

Abstract
The efficacy of oncolytic viruses (OVs), such as reovirus, is dictated by host immune responses, including those mediated by the pro-versus anti-inflammatory macrophages. As such, a detailed understanding of the interaction between reovirus and different macrophage types is critical for therapeutic efficacy. To explore reovirus–macrophage interactions, we performed tandem mass tag (TMT)-based quantitative temporal proteomics on mouse bone marrow-derived macrophages (BMMs) generated with two cytokines, macrophage colony stimulating factor (M-CSF) and granulocytic–macrophage colony stimulating factor (GM-CSF), representing anti- and proinflammatory macrophages, respectively. We quantified 6863 proteins across five time points in duplicate, comparing M-CSF (M-BMM) and GM-CSF (GM-BMM) in response to OV. We find that GM-BMMs have lower expression of key intrinsic proteins that facilitate an antiviral immune response, express higher levels of reovirus receptor protein JAM-A, and are more susceptible to oncolytic reovirus infection compared to M-BMMs. Interestingly, although M-BMMs are less susceptible to reovirus infection and subsequent cell death, they initiate an antireovirus adaptive T cell immune response comparable to that of GM-BMMs. Taken together, these data describe distinct proteome differences between these two macrophage populations in terms of their ability to mount antiviral immune responses.
Keywords: antigen presentation/processing, antiviral immune response, cell differentiation, macrophages, oncolytic viruses, quantitative proteomics, reovirus, viral defense
Graphical Abstract
■ INTRODUCTION
Oncolytic viruses (OVs) have been shown to preferentially target and kill cancer cells.1 One such OV, reovirus, is currently undergoing phase I, II, and III clinical trials for a variety of cancers.2,3 Following their therapeutic administration, OVs promptly initiate a cascade of antiviral immune responses. A major part of this antiviral immune response is constituted by macrophages, which make up a substantial proportion of the tumor microenvironment (TME).4–6 As such, macrophages play a key role in OV-induced antitumor immunity.7–10
In the TME, infiltrating monocytes differentiate into two putative subsets11–14 and are formally referred to as classically (Ml) and alternatively (M2) activated macrophages.15,16 These subsets represent opposing ends of a spectrum of macrophage functions, where M1 macrophages are proinflammatory and associated with a Th1 immune response, while M2 macrophages are anti-inflammatory and are associated with wound repair and a Th2 immune response.17–20 In the context of cancer, M1 and M2 macrophages have been found to bear anti- and protumor functions, respectively.4,7,21,22 Thus, it is possible that these distinct macrophage phenotypes play a differential role during OV-based cancer therapy. Yet, direct interactions between OVs, such as oncolytic reovirus, and pro-versus anti-inflammatory macrophages are poorly understood.
To better understand OV-macrophage interactions, we compared tandem mass tag (TMT)-based quantitative proteomics of M1-like bone marrow-derived macrophages (BMMs) generated with granulocytic–macrophage colony stimulating factor (GM-CSF) (GM-BMMs) to that of M2-like BMMs generated with macrophage colony stimulating factor (M-CSF) (M-BMMs) following exposure to reovirus. We observed a lower abundance of many key antiviral defense proteins (e.g., ISG20 and OASL1) and a higher abundance of reovirus infection proteins (e.g., JAM-A) in GM-BMMs. Validation experiments confirmed that GM-BMMs are more susceptible to infection and increase inflammatory markers [IFNα/β and nitric oxide (NO)] than M-BMMs. These findings capture differential antiviral responses within M1- and M2-like macrophages and put forward direct implications for macrophage plasticity in OV-based cancer therapies.
■ EXPERIMENTAL SECTION
Reovirus Production
Type 3 Dearing reovirus was cultured and isolated according to a previously established protocol.23 Titration for reovirus was done using L929 cells (American Type Culture Collection, Manassas, VA) by standard plaque assay as described previously.24
Antibodies and Reagents
The following antibodies were purchased from BioLegend (San Diego, CA): PerCP/Cy5.5-anti-mouse CD11b (M1/70), APC-anti-mouse F4/80 (BM8), APC-anti-mouse CD40 (3/23), APC-anti-mouse CD80 (16-10A1), PE-anti-mouse CD86 (GL-1), PE-anti-mouse CD3ε (145-2C11), fluorescein isothiocyanate (FITC)-anti-mouse CD4 (RM4-5), PerCP/Cy5.5-anti-mouse CD8a (53-6.7), purified anti-IRF3 (12A4A35). Anti-mouse CD16/32 was purchased from BioXCell (West Lebanon, NH). Alexa 488-conjugated annexin V was purchased from Invitrogen (Carlsbad, CA). Anti-reovirus polyclonal rabbit antibody was produced in house and used as previously described.25 Monoclonal sigma 3 antibody (clone 4F2) was obtained from Developmental Studies Hybridoma Bank. FITC–dextran 70 was purchased from Sigma-Aldrich. Ovalbumin peptide-SIINFEKL (ova257-264) was purchased from GenScript (Piscataway, NJ). Granulocyte macrophage colony stimulating factor (GM-CSF) was purchased from Cedarlane (Burlington, ON). Macrophage colony stimulating factor (M-CSF) was purchased from Shenandoah (Warwick, PA).
