

Abstract
Neuromuscular circuits (NMCs) are vital for voluntary movement, and effective models of NMCs are needed to understand the pathogenesis of, as well as to identify effective treatments for, multiple diseases, including Duchenne’s muscular dystrophy and amyotrophic lateral sclerosis. Microfluidics are ideal for recapitulating the central and peripheral compartments of NMCs, but myotubes often detach before functional NMCs are formed. In addition, microfluidic systems are often limited to a single experimental unit, which significantly limits their application in disease modeling and drug discovery. Here, we developed a microfluidic platform (MFP) containing over 100 experimental units, making it suitable for medium-throughput applications. To overcome detachment, we incorporated a reactive polymer surface allowing customization of the environment to culture different cell types. Using this approach, we identified conditions that enable long-term co-culture of human motor neurons and myotubes differentiated from human induced pluripotent stem cells inside our MFP. Optogenetics demonstrated the formation of functional NMCs. Furthermore, we developed a novel application of the rabies tracing assay to efficiently identify NMCs in our MFP. Therefore, our MFP enables large-scale generation and quantification of functional NMCs for disease modeling and pharmacological drug targeting.
Keywords: Microfluidics, poly(ethylene-alt-maleic anhydride), Neuromuscular circuit, Skeletal muscle, Rabies viral tracing, Motor unit
1. Introduction
Neuromuscular circuits (NMCs), consisting of motor neurons (MNs) innervating skeletal muscles, are critical for voluntary movement. Degeneration of the NMC plays a critical role in several disorders, including amyotrophic lateral sclerosis, spinal muscular atrophy, and Duchenne muscular dystrophy. It is thought, moreover, that the deteriorating connection between MNs and myotubes is one of the earliest stages of amyotrophic lateral sclerosis, suggesting that therapeutics focusing on protecting MNs alone might not prevent motor dysfunction in patients [1]. Instead, drugs must also preserve NMCs in order to be effective. Additionally, NMCs are used by pathogens, such as rabies virus, to invade the central nervous system (CNS). Once in the CNS, these pathogens cause acute flaccid paralysis and death and effective drugs are needed to prevent viral spreading. Therefore, models of human NMCs are critical for the development of drugs for a variety of disorders.
Although transgenic mouse models have been widely used to study diseases involving NMCs [2–5], there are important species-specific differences, and drugs identified using these models have often failed in clinical trials. In comparison to animal models, cell culture has the advantage of being faster, more cost-effective and reproducible. However, cell culture often relies on transformed cell lines that do not accurately model NMCs. Induced pluripotent stem cells (iPSCs) are ideal tools for generating functional skeletal muscle and MNs. Through reprogramming, iPSCs can be obtained from a patient with a specific phenotype and genotype to recapitulate disease pathogenesis. Thus, in principle, iPSC-derived models can be used to generate NMCs, and two-dimensional (2D) in vitro culture models have been developed to study human iPSC-derived NMCs [6–8].
Microfluidic systems are well suited for generating models of human NMCs as they have the potential to physically compartmentalize different cell types. Taylor et al. previously proposed a polydimethylsiloxane (PDMS)-based microfluidic separation chamber, which enables the investigation of axonal biology [9]. A similar concept was adapted for studying iPSC-derived NMCs [10]. Recently, a compartmentalized microfluidic system was used to establish a physiological model of the NMC using iPSC-derived MN spheroids and three-dimensional (3D) skeletal muscle bundles [11]. However, skeletal myotubes detach from the glass surface after approximately one week [12–15], which is not enough time to form functional NMCs. Overcoming myotube detachment is one of the most important challenges in being able to generate 2D microfluidic models of NMCs. In addition, current microfluidic models of the NMCs are often limited to one experimental unit at a time, and the assays used to evaluate NMC function are complicated and laborious, considerably restricting their application in disease modeling and drug discovery [16]. For example, recent reports used live cell calcium imaging and muscle contraction to assess the function of NMCs in microfluidic models [11,17], a procedure that is extremely cumbersome even for small-scale studies.
Here, we designed and manufactured a novel microfluidic platform (MFP) for modeling human NMCs. Each experimental unit contains a CNS and peripheral chamber connected by micro-channels for MN axons, and each MFP contains over 100 individual experimental units, making it suitable for medium-throughput applications, such as disease modeling and drug discovery. By introducing a thin film of the reactive polymer poly(ethylene-alt-maleic anhydride) (PEMA) inside the MFP, we were able to create a customized micro-environment enabling cultivation of iPSC-derived MNs and skeletal myotubes for at least 3 weeks, resulting in the formation of NMCs. Optogenetics was used to demonstrate that functional NMCs were formed in our MFP. Furthermore, we showed the first proof-of-principle for using monosynaptic rabies tracing to detect NMCs in our MFP. Therefore, our MFP and rabies tracing system make it possible to model human NMCs for medium-throughput applications.
2. Materials and methods
2.1. Master fabrication
The master used for the fabrication of the microfluidic platforms includes structures with different heights, exactly 100 lines with a height of 3 μm, width of 6 μm and length of 900 μm connecting two squared pillars with a height of 400 μm and side of 3.5 mm.
The master was produced by a multi photolithography process using photoresists with different viscosity to create two layers with different heights. The first layer of SU8 3025 diluted in thinner (1:1 w/w) was spin coated at 6000 rpm for 30 s on a 4” silicon wafer and then exposed to UV light (200 mJ/cm2) through a chromium mask containing both the chambers and channels geometry. After post baking, a second layer of SU8 2150 was spin coated on top of the first layer at 1000 rpm for 30 s. By using a mask aligner (EVG 620 UV-NIL and μCP tools) the structures on the first layer were aligned with a photo mask containing only the chambers geometry and exposed to UV light (400 mJ/cm2). After baking, the photoresist was developed in Mr600 Developer for 10 min.
2.2. PDMS replica molding
The structures from the master were replicated on PDMS by soft lithography process. The base (Sylgard 184) and curing agent were mixed in ratio 10:1 (w/w), degassed, poured on the master surface and polymerized in an oven at 75 °C for 1.5 h. The cured PDMS replica was peeled off the master surface and the features corresponding to wells were punched out with a 3.5 mm biopsy punch.
2.3. PLO coating and PLO-PDMS device
In order to achieve the PLO coating on glass coverslip, first it was cleaned with ethanol and acetone by sonication. The cleaned glass coverslip was incubated in 30% PLO commercial stock (Sigma P4957, in PBS) at 37 °C in a 5% CO2 incubator overnight. After the incubation, the coverslip was washed with sterile water and was dried and sterilized under UV for 20 min. The punched PDMS replica was cleaned with water followed by ethanol under sonication and then air-dried. It was incubated in DMEM/F-12 and Neurobasal medium (1:1) supplemented with 100 μg/ml penicillin-streptomycin (Biochrom) and 2 mM l-gluta-mine (Biochrom) (PSG), N2 and B27 supplement (N2B27, Thermo Fisher) at 37 °C in a 5% CO2 incubator for two days. After the incubation, the PDMS replicas were washed under sonication with sterile water and dried and sterilized under UV for 20 min. The dried PDMS replica was placed and gently pressed on the PLO-coated glass cover-slip. Subsequently, Laminin (5 μg/ml, LN521, biolamina) (PLO-Laminin), was applied inside the chambers. The prepared assembly was placed under desiccation to remove air bubbles from the micro-channels. Afterward, the assembly was incubated at 37 °C for 2 h or at room temperature overnight.
2.4. PEMA coating
In order to coat glass cover slips with PEMA, they were first cleaned using ultrasonication for 30 min in milliQ H20 and ethanol (Fisher Chemicals). Subsequently, cover slips were cleaned with RCA solution (volume ratio = milliQ H20: H202 (35%, AppliChem): NH3 (28–30 wt%, Acros Organics) = 5 : 1: 1) for 10 min at 70 °C, rinsed with milliQ and dried with N2-gas. Silanization was performed by incubating cover slips for 2 h in 20 mM 3-Aminopropyltriethoxysilane in isopropanol/water (9:1) (M = 221 g/mol). Subsequently, cover slips were rinsed in isopropanol and dried with N2-gas. After further drying the cover slips at 120 °C for 1 h, they were spin-coated with PEMA copolymer solution (0.3 wt% PE-MSA (Sigma, M = 125,000) in acetone (Fisher Chemicals): Tetrahydrofurane (Acros) = 1 : 2, filtered) for 30 s at 4000 rpm (1500 rpm/s). The spin-coated slides were incubated for 2 h at 120 °C. To remove excess polymer, slides were immersed for 15 min, rinsed using acetone and dried at 120 °C.
2.5. PEMA-PDMS device bonding
The punched PDMS replica (with structures facing up) was treated with oxygen plasma (200 W, 30 s) and immediately used for silanization process by APTES under desiccation for 1 h. Meanwhile, the PEMA-coated glass coverslip was annealed at 120 °C on a hot-plate. The silanized PDMS replica was placed (with structures facing down) on an annealed PEMA-coated coverslip and pressed gently. The assembly was allowed to bond at 120 °C on a hot-plate for 2 h with approximately 650 g of weight on top of the assembly. Afterward, the PEMA-PDMS bonded device was treated with air plasma (50 W, 120 s) to render the micro-channels hydrophilic in order to facilitate the filling of the channels with cyclo(Arg-Ala-Asp-D-Tyr-Lys) (cRGD) (Peptides International, 50 μg/ml) in borat buffer (pH = 8) (PEMA-cRGD Laminin) or BME (biotechne, 12–18 μg/ml) (PEMA-BME). PEMA-BME MFPs were incubated at room temperature overnight. PEMA-cRGD-Laminin MFPs were washed after 2 h incubation at room temperature and incubated with Laminin overnight. Laminin and BME were washed two times with PBS before plating the cells.
2.6. Preparation of the glass cover slips
Glass cover slips were cleaned in ethanol, washed two times with sterile water and incubated under UV for 20 min. Afterward, they were incubated at room temperature over night with Laminin (Laminin) or BME (BME). Glass cover slips with PLO functionalization were coated with PLO as described above, incubated at room temperature over night with Laminin (PLO-Laminin), BME (PLO-BME) or PBS (PLO). PEMA glass cover slips were prepared as described above. After heat activation at 120 °C in a dry-oven, PEMA slides were cooled and subsequently incubated with Laminin (PEMA-Laminin) or BME (PEMA-BME) overnight at room temperature, or with cRGD for 2 h at room temperature. cRGD was washed off after incubation using PBS. PEMA-cRGD-Laminin slides were then incubated with Laminin overnight at room temperature. Glass cover slips were applied inside a 12-well culture plate before seeding the cells.
