains expressing GCaMP6f and LAR-GECO1.2 (low affinity red GECI) [22] targeted to the mitochondria (Fig. 1). In order to target a GECI to the endoplasmic reticulum the coding sequence of the P30 gene (major Surface Antigen 1 of Toxoplasma) is fused to the 5’-end and the ER retention signal HDEL to the 3’ end [16]. We suggest using LAR-GECO1.0, a red GECI whose kinetics favor expression within the endoplasmic reticulum [22]. We have observed expression of GECIs in the ER only transiently (unpublished). Parasites do not appear to tolerate the expression of these indicators in the ER and we have not been able to isolate clones. The addition of the ferredoxin NADP+ reductase (FNR) targeting sequence at the 5’-end will confer localization to the apicoplast, and we have successfully constructed a strain expressing GCaMP6f localized to the apicoplast [23] (unpublished). For expression of GECIs in the PV the vector ptub_SAG1-IE_DsRed_dhfr_sag1CATsag1 can be used and the GECI of interest like GCaMPs or GECOs can be cloned at the SmaI (5’)/SnaBI (3’) restriction sites, substituting the DsRed gene. Selection can be done using antibiotic resistance or alternatively through enrichment and subclone of a population previously sorted by Fluorescence Activated Cell Sorting (FACS) [16, 23]. We have modified the plasmid pCTH3 [24] by removing the chloramphenicol cassette to generate pTH3. This plasmid has successfully been used to generate stable cell lines expressing GCaMP6f/s alone or in tandem with a (P)2A cleavage site (see below section 8) preceding a red fluorescence protein for ratiometric measurements [25]. 2A peptides (P2A, T2A, F2A, and E2A) are highly conserved peptides sequences that fail to form a peptide bond between a glycine and a proline residue at the carboxy terminus of the consensus sequence, through a mechanism termed ribosomal skipping. Translation of the proline and downstream gene continues resulting in two, separate peptide products originating from a single mRNA [26].
Figure 1: Images of Toxoplasma gondii parasites expressing GCaMP6 in the cytoplasm, mitochondria and parasitophorous vacuole.
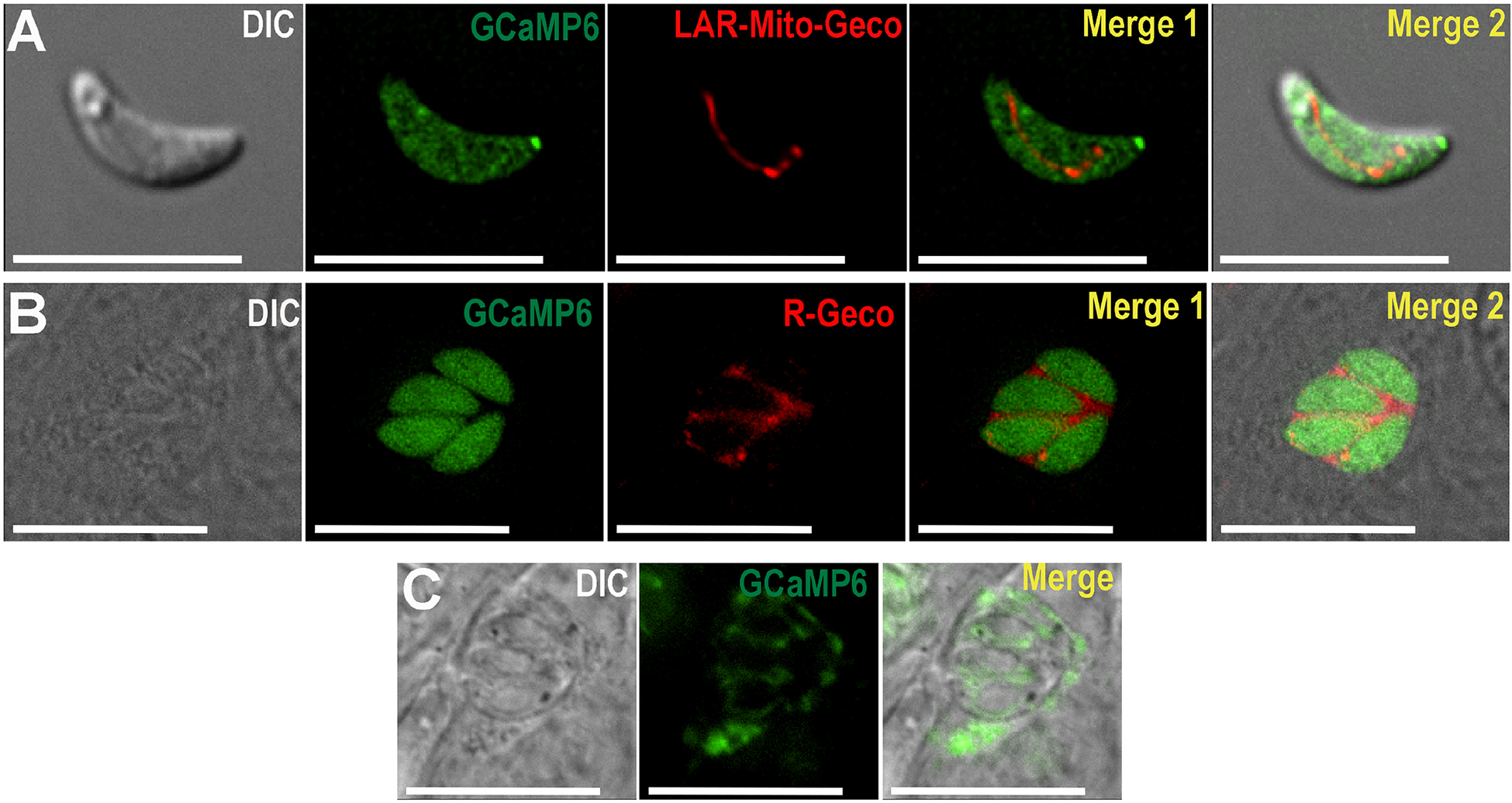
A, Live cell imaging of extracellular tachyzoites of the RH strain expressing GCaMP6f (pTUBGCaMP6f) (green) in the cytosol and LAR-Geco (pDT7S4H3-SOD2-Mito-LAR-Geco) in the mitochondria (red). B, Live cell imaging of intracellular parasites expressing GCaMP6f (pTUBGCaMP6f) in the cytosol (green) and R-Geco (ptub_SAG1-IEα-R-GECO_dhfr_sag1CATsag1) in the PV (red). C, Live cell imaging of intracellular RH parasites expressing GCaMP6f (pDT7S4H3-P30-GCaMP6f) in PV.
3.2. Transfection, selection and subcloning
Transfection of T. gondii tachyzoites with GECI plasmids is performed using a published protocol [27]. The DNA used for transfection is resuspended in 100 μl of cytomix solution (120 mM KCl, 0.15 mM CaCl2 10 mM K2HPO4/KH2PO4, 25 mM HEPES pH 7.6, 2 mM EGTA, 5 mM MgCl2). Freshly isolated and purified parasites are resuspended in the same solution at a concentration of 3.3 × 107/ml. 300 μl of cell suspension and 100 μl of DNA (20–100 μg) is mixed in a 0.4 cm electroporation cuvette. We use a BioRad electroporator with an exponential decay protocol set at 1.5 kV, 25 μF. Pulse time is usually between 0.4–0.6 ms.