Isolation of BMMCs, Macrophage Differentiation, and Tissue Culture
All mice used in the described experiments were female wild-type C57BL/6 strain, age 6–8 weeks, purchased from Charles River Laboratories (Montreal, QC, Canada). BMMCs were isolated from the femur and tibia bones and briefly treated with red blood cell-lysing ammonium chloride potassium [ACK; 150 mM ammonium chloride, 10 mM potassium bicarbonate, 10 μM ethylenediaminetetraacetic acid (EDTA)] prior to being cultured (37 °C at 5% CO2 concentration) in Roswell Park Memorial Institute (RPMI)-1640 complete medium [10% fetal bovine serum (FBS), 1 mM glutamate, 1 mM sodium pyruvate, 1 mM nonessential amino acids, 100 units/mL penicillin, 100 μg/mL streptomycin, and 0.25 ĝ/mL amphotericin B (all purchased from Invitrogen, Carlsbad, CA)]. Cultures were then supplemented with either GM-CSF (20 ng/mL) or M-CSF (100 ng/mL) for 7 days to generate M1 and M2 macrophages, respectively, as per standard protocols.26
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
RNA extractions were performed using the TRIzol procedure. The purified RNA was diluted to a concentration of 2 μg and used for the synthesis of complementary DNA (cDNA) using Superscript II (Invitrogen, Carlsbad, CA). cDNA was amplified and quantified using the CFX96 touch real-time PCR (RT-PCR) instrument (BioRad), and gene-specific primers for murine Cd40, Cd80, Cd86, Ifna, Ifnb, Lambdal, and Sigmal were purchased from Invitrogen. qPCR data were collected and calculated using the Livak and Schmittgen’s 2−ΔΔCT method27 and normalized to the 3-phosphate dehydrogenase (GAPDH) reference gene, followed by a comparison against respective controls.
Western Blotting
Cell lysis was achieved using RIPA buffer [50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% Triton, and 0.5% deoxycholate]. Cell lysates were sonicated for 15 s on ice, and debris was removed by centrifugation at 13000g for 5 min. Total protein content was measured using a bicinchoninic acid (BCA) assay (Thermo Scientific). Extracts of 20 μg of protein were separated using SDS-polyacrylamide gel electrophoresis (PAGE), transferred to nitrocellulose membranes, and immunoblotted with target-specific antibodies.
T Cell Activation Assay
For T cell analysis, cells were initially prepared by first filtering a crushed spleen from a female C57BL/6 mouse (age 6–8 weeks) using a 40 μm filter. Red blood cells were lysed with ACK. Prepared splenocytes (1 × 106 cells per well in a 96-well plate) were incubated in RPMI-1640 (Gibco) complete media for 24 h with treatments as indicated. After 24 h of incubation, samples were processed for flow cytometry. First, splenocytes were washed and incubated with CD16/CD32 antibodies to block Fc antibody receptors, followed by incubation with fluorophore-labeled primary antibodies, and finally were fixed in 4% paraformaldehyde (PFA). Data acquisition was done with BD FACSCalibur, where a minimum of 10 000 events were collected.
NO and Reactive Oxygen Species (ROS) Quantification
After 24 h of virus treatment, nitric oxide was quantified using the Griess Reagent System as described by the manufacturer (Promega, Madison, WI). The absorbance was measured at 530 nm using a SpectraMax M2 spectrophotometer (Molecular Devices, San Jose, CA). ROS production was analyzed by flow cytometry detection of 2’,7’-dichlorofluorescin diacetate (DCFDA) (Thermo Scientific), as described by the manufacturer.
Confocal Microscopy
Macrophages were grown on poly-D-lysine-coated coverslips (VWR) for optimal fluorescent imaging. Cells were fixed in 4% PFA, permeabilized with 0.25% Triton X-100, and blocked in 1% BSA at room temperature. Cells were incubated with anti- CD11b (clone 5C6; 1:200 dilution, 488 nm; purchased from BioRad) and anti-reovirus (1:5000 dilution, 550 nm) antibodies overnight at 4 °C. Cells were also stained with the nuclear stain 4’,6-diamidino-2-phenylindole (DAPI, dihydrochloride, 405 nm) at a concentration of 1 μg/mL. Coverslips were mounted using a ProLong Gold antifade mounting medium (Invitrogen) to prevent signal fading. Slides were imaged using the Zeiss LSM 710 laser scanning confocal microscope (Carl Zeiss Canada Ltd., ON).
Flow Cytometry and Statistical Analysis
Flow cytometry data were acquired using the BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and subsequently analyzed using Flowing Software or FCS Express V5 software (DeNovo Software, Los Angeles, CA). Depending on the indicated experiment, one-way analysis of variance (ANOVA) tests were performed to compare between multiple groups, and p values of <0.05 were considered significant. Statistical significance is represented by asterisks above the bar graphs (****p ≤ 0.0001, ***p ≤ 0.0005, **p ≤ 0.001, *p ≤ 0.01, n.s. = p > 0.05).
FITC–Dextran Assay
Cellular uptake of FITC–dextran (Sigma) was determined using flow cytometry. Here, the culture medium was supplemented with FITC–dextran (1 mg/mL) and cells were then treated with reovirus at indicated concentrations. After 24 h, cells were collected and analyzed via flow cytometry.
Plaque Assay
Macrophages were plated at 1 × 106 cells/well in 12-well plates, treated with reovirus at MOI 1 or 10. Following the 4 h incubation, media containing free-virus was removed, cells were washed with phosphate-buffered saline (PBS), and fresh RPMI complete media supplemented with GM-CSF (20 ng/mL) or M-CSF (100 ng/mL) was added to the cultures. Cells were then cultured for an additional 20 and 44 h (24 and 48 h total, respectively). The supernatant from GM- and M-BMMs was collected and centrifuged to obtain reovirus progeny virions and tittered on L929 cells.