2.7. Differentiation of motor neurons
iPSCs used in this project were previously generated and characterized [18]. For the derivation of MNPs, iPSCs were cultured on a 6-well plate until they reached 80% confluency. The cells were incubated with collagenase IV at 37 °C. Pieces of colonies were collected by sedimentation and resuspended in hESC medium (KO-DMEM supplemented with 20% serum replacement, PSG, 1% non-essential amino acids and 0.1 mM β-mercaptoethanol) supplemented with 200 μM ascorbic acid (AA, Sigma), 3 μM CHIR99021 (CHIR, Axon), 0.5 μM dorsomorphin (Selleck Chem), 10 μM SB-431542 (SB, Biomol) and 5 μM Y-27632 dihydrochloride (Y, Abcam) and plated in a 10 cm petri dish. After four days, medium was replaced with N2B27 supplemented with 200 μM AA, 3 μM CHIR, 0.5 μM purmorphamine (PMA, Cayman), 0.1 μM retinoic acid (RA, Sigma), 2 μM SB (Cayman), 2 μM DMH-1 (Tocris) and maintained for another two days. The embryoid bodiess formed in the suspension culture were disaggregated and plated on Matrigel (Corning) coated 12-well dishes. After four to six days, medium was replaced by motor neuron precursor cell medium (MNP medium) which additionally contained 0.5 mM valproic acid (VPA, Cayman). Motor neuron precursor cells (MNPs) were plated at a density of 3 × 105 cells on a 12-well plate and cultured in N2B27 supplemented with 200 μM AA, 1 μM RA, 2 pg/μl glia-derived growth factor (GD, Peptrotech), 1 pg/μl brain-derived growth factor (BD, Peprotech) and 0.5 μM smoothened agonist (Biomol) for six days. Afterward, medium was exchanged to maturation medium (MAT) consisting of N2B27 supplemented with 200 μM AA, 0.1 mM dibutyryl cyclic-AMP sodium salt (Sigma), 1.5 pg/μl transforming growth factor beta 3 (Peprotech), 4 pg/μl GD, 2 pg/μl BD and 5 pg/μl activin A (aA, eBioscience). After 48 h, pre-differentiated MNs were replated inside the MFP (6 × 104 cells) or onto 24-mm glass cover slips (5 × 105 cells). MNs were matured by changing MAT without aA and with 0.1 μM γ-secretase inhibitor (compound E, Merck) every second day.
2.8. Differentiation of skeletal myotubes
iPSC-derived myoblasts (MBs) were obtained from the laboratory of April Pyle (UCLA, USA), using a protocol previously described [19]. In brief, iPSCs (S21 line) were directed to differentiate for 50 days and myogenic progenitor cells enriched by flow cytometry using cell surface markers HNK1-ERBB3+NGFR+. iPSC MBs were then maintained in SkBM-2 supplemented with 20 ng/ml human fibroblast growth factor 2 (Peprotech). For differentiation into skeletal myotubes, MBs were plated into the MFP (1 × 104 cells) or onto glass cover slips inside a 12-well plate. After 1 day, they achieved 70–80% confluency and were then cultured in DMEM-F12 (Thermo Fisher) supplemented with N2 supplement (1:100, Thermo Fisher), ITS-G (Thermo Fisher), insulin-like growth factor 10 ng/ml (Peptrotech), 10 μM SB and PSG. Six days later, the medium was supplemented with 10% FBS (Thermo Fisher), 100 ng/ml recombinant rat agrin (R&D), 4 pg/μl GD (PeproTech).
2.9. Immunocytochemistry
Cells were fixed for 20 min at room temperature in 4% paraformaldehyde in PBS. Permeabilization and blocking of nonspecific epitopes was performed simultaneously using 0.1% Triton X-100, 1% BSA and 10% FBS in PBS for 45 min. Subsequently, the primary antibodies (mouse anti tubulin beta 3 (TUBB3) (1:1000, Covance), monoclonal Anti-Myosin (MY-32) (1:500, Sigma), microtubule-associated protein 2 (MAP2) (1:5000, Abcam), neurofilament heavy polypeptide (SMI-32) (1:500, Millipore), choline Acetyltransferase (CHAT) (1:400, Millipore), Islet-1 (1:1500, Abcam), titin (T11) (1:1000, Bio-Techne), sarcomeric alpha Actinin antibody (EA-53) (1:200, GeneTex) were applied overnight at 4 °C in 0.1% BSA in PBS. The next day, the cells were washed with 0.1% BSA in PBS and incubated with the secondary antibody for 1 h at room temperature. Conjugated bungarotoxin (α-Bungarotoxin, CF®640R, biotium, 1:500) was included in the secondary antibody incubation step. Finally, cells were washed three times with 0.1% BSA in PBS-T (0.005% Tween-20), including Hoechst counter-staining for nuclei in the second washing step. Cells were imaged either with a Zeiss ApoTome or a laser scanning confocal microscope (Zeiss LSM780/FCS) and, if necessary, pictures from individual channels were merged using Fiji.
2.10. Axonal outgrowth
MNs were seeded inside one compartment of the MFP and channel exits of the opposed compartment were captured every 12 h using the IncuCyte live cell imaging system. Neurite outgrowth was quantified using the neurite analysis assay on phase contrast images provided by the IncuCyte NeuroTrack Software Module or by counting the number of neurites per frame. Time of axonal outgrowth was measured by monitoring microchannel exits of the peripheral compartment daily and noting down at which day the first axons grew through the micro-channels.
2.11. RNA isolation and qPCR analysis
RNA samples were harvested using the NucleoSpin RNA kit (Macherny Nagel) according to the manufacturer’s instructions. Complementary DNA was produced by reverse transcription using MMLV RT (Promega). Quantitative polymerase chain reaction (qPCR) was performed using iTaq Universal SYBR Green mix (Biorad) and primer at a concentration of 2.5 μM (MYH-8 fw ACATTACTGGCTGGCTGGAC, hMYH-8 rev TTCGCGCTGCTATCTGCTTC; MYOG fw CCAGC GAATGCAGCTCTCAC, MYG rev GCAGATGATCCCCTGGGTTG; ACTA1 fw AAGATCAAGATCATCGCCCCG, ACTA1 rev CCTCGTCGTACTCCTG CTTG; GAPDH fw CTCCTGTTCGACAGTCAGCC, GAPDH rev ACCAAA TCCGTTGACTCCGAC; HPRT1 fw CCCTGGCGTCGTGATTAGTG, HPRT1 rev TCGAGCAAGACGTTCAGTCC) using the Lightcycler 480 (Roche). Cycling conditions were set as follows: denaturation for 10 min at 95 °C, 40 cycles alternating 15 s at 95 °C and 60 s at 60 °C of 15 s. Crossing points were determined by LightCycler 480 software. Relative expression levels were quantified using the 2−2Δ method and normalized to housekeeping genes glycerinaldehyd-3-phosphat-dehydrogenase (GAPDH) and Hypoxanthine-Phosphoribosyl-Transferase 1 (HPRT1) and to the undifferentiated MBs.
2.12. Optogenetics
Optogenetic competent skeletal myotubes and MNs were derived by infecting them with pLV- EF1α-ChR2-EYFP (plasmid backbone pLV_TRET_hNgn2_UBC_Puro was a gift from Ron Weiss (Addgene plasmid # 61,474) [20] and the EF1α promoter as well as ChR2-EYFP were taken from pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA, a gift from Karl Deisseroth (Addgene plasmid # 20,298)) and pLenti-hSyn-CatCh-EYFP-UBC-puro (the plasmid backbone was a gift from Karl Deisseroth (Addgene plasmid # 26,775) [21], CatCh-EYFP [22], a kind gift of Ernst Bamberg and the UBC-puro cassette [20] was a gift from Ron Weiss (Addgene plasmid # 61,474)), respectively. After 10 days, cells were used for optogenetic stimulation. Myotubes were stimulated using blue light pulses (475 nm) of 20 ms at a frequency of 0.2 Hz. To detect functional NMCs, MNs were stimulated using blue light pulses (475 nm) of 200 ms at a frequency of 0.2 Hz. Cells were video captured while recording using a Zeiss Axiovert 200 4D-System. Videos were processed using Metamorph and Fiji and contraction was quantified by change in pixel as a result of muscle movement.
2.13. Generation of the myoblast starter cells
The construct for the generation of the starter population was purchased from Addgene (number 30,195) and the human Synapsin promotor was replaced by the human Elongation factor-1 alpha (hEF1α) promotor. The plasmid was transfected into HEK293T cells together with VSVG and Gag/Pol vector (provided by the Calegari group, CRTD, Germany). Supernatant was harvested after 48 and 96 h and concentrated by ultracentrifugation at 32,500 rpm for 3 h 30 min and resuspended in PBS. MBs were infected and sorted against green fluorescent protein (GFP) using the BD FACS AriaIII.
2.14. RABVΔG-mCherry production and infection of myoblast starter cells
ENVA pseudotyped RABVΔG-mCherry was produced according to Osakada and Callaway [23] using the pSADdeltaG-mCherry vector (Addgene, 32,636). Production cell lines were provided by John Naughton (Salk Institute, USA). The protocol was stopped after step 60 and the virus was concentrated via ultracentrifugation only once at 32.500 rpm for 3 h 30 min. Tittering was performed using TVA-expressing HEK293T cells (provided by John Naughton at the Salk Institute, USA) as described [23]. Titers were 2–3×107 TU/ml and the myotube starter population was infected by adding 0.5 μl pseudotyped virus to the peripheral compartment.
2.15. Image analysis
To assess the adhesion time on glass cover slips with different substrates, MNs or myotubes were monitored daily using a cell culture microscope and the day of detachment was noted for each condition. Detachment experiments were stopped after 21 days of culturing and repeated three times. The adhesion time of the three replicates was averaged. Glass cover slips were immunostained with MY-32 and TUBB3. Images were captured and analyzed regarding cluster size, axonal growth and number of myotubes. Cluster size of MN cultures was determined by measuring the diameter of the MN clusters using ImageJ. Axonal growth was evaluated by measuring the percentage area of TUBB3 positive neurites using Fiji. Numbers of myotubes were generated by counting MY-32 positive myotubes per image.
2.16. Electrophysiology
Standard MEA chambers (60MEA200/30iR-Ti-gr, Multichannel Systems) were coated with Poly-d-lysine (PDL, 1 mg/ml stock, 50 μl per electrode area) and incubated overnight at 37 °C. The arrays were washed three times with sterile ddH2O and dried. Laminin (5 μg/ml, LN521, biolamina) was added to the electrode area (50 μl) and incubated overnight at room temperature. After eight days differentiation, MNs were dissociated with Accutase seeded on each MEA chip (200,000 cells per MEA in total). The cells were cultured in conditional medium including 75% N2B27 supplemented with 200 μM AA, 0.1 mM dibutyryl cyclic-AMP sodium salt (Sigma), 1.5 pg/μl transforming growth factor beta 3 (Peprotech), 4 pg/μl GD, 2 pg/μl BD and 5 pg/μl activin A (aA, eBioscience) and 25% astrocyte medium (DMEM plus N2 Supplement, 10% One Shot™ Fetal Bovine Serum and 1% penicillin-streptomycin) collected from cultured rat primary cortical astrocytes. Half of the media was exchanged weekly with fresh media. Spontaneous neural network activities of motor neurons were recorded by using MEA1060-Inv-BC (sampling rate 25 K Hz) and software user interface (MC_Rack) provided by Micro Channel Systems (MCS). The spontaneous activities of the differentiated motor neural networks were collected 15 and 18 days after replating. To be sure about spontaneous action potentials generated from MNs, spontaneous electrical activities were evaluated in the presence of 1 μM TTX (abcam). Additionally, the neuronal action potentials were recorded after washing out TTX. Recorded data were replayed, and filtered (Butterworth 2nd order, high pass filter cut-off at 100 Hz) and timestamps of the action potential were detected by a negative threshold (−5 standard deviation of the peak-to-peak noise). The shape and timing of the action potential were also considered in this analysis. The definition of active electrodes in the analysis was the electrodes that recorded more than three action potentials per minute (0.05 Hz). The average spike frequency (action potentials per second) was measured. Action potential frequency differences under different conditions were statically tested for significance (Student’s t-test). For all analyses, p < 0.05 was considered as significant.