With the aim of isolating a clonal population, GECI expressing parasites are first selected with drugs if they were transfected with plasmids containing known selectable marker genes like dihyrofolate reductase (DHFR) for pyrimethamine selection and chloramphenicol acetyl transferase for chloramphenicol selection. For GECI genes introduced into the UPRT locus, the pro-drug 5’-fluo-2’-deoxyuridine (FUDR) is used for selection. After selection, drug resistant parasites are serially diluted in 96-well plates and allowed to grow for 6–7 days. Wells with one small plaque are selected for further analysis. When using plasmids devoid of selectable markers, parasites are sorted and enriched by FACS. In this case, parasites can be FACS sorted two independent times to obtain single clones of GECI expressing parasites. Following electroporation, parasites are dispensed into a small T25 flask previously prepared with confluent fibroblasts and allowed to grow until they lyse out. The first FACS sorting can be done with the parasites released from this culture, producing approximately 500–5000 parasites likely expressing the GECI gene. This enriched population is dispensed into a new T25 flask and allowed to grow until parasites egress naturally. These parasites can be subjected to a second (and third) round of FACS sorting. In the second sorting, parasites are categorized in weak, medium or strong expression of the GECI gene. We typically use parasites with a medium level of GECI expression, because highly fluorescent parasites will likely have higher fluorescence background resulting in a lower functional dynamic range of the GECI fluorescence. After this final FACS sorting, the desired enriched population is diluted into a 96 well plate at 1 parasite/well in order to obtain single clones. It is possible to express GECIs (GCaMPs or GECOs) at specific compartments like the cytosol (Figure 1A), mitochondria (Fig. 1A), apicoplast (not shown) or the parasitophorous vacuole (Figure 1B–C) by using specific targeting signals like the mitochondrial targeting signal of the superoxide dismutase [21], the apicoplast targeting signal of the ferredoxin oxidoreductase [28, 29] and the vector ptub_SAG1-IE_ GECI_dhfr_sag1CATsag1 for PV labeling [18]. Transfection, selection and sub-cloning is done as described above and these clones expressing rGECO or GCaMP6 in the PV (Fig. 1B–C) are further characterized for their egress phenotype as described below.
3.3. Selecting an isolate with ideal fluorescence properties (Note 2)
We usually analyze 10–12 clones for their responses to ionophores, inhibitors and extracellular calcium. Figure 2A–B shows the characterization of 2 clones transfected with GCaMP6f and Figure 2C–D shows the characterization of two clones expressing GCaMP6s (see Table 1 for the characteristics of each indicator). GCaMP6s-expressing parasites show larger fluorescence increase in response to agonists and we attribute this to the slower Ca2+ dissociation kinetics of the indicator (compare the response of GCaMP6f to the response of clones 7 and 9 that express GCaMP6s in Fig. 2B).
Figure 2: Characterization of clones in a plate reader:
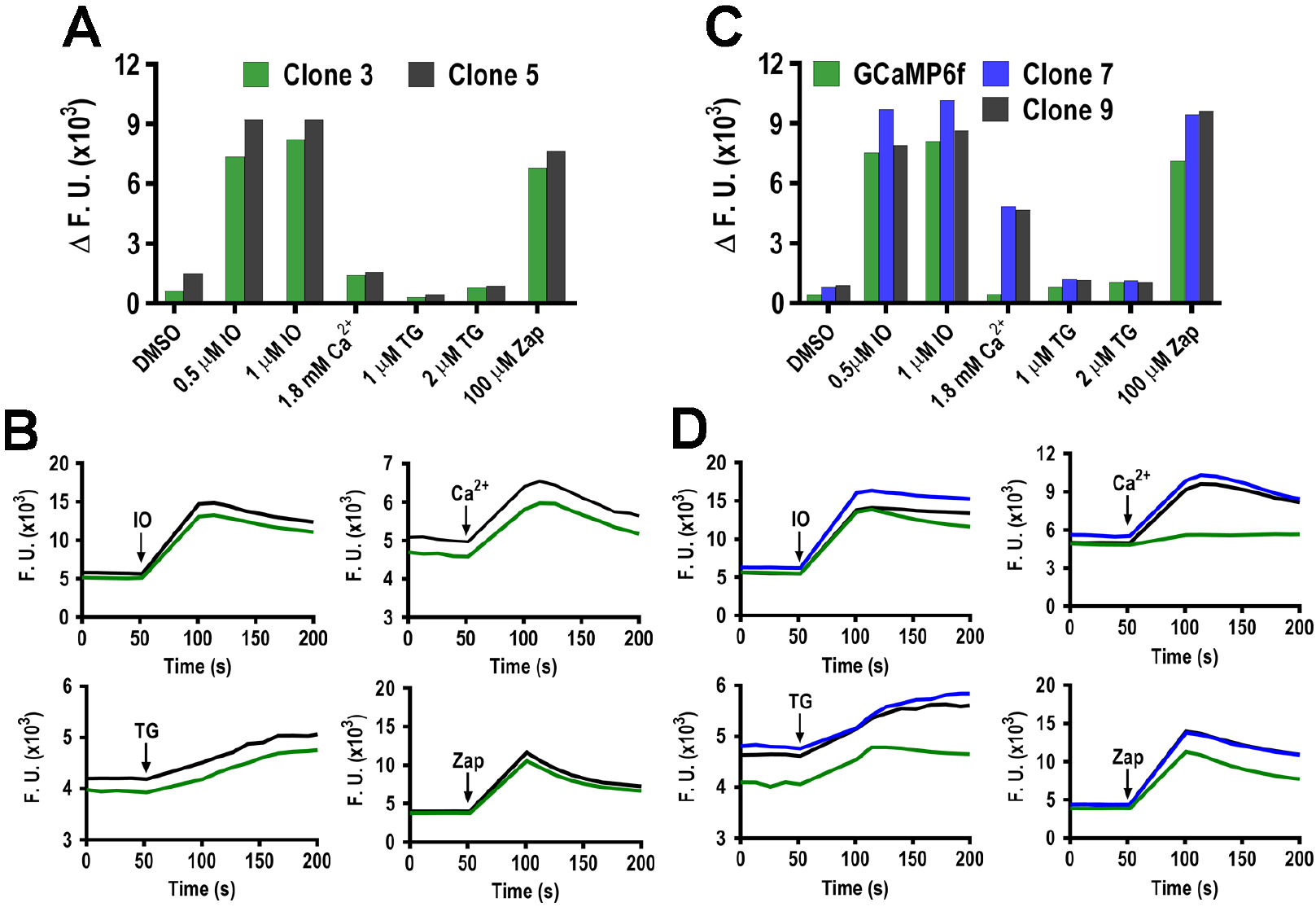
A suspension of 5 × 106 tachyzoites/ml in Ringer’s buffer with 100 μM of EGTA is dispensed into each well of a 96 well plate after shaking for 3 min the fluorescence is measured. Reagents are added at 60 seconds (method 3.3). A, Fluorescence changes before and after the addition of the indicated reagents with two Toxoplasma clones (3 and 5). DMSO is used as a control. B, kinetic measurements for clones 3 and 5 showing changes in the fluorescence of GCamP6f in function of time. IO, ionomycin; TG, thapsigargin, Zap, zaprinast, Ca2+, extracellular Ca2+, 1.8 mM. C, same experiment to the one presented in A with clones 7 and 9 that express GCamP6s and compare with the reference cell line expressing GCamp6f. D, kinetic measurements with clones 7 and 9. Same conditions as B.