Quantitative Multiplexed Proteomics
Macrophages were washed in phosphate-buffered saline (PBS) and removed from plates by gentle scraping in 1 mL of lysis buffer [2% SDS, 150 mM NaCl, 5 mM dithiothreitol (DTT), and 50 mM Tris (pH 8.5)] and one protease inhibitor tablet (Sigma). Immediately following, cell samples were homogenized using a Tissue Ruptor (Qiagen, Germany) proceeded by brief sonication. Samples were then heated to 60 °C for 30 min, cooled, and supplemented with iodoacetamide (14 mM) to alkylate cysteine residues before being subjected to methanol–chloroform precipitation and dried down using a vacuum centrifuge (Thermo Scientific). Next, samples were resuspended in 2 mL of urea buffer (8 M urea, 50 mM Tris, pH 8.8) by gentle drilling, followed by brief sonication, and kept on ice. Total protein content was measured using a BCA assay (Thermo Scientific). An aliquot of 100 μg of protein was diluted to 1.5 M urea and digested in sequencing grade trypsin (Promega, Madison, WI) overnight at 37 °C at a ratio of 1:100 trypsin/protein. The digest (peptides) was adjusted to <3 pH using formic acid, was desalted using 60 mg of solid-phase 3 cc extraction cartridges (Waters, Milford, MA), and dried down. Dried peptides were resuspended in 100 mM N-(2-hydroxyethyl)piperazine-N’-ethanesulfonic acid (HEPES), 30% acetonitrile, and 10 μL of TMT10 reagents (Thermo Scientific) prealiquoted at a concentration of 20 mg/mL in anhydrous acetonitrile. The reaction was quenched with 0.5% hydroxylamine (Sigma), and the contents were mixed equally, desalted using a 60 mg of solid-phase 3 cc extraction cartridge (Waters, Milford, MA), and dried down. Peptides were fractionated using an Agilent 300-Extend, 4.6 mm Δ 250 mm, 5 mm particle size C18 column (Agilent). A gradient of 5–40% acetonitrile (10 mM ammonium formate, pH 8) was applied at a flow rate of 800 mL/min using an Agilent 1100 pump. Fractions were collected every 0.38 min, beginning at 10 min; then, every 11th fraction was combined to a single sample to create 11 fractions, which were then desalted using home-made Stage-tips packed with Empore C18 extraction material (Sigma) as previously described and28 then dried down and subjected to LC-SPS-MS3.
2D-LC-MS3 and Data Processing
Mass spectrometry was performed using an Orbitrap Fusion, and the TMT reporter ions were quantified from the MS3 scan using the SPS-MS3 method as previously described.29,30 Peptides were then identified and subsequently quantified using a Sequest-based software pipeline. Spectra were searched against a concatenated mouse-reovirus database, and peptide-spectral matches (PSMs) were filtered to a 1% false discovery rate (FDR) using linear discriminant analysis in combination with the target-decoy method as previously described.31 TMT10 reporter ion S/N values from the MS3 scans were used to quantify the matching peptides. Protein quantification was done by using the sum of reporter ion peptide S/N values for each protein. Protein levels are expressed as a percent of the total S/N for all samples. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE32 partner repository with the data set identifier PXD015203.
All quantified proteins in the data set were subjected to K- means clustering with Euclidean distance using MultiExperiment Viewer (MeV).33 Clusters were then analyzed using DAVID Bioinformatics Database34 to determine enriched biological process GO terms.
■ RESULTS
Comparison of Proteome Remodeling within GM-BMMs and M-BMMs Following Exposure to Reovirus
First, we employed TMT-based multiplexed proteomics to characterize proteome remodeling in BMMs in response to oncolytic reovirus. For this, BMMs17–19,26,35 differentiated with M-CSF or GM-CSF for 7 days were treated with reovirus (MOI 1) over a time course of 48 h (Figure 1A). Temporal proteomics was performed using TMT10 and 2D-LC-SPS-MS329 analysis, resulting in a data set of 6863 quantified mouse proteins across all samples. These data included four reovirus proteins (Mu-NS, Lambda1, Mu-1, and Sigma-NS), revealing that GM-BMMs contained higher viral protein expression than M-BMMs (Figure 1B). This observation was further confirmed by western blotting (Figure S1A,B) and flow cytometry for annexin V at 24 and 48 h of infection (Figure S1C). Principal component analysis (PCA) revealed that most of the variance in the data set arose between GM-BMMs and M-BMMs rather than from infection (Figure 1C). Contributing to these differences, 674 and 627 proteins were at least 2fold more abundant in GM-BMMs and M-BMMs (without infection), respectively (Figures 1D and S2A), with many of these proteins correlating with the phenotype of each macrophage. For example, interferon regulatory factor 4 (IRF4,36 Figure 1D) and S100A837,38 (Figure S2A), shown to be critical in the development and function of inflammatory macrophages, were more abundant in GM-BMMs. Conversely, stabilin-1 (Stab1)39 and allograft inflammatory factor 1 (AIF1),40 known for homeostatic function and tumor-associated polarization, respectively, of M2-like macrophages were found to be more abundant in M-BMMs (Figure S2A,B). Functional annotation using DAVID34 revealed enrichment of “lipid metabolism” and “transport” GO terms in the 674 GM-BMM-specific proteins (Figure 1E). These included monoglyceride lipase (MGLL), long-chain fatty-acid-CoA ligase 1 (ACSL1), and several transport proteins (Figure 1F). Alternatively, “viral defense” and “immune system processes” GO terms were enriched in the 627 M-BMM-specific proteins (Figure 1E). These include the 2’-5’-oligoadenylate synthaselike protein 2 (OASL2) and protein-tyrosine kinase 1-β (PTK2b), among other key viral response proteins (Figure 1F). Overall, our data set correlates well with that reported by Na et al.41 (Figure S2B).
Figure 1.