2.17. PEMA-MFP characterization
The bonding strength was tested by activating a pressure-driven flow inside the MFP micro-channels by using the Fluigent MFCS™-EX microfluidic controller. One vial containing dH2O was connected to one of the MFP chambers via FEP tubing (I.D. 250 μm, O.D. 1.5 mm, Postnova Analytics GmbH, Germany) and an in-line flow sensor was included between the vial and the MFP chamber (Fluigent Flow Unit M). The flow, through the tubing towards the MFP, was activated by pressurizing the liquid inside the vial via the microfluidic controller and the corresponding flow rate was measured through the flow sensor. The pressure was increased in magnitude of 10 mbar with a lag time of 5 min until it reached the critical pressure (Pc), at which delamination of PDMS from the PEMA-coated cover slip was observed under inverted microscope (Zeiss, Axio Observer. A1). The MFP was treated with air plasma (120 s, 50 W) to favor spontaneous capillary motion of the liquids inside the micro-channels. After plasma treatment, one of the MFP wells was filled, pipetting 40 μl of dH2O, and the penetration dynamics inside the micro-channels was recorded at 1000 fps by using an inverted microscope (Zeiss, Axio Observer. A1) equipped with a fast camera (Mikroton, EoSens CL 1362). The recorded video was analyzed by Fiji to reconstruct the penetration dynamics, measuring the length covered by the liquid, z, with time inside the MFP micro-channels. The experimental data reported in Fig. 6E were fitted by using the Lucas-Washburn equation (z = A∙(time)0.5), which is usually employed to describe the advancement motion of a fluid front in a capillary [24].
Fig. 6.
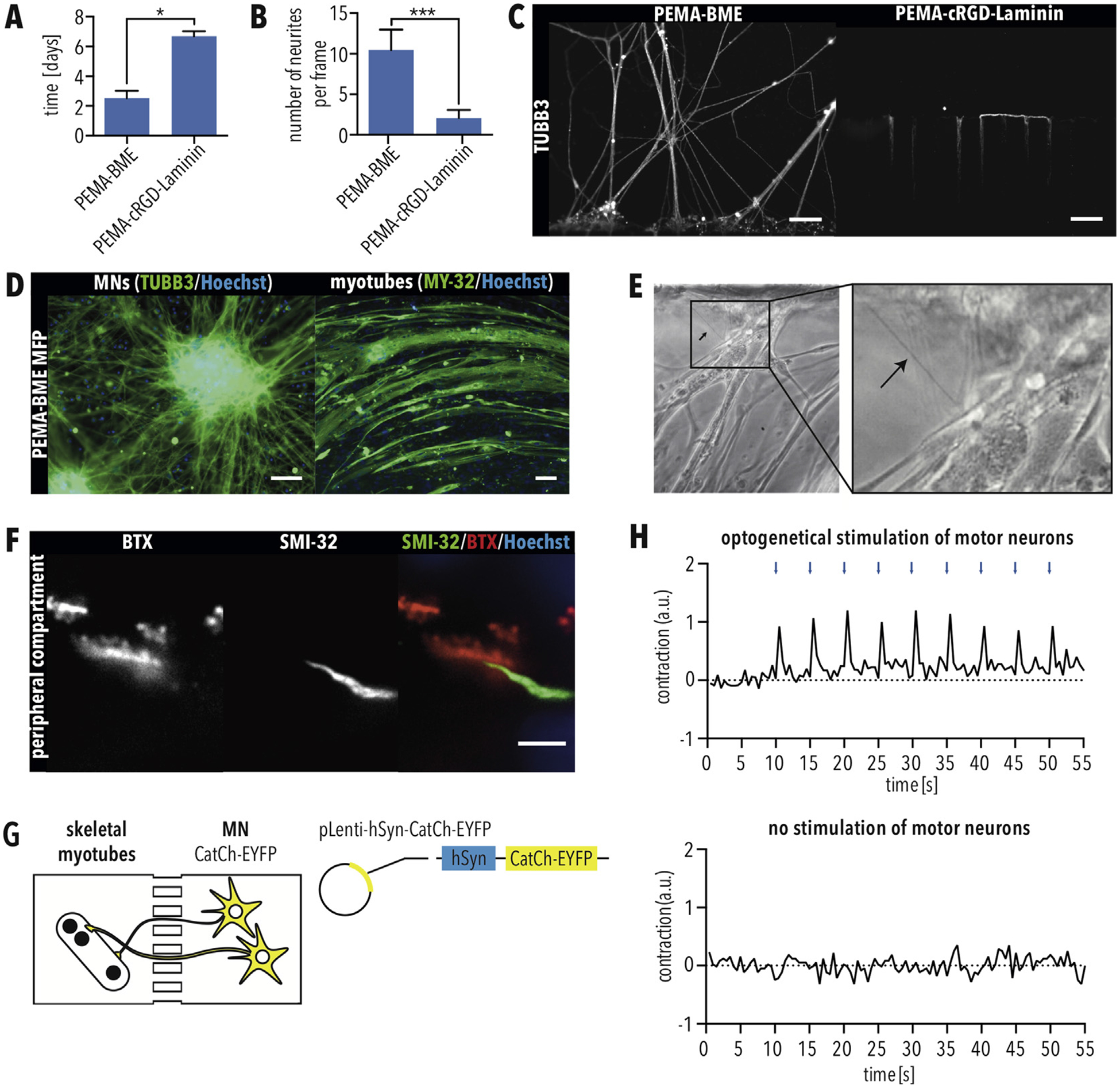
Formation of functional motor units using MFPs with PEMA-BME (A–B) Quantification of (A) days required for MN axons to grow through the micro-channels and (B) number of MN axons per image. Error bars indicate SEM. * and *** indicate p < 0.05 and 0.001, respectively, according to unpaired T-test with Welch’s correction. (C) Immunostaining for TUBB3 of MNs axons exiting the micro-channels when cultured as indicated. Scale bar = 50 μm. (D) Immunostaining of MNs and myotubes for TUBB3 and MY-32, respectively, when cultured in MFPs with PEMA-BME for 21 days. Scale bar = 50 μm. (E) Phase contrast image showing axons of MNs exiting micro-channels and attaching to myotubes in the peripheral compartment. (F) Immunostaining of neuromuscular junctions (NMJs) using the MN marker SMI-32 as well as fluorescently conjugated BTX, which labels acetylcholine receptors. Scale bar = 5 μm. (G) Schematic of optogenetic set up. MNs and myotubes were cultivated in the PEMA-BME MFP, and MNs were infected with a lentivirus carrying CatCh-EYFP under a Synapsin promotor (SynP). (H) 10 days after infection cultures were illuminated with 200 ms blue light (475 nm) pulses every 5 s (0.2 Hz). Contraction was quantified by change in pixel. Myotubes showed contraction in response to light pulses. Contraction was not recorded without stimulation. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3. Results
3.1. Differentiation of iPSCs into functional MNs and myotubes
To model the CNS component of the NMC, we derived MNs from iPSCs via expandable motor neuron precursors (MNPs) (Fig. 1A). MNs displayed expression of the neuronal markers, TUBB3 and MAP2 as well as the MN marker CHAT and SMI-32 (Fig. 1B). MNs differentiated into Islet-1-positive MNs with an efficiency of approximately 50% (Fig. 1B and C). Multielectrode array analysis demonstrated that MNs spontaneously fired action potentials (Fig. 1D and E), demonstrating that they were electrophysiologically functional.
Fig. 1.
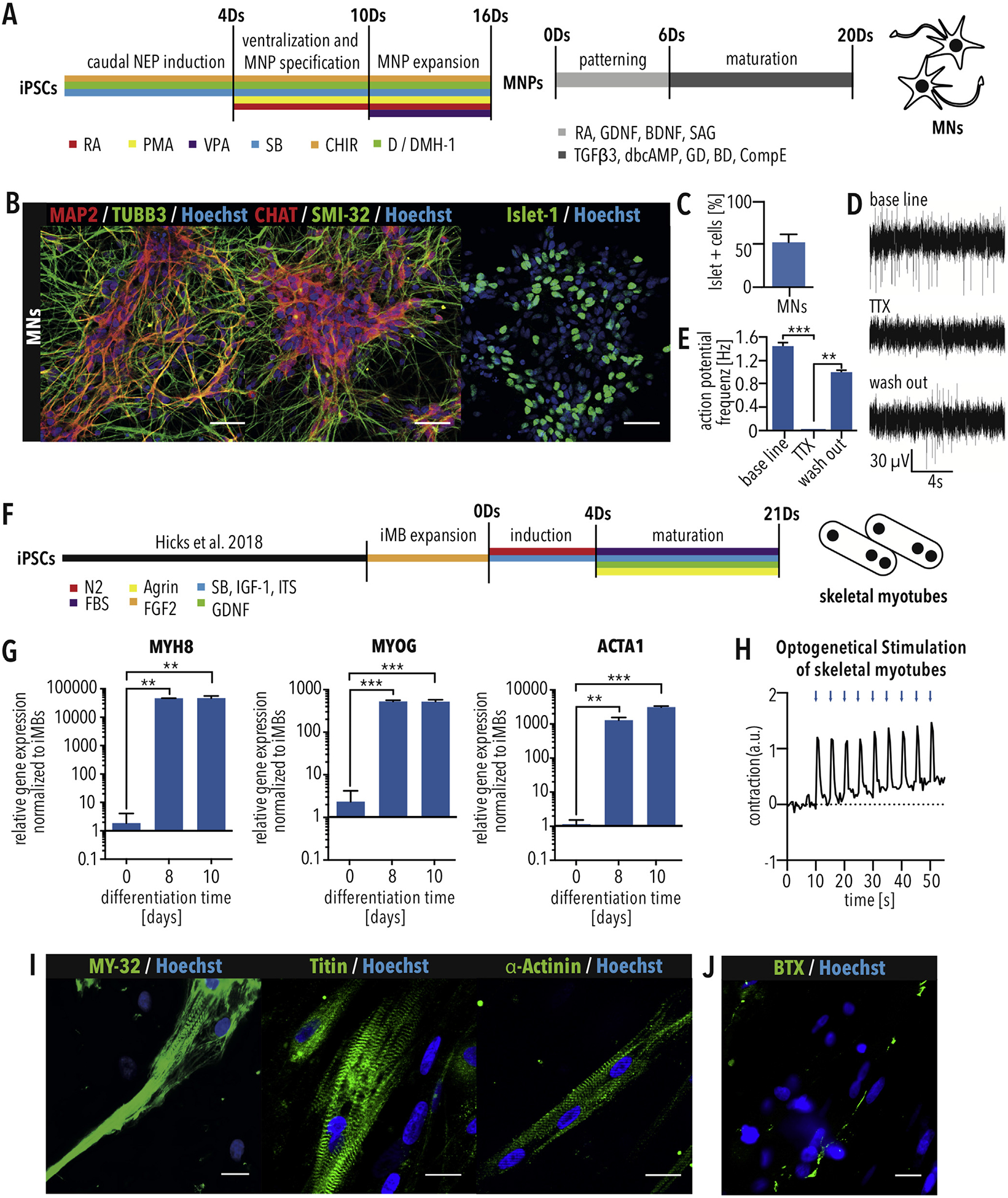
Derivation of MNs and skeletal myotubes from iPSCs (A) Schematic of MN differentiation protocol. MNPs = motor neuron progenitors. (B) Immunostaining of iPSC-derived MNs for the neuronal markers MAP2 and TUBB3 and the MN markers CHAT, SMI-32 and Islet-1. Scale bar = 50 μm. (C) Quantification of MN differentiation efficiency using Islet-1 immunostaining. n = 3. Error bars show SEM. (D) Multi-electrode array recording of MNs showing spontaneous activity (base line), which can be blocked using 1 μM tetrodotoxin (TTX) treatment and recovered by washing out (E) Quantification of action potential frequency shows significant effect of TTX, which proves electrical activity of neurons. (F) Schematic of skeletal myotube differentiation protocol. Bars indicated SEM. ** and *** indicate p < 0.01 and 0.001, respectively, according to t-test. (G) Quantitative RT-PCR showing increased expression of the indicated skeletal myotube markers. Bars indicated SEM. * and ** indicate p < 0.05 and 0.01, respectively, according to one-way ANOVA and Dunnett’s multiple comparisons test. (H) Myotube contraction after contraction optogenetic stimulation using ChR2-EYFP with 20 ms blue light pulses (475 nm) at 0.2 Hz. (I) Skeletal myotubes differentiated from myoblasts show skeletal muscle marker expression (MY-32, titin, α-actinin) and striation. Scale bar = 25 μm. (J) Skeletal myotubes display acetylcholine receptor clusters visualized with fluorescently conjugated α-bungarotoxin (BTX). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, skeletal myotubes were differentiated from iPSC-derived myoblast cells. After an initial induction phase, myotubes were matured in the presence of FBS, Agrin, and GDNF (Fig. 1F). After 8 days, quantitative RT-PCR showed that myotubes expressed the skeletal muscle markers MYH8, MYOG and ACTA1 (Fig. 1G). Culturing for 21 days resulted in myotubes that could be stimulated to contract using optogenetics (Fig. 1H). Immunostaining confirmed expression of fast myosin skeletal heavy chain (MY-32), titin and skeletal α-actinin, which showed patterns of striation (Fig. 1I), and α-Bungarotoxin (BTX) staining identified formation of dense acetylcholine receptor clusters (Fig. 1J). These results demonstrate the formation of terminal differentiated skeletal myotubes.