The procedure to select the fittest clone with maximum response is described:
Clonal lines are grown in flasks with confluent hTERT fibroblasts to yield approximately 5 × 108 parasites. Tachyzoites are collected 48 h after infection and when the culture shows 70–80% host cell lysis. Parasites are gently scraped off, purified by filtration through an 8 μM nucleopore membrane and centrifuged at 706 g for 10 min. The parasites are washed twice and resuspended to a final concentration of 6 × 107 parasites/ml in extracellular buffer: 155 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 3 mM NaH2PO4, 10 mM Hepes, pH 7.3, and 10 mM glucose.
100 μl of the parasite suspension prepared in 1 is dispensed into each well of a 96 well and 100 μM EGTA is added to each well to chelate extracellular Ca2+ (Note 3).
The plate reader is set to record Green and Red Fluorescence changes using monochromators set to the respective channels: GCaMP6f/s 487 excitation and 508 emission and mScarletI 569 excitation and 593 emission. We use a BioTek Synergy H4 Plate Reader with a tungsten halogen and xenon flash as the light source.
Fluorescence changes in response to the addition of: DMSO as control for solvent (0.4%), thapsigargin (TG) (0.5 and 1 μM), ionomycin (IO) (0.5 and 1 μM), zaprinast (Zap) (100 μM) and Ca2+ (1.8 mM) are measured (Figure 2).
In a standard measurement, three min are allowed to establish a baseline followed by the addition of the ionophore or inhibitor. The concentration of the solution of the reagent is adjusted so that the volume added is 20 μl per well. Fluorescence is measured for four min after addition of the reagent. Representative tracings are shown in Fig. 2B and D.
The average fluorescence prior to addition of the reagent is subtracted from the maximum peak fluorescence attained after addition of the reagent to calculate the ΔF shown in Fig. 2A and C of 3 replicative wells for each condition. We select two clones that show largest ΔF indicating larger response of cytosolic Ca2+ (Figure 2).
3.4. Characterization of isolates - Ensuring isolate retains WT calcium signaling properties
These control experiments will measure cytosolic calcium of the GECI expressing mutants, by loading the GECI-expressing tachyzoites with the chemical fluorescent calcium indicator, Fura2-AM, and measuring fluorescence changes from both indicators using a high sensitivity, fast switching, fluorescence spectrophotometer (Notes 3 and 4). In addition, if cells express the ratiometric version of GCaMP6 (GCaMP6-mScarlett for example) both the fluorescence of GCaMP and the fluorescence of the reference protein can be recorded for calculation of the fluorescence ratio.
The loading of the chemical indicator to carry out these controls can be performed as follows:
-
Host cell Preparation (Note 5)
Confluent T75 flasks of hTERT cells (the surface area of the flask is completely covered by the cell monolayer) are detached and re-suspended to a concentration of 5×107 host cells/ml in DMEM-HG media with 10% calf serum and aliquoted into 3–5 T75 flasks. These cells should be allowed to grow to complete confluency (typically 5–7 days).
-
Toxoplasma Infection (Note 6)
Prior to parasite infection the media of the hTERT flasks (T75s) is replaced with fresh DMEM-HG media containing 1% calf serum pre-warmed at 37 °C. 4–6 T75 flasks should be infected with 1.5–2.5×107 parasites per flask 48 h prior to the desired time of collection. The media of these flasks is replaced with the same media 12–24 h after infection to remove any dead parasites and host cell debris.
-
Collecting Parasites
Parasites are collected ~48 h after infection. Ideally, at the time of collection, flasks should contain approximately 50% of host cells lysed and 50% heavily infected. A cell scraper is utilized to detach the entire monolayer, as it should only be partially lysed. This parasite/host cell solution should be transferred and filtered through a vacuum filter holder with receiver, housing a 47 mm, 8.0 μM nuclepore Track-Etch Membrane. This filtered suspension is then transferred to a 50 ml conical tube and centrifuged at RT for 10 min at 705 g. The pellet is gently re-suspended in 10 ml of Ringers buffer and a small aliquot diluted for counting. The suspension is centrifuged again at 705 g for 10 min. The resulting pellet is resuspended at 1×109 tachyzoites/ml in Ringers buffers with 1.5 % sucrose (loading buffer) for Fura2-AM loading.
-
Loading of Fura2-AM
Fura2-AM is added to this cell suspension (or to the loading buffer) to a final concentration of 5.15 μM and is allowed to incubate at 26° C for 27 min, protected from light (Note 7). Parasites are then centrifuged at 875 g for 2 min and washed twice in 1 ml of Ringers buffer and resuspended at 1×109 tachyzoites/ml. Parasites are viable for fluorometric experiments for approximately 2–3 h if kept on ice and covered from light.
-
Fluorometric Analysis (Figure 3)
Fluorescence measurements are carried out in a cuvette containing 5 × 107 tachyzoites in suspension in 2.5 ml of the appropriate reaction buffer. The parasites have been previously loaded with a fluorescent indicator or are expressing a genetic calcium indicator. The cuvette is placed in a Hitachi F-7000 or F-4500 fluorescence spectrophotometer, which allows kinetic measurements. These experiments are typically performed in extracellular buffer with 100 μM EGTA to chelate contaminating Ca2+. GCaMP6f excitation is 488 nm with emission at 510 nm. If two fluorescent proteins are expressed, such as GCaMP6 and mScarlet, an RFP, red fluorescence can also be measured. mScarlet excitation is 569 nm with emission at 594 nm [30]. Fura2-AM has excitations of 380 nm for its Ca2+ un-bound state, and 340 nm for its Ca2+-bound state with emission at 510 nm. The instrument’s parameters need to be adjusted to accommodate the consecutive measurements of two reagents (Fura2-AM and GCaMP6f) with appropriate excitation and emission wavelengths selected. The shortest cycle time allowable on the instrument should be selected.
The cycle time parameter will determine how fast the instrument switches from excitation and emissions from the first reagent to the second. Drugs and reagents are added via an appropriately sized Hamilton syringe during fluorometric readings, whereby changes in fluorescence of our indicators are detected in real time.
Figure 3: Ratiometric measurements with a clone expressing GCaMP6s-mScarlet.