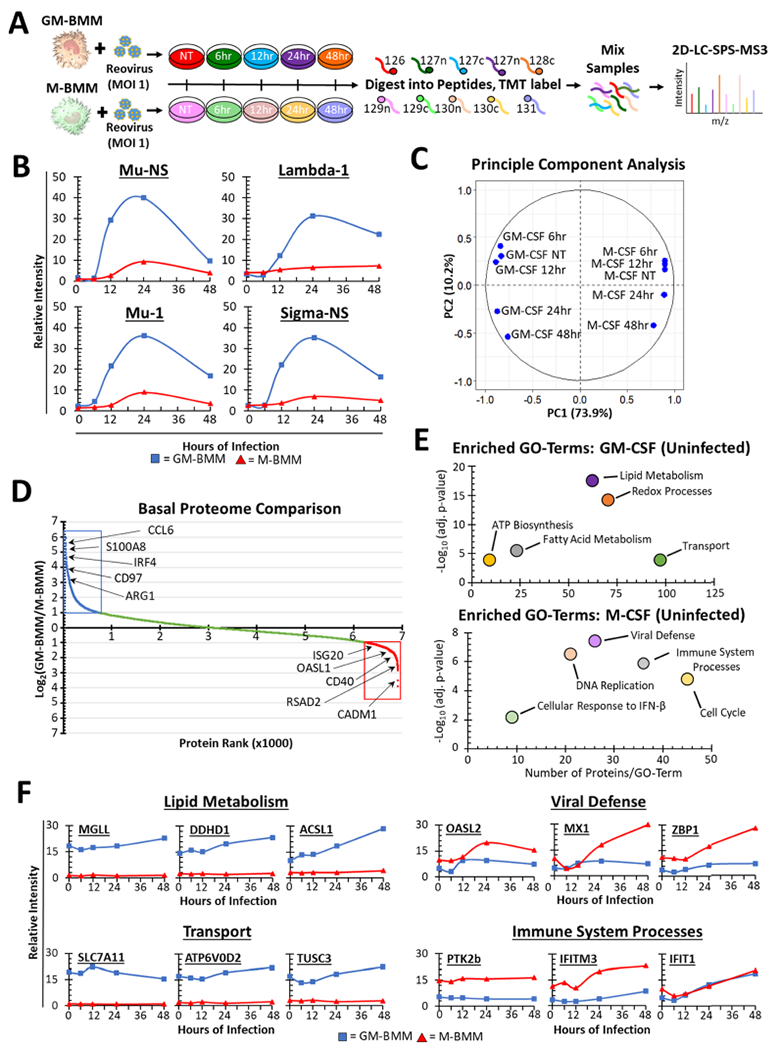
Quantitative proteomic analysis of GM-BMMs and M-BMMs during reovirus infection. (A) Experimental design of the quantitative temporal proteomics of reovirus-infected GM-BMMs and M-BMMs. After differentiation for 7 days, cultures were treated with reovirus (MOI 1) for 0, 6, 12, 24, or 48 h and subjected to multiplexed proteomics. Bone marrow from five naive C57BL/6 mice were pooled together for each time point. (B) Induction of reovirus proteins measured in GM-BMMs and M-BMMs. (C) Principal component analysis (PCA) of proteomics data from reovirus-infected GM-BMMs and M-BMMs. (D) Basal log2(GM-BMM/M-BMM) ratios in uninfected cells. (E) DAVID GO-term enrichment in GM-BMM and M-BMM specific proteins (basal). Shown are the –log(p-value) (y axis) versus the number of proteins identified in each GO term (x axis). (F) Representative proteins from GO terms identified in (E). *adj. p-value = Benjamini–Hochberg value.
Pavlidis template matching33 revealed multiple cellular proteins sharing similar expression profiles (p-value <0.05) to those of the viral protein Mu-1. These include triggering receptor expressed in myeloid cells 4 (TREM4) and GM20547, which have not previously been associated with viral responses, suggesting that they have a role in virus uptake or infection (Figure S2C). Conversely, MS4A4C increased similarly in GM-BMMs and M-BMMs (Figure S2C). Altogether, our comprehensive data set is thus a useful resource in identifying known and yet uncharacterized proteins in macrophages induced following exposure to the virus. The quantitative proteomics data set was submitted to Mendeley Data and is available with the DOI: 10.17632/tnbth6sn7s.1
Oncolytic Reovirus Induces Distinct Patterns of Protein Expression in GM-BMMs and M-BMMs
To determine temporal patterns of virus-induced proteins, we employed K-means clustering of the data set (n = 10 clusters), revealing early versus late patterns of proteins differing between GM-BMMs and M-BMMs (Figure 2A). Enrichment analysis of clusters using DAVID34 revealed functional enrichments of “inflammatory response”, “MHC class-II antigen presentation”, and “chemotaxis” among the early-induced proteins in GM-BMMs (cluster 2 in Figure 2A) (Figure 2B–i). Alternatively, “antigen presentation via class I”-type GO terms were enriched among late-induced proteins in GM-BMMs (cluster 3 in Figure 2A) (Figure 2B–ii). We observed GO-term enrichment of “viral defense” and “immune system processing” (Figure 2B–iii) among late-induced proteins in the M-BMM proteins (cluster 10 in Figure 2A). Specific examples of proteins that belong to GO terms identified throughout Figure 2B include CD74, CCL2, H2-K1, H2-D1, IFIT3, CASP4, IST1, and SERPING1 (Figure 2C). These data suggest that GM-BMMs initiate an early response to reovirus that after 24 h transitions to antigen processing and presentation.
Figure 2.