3.2. Designing a medium-throughput MFP
Since the NMC consists of two different compartments, the CNS and the peripheral skeletal muscle, we designed and manufactured an MFP with two compartments interconnected by 100 micro-channels (Fig. 2A). The micro-channels had lengths of 900 μm (Fig. 2B), which were shown to exclusively allow axonal growth into the separated compartment [9]. By plating MN somas in one compartment and myotubes in the other compartment, it is possible to generate a miniature NMC with MN somas in a compartment representing the CNS and the peripheral myotube compartment representing the periphery (Fig. 2A). The MFP is made up of 126 experimental units in a 384-well microtiter format (Fig. 2A), facilitating the usage of liquid handling devices and automated imaging systems. Each MFP was prepared by curing PDMS on a photoresist master, peeling it off, punching holes and attaching it against a glass cover slip (Fig. 2B).
Fig. 2.
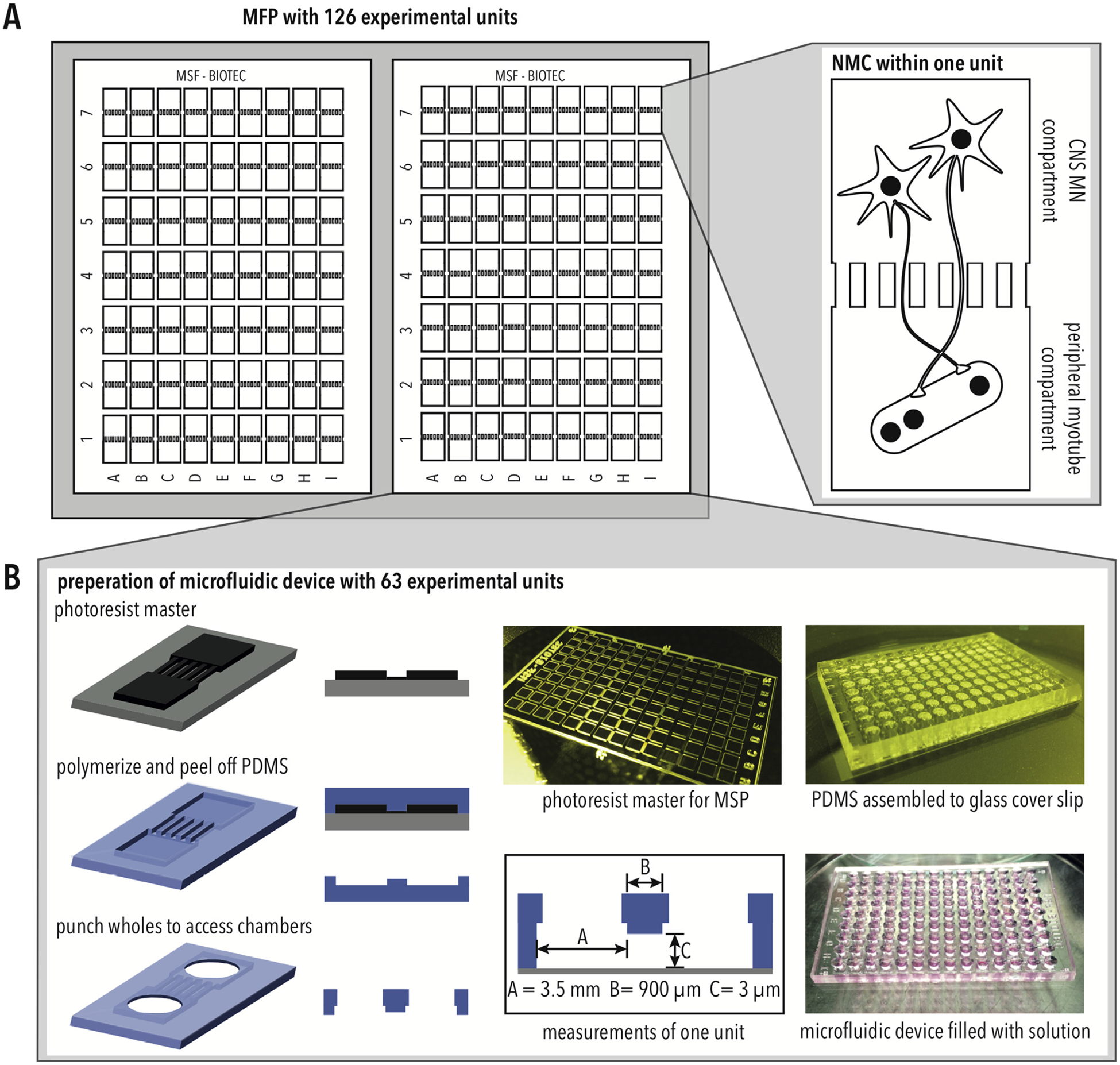
Microfluidic platform for modeling NMCs (A) Schematic of MFP design, including 126 experimental units each with two chambers. (B) The MFP is manufactured by polymerizing PDMS on a photoresist master, punching to access chambers, and assembled with a glass cover slip.
3.3. Coating with poly-l-ornithine (PLO)-Laminin is not sufficient for culturing iPSC-derived myotubes
To model the CNS component of an NMC, we assembled the MFP using a PLO-Laminin coated glass cover slip, and iPSC-derived MNs were applied to the CNS compartment (Fig. 3A). Axonal outgrowth was tracked inside the peripheral compartment. After 12 days, MFPs were immunostained for the neuronal-specific marker β3-tubulin (TUBB3), showing that MNs effectively attached inside the CNS compartment of the MFP (Fig. 3B). Furthermore, live-cell imaging demonstrated that axons extended into the peripheral compartment four days after plating the dissociated MNs into the MFP (Fig. 3C). Axonal growth was continuous for at least 10 days (Fig. 3C). Therefore, we could show that our PLO-Laminin coated MFP is sufficient for culturing MNs inside the CNS compartment and that these neurons can access the peripheral compartment.
Fig. 3.
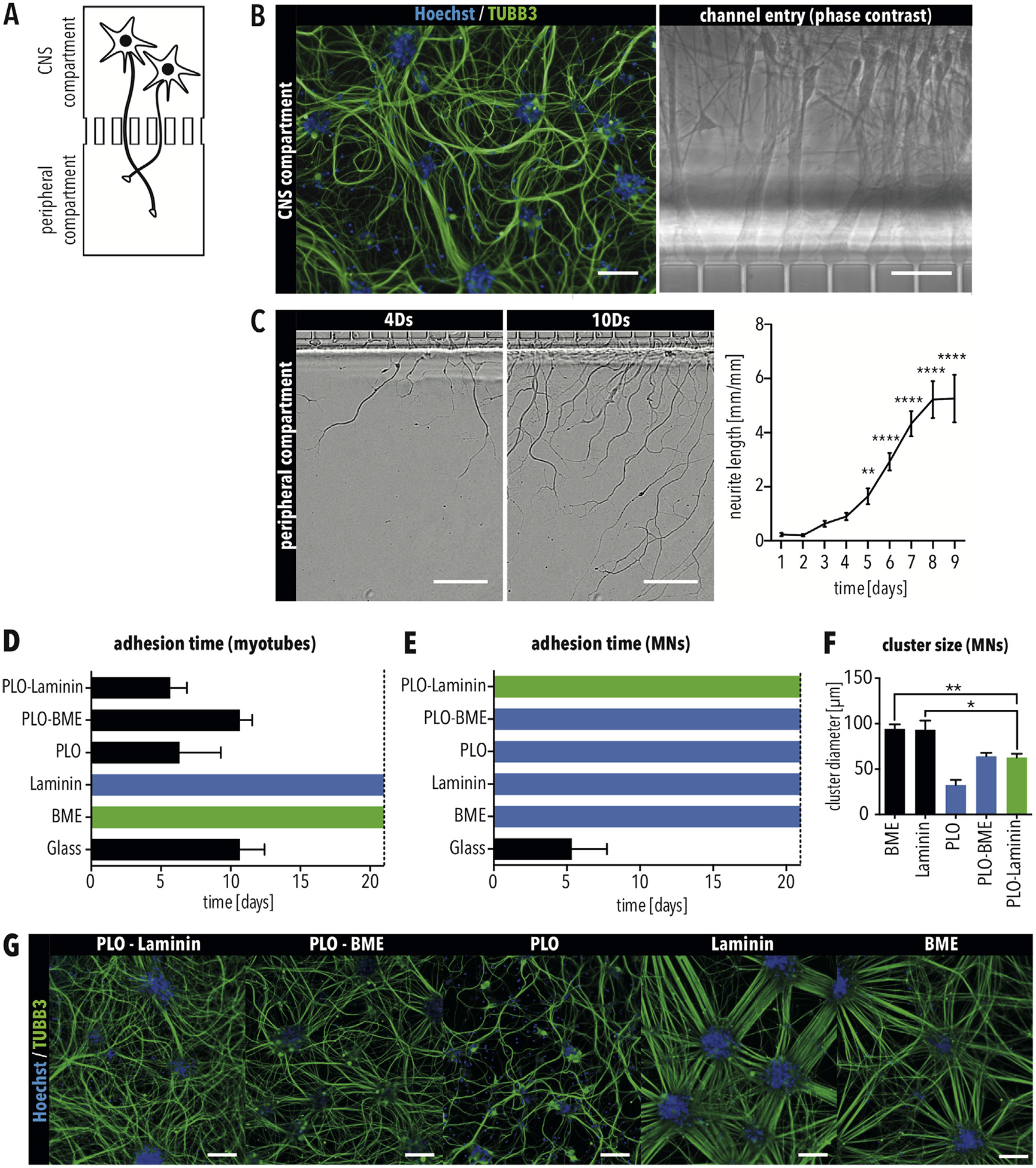
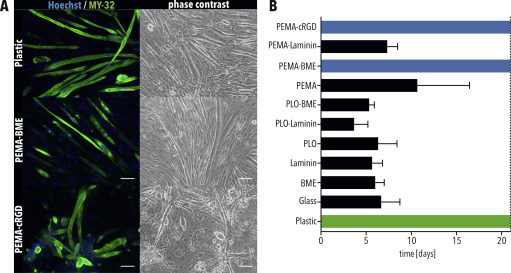
Motor neurons and skeletal myotubes have different adhesion requirements (A) Schematic of the CNS compartment inside one experimental unit of the MFP. (B) Immunostaining for TUBB3 of MNs inside the CNS compartment. Scale bar = 100 μm. Axons enter the micro-channels towards the peripheral compartment. Scale bar = 25 μm. (C) Axons of MNs exit the micro-channels into the peripheral compartment four days (4Ds) after plating in the CNS compartment. Scale bar = 50 μm. (D–E) Number of days that (D) skeletal myotubes and (E) MNs were attached on glass cover slips functionalized as indicated. (F) Size of clusters formed by MNs in MFPs using glass cover slips functionalized as indicated. Bars in all graphs indicate SEM. Green bars indicate the positive control condition. Black bars indicate coatings resulting in cell detachment. Blue bars indicate conditions selected for further testing. * and ** indicate p < 0.05 and 0.01, respectively, according to ordinary one-way ANOVA and Dunnett’s multiple comparisons test. (G) Immunostaining for TUBB3 of MNs in the CNS compartment using the indicated condition. Scale bar = 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To generate a model of an NMC, iPSC-derived myoblast cells were seeded and differentiated in the peripheral compartment. However, differentiating cells started detaching from the PLO-Laminin coating after only five days (Fig. 3D), which is not enough time for a co-culture to form an NMC [11,25].