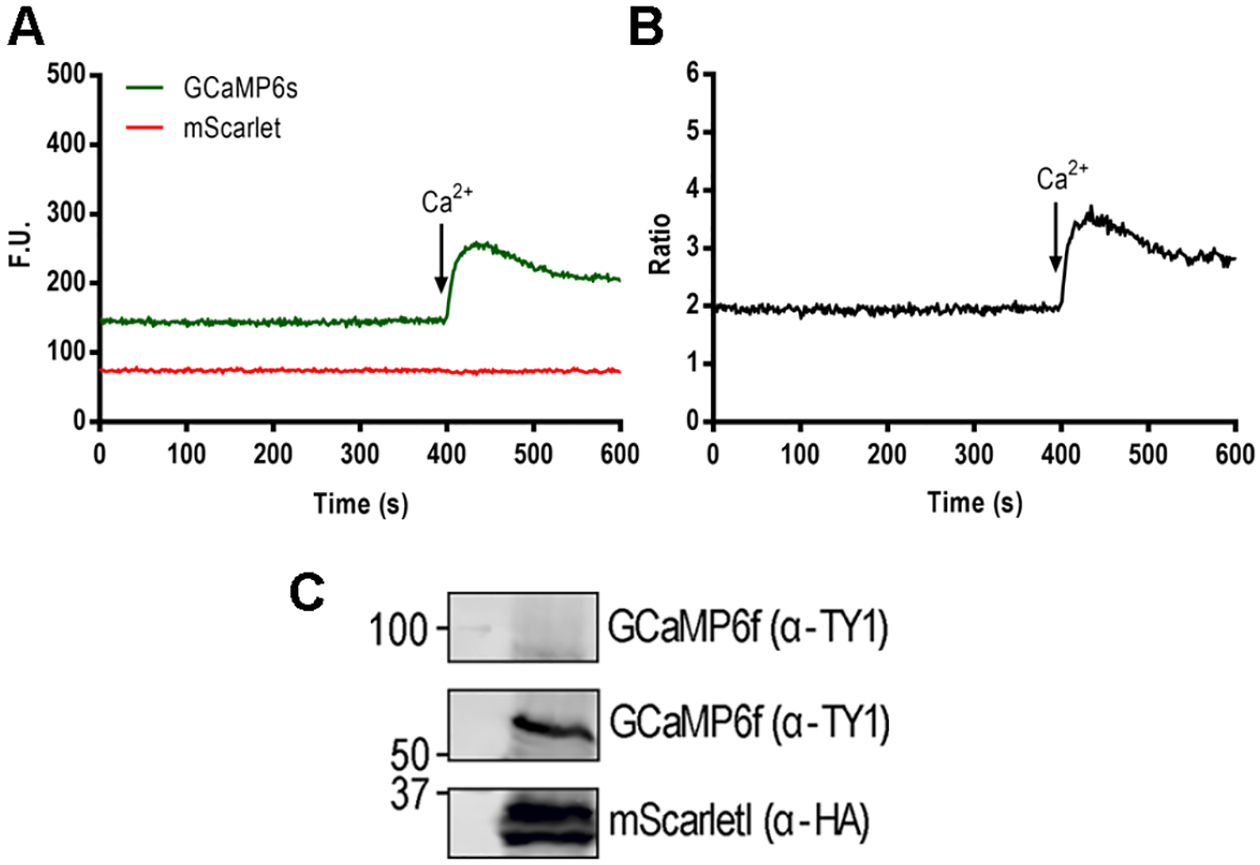
A, GCaMP and mScarlet fluorescence changes in function of time measured in the F-7000 fluorescence spectrophotometer. A, Parasites at a concentration of 2 × 107 tachyzoites/ml were resuspended in Ringer’s buffer with 100 μM of EGTA at a final volume of 2.5 ml. 1.8 mM of Ca2+ is added at 400 seconds. B, ratio calculation of the fluorescence tracings shown in A. C, Western blots showing the expression of GCaMP6f-Ty1 and mScarletI-3xHA. Antibodies: Mouse α-Ty1 1:2000 (a generous gift from Dr. Etheridge) and Rat α-HA 1:200 (Roche). Secondary: Goat α-mouse IRDye 800 (LI-COR) and Goat α-rat IRDye 680 (LI-COR).
3.5. Time Lapse Microscopy of GCaMP6f-expressing Parasites. (Notes 8–9)
After successful characterization and isolation of the ideal clone, the clonal line can be used to study cytosolic Ca2+ oscillations across the lytic cycle. The methodologies described below are for egress and extracellular motility. These protocols represent a general template and can be tailored (for example, host cell choice, imaging dish, laser exposure, and laser intensity) to match the needs/equipment of the user:
-
Plating HeLa Cells for Microscopy
A confluent T25 flask of HeLa cells is split and plated onto 6–8 35 mm glass bottom MatTek dishes and grown overnight using DMEM-HG with 10% FBS. Ideally, the cells should be 60–70% confluent by the next day.
-
Transient Transfection of Host Cells
For transient transfection of HeLa cells we typically use 3 μL of PolyJet in Vitro DNA transfection Reagent to 1 μg of DNA, as per the manufacturer’s instructions.
-
Infecting for Microscopy
From a lysed culture of tachyzoites expressing GCaMP6f we infect with 1 × 106 parasites per MatTek dish of HeLa cells and let them grow overnight. We image these dishes 24 h after the infection and we select parasitophorous vacuoles (PVs) with 2–4 tachyzoites because are the ones that will allow us to analyze single cell fluctuations without interference of the fluorescence of the neighboring parasite. Parasites in PVs with 8 or more parasites are difficult to single out.
-
Time Lapse Egress Protocol
We image cells at 37 °C using a Deltavision Elite system. First, the DMEM-HG is replaced with extracellular (Ringer) buffer. Because this buffer is more defined and does not contain phenol red, which would quench fluorescence, it is the preferred buffer to use for egress experiments. As per settings, we generally use 32% laser intensity for the FITC (Fluorescein Isothiocyanate), DIC (Differential Interference Contrast), and 10% laser intensity for TRITC (Tetramethylrhodamine) filter sets with an acquisition rate of 1 frame per second per each channel with 0.150 ms exposure. A typical movie for egress lasts about 10–20 min including 1 min of baseline prior to Ca2+ or other pharmacological drug addition; the remaining 9–19 min are used to record the response and recovery from the stimuli. The final drug concentrations that can be used are: ionomycin (0.1–1 μM), histamine (5 and 100 μM), thapsigargin (1 μM), DTT (5 mM) saponin (0.01%), nifedipine (10 μM). For Ca2+ free experiments, we use Ringer buffer supplemented with 100 μM or 1 mM EGTA. An example of a typical egress video with Thapsigargin stimulation in which the host cells are transiently transfected with R-GECO and infected with GCaMP6f expressing parasites can be seen in Fig. 4. Egress videos are quantified using the FIJI ImageJ plugin suite [31].
-
Long Term Time-Lapse Microscopy
For long-term time-lapse experiments, we typically use Ibidi 35 mm μ-Dish. These are designed with lid that locks into place in order to minimize evaporation. The main concern when performing a long-term time-lapse experiments is photobleaching and phototoxicity from continual laser exposure. While new microscopes are becoming available that help mitigate these two issues, such as light-sheet microscopy, we adjust our exposure settings on the Deltavision to image each channel every two min [32].