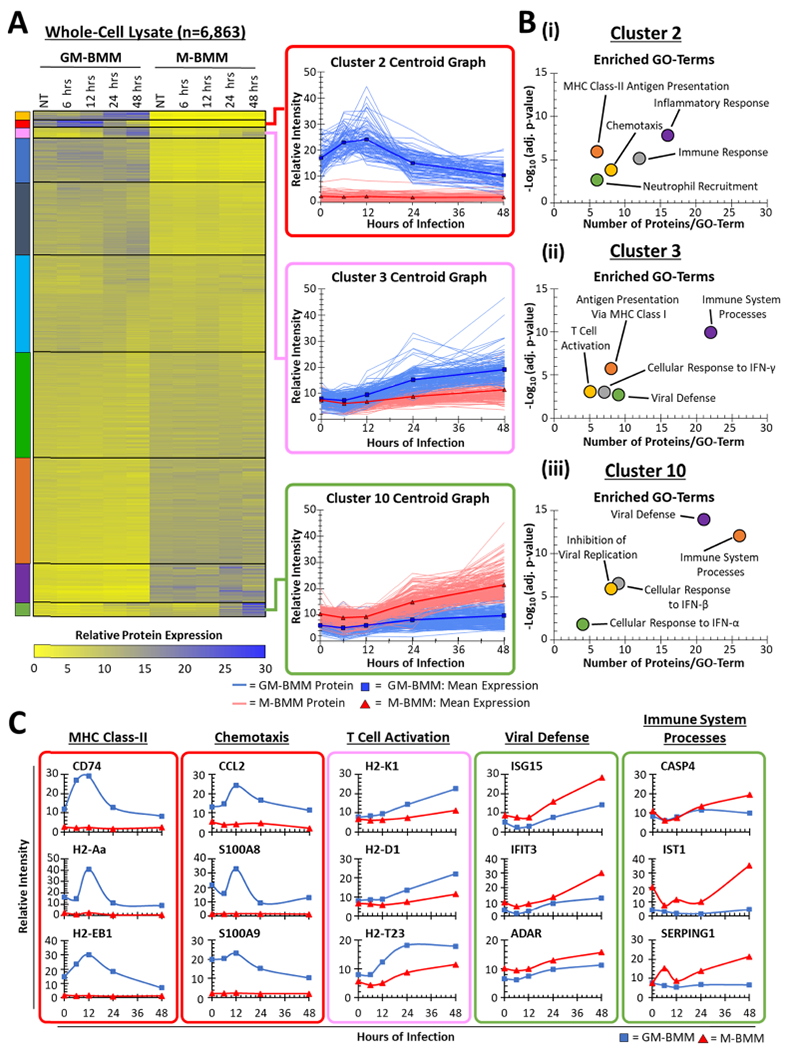
K-means clustering of GM-BMM and M-BMM proteome data. (A) Heat-map of all 6863 proteins identified within the proteome clustered using K-means with 10 classes. Colored boxes indicate the cluster number. All proteins in each cluster are shown in the inset. (B) DAVID GO-term analysis of individual K-means clusters from (A). (C) Representative proteins within selected GO terms from (B). Colored boxes indicate the associated cluster.
GM-BMMs and M-BMMs Differ in the Expression of Proteins Involved in Reovirus Entry and Antiviral Defense
Since reovirus infection requires specific host proteins42–45 (Figure 3A), we examined several of these key proteins known for their role in reovirus binding and viral assembly. Interestingly, we observed higher expression of the reovirus receptor protein JAM-A and the lysosomal tracking protein Src-kinase (SRC)46 in GM-BMMs than in M-BMMs (Figure 3B). Induction of the early-endosome associated markers Rab GTPase (Rab)-5c and Rab7a also occurred only in the GM-BMMs (Figure 3B). Proteolytic viral disassembly proteins cathepsin (CTS)-L, -B, and -S47–52 were all induced or basally 5-fold higher in GM-BMMs than in M-BMMs (Figure 3B). Opposingly, the expression of interferon-inducible transmembrane protein 3 (IFITM3), an inhibitory protein that restricts reovirus infection by modulating late-endosome function,53 in GM-BMMs was more than 2-fold lower than that in M-BMMs (Figure 3B).
Figure 3.
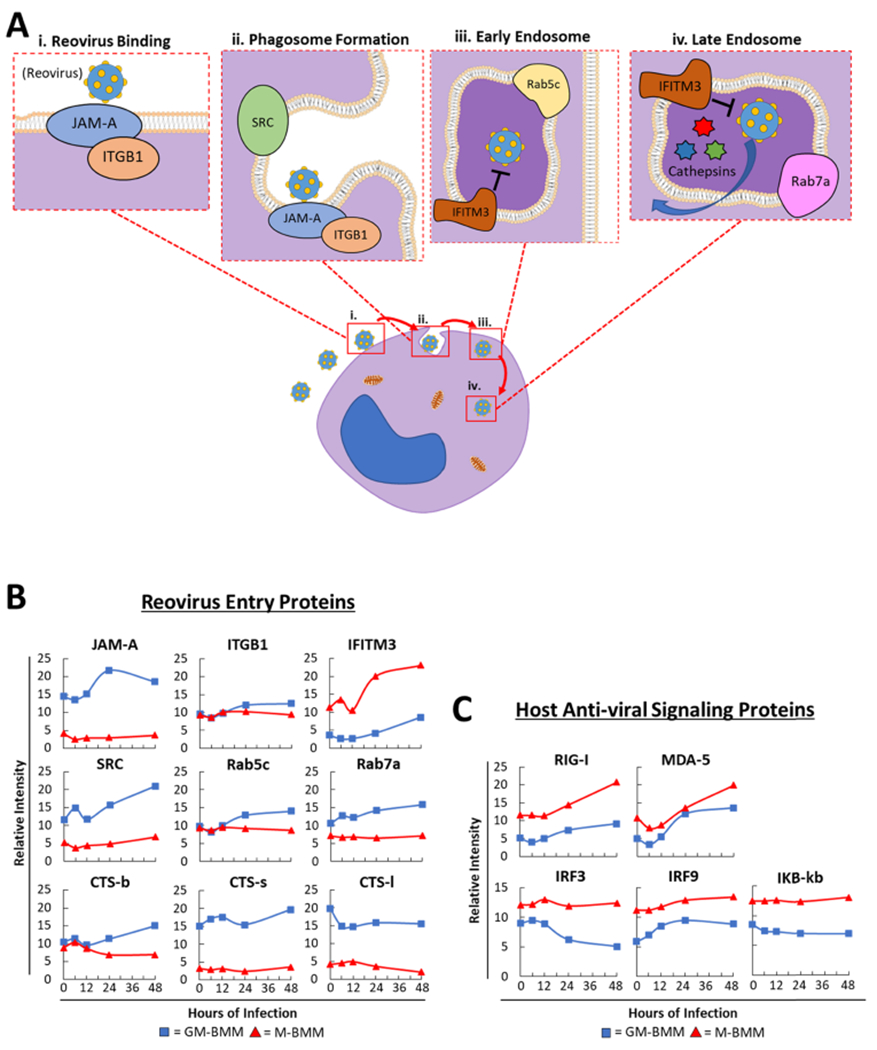
Differential viral entry and antiviral proteins between GM-BMMs and M-BMMs. (A) (i-iv) Schematic representation of reovirus infecting a host cell. Cartoon representation depicts proteins involved in proper reovirus binding, phagosome formation, and early- and late-endosome composition. (B) Relative abundance of different proteins involved in reovirus binding [from (A)], endocytosis, and uncoating between GM-BMMs and M-BMMs. (C) Relative abundance of key host cell antiviral signaling proteins.