As MB-derived myotubes are generally cultured on Matrigel-coated plastic dishes [19], we tested different surface coatings on glass cover slips to identify a coating compatible with culturing both MNs and myotubes. As positive controls, myotubes and MNs were cultured on glass cover slips coated with basal membrane extract (BME), a Matrigel replacement, and PLO-Laminin, respectively. As negative controls, cells were cultured on untreated glass cover slips. Unfortunately, myotubes detached from all conditions using PLO-functionalized glass (Fig. 3D). Although myotubes attached to BME as well as to Laminin alone (Fig. 3D), MNs did not (Fig. 3E). Instead, MNs showed prominent clustering (Fig. 3F and G). The cluster diameter of MNs cultured on BME and Laminin was significantly larger in comparison to the previously used PLO-Laminin coating (Fig. 3F), demonstrating that MNs prefer to attach to each other instead of the substrate [26]. Therefore, an alternative substrate is needed to culture both MNs and myotubes in our MFPs.
3.4. PEMA coated glass slides are suitable for culturing human MNs and myotubes
The properties of maleic anhydride copolymers are ideal for functionalizing glass surfaces for cell culture because they can be covalently bound to glass surfaces as well as amino-terminated molecules such as lysine or the N-terminus of proteins. For this reason, maleic anhydride copolymers have previously been used to create customized extra-cellular matrix (ECM) microenvironments [27–29]. We hypothesized that functionalization of the glass surface inside the MFP could be used for culturing MNs as well as myotubes.
PDMS presents a hydrophobic surface and wetting of the micro-channels requires a hydrophilic environment. Thus, more commonly used maleic anhydride copolymers, such as poly(maleic anhydride-alt-1-octadecene), were ruled out due to their hydrophobic behavior resulting from long alkyl chains. The short ethylene unit of PEMA renders the copolymer water soluble [27]. Therefore, PEMA functionalization was used to create MFPs compatible with culturing MNs and myotubes.
Since PEMA can reactively bind to a variety of proteins, we tested different substrates using both myotubes and MNs, including PEMA-BME, PEMA-Laminin, PEMA-cyclic RGD, and PEMA-cRGD-Laminin. As positive controls, myotubes and MNs were cultured on BME and PLO-Laminin, respectively. Untreated PEMA functionalized glass was used as a negative control. After 21 days of cultivation, MNs and myotubes adhered to all substrates except PEMA-cRGD and untreated PEMA glass slides (Fig. 4A–C and E). The number of myotubes on PEMA-BME, PEMA-Laminin, and PEMA-cRGD-Laminin were similar to BME, the positive control (Fig. 4C and F). Skeletal myotubes showed patterns of striation for all conditions, indicating maturation (Fig. 4D). However, when cultured on PEMA-Laminin slides, MNs formed significantly larger clusters compared to the standard coating, PLO-laminin, demonstrating that MNs adhere poorly to PEMA-Laminin (Fig. 4E and G). Immunostaining for TUBB3 showed that axonal growth was impaired on PEMA-cRGD coated slides compared to PLO-Laminin (Fig. 4H). Since axonal outgrowth is critical for MNs to innervate myotubes in the adjacent chamber, PEMA-cRGD was excluded from future use. PEMA-BME and PEMA-cRGD-Laminin gave the best results for MN attachment and axonal outgrowth. We also found that C2C12-derived myotubes attached when using PEMA-BME, PEMA-cRGD and PEMA-cRGD-Laminin functionalized glass, demonstrating the robustness of our approach (Fig. S1). Taken together, these results demonstrate that MNs as well as myotubes can be cultured on PEMA-BME and PEMA-cRGD-Laminin functionalized glass cover slips. Thus, PEMA-BME and PEMA-cRGDLaminin were selected for further development of our MFP.
Fig. 4.
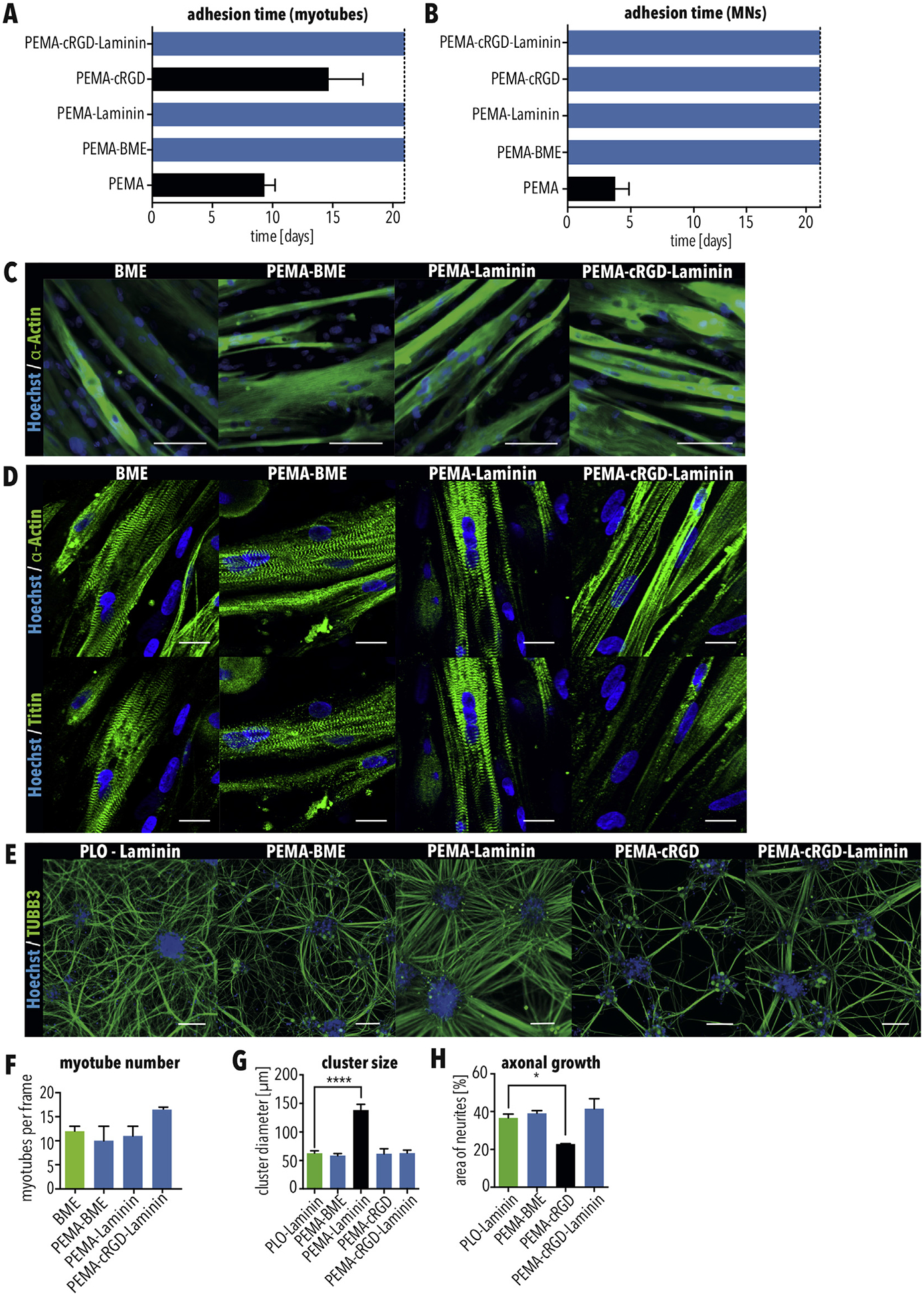
PEMA-BME and PEMA-cRGD-Laminin are suitable coatings for culturing MNs and skeletal myotubes (A–B) Quantification of how long (A) myotubes and (B) MNs were attached when cultured as indicated. (C–D) Immunostaining of (C) myotubes for MY-32 (MyHC) (scale bar = 100 μm), which marks skeletal muscle, and (D) titin and α-actinin (scale bar = 25 μm) which marks sarcomeres which were cultured on glass cover slips functionalized as indicated. (E) Immunostaining of MNs for TUBB3, which were cultured on glass cover slips functionalized as indicated. Scale bar = 100 μm. (F) Quantification of myotubes after 21 days of culture as indicated. (G–H) Quantification of (G) cluster size and (H) axonal growth of MNs when cultured as indicated. Bars in all graphs indicate SEM. Green bars indicate the positive control condition. Black bars indicate coatings resulting in cell detachment. Blue bars indicate conditions selected for further testing. * and **** indicate p < 0.05 and 0.0001, respectively, according to ordinary one-way ANOVA and Dunnett’s multiple comparisons test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. PEMA MFP device preparation using PDMS silanization and plasma treatment
Next, we aimed to incorporate the PEMA copolymer surface inside our MFP. At first, a reversible sealing based on the conformal contact of the PDMS replica [30] on a PEMA-coated cover slip was used. The conformal contact between the PDMS and PEMA-coated cover slip was not enough to confine cell and axon growth respectively inside the chambers and micro-channels. Cells were able to grow at the PDMS/PEMA interface delaminating the device with a consequent leakage of the culture medium (Fig. 5B, left).
Fig. 5.