-
Motility experiments
For extracellular motility experiments, we use 35 mm glass bottom coverslip dishes (MatTek) that are treated overnight with 2 ml of a 10% FBS solution in a phosphate-buffered saline solution (PBS) at 4 °C. The dishes are washed once with PBS prior to use. Freshly egressed tachyzoites expressing GCaMP6f are collected, purified, and resuspended in 1 ml of Ringer buffer supplemented with 100 μM EGTA. We usually load ~2.5 × 107 parasites divided evenly among 6–8 dishes and incubate the dishes on ice for ~15 min to allow for the parasites to adhere. We use a small tissue culture cloning cylinders (Bel-Art, Cat # 378470100) to keep parasites concentrated in the middle of the dish to increase the efficiency of imaging. After removing the cell cylinder, we wash very gently with 1 ml of Ringer Buffer and bring the final volume back up to 2 ml with the same butter supplemented with 100 μM EGTA. We image using a Zeiss LSM 710 confocal microscope set at 37 °C Prior to imaging, we allow for cells to warm up for ~5 min at 37 °C. After establishment of a 1.5–2 min baseline for imaging, we stimulate gliding motility using pharmacological agents (see Time Lapse Egress Protocol for drug concentrations), and approximately an additional 900 frames are recorded. MTrackJ is a FIJI/ImageJ plugin that can manually track objects within a given movie (https://imagescience.org/meijering/software/mtrackj/). MTrackJ interface is simple and user friendly and has successfully been used to track parasites[33]. Typically, our lab uses a custom-made motility algorithm to track calcium oscillations in extracellular parasites and we have successfully applied the algorithm to track egressing parasites[34, 35]. This algorithm uses optical flow and fluorescence intensities to robustly track Ca2+ oscillations in extracellular parasites. For this algorithm, images must be input in dimensions of 400×400 at a frame rate of 20 fps.
Figure 4. In Vivo Dual Imaging of Host Cells Infected with GCaMP6f Expressing Parasites.
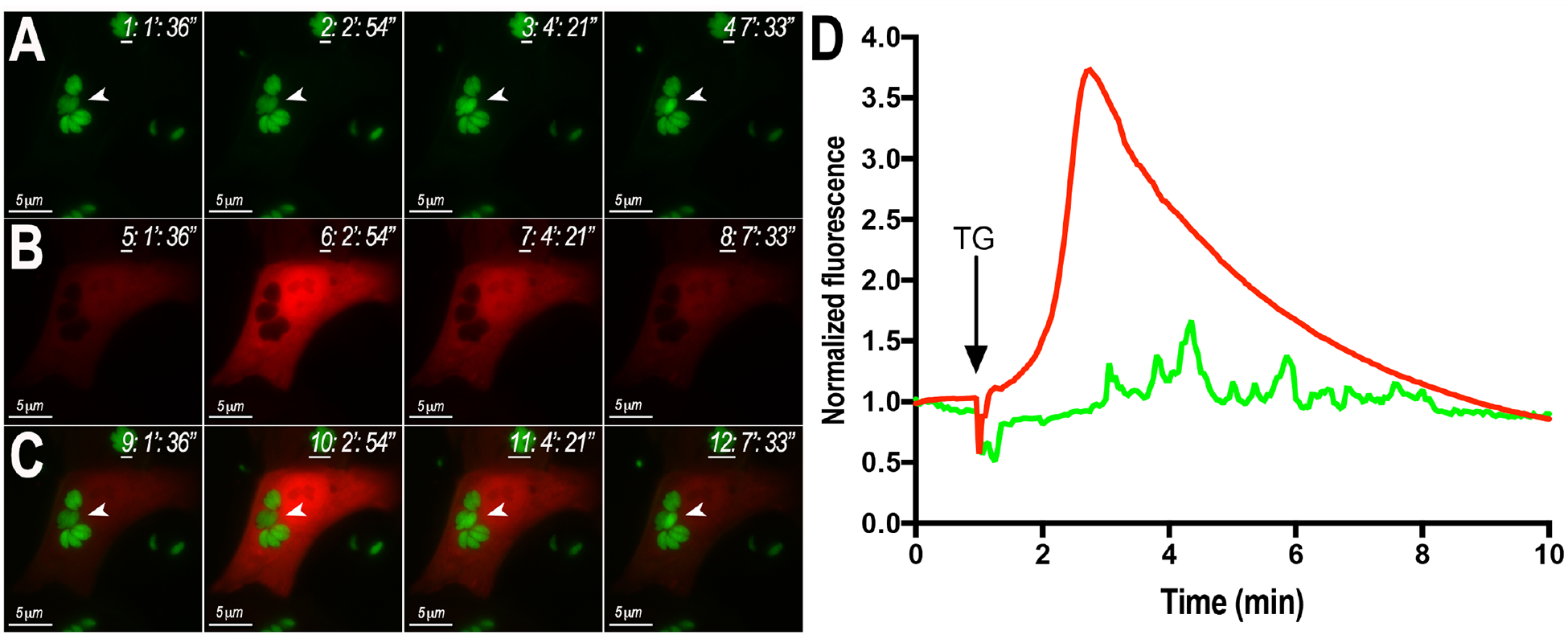
A, B, still-images obtained from videos showing changes of GCaMP (tachyzoites) and GECO (Hela cells) fluorescence after exposure to 2 μM thapsigargin (TG). C) Overlay of A and B. D) Fluorescence Tracings of Host Cells (red) and Parasites (green) after exposure to thapsigargin (TG). Video taken by Christina Moore.
4. NOTES
Plasmid Construction
-
1
The disadvantage of random integration of the GECI gene in an unspecified location of the genome, is that it could result in additional effects on the fitness of the parasites. The expression level of the GECI gene varies within different clones because it depends where it is inserted in the genome. Since calcium levels are tightly regulated in cells, and the GECIs can bind Ca2+ to affect the free calcium level, we reasoned that different expression level of the GECIs may have different effect on Ca2+ homeostasis. Random integration allows us to select clones with different expression level of the GECIs for different experiments.
Characterization of Isolates
-
2
GECIS are useful tools to study Ca2+ content of organelles and potential microdomains in Toxoplasma. GCaMPs have been designed to detect Ca2+ via a Ca2+ binding protein, calmodulin (CaM). Calmodulins are Ca2+ binding proteins expressed in all eukaryotic cells that participate in a number of signaling pathways that control important cellular functions [36]. Introducing extra copies of CaM, by way of a GECI, can potentially alter the Ca2+ dynamic in the cell [37]. It is highly recommended to perform control experiments with all GECI-expressing cell lines to determine if the transgenic line still retains the calcium signaling properties of a non-GECI expressing parasite (WT). We usually perform growth assays (Fig. 5A), egress experiments of intracellular parasites (Fig 5 B), response to extracellular Ca2+ (Fig. 5C) and test cytosolic Ca2+ responses with Fura2-am loaded parasites (Fig. 5D and E). We check if changes in GECI’s fluorescence are concurrent with the changes observed with chemical indicators. The experiment shown in Fig. 5D and E compare the Ca2+ response between RH and a clonal line expressing GCaMP6s (compare black and blue tracings). It is evident that the responses are diminished in the GECI expressing parasites and we attribute this to the Ca2+- binding effect of the genetic indicator. It is very likely that the expression of an exogenous copy of GCaMP6 would alter the Ca2+ dynamics.