We also examined our data set for essential host antiviral signaling proteins and observed 2-fold lower expression of key dsRNA viral sensing proteins retinoic-acid-inducible gene I (RIG-I) and melanoma-differentiation-associated gene-5 (MDA-5) in GM-BMMs than in M-BMMs (Figure 3C). Key antiviral transcription factors interferon regulatory factors 3 and 9 (IRF3 and IRF9) and IKB-B were also less abundant in GM-BMMs (Figure 3C). Collectively, these data showed that GM-BMMs and M-BMMs are differentially equipped with the cellular proteins essential for reovirus entry and mounting antiviral response, respectively.
GM-BMMs Are More Susceptible to Oncolytic Reovirus Infection Than M-BMMs
As a result of differential reovirus protein abundance between GM-BMMs and M-BMMs, we next directly compared their susceptibility to reovirus infection. To determine population-specific reovirus infection among the BMMs, we performed intracellular flow-cytometry-based analysis of reovirus proteins and observed significantly higher infection of CD11b+ F4/80+ cells at both 24 and 48 h in GM-BMMs as compared to M-BMMs (Figure 4A). As a further validation, we measured levels of reoviral mRNA and observed higher copy numbers of reoviral Lambdal and Sigmal mRNA in GM-BMMs after 24 h (Figure 4B). These results were also confirmed using confocal microscopy, which demonstrated higher reoviral staining in GM-BMMs (Figure S3). A dextran assay also demonstrated the overall increased uptake in M-BMMs as compared to GM-BMMs (Figure 4C). Finally, more viable extracellular viral progeny were observed in media from GM-BMMs than in M-BMMs, as measured using a plaque assay at both 24 and 48 h postinfection (Figure 4D). Together, these data validate the greater susceptibility to reovirus infection of GM-BMMs than M-BMMs that was suggested by the proteomics data.
Figure 4.
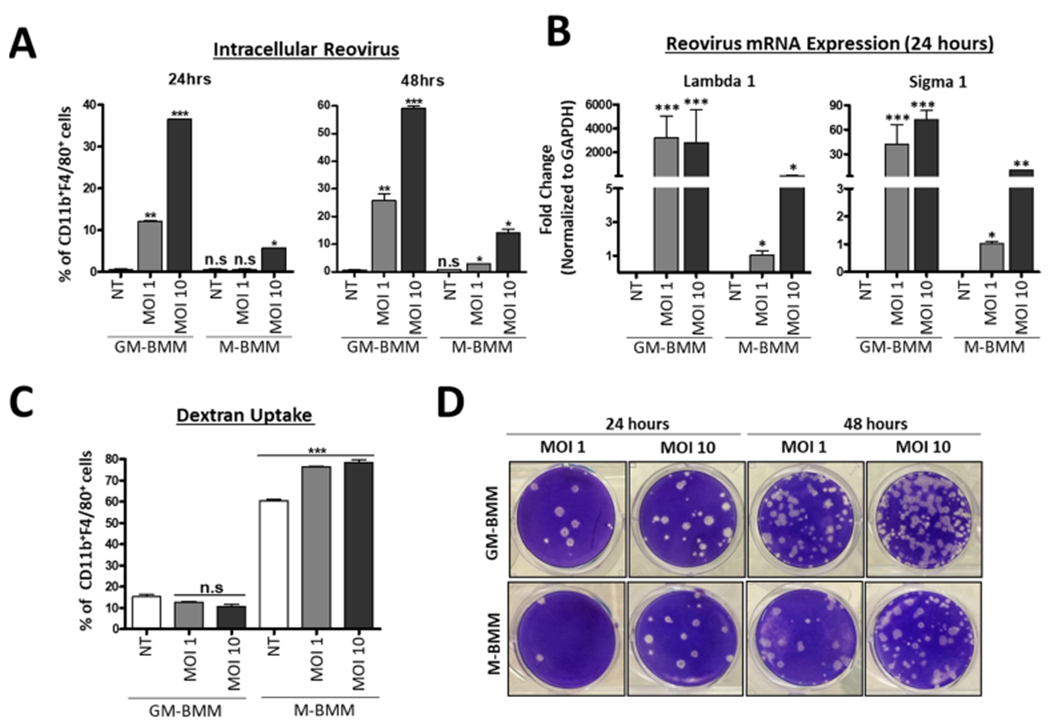
GM-BMMs display higher susceptibility to reovirus infection than M-BMMs. (A) Flow cytometric analysis of CD11b+ F4/80+ Reovirus+ cells from GM-BMMs and M-BMMs after 24 and 48 h of reovirus infection. (B) GAPDH-normalized RT-PCR quantitation of reovirus Lambda1 and Sigma1 following 24 h of infection. (C) Flow cytometric analysis of CD11b+ F4/80+ FITC–dextran+ uptake between GM-BMMs and M-BMMs after 24 h of infection. (D) Infectivity of virus progeny from GM-BMMs and M-BMMs after 24 and 48 h of infection (1:100 dilution). Statistics were performed using a one-way ANOVA (****p ≤ 0.0001, ***p ≤ 0.0005, **p ≤ 0.001, *p ≤ 0.01, and n.s. = p > 0.05).