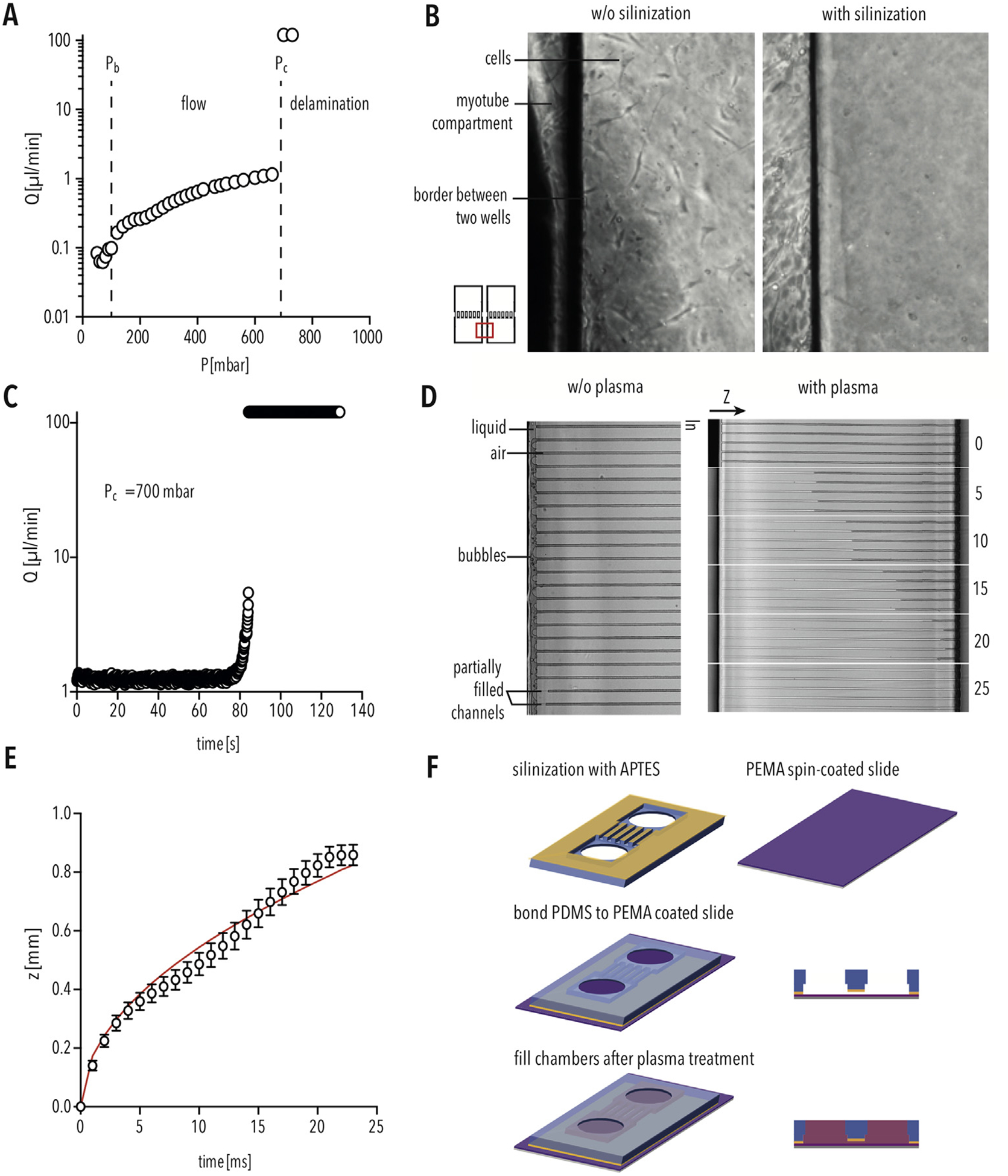
PEMA MFP device preparation using PDMS silanization and plasma treatment (A) Flowrate (μl/min) inside the PEMA-PDMS device corresponding to the applied pressure (mbar). Pb indicates capillarity back-pressure, the pressure required to fill the well. Pc indicates the critical pressure, the pressure at which the PDMS delaminates from the PEMA-coated cover slip. (B) Silanization of PDMS results in sufficient bonding to prevent cell growth between the compartments. (C) Flowrate (μl/min) inside the PEMA-PDMS device over the time period (s) at constant critical pressure (Pc) of 700 mbar. (D) Bright-field image of filling the micro-channels without (left) and with (right) air plasma treatment. (E) Covered length (z) for water penetration inside the air plasma treated micro-channels over a time period (ms). The solid red line is the best fit curve by z = A∙(time)0.5 [24]. Bars indicate SD. (F) Schematic of PEMA MFP preparation. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Hence, it was necessary to attain an irreversible seal between the covalent bonding of PEMA and PDMS. To enable it, primary amino groups are required at the PDMS surface. Modification of the PDMS surface using plasma treatment, followed by silanization was shown to create primary amino functional groups on the PDMS surface [31,32]. We silanized the PDMS device using APTES and heat activated the PEMA coated glass slides before assembly. In this way primary amino groups of the PDMS react with the anhydride groups of the PEMA co-polymer, forming imide bonds [32]. Bonding strength between silanized PDMS and PEMA was characterized by connecting the MFP to a microfluidic pressure controller in order to activate pressure-driven flow of dH2O inside the micro-channels. The pressure was varied between 50 and 730 mbar. Initially, a pressure of 50 mbar was applied to fill the chamber. For 50 mbar < P < 100 mbar, the flow rate did not vary and the liquid was not able to enter the micro-channels due to the capillarity backpressure (Pb). For Pb = 100 mbar < P < 700 mbar, the liquid started to move into the channels with a gradual increase of the flow rate from 0.2 μl/min up to 1.2 μl/min (Fig. 5A). For P = 700 mbar, the device started to delaminate after 80 s with a drastic increase of the flow rate (Fig. 5C). After the irreversible sealing of the MFP the MBs were seeded and we observed that, as expected, cell growth was restricted to the respective compartment (Fig. 5B, right). However, when applying solution inside the chambers, we did not detect spontaneous filling of the micro-channels as previously found for the PLO functionalized MFP (Fig. 5D, left panel). The small size of the channels (width = 6 μm, height = 3 μm) and the hydrophobic nature of PDMS in combination with the less hydrophilic PEMA surface compared to glass surface, disfavored the spontaneous penetration of the liquid with a capillarity backpressure. The capillarity pressure is expressed by the Young-Laplace equation, namely Pb = 2σcos(ϑ)/R, where σ is the surface tension of the liquid, ϑ the effective contact angle and R the hydraulic radius [33]. For our hybrid micro-channel geometry, characterized by three PDMS walls (ϑPDMS ≈ 115°) [34] and one PEMA wall (ϑPEMA ≈ 57°) [35], cos(ϑ) < 0, the liquid cannot penetrate in the channel. In order to coat the PEMA surface in the micro-channel region of the MFP, it is necessary to make the PDMS walls of the microchannel hydrophilic in order to apply BME and cRGD inside the micro-channel. Plasma treatment can be used to change the PDMS wettability after the MFP sealing [36], enabling easy filling of the micro-channel, without using an external pump system (Fig. 6C, right panel). We found that air plasma treatment (50 W) for 120 s is sufficient to achieve proper channel filling (Fig. 5D and E). The liquid pipetted in the MFP chamber filled the micro-channels in about 25 ms, with penetration dynamics in good agreement with the Lucas Washburn equation (Fig. 5E) [24]. By silanization of the PDMS and plasma treatment of the assembled device, we are now able to use PEMA-functionalized glass surfaces inside the MFP (Fig. 5F).
3.6. Formation of human NMCs using our MFP
To form NMCs within the MFP, MNs must extend their axons through the micro-channels to the peripheral compartment. For this reason, we evaluated axonal growth through the micro-channels of the PEMA-BME and PEMA-cRGD-Laminin coated MFPs. iPSC-derived MNs were plated in one chamber of the MFP, and immunostaining for TUBB3 was used to quantify axons exiting into the peripheral compartment. When cultured in MFPs with PEMA-cRGD-Laminin, MN axonal outgrowth was significantly slower than in MFPs with PEMA-BME (Fig. 6A). Furthermore, we could show that PEMA-cRGD-Laminin coated MFPs showed a significantly lower number of axons exiting the micro-channels compared to MFPs with PEMA-BME (Fig. 6B and C), suggesting impaired axonal growth inside the peripheral compartment. In contrast, using MFPs with PEMA-BME, we observed that MNs extended their axons into the peripheral compartment after only two to three days (Fig. 6A), which is comparable with MFPs coated with the positive control, PLO-Laminin (Fig. 3C). Therefore, we selected MFPs with PEMA-BME as the most suitable coating to model the NMCs.
Next, we cultured both MNs and myotubes using the PEMA-BME coated MFP. Differentiated MNs were applied inside the CNS compartment of the MFP and MBs were plated in the peripheral compartment to form myotubes. After co-culturing for 21 days, immunostaining demonstrated that TUBB3-positive MNs as well as MY-32 - positive myotubes were successfully attached, differentiated, and cultured inside the MFP (Fig. 6D). MNs extended their axons inside the opposed peripheral compartment, where they attached to myotubes (Fig. 6E), and immunostaining showed the accumulation of fluorescently conjugated bungarotoxin at the distal ends of SMI-32-positive MN axons (Fig. 6F), consistent with the formation of neuromuscular junctions. We detected muscle twitching when myotubes were co-cultured with MNs (Video S1). To validate the formation of functional NMCs in our MFP, a lentiviral vector was used to overexpress the ChR2 derivate CatCh [22] specifically in iPSC-derived MNs, which were co-cultured with myotubes in our MFP (Fig. 6G). In response to the stimulation of MNs with 200 ms pulses of blue light (475 nm) every 5 s (0.2 Hz), myotube contraction was observed (Fig. 6H). Control cells were recorded without stimulation (Fig. 6H). These data demonstrate that functional NMCs are formed in our MFP.
Supplementary data related to this article can be found at https://doi.org/10.1016/j.biomaterials.2019.119537.
To assess NMCs formation in our MFP, we applied monosynaptic tracing using a recombinant rabies virus (RABV) [37,38]. First, we generated an MB “starter” population, which expressed a histone-tagged GFP, the avian ASLV type A (EnvA) receptor (TVA), and rabies glycoprotein (G) (Fig. 7A and B). After co-culturing starter MBs and MNs for 12 days in the MFP, differentiated starter myotubes were infected with an RABV deletion mutant, in which the gene coding for G is replaced by mCherry (RABVΔG-mCherry) (Fig. 7C). EnvA pseudotyped RABVΔG-mCherry requires TVA for infection, which is only expressed by the starter myotubes (Fig. 7C). Since the starter cells express the glycoprotein, RABVΔG-mCherry is genetically complemented, leading to the production of infectious RABVΔG-mCherry particles that enter MNs connected via a functional NMC (Fig. 7C). However, since MNs do not express G, no additional infectious virus is produced. After seven days, the CNS and peripheral chambers were imaged using confocal microscopy. We found that HTB - myotubes showed mCherry expression 3 days following infection with RABVΔG-mCherry-EnvA (Fig. 7D), demonstrating that the starter myotubes were successfully infected, and seven days following infection, most of the HTB-myotubes expressed the mCherry reporter (Fig. 7D and E). Furthermore, mCherry-positive and GFP-negative MNs were detected in close proximity to the micro- channels 5 days following infection of the starter myotubes (Fig. 7F), and the number of traced MNs was significantly increased at 7 days following infection (Fig. 7F and G). Although skeletal myotubes show already mCherry expression 3 days following infection, MNs did not, suggesting that mCherry-positive cells are a result of RABV tracing instead of direct infection. To demonstrate that MNs are the result of tracing, pure MN cultures were exposed to RABVΔG-mCherry-EnvA particles and analyzed using flow cytometry. Similar to uninfected controls, no mCherry-positive cells were detected in MNs following exposure to RABVΔG-mCherry-EnvA particles (Fig. S2). To further validate that mCherry-positive MNs are a result of retrograde RABV transmission across neuromuscular junctions (NMJs), MNs were co-cultured with skeletal myotubes not expressing TVA, hisGFP and G (HTB-negative). As expected, no traced MN were detected when co-cultured with HTB-negative skeletal muscle cells (Fig. S2). This demonstrates the formation of an NMC in the CNS and peripheral compartments of our MFP using PEMA-BME coating. This is the first time that monosynaptic RABV tracing was adapted and successfully used to detect NMCs, which can be used for disease modeling in combination with our MFP.
Fig. 7.