-
3
For plate reader measurements, we test several concentrations of parasites per well. The number of parasites needs to be adjusted because of the lower sensitivity of the plate reader compared to the fluorescence spectrometer. Too few parasites may not be sufficient to produce a response for thapsigargin or Ca2+. These two triggers usually lead to an increase of cytosolic Ca2+ of 200–300 nM and it is possible that this change could be unnoticed if the number of parasites is too low. If the number of parasites is too high it can oversaturate the reading. We found that 6 × 107 is the ideal number for these experiments.
-
4
Clones with high background fluorescence are not ideal because they could potentially be unfit. We select clones with the largest range in response to ionophores and inhibitor additions and in general these clones show low background fluorescence.
Figure 5. Physiological characteristics of tachyzoites expressing GECIs.
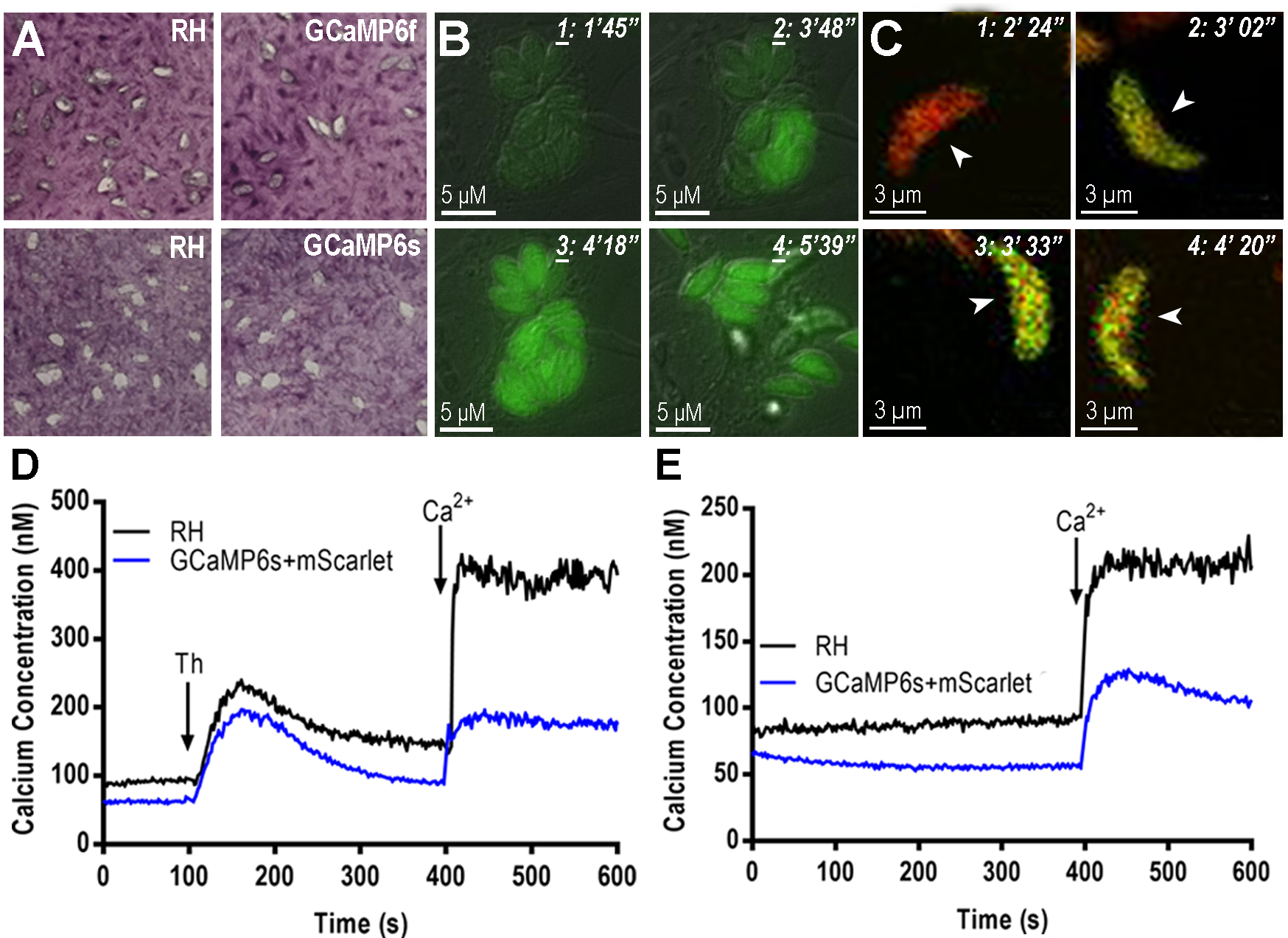
A, Plaques formed after 7 days of growth of 200 freshly isolated and purified tachyzoites per well. The host cells are hTert previously grown to confluency in 6 well plates. Infected host cell monolayer was fixed with 70% EtOH and stained with crystal violet. Plaque sizes were measured using FIJI. The plaques formed by tachyzoites expressing GCaMPf and GCaMPs were similar to the ones formed by tachyzoites of the RH strain (WT). B, egress experiments of tachyzoites expressing GCaMP6f. Hela cells were grown in MatTek dishes for 24 hrs and infected with 5 × 105 tachyzoites. Egress was stimulated with 100 μM of Zaprinast which was added 1 min after the start of the recording. C, GCaMP6f-Ty1-P2A-mScarletI expressing parasites were loaded in media supplemented with 100 μM EGTA. 1.8 mM Ca2+ was added (at ~3 min) leading to an increase in the GCaMP6f fluorescence signal. The signal from the mScarletI is Ca2+ insensitive thus allowing for ratiometric analysis. D, tachyzoites expressing GCaMP6s and mScarlet were loaded with FURA2-AM (see method 3.4) to measure cytosolic calcium in response to 1 μM thapsigargin, which was added at 100 s and 1.8 mM calcium, added at 400 s. E, same as C but only 1.8 mM of calcium was added at 400 s. Cytosolic calcium responses were compared between RH strain (black tracing) and the GCaMP6s expressing parasites (blue tracing).
Parasite Growth, Purification, and Loading
-
5
Flasks with 75cm2 of growth area (T75) produce the highest yield of Toxoplasma tachyzoites within a reliable timeframe as long as they are infected with a specific number of parasites per flask (Note 6) and the media is changed 8–24 h after the initial infection. Larger flasks, like 175 cm2 provide a larger surface but the total yield of parasites per flask is not correlative to the size and volume used. We find that the progression of the infection tends to be uneven in larger flasks and the collected time and number of parasites is unpredictable.
-
6
The infection of the T75 flasks is done with a fixed number of parasites (1–2.5×107 total parasites). This number depends on the fitness of the parasites used for infection. Usually, a completely lysed culture will contain a larger proportion of unhealthy parasites and will require a higher infection number.
Fluorometric Experimentation with GECI cell lines
-
7
For more efficient loading it is a better procedure to add the Fura2-AM to the loading buffer, which is used to resuspend the parasites instead of adding it to the suspension. Parasite loading is more efficient and result in healthier parasites when using volumes larger than 300 μl.