GM-BMMs and M-BMMs Mount Differential Antiviral Innate and Adaptive Immune Responses
As our proteomics data set revealed differential expression of proteins involved in immune response, we performed several immunological assays to confirm these effects in GM-BMMs and M-BMMs. Interestingly, while IRF3 (which directly binds to the IFN-β promoter54) expression was lower in GM-BMM as compared to M-BMM cells, as identified by proteomics (Figure 3C) and confirmed by western blotting (Figure S4), we observed significantly greater increase in transcript levels of the inflammatory cytokines IFN-Å and -β in GM-BMMs than in M-BMMs with reovirus at MOI 1 and 10 (Figure 5A). This discrepancy between IRF3 expression and type I IFN production is likely due to differences in the activation of IRFs, which is regulated by phosphorylation events rather than protein expression,54 throughout reovirus infection. Similarly, reovirus-infected and LPS-treated GM-BMMs produced higher levels of NO and ROS than M-BMMs (Figure 5B). Together, these findings show that GM-BMMs induce a greater interferon, NO, and ROS response, when infected with reovirus in line with their relatively higher susceptibility to reovirus infection, compared to M-BMMs (Figure 5A,B).
Figure 5.
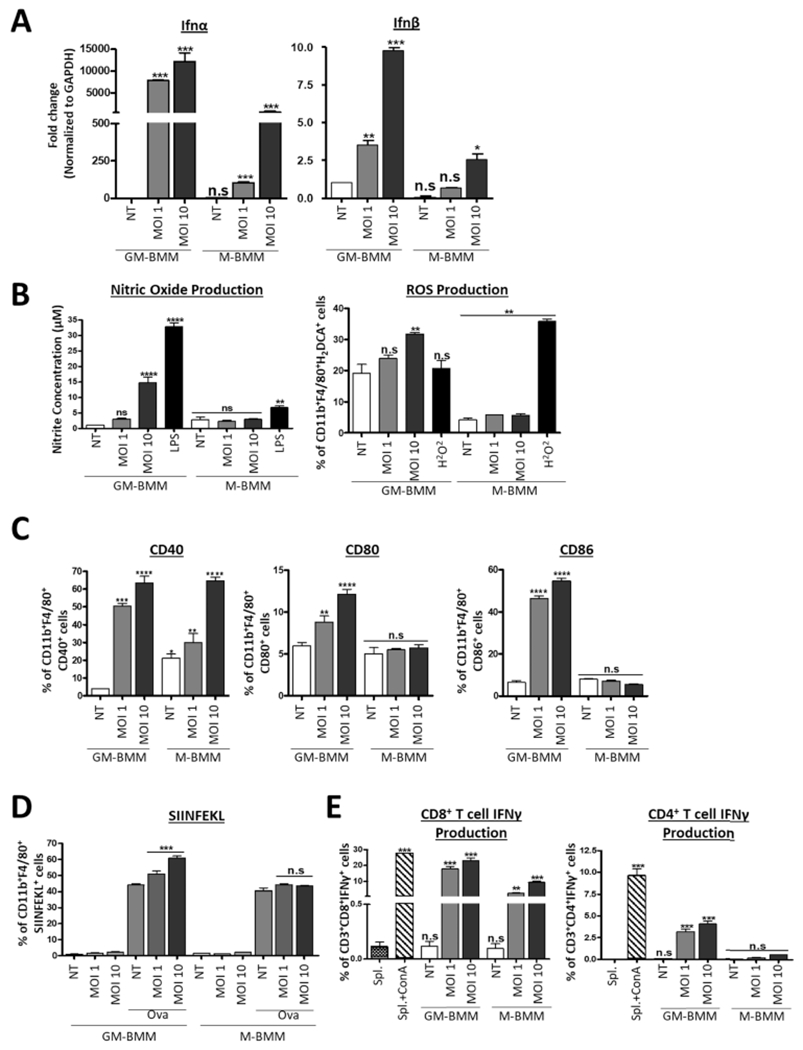
Immunological differences between reovirus-infected GM-BMMs and M-BMMs. (A) qRT-PCR of interferon (IFN)-α and IFN-β in GM-BMMs and M-BMMs after 24 h of reovirus infection. (B) Nitric oxide concentration in GM-BMMs and M-BMMs quantified using the Griess Reagent System and ROS production determination using flow cytometric analysis of DCFDA fluorescence of CD11b+F4/80+ cells. (C) Flow cytometric analysis of co-stimulatory molecule expression on GM-BMMs and M-BMMs after 24 h of infection. Primary gating was on CD11b+F4/80+ cells. (D) Flow cytometric analysis of H2Kb-SIINFEKL in GM-BMMs and M-BMMs cultured in the presence of SIINFEKL after 24 h of reovirus infection. Primary gating was on CD11b+ F4/80+ cells. (E) Flow cytometric analysis of IFNγ+ cells of CD4+ and CD8+ splenic T cell populations from reovirus-infected mice following co-culture with GM-BMMs and M-BMMs. ConA was used as a positive control. Statistical significance was determined using a one-way ANOVA (****p ≤ 0.0001, ***p ≤ 0.0005, **p ≤ 0.001, *p ≤ 0.01, and n.s. = p > 0.05).
Based on differences in antigen presentation proteins in the proteomics data, we also assessed the expression of co-stimulatory molecules CD40, CD80, and CD86 and observed that that GM-BMMs exhibit significantly higher surface expression of all three proteins in response to reovirus compared to M-BMMs (Figure 5C). We also compared the antigen presentation capacity of the CD11b+F4/80+CD40+ population in GM-BMMs and M-BMMs using the ovalbumin CD8 T cell epitope peptide (SIINFEKL), a commonly used surrogate antigen.55 SIINFEKL was presented in both GM-CSF and M-CSF CD11b+F4/80+CD40+ BMMs; however, reovirus enhanced presentation of SIINFEKL only in GM-BMMs (Figure 5D).
Finally, we evaluated how GM-BMMs and M-BMMs infected with reovirus influence the adaptive immune response. Here, BMMs were treated with reovirus (MOI 1 or 10) for 24 h and were then co-cultured for an additional 24 h with splenocytes that were isolated from separate C57BL/6 mice that had been injected with reovirus 7 days prior. By quantifying IFNγ production in T cells, we found that co-culture with GM-BMMs stimulated significantly more IFNγ production by both CD8+ and CD4+ T cells (Figure 5E). Taken together, these data confirm greater adaptive immune responses in GM-BMMs in response to reovirus, which may be extrapolated to other models of inflammatory macrophages important in virus infection. Most importantly, these analyses further validate our proteomics analysis, suggesting that GM-BMMs and M-BMMs mount differential immune responses following exposure to oncolytic reovirus.