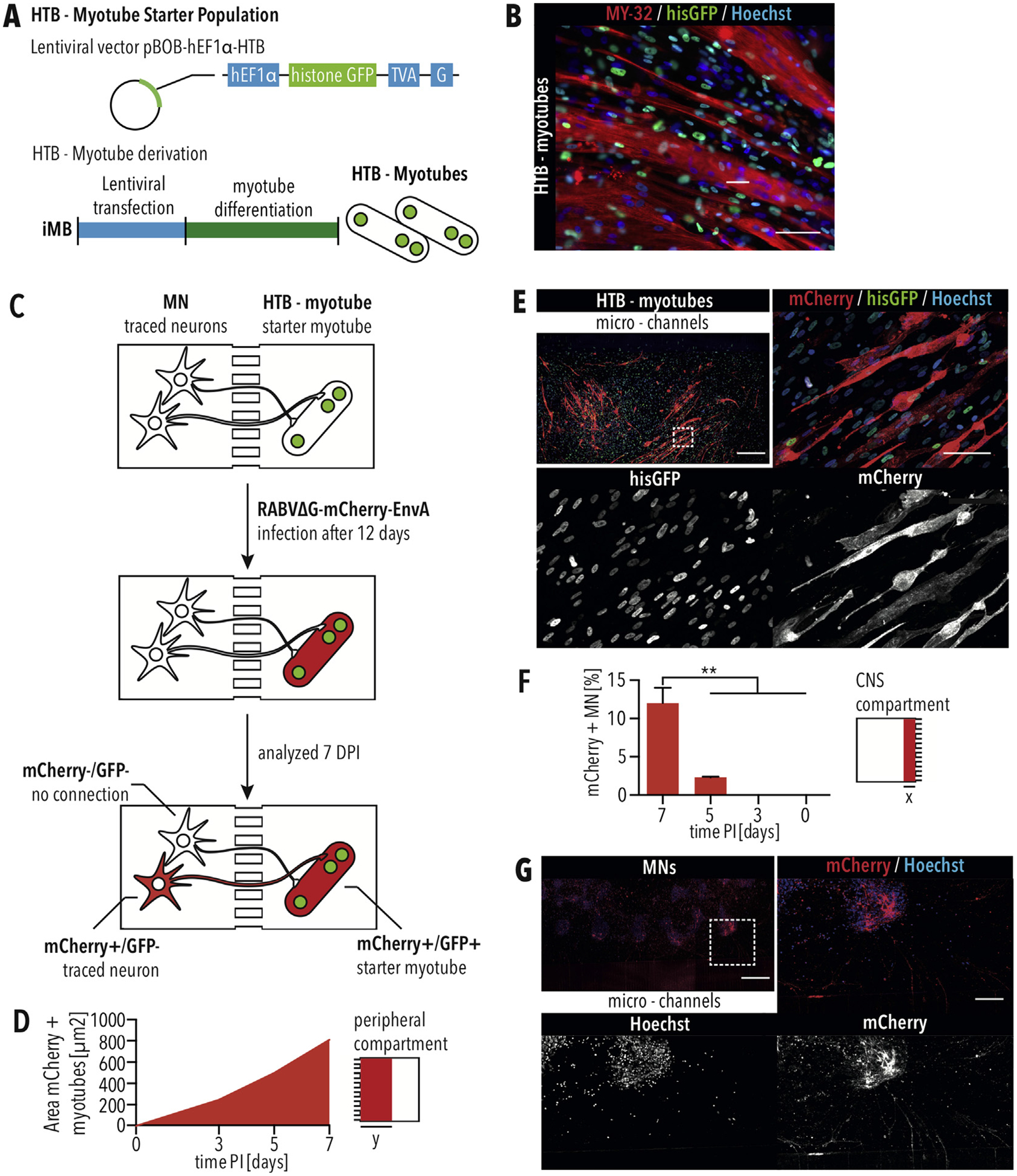
Rabies tracing of NMCs using MFPs with PEMA-BME (A) The starter skeletal myotubes are derived by transfecting myoblast cells with the pBOB-hEF1α-HTB lentivector carrying the EnvA receptor (TVA), histone GFP (hisGFP), and the RABV G protein under the control of a hEF1α promotor followed by differentiation into skeletal myotubes. (B) The HTB-myotube starter population shows nuclear GFP and expresses the skeletal muscle marker MY-32. Scale bar = 50 μm. (C) Diagram of retrograde monosynaptic tracing inside the microfluidic platform. (D) Quantification of the mCherry-positive area within the peripheral compartment of 1700 μm (y) from the micro-channels for myotubes expressing the mCherry reporter. (E) mCherry-positive myotubes within the peripheral compartment seven days following infection. Scale bar of tile scan = 500 μm. Dashed square marks position of close up. Scale bar of close up = 100 μm. (F) Quantification of traced MNs, which are mCherry-positive, in an area of 725 μm (x) from the micro-channels. The greatest number of traced MNs were detected seven days following infecting the HTB-myotubes with RABVΔG-mCherry-EnvA. Error bars indicate standard error of the mean (SEM). ** indicates p < 0.01, according to one-way ANOVA and Dunnett’s multiple comparisons test. (G) mCherry-positive traced MNs in the CNS compartment seven days following infection. Scale bar of tile scan = 500 μm. Dashed square marks position of close up. Scale bar of close up = 100 μm.
4. Discussion
Degeneration of NMCs plays a critical role in multiple diseases, including amyotrophic lateral sclerosis, spinal muscular atrophy, and Duchenne muscular dystrophy. NMCs can be used by pathogenic viruses to invade the CNS. Models of human NMCs are urgently needed to better understand pathogenesis of neuromuscular diseases as well as identify novel drugs to counteract disease progression, but this has been challenging because NMCs involve both CNS and peripheral compartments. Although microfluidics can compartmentalize cell cultures, these devices have been limited to a single experimental unit, making their use in disease modeling and drug discovery extremely cumbersome. Here, we report the development of a medium-throughput MFP that can generate up to 126 NMCs in parallel. In addition, the MFP is designed to be compatible with automated liquid handling and imaging systems, making it a suitable platform for compound screening.
One of the critical challenges with generating in vitro 2D models of NMCs is that myotubes delaminate before functional circuits can be efficiently formed. We overcame this issue by developing an MFP with a reactive polymer surface of the maleic anhydride PEMA. One of the major advantages of using PEMA inside the MFP is that it can be crosslinked to a variety of ECM substrates as well as growth factors to glass surfaces. Thus, PEMA functionalization can create a customized microenvironment for each cell type in the respective microfluidic compartment. We identified specific conditions, including PEMA-BME and PEMA-cRGD-Laminin, which enable long-term cultivation of MNs as well as myotubes in our MFPs.
Maleic anhydride copolymers, such as PEMA, have never been used inside microfluidic systems. To achieve long-term NMC cultures inside a PEMA functionalized MFP, we had to address specific aspects of fabrication, including bonding and surface chemistry inside the micro-channels. To obtain an irreversible seal between PEMA and PDMS, silanization of PDMS was carried out using oxygen plasma treatment and APTES. We could show that functionalizing PDMS with primary amino groups by silanization enables sufficient bonding of PDMS and PEMA to prevent medium leakage and cell growth between the boundaries (Fig. 5A–C). It is crucial to be able to fill micro-channels in order to be able to use the MFP. Glass surfaces coated with PEMA are more hydrophobic, and, thus, spontaneous filling of the micro-channels with aqueous solution was prevented. We were able to overcome the capillarity backpressure by using air plasma treatment to maintain an overall balance of surface tensions that favors the spontaneous penetration of liquid inside the micro-channels. In this way, we achieved channel filling and were able to use the PEMA coated MFP for the in vitro NMC model. This is the first time a microfluidic system with a reactive polymer surface was established. Our fabrication approach is not only suitable to generate an MFP to mimic NMCs in vitro, but it is of interest for other lab-on-a-chip applications where an easy, flexible and stable surface functionalization with proteins or peptides is needed and capillarity or pressure-driven flow is required.
Our optogenetics data demonstrate that functional NMCs were formed in our MFP. However, simply producing NMCs in parallel is not enough; an assay platform is needed that can be applied to quantify NMC formation. We addressed this issue by developing a novel application of RABV tracing. Monosynaptic RABV tracing was originally developed to map the connectivity between specific neuronal populations inside the brain [38–40]. RABV tracing has proven to be very adaptable and was subsequently used to map the sensory and premotor circuit in mice [41,42]. To date, RABV has not been applied to skeletal muscle to trace directly connected MNs, and we demonstrated the first proof-of-principle that monosynaptic RABV tracing can be used on NMCs in vitro. In contrast to patch clamping, calcium imaging and muscle contraction assays, the RABV tracing assay can be automated relatively easily, making it suitable for drug discovery. Using our approach, multiple conditions could be tested in parallel, and lower-throughput approaches such as patch clamping, could be used for validation. RABV tracing could even facilitate patch clamping by identifying specific MNs that are connected to myotubes, which could save the time normally required to test many different MNs before a functional NMC is identified.
One of the advantages of our MFP is that it can be modified to incorporate additional cell types. For example, Schwann cells play an important role in forming NMCs [43,44] and have been implicated in the pathogenesis of amyotrophic lateral sclerosis and spinal muscular atrophy [45]. Thus, our MFP could be modified to incorporate Schwann cells in addition to MNs and myotubes. A critical advantage of using PEMA functionalization in the MFP is that additional factors could be incorporated to promote survival and maturation including, for example, VEGF and GDNF, which stimulate the growth cone and regeneration of neuromuscular junctions [46,47].
Our MFP is well suited to identifying novel small molecule compounds for a variety of disorders. Using iPSCs, it is possible to generate theoretically limitless quantities of MNs and myotubes from specific patients with known phenotypes and genotypes. For example, iPSC-derived MNs and myotubes can be generated from amyotrophic lateral sclerosis patients and cultured inside the medium-throughput MFP. Using the RABV tracing assay, changes in the numbers of NMCs can be detected by comparing the number of MNs with functional neuromuscular junctions to myotubes. In this manner, NMCs derived from patients could be compared with isogenic wild type controls. In addition, drug candidates could be screened for their ability to protect NMCs from degeneration. Development of an automated liquid handling as well as an imaging workflow would facilitate this application.
Therefore, our study significantly advances the use of microfluidics to generate functional NMCs for medium-throughput applications, including disease modeling and drug discovery.
Supplementary Material
{kind=link}
{kind=link}
HIGHLIGHTS.
Manufacture of a microfluidic platform (MFP) for medium-throughput applications.
Silanization and plasma treatment enables incorporation of PEMA film into MFP.
PEMA based coatings enable culture of iPSC-derived neurons and myotubes in MFPs.
RABV tracing and optogenetics showed neuromuscular circuit formation in MFPs.
Acknowledgements
This work was aided by the CRTD Light Microscopy facility. We thank T. Levin for assisting with revising the manuscript. We thank N. Rein, M. Grimmer, A. Sójka and M. Dimaki for the preparation of the PEMA coated glass cover slips. We thank J. Naughton for providing us with the RABV production cell lines and for advice on cell handling. We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) and the CRTD, which is part of the Technische Universität Dresden. This work was financed by the DFG Research Center (DFG FZT 111) and Cluster of Excellence (DFG EXC 168), including a seed grant. JB was sponsored by the Hans und Ilse Breuer Stiftung. JS was supported by the European Union’s Horizon 2020 research and innovation program (643417) and the Bundesministerium für Bildung und Forschung (01EK1606A and 01ED1601B). This is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project supported by the following funding organizations under the aegis of JPND-www.jpnd.eu: Germany, Bundesministerium für Bildung und Forschung; Israel, Ministry of Health; Italy, Ministero dell’Istruzione dell’Università e della Ricerca; Sweden, Swedish Research Council; and Switzerland, Swiss National Science Foundation.