Imaging/Microscopy
-
8
Ca2+ dynamics in T. gondii parasites expressing GECIs can be studied by video microscopy along its lytic cycle (Figure 4). In Toxoplasma GECI’s have been successfully used to visualize Ca2+ fluctuations during invasion, egress and gliding [14]. GECI expressing parasites were also used for genetic screens to identify regulators of Ca2+ signaling, and probe how pharmacological stimulation induces increases in cytosolic Ca2+ levels [15]. Recent work combining Hela cells expressing cytosolic R-GECO infected with parasites expressing GCaMP6f demonstrated that an increase in host cytosolic Ca2+ preceded Ca2+ oscillations in the parasite [14]. Expression of the B-GECO indicator in the cytosol of Hela and infection with tachyzoites expressing GCaMP3 or GCaMP6 in their cytosol and a R-GECO in the PV it is possible to simultaneously follow Ca2+ fluctuations (blue) in the host cytosol, the PV (red) and the parasite cytosol (green) [14]. This was only possible because of specific expression of genetic indicators in the host cytoplasm, the parasitophorous vacuole and the Toxoplasma cytosol. Additionally, GECI’s have been applied to extracellular parasites as well in which parasites clones expressing GCaMP6f/s were used to track Ca2+ oscillations in motile parasites and link them to motility patterns [14, 34, 35, 38]. GCaMPs are single-wavelength and prone to artifacts such as loss of focus, bleaching, and imprecise initial detection due to low baseline. A partial solution to this problem is the simultaneous expression of a “reference” indicator insensitive to calcium that will help correct for these issues. Ratiometric studies in T. gondii using parasites expressing GCaMP6s and mCherry, both driven by separate promoters, but in the same genomic location has been employed by Stewart et al [20]. An alternative mechanism to produce two separate proteins is to use the well-characterized 2A system. In this strategy, a short peptide is introduced between the transcripts corresponding to two proteins in the same mRNA. During translation, the ribosome fails to form a peptide bond between a conserved glycine and proline residue. This system has been successfully utilized in Plasmodium and Toxoplasma [25, 39]. Additionally, our lab has successfully constructed ratiometric strains expressing GCaMP6f-(P)2A-mScarletI [30], and the expression validated by Western Blots (Figure 3 and 5C), which show that the 2A functions correctly to produce two separate proteins. Fluorometric analysis confirms mScarlet’s insensitivity to calcium. In frame fusion of a Ca2+ insensitive reference fluorescence protein is not advised due to the potential risk for FRET interaction [40]. These ratiometric strategies enable calibration of the GECI using digitonin or ionomycin, though these studies have been performed in mammalian cells and not in parasites [41]. As GECI’s are fluorescent proteins, they are prone to intrinsic factors that should be taken into consideration. GECI performance is susceptible to brightness (extinction coefficient × quantum yield), pH, folding/stability, and photobleaching, and these factors should be considered when imaging with GECI’s [6].
-
9
GECI’s enable for long-term imaging, but care must be taken to avoid phototoxicity. As a general rule, we prefer to use approximately one frame per second for egress experiments and approximately two frames per second for extracellular motility. We also prefer to use GCaMP-expressing parasites over R-GECO and jRGECO1a expressing parasites. GCaMP’s are further developed and exhibit a larger dynamic range and less photobleaching [12, 42]. A relatively universal standard for comparing GECI performance is the signal-to-noise ratio (SNR). The SNR is defined as the ratio of the fluorescence signal change (ΔF = Fobs − F0, where Fobs is sensor fluorescence at peak [Ca2+] and F0 is sensor fluorescence at baseline [Ca2+]) to the baseline fluorescence (F0N−1/2, where N is the number of photons detected by the sensor) [43]. For extracellular motility experiments, we prefer to use a Zeiss LSM 710 confocal microscope. Confocal microscopy offers enhanced Z-resolution and appears to offer improved sensitivity in detecting Ca2+ oscillations in motile parasites. Additionally, since the light is more focused in a confocal microscope (and coupled to the small size of a T. gondii cell (~5 μm)), our cells seem to require less excitation; and thus, we see less phototoxicity versus our widefield microscope. On the contrary, when working with intracellular parasite and egress experiments we prefer to use the Deltavision Elite widefield microscope. Though sacrificing Z-resolution versus confocal microscopy, widefield microscopy offers improved area of focus through imaging larger surface area. While imaging both fluorescent host cells as well as parasites, this feature enables for a broader image that encompasses both the entire host cell as well as the parasite to provide for a sharper image in comparison to the LSM710. Though improvements have been made to red GECI’s their brightness and photostability are not comparable with the GCaMP series, thus brightness is an important consideration. Because widefield microscope does not attenuate the signal through a pinhole, signal strength is stronger and requires less excitation for visualization. Additionally, the Deltavision also has a “Fast Acquisition” mode that rapidly switches between channels and can be ideal for fast, sensitive multi-color Ca2+ imaging experiments to detect Ca2+ oscillation, though it should be noted that a confocal is capable of capturing the signaling from 2–4 fluorophores but at a slow speed. If a phase-contrast image is desired, we prefer to use the Deltavision’s differential interference contrast. While DIC is possible on the LSM710, it is possible that the Deltavision’s use of a more efficient charge-coupled device (CCD) versus a photomultiplier tube (PMT) on the confocal provides for a sharper, crisper image, as a CCD, generally speaking, has a higher quantum yield over a PMT.
ACKNOWLEDGMENTS
This work was partially funded by NIH grants AI096836, AI128356 and AI110027 to SNJM.
A.C. was supported by an NIH diversity supplement to AI128356. S.V. was partially supported by a fellowship of the Office of the Vice-president for Research, UGA. We would also like to thank Dr. Muthugapatti Kandasamy from the Biomedical Microscopy Core and Julie Nelson from the Cytometry Shared Resource Laboratory of the University of Georgia. Eric Dykes helped performed the westerns of the cells expressing GCAMP-mScarlet and Christina Moore made the videos used for Figure 4.