Footnotes
Declaration of competing interest
The authors have no competing interests to declare.
Data availability statement
The raw/processed data that support the findings of this study are available from the corresponding author, JS, upon reasonable request.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2019.119537.
References
- [1].Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW, Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis, J. Neurosci 33 (11) (2013) 4657–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kanning KC, Kaplan A, Henderson CE, Motor neuron diversity in development and disease, Annu. Rev. Neurosci 33 (2010) 409–440. [DOI] [PubMed] [Google Scholar]
- [3].McGreevy JW, Hakim CH, McIntosh MA, Duan D, Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy, Dis. Model Mech 8 (3) (2015) 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wells DJ, Tracking progress: an update on animal models for Duchenne muscular dystrophy, Dis. Model Mech 11 (6) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Edens BM, Ajroud-Driss S, Ma L, Ma YC, Molecular mechanisms and animal models of spinal muscular atrophy, Biochim. Biophys. Acta 1852 (4) (2015) 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Demestre M, Orth M, Fohr KJ, Achberger K, Ludolph AC, Liebau S, Boeckers TM, Formation and characterisation of neuromuscular junctions between hiPSC derived motoneurons and myotubes, Stem Cell Res. 15 (2) (2015) 328–336. [DOI] [PubMed] [Google Scholar]
- [7].Puttonen KA, Ruponen M, Naumenko N, Hovatta OH, Tavi P, Koistinaho J, Generation of functional neuromuscular junctions from human pluripotent stem cell lines, Front. Cell. Neurosci 9 (2015) 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Steinbeck JA, Jaiswal MK, Calder EL, Kishinevsky S, Weishaupt A, Toyka KV, Goldstein PA, Studer L, Functional connectivity under optogenetic control allows modeling of human neuromuscular disease, Cell stem cell 18 (1) (2016) 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL, A microfluidic culture platform for CNS axonal injury, regeneration and transport, Nat. Methods 2 (8) (2005) 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santhanam N, Kumanchik L, Guo X, Sommerhage F, Cai Y, Jackson M, Martin C, Saad G, McAleer CW, Wang Y, Lavado A, Long CJ, Hickman JJ, Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics, Biomaterials 166 (2018) 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Osaki T, Uzel SGM, Kamm RD, Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons, Sci. Adv 4 (10) (2018) eaat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang PY, Thissen H, Tsai WB, The roles of RGD and grooved topography in the adhesion, morphology, and differentiation of C2C12 skeletal myoblasts, Biotechnol. Bioeng 109 (8) (2012) 2104–2115. [DOI] [PubMed] [Google Scholar]
- [13].Lam MT, Sim S, Zhu X, Takayama S, The effect of continuous wavy micro-patterns on silicone substrates on the alignment of skeletal muscle myoblasts and myotubes, Biomaterials 27 (24) (2006) 4340–4347. [DOI] [PubMed] [Google Scholar]
- [14].Sun Y, Duffy R, Lee A, Feinberg AW, Optimizing the structure and contractility of engineered skeletal muscle thin films, Acta Biomater. 9 (8) (2013) 7885–7894. [DOI] [PubMed] [Google Scholar]
- [15].Duffy RM, Sun Y, Feinberg AW, Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle, Ann. Biomed. Eng 44 (6) (2016) 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rothbauer M, Zirath H, Ertl P, Recent advances in microfluidic technologies for cell-to-cell interaction studies, Lab Chip 18 (2) (2018) 249–270. [DOI] [PubMed] [Google Scholar]
- [17].Zahavi EE, Ionescu A, Gluska S, Gradus T, Ben-Yaakov K, Perlson E, A compartmentalized microfluidic neuromuscular co-culture system reveals spatial aspects of GDNF functions, J. Cell Sci 128 (6) (2015) 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, Wu G, Muller S, Brockmann K, Kluba T, Maisel M, Kruger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki O-E, Klingauf J, Kuhlmann T, Klewin M, Muller H, Gasser T, Scholer HR, Sterneckert J, Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression, Cell stem cell 12 (3) (2013) 354–367. [DOI] [PubMed] [Google Scholar]
- [19].Hicks MR, Hiserodt J, Paras K, Fujiwara W, Eskin A, Jan M, Xi H, Young CS, Evseenko D, Nelson SF, Spencer MJ, Handel BV, Pyle AD, ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs, Nat. Cell Biol 20 (1) (2018) 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Busskamp V, Lewis NE, Guye P, Ng AH, Shipman SL, Byrne SM, Sanjana NE, Murn J, Li Y, Li S, Stadler M, Weiss R, Church GM, Rapid neurogenesis through transcriptional activation in human stem cells, Mol. Syst. Biol 10 (2014) 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K, Molecular and cellular approaches for diversifying and extending optogenetics, Cell 141 (1) (2010) 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kleinlogel S, Feldbauer K, Dempski RE, Fotis H, Wood PG, Bamann C, Bamberg E, Ultra light-sensitive and fast neuronal activation with the Ca(2) +-permeable channelrhodopsin CatCh, Nat. Neurosci 14 (4) (2011) 513–518. [DOI] [PubMed] [Google Scholar]
- [23].Osakada F, Callaway EM, Design and generation of recombinant rabies virus vectors, Nat. Protoc 8 (8) (2013) 1583–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Washburn EW, The dynamics of capillary flow, Phys. Rev 17 (3) (1921) 273–283. [Google Scholar]
- [25].Martin NR, Passey SL, Player DJ, Mudera V, Baar K, Greensmith L, Lewis MP, Neuromuscular junction formation in tissue-engineered skeletal muscle augments contractile function and improves cytoskeletal organization, Tissue Eng. A 21 (19–20) (2015) 2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ojovan SM, McDonald M, Rabieh N, Shmuel N, Erez H, Nesladek M, Spira ME, Nanocrystalline diamond surfaces for adhesion and growth of primary neurons, conflicting results and rational explanation, Front. Neuroeng 7 (2014) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pompe T, Zschoche S, Herold N, Salchert K, Gouzy M-F, Sperling C, Werner C, Maleic anhydride copolymers–a versatile platform for molecular biosurface engineering, Biomacromolecules 4 (4) (2003) 1072–1079. [DOI] [PubMed] [Google Scholar]
- [28].Prewitz MC, Seib FP, von Bonin M, Friedrichs J, StiSsel A, Niehage C, Muller K, Anastassiadis K, Waskow C, Hoflack B, Bornhauser M, Werner C, Tightly anchored tissue-mimetic matrices as instructive stem cell microenvironments, Nat. Methods 10 (8) (2013) 788–794. [DOI] [PubMed] [Google Scholar]
- [29].Muller E, Wang W, Qiao W, Bornhauser M, Zandstra PW, Werner C, Pompe T, Distinguishing autocrine and paracrine signals in hematopoietic stem cell culture using a biofunctional microcavity platform, Sci. Rep 6 (2016) 31951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ng JM, Gitlin I, Stroock AD, Whitesides GM, Components for integrated poly (dimethylsiloxane) microfluidic systems, Electrophoresis 23 (20) (2002) 3461–3473. [DOI] [PubMed] [Google Scholar]
- [31].Roth J, Albrecht V, Nitschke M, Bellmann C, Simon F, Zschoche S, Michel S, Luhmann C, Grundke K, Voit B, Surface functionalization of silicone rubber for permanent adhesion improvement, Langmuir 24 (21) (2008) 12603–12611. [DOI] [PubMed] [Google Scholar]
- [32].Cordeiro AL, Zschoche S, Janke A, Nitschke M, Werner C, Functionalization of poly(dimethylsiloxane) surfaces with maleic anhydride copolymer films, Langmuir 25 (3) (2009) 1509–1517. [DOI] [PubMed] [Google Scholar]
- [33].Delamarche E, Bernard A, Schmid H, Bietsch A, Michel B, Biebuyck H, Microfluidic networks for chemical patterning of substrate: design and application to bioassays, J. Am. Chem. Soc 120 (3) (1998) 500–508. [Google Scholar]
- [34].Girardo S, Cecchini M, Beltram F, Cingolani R, Pisignano D, Polydimethylsiloxane-LiNbO3 surface acoustic wave micropump devices for fluid control into microchannels, Lab Chip 8 (9) (2008) 1557–1563. [DOI] [PubMed] [Google Scholar]
- [35].Freudenberg U, Zschoche S, Simon F, Janke A, Schmidt K, Behrens SH, Auweter H, Werner C, Covalent immobilization of cellulose layers onto maleic anhydride copolymer thin films, Biomacromolecules 6 (3) (2005) 1628–1634. [DOI] [PubMed] [Google Scholar]
- [36].Kim SC, Sukovich DJ, Abate AR, Patterning microfluidic device wettability with spatially-controlled plasma oxidation, Lab Chip 15 (15) (2015) 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Miyamichi K, Amat F, Moussavi F, Wang C, Wickersham I, Wall NR, Taniguchi H, Tasic B, Huang ZJ, He Z, Callaway EM, Horowitz MA, Luo L, Cortical representations of olfactory input by trans-synaptic tracing, Nature 472 (7342) (2011) 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N, Whole-brain mapping of direct inputs to midbrain dopamine neurons, Neuron 74 (5) (2012) 858–873. [DOI] [PubMed] [Google Scholar]
- [39].Yetman MJ, Washburn E, Hyun JH, Osakada F, Hayano Y, Zeng H, Callaway EM, Kwon HB, Taniguchi H, Intersectional monosynaptic tracing for dissecting subtype-specific organization of GABAergic interneuron inputs, Nat. Neurosci 22 (3) (2019) 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Deshpande A, Bergami M, Ghanem A, Conzelmann KK, Lepier A, Gotz M, Berninger B, Retrograde monosynaptic tracing reveals the temporal evolution of inputs onto new neurons in the adult dentate gyrus and olfactory bulb, Proc. Natl. Acad. Sci. U. S. A 110 (12) (2013) E1152–E1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stanek E.t., Cheng S, Takatoh J, Han BX, Wang F, Monosynaptic premotor circuit tracing reveals neural substrates for oro-motor coordination, Elife 3 (2014) e02511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zampieri N, Jessell TM, Murray AJ, Mapping sensory circuits by anterograde transsynaptic transfer of recombinant rabies virus, Neuron 81 (4) (2014) 766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Son YJ, Thompson WJ, Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells, Neuron 14 (1) (1995) 133–141. [DOI] [PubMed] [Google Scholar]
- [44].Auld DS, Robitaille R, Glial cells and neurotransmission: an inclusive view of synaptic function, Neuron 40 (2) (2003) 389–400. [DOI] [PubMed] [Google Scholar]
- [45].Santosa KB, Keane AM, Jablonka-Shariff A, Vannucci B, Snyder-Warwick AK, Clinical relevance of terminal Schwann cells: an overlooked component of the neuromuscular junction, J. Neurosci. Res 96 (7) (2018) 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Olbrich L, Foehring D, Happel P, Brand-Saberi B, Theiss C, Fast rearrangement of the neuronal growth cone’s actin cytoskeleton following VEGF stimulation, Histochem. Cell Biol 139 (3) (2013) 431–445. [DOI] [PubMed] [Google Scholar]
- [47].Magill CK, Moore AM, Yan Y, Tong AY, MacEwan MR, Yee A, Hayashi A, Hunter DA, Ray WZ, Johnson PJ, Parsadanian A, Myckatyn TM, Mackinnon SE, The differential effects of pathway-versus target-derived glial cell line-derived neurotrophic factor on peripheral nerve regeneration, J. Neurosurg 113 (1) (2010) 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.