Literature
- [1].Endo T, Sethi KK, Piekarski G (1982). Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Experim Parasitol 53(2) 179–88. [DOI] [PubMed] [Google Scholar]
- [2].Berridge MJ, Bootman MD, Roderick HL (2003). Calcium signalling: dynamics, homeostasis and remodelling, Nat Rev Mol Cell Biol 4(7) 517–29. [DOI] [PubMed] [Google Scholar]
- [3].Grynkiewicz G, Poenie M, Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260(6) 3440–50. [PubMed] [Google Scholar]
- [4].McCombs JE, Palmer AE (2008). Measuring calcium dynamics in living cells with genetically encodable calcium indicators. Methods 46(3) 152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Meth 6(12) 875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tian L, Hires SA, Looger LL (2012). Imaging neuronal activity with genetically encoded calcium indicators. Cold Spring Harbor protocols (6) 647–56. [DOI] [PubMed] [Google Scholar]
- [7].Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE (2011). An expanded palette of genetically encoded Ca(2)(+) indicators. Science 333(6051) 1888–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schuler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, Kugler S, Lagnado L, Hegemann P, Gottschalk A, Schreiter ER, Looger LL (2013). Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Suzuki J, Kanemaru K, Iino M (2016). Genetically Encoded Fluorescent Indicators for Organellar Calcium Imaging. Biophys J 111(6) (2016) 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499(7458) 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Deo C, Lavis LD (2018). Synthetic and genetically encoded fluorescent neural activity indicators. Curr Opin Neurobiol 50 101–108. [DOI] [PubMed] [Google Scholar]
- [12].Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, Patel R, Macklin JJ, Bargmann CI, Ahrens MB, Schreiter ER, Jayaraman V, Looger LL, Svoboda K, Kim DS. (2016) Sensitive red protein calcium indicators for imaging neural activity. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bassett JJ, Monteith GR (2017). Genetically Encoded Calcium Indicators as Probes to Assess the Role of Calcium Channels in Disease and for High-Throughput Drug Discovery Adv Pharmacol, Elsevier, pp. 141–171. [DOI] [PubMed] [Google Scholar]
- [14].Borges-Pereira L, Budu A, McKnight CA, Moore CA, Vella SA, Hortua Triana MA, Liu J, Garcia CR, Pace DA, Moreno SN (2015). Calcium Signaling throughout the Toxoplasma gondii Lytic Cycle: A STUDY USING GENETICALLY ENCODED CALCIUM INDICATORS. J Biol Chem 290(45) 26914–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sidik SM, Hortua Triana MA, Paul AS, El Bakkouri M, Hackett CG, Tran F, Westwood NJ, Hui R, Zuercher WJ, Duraisingh MT, Moreno SN, Lourido S (2016). Using a Genetically Encoded Sensor to Identify Inhibitors of Toxoplasma gondii Ca2+ Signaling. J Biol Chem 291(18) 9566–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Striepen B, He CY, Matrajt M, Soldati D, Roos DS (1998). Expression, selection, and organellar targeting of the green fluorescent protein in Toxoplasma gondii. Mol Biochem Parasitol 92(2) 325–38. [DOI] [PubMed] [Google Scholar]
- [17].Brooks CF, Johnsen H, van Dooren GG, Muthalagi M, Lin SS, Bohne W, Fischer K, Striepen B (2010). The Toxoplasma apicoplast phosphate translocator links cytosolic and apicoplast metabolism and is essential for parasite survival. Cell Host Microbe 7(1) 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kafsack BF, Pena JD, Coppens I, Ravindran S, Boothroyd JC, Carruthers VB (2009). Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 323(5913) 530–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brown KM, Lourido S, Sibley LD (2016). Serum Albumin Stimulates Protein Kinase G-dependent Microneme Secretion in Toxoplasma gondii. J Biol Chem 291(18) (2016) 9554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stewart RJ, Whitehead L, Nijagal B, Sleebs BE, Lessene G, McConville MJ, Rogers KL, Tonkin CJ (2017). Analysis of Ca(2)(+) mediated signaling regulating Toxoplasma infectivity reveals complex relationships between key molecules. Cell Microbiol 19(4). [DOI] [PubMed] [Google Scholar]
- [21].Brydges SD, Carruthers VB (2003). Mutation of an unusual mitochondrial targeting sequence of SODB2 produces multiple targeting fates in Toxoplasma gondii. J Cell Sci 116(Pt 22) 4675–85. [DOI] [PubMed] [Google Scholar]
- [22].Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SR, Usachev YM, Taylor CW, Campbell RE (2014). Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem J 464(1) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pino P, Foth BJ, Kwok LY, Sheiner L, Schepers R, Soldati T, Soldati-Favre D (2007). Dual targeting of antioxidant and metabolic enzymes to the mitochondrion and the apicoplast of Toxoplasma gondii. PLoS Pathog 3(8) e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van Dooren GG, Tomova C, Agrawal S, Humbel BM, Striepen B (2008). Toxoplasma gondii Tic20 is essential for apicoplast protein import. Proc Nat Acad Sci, USA 105(36) 13574–13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wagner JC, Goldfless SJ, Ganesan SM, Lee MC, Fidock DA, Niles JC (2013). An integrated strategy for efficient vector construction and multi-gene expression in Plasmodium falciparum. Malaria J 12(1) 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Szymczak-Workman AL, Vignali KM, Vignali DA (2012). Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb Protoc 2012(2) 199–204. [DOI] [PubMed] [Google Scholar]
- [27].Jakot D, Meisner M, Sheiner L, Soldati-Favre D, Striepen B (2014). Genetic Manipulation of Toxoplasma gondii. Second ed., Academic Press. Elsevier. [Google Scholar]
- [28].Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, Cowman AF, Besra GS, Roos DS, McFadden GI (1998). Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Nat Acad Sci, USA 95(21) 12352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Harb OS, Chatterjee B, Fraunholz MJ, Crawford MJ, Nishi M, Roos DS (2004). Multiple functionally redundant signals mediate targeting to the apicoplast in the apicomplexan parasite Toxoplasma gondii. Euk Cell 3(3) 663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, Gadella TW Jr. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14(1) 53–56. [DOI] [PubMed] [Google Scholar]
- [31].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7) 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Santi PA (2011). Light sheet fluorescence microscopy: a review. J Histochem Cytochem 59(2) 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Williams MJ, Alonso H, Enciso M, Egarter S, Sheiner L, Meissner M, Striepen B, Smith BJ, Tonkin CJ (2015).Two Essential Light Chains Regulate the MyoA Lever Arm To Promote Toxoplasma Gliding Motility. MBio 6(5) e00845–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fazli MS, Vella SA, Moreno SN, Quinn S (2017). Computational motility tracking of calcium dynamics in Toxoplasma gondii. arXiv preprint arXiv:1708.01871 [Google Scholar]
- [35].Fazli MS, Vella SA, Moreno SN, Quinn S (2018). Unsupervised Discovery of Toxoplasma gondii Motility Phenotypes. arXiv preprint arXiv:1801.02591. [Google Scholar]
- [36].Chin D, Means AR (2000). Calmodulin: a prototypical calcium sensor. Trends Cell Biol 10(8) 322–8. [DOI] [PubMed] [Google Scholar]
- [37].Yang Y, Liu N, He Y, Liu Y, Ge L, Zou L, Song S, Xiong W, Liu X (2018). Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat Commun 9(1) 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nebl T, Prieto JH, Kapp E, Smith BJ, Williams MJ, Yates JR 3rd, Cowman AF, Tonkin CJ (2011). Quantitative in vivo analyses reveal calcium-dependent phosphorylation sites and identifies a novel component of the Toxoplasma invasion motor complex, PLoS Path 7(9) e1002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC, Lourido S (2016). A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166(6) (2016) 1423–1435 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cho JH, Swanson CJ, Chen J, Li A, Lippert LG, Boye SE, Rose K, Sivaramakrishnan S, Chuong CM, Chow RH (2017). The GCaMP-R Family of Genetically Encoded Ratiometric Calcium Indicators. ACS Chem Biol 12(4) 1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park JG, Palmer AE (2015). Measuring the in situ Kd of a genetically encoded Ca2+ sensor. Cold Spring Harb Protoc 2015(1) pdb prot076554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lin MZ, Schnitzer MJ (2016). Genetically encoded indicators of neuronal activity. Nat Neurosci 19(9) 1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K (2004). Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE 2004(219) pl5. [DOI] [PubMed] [Google Scholar]
- [44].Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32(40) 13819–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kuchipudi A, Arroyo-Olarte RD, Hoffmann F, Brinkmann V, Gupta N (2016). Optogenetic monitoring identifies phosphatidylthreonine-regulated calcium homeostasis in Toxoplasma gondii. Microb Cell 3(5) (2016) 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]