■ DISCUSSION AND CONCLUSIONS
Here, we present a comprehensive proteomic data set revealing differential antiviral responses between GM-BMMs and M-BMMs, often used to model proinflammatory (M1-like) and anti-inflammatory (M2-like) macrophages, respectively. The accuracy and depth of SPS-MS3 allowed the creation of a comprehensive data set that will serve as a valuable resource for understanding the role of GM-BMMs and M-BMMs in viral infection. These data expand on previous work by Na et al.41 that compared GM-BMMs and M-BMMs to a depth of 3990 proteins. Additionally, we have identified numerous virus-induced host proteins for which a previous role in viral response has not been described, representing tantalizing targets for future investigations.
Importantly, our data demonstrate that inherent antiviral pathways may make M1-like macrophages more susceptible to oncolytic virus infection than M2-like macrophages, having direct implications for OV therapy. Alternatively, M2-like macrophages more readily initiate antiviral innate and adaptive immune responses following exposure to oncolytic reovirus.
Our observation of differential viral susceptibility and phagocytic differences between macrophages is in accordance with the previous literature,41 but we here shed light on the proteins involved. For example, we show that a key reoviral entry protein, JAM-A, is more abundant in GM-BMMs. Moreover, the low expression of IFITM3 and high expression of cathepsin proteins (CST-S, CST-L) in GM-BMMs suggest that reovirus can escape endosomal degradation and sustain replication relatively better in M1-like macrophages (GM-BMMs).
Clinical data clearly show that the polarization states of macrophages (M1- or M2-like) in the TME affect tumor grade and treatment outcomes.21,56–58 Although it is unclear how well GM-BMMs and M-BMMs reflect M1- and M2-like macrophages found in vivo, GM-CSF and M-CSF are produced by a variety of cells including endothelial cells59 and fibroblasts,60 relevant in models of both cancer and infection.61 BMMs cultured in the presence of GM-CSF or M-CSF may indeed reflect, at least in part, the biology of polarized TAMs that are either anti- (M1-like) or protumorigenic (M2-like). Of note, the use of GM-CSF in combination with reovirus and other oncolytic viruses has been shown to enhance antitumor immunity. Indeed, the FDA-approved OV T-VEC (and other OV formulations like Pexa-Vec) has been genetically modified to overexpress GM-CSF.62–67
In summary, our results provide multiple insights into basal macrophage functions, potential therapeutic targets, and a global overview of how M1-and M2-like macrophage biology is affected by oncolytic reovirus. The data set puts forward an invaluable hypotheses-generating resource that should be of importance when designing an immunotherapeutic regimen using oncolytic reovirus.
Supplementary Material
Temporal reovirus infection in GM-BMM and M-BMM cells (Figure S1); characterization of the GM-BMM and M-BMM proteomic data set (Figure S2); fluorescence microscopy of GM-BMM and M-BMM cells infected with reovirus (Figure S3); IRF3 expression in GM-BMM compared to M-BMM cells (Figure S4) (PDF)
■ ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Cancer Society Research Institute (CCSRI), Canadian Institutes of Health Research (CIHR), and the Terry Fox Research Institute (TFRI). M.A.G. is a trainee in the Cancer Research Training Program (CRTP) of the Beatrice Hunter Cancer Research Institute (BHCRI), with funds provided by the QEII Health Sciences Centre Foundation and the GIVETOLIVE Becky Beaton Award. J.P.M. and B.E.K. are supported through the BHCRI. D.R.C. and Y.K. are supported by CIHR. S.G. is supported by the Dalhousie Medical Research Foundation (DMRF).
■ ABBREVIATIONS
- BMM
bone marrow-derived macrophage
- GM-CSF
granulocytic–macrophage colony stimulating factor
- M-CSF
macrophage colony stimulating factor
- OV
oncolytic virus
- ROS
reactive oxygen species
- TME
tumor microenvironment
- TAM
tumor-associated macrophage
- TMT
tandem mass tag
- ACK
ammonium chloride potassium
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00583.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.9b00583
The authors declare no competing financial interest.
Contributor Information
Michael A. Giacomantonio, Dalhousie University, Halifax, Canada
Andra M. Sterea, Dalhousie University, Halifax, Canada
Youra Kim, Dalhousie University, Halifax, Canada.
Joao A. Paulo, Harvard Medical School, Boston, Massachusetts
Derek R. Clements, Dalhousie University, Halifax, Canada
Barry E. Kennedy, Dalhousie University, Halifax, Canada
Moamen J. Bydoun, Dalhousie University, Halifax, Canada
Ge Shi, Dalhousie University, Halifax, Canada.
David M. Waisman, Dalhousie University, Halifax, Canada
Steven P. Gygi, Harvard Medical School, Boston, Massachusetts.
Carman A. Giacomantonio, Dalhousie University, Halifax, Canada
J. Patrick Murphy, Dalhousie University, Halifax, Canada, and University of Prince Edward Island, Charlottetown, Canada.
Shashi Gujar, Dalhousie University, Halifax, Canada, Beatrice Hunter Cancer Research Institute, Halifax, Canada, and Dalhousie University, Halifax, Canada.
■ REFERENCES
- (1).Gong J; Sachdev E; Mita AC; Mita MM Clinical Development of Reovirus for Cancer Therapy: An Oncolytic Virus with Immune-Mediated Antitumor Activity. World J. Methodol 2016, 6, 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Maitra R; Ghalib MH; Goel S Reovirus: A Targeted Therapeutic-Progress and Potential. Mol. Cancer Res 2012, 10, 1514–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Clements D; Helson E; Gujar SA; Lee PW Reovirus in Cancer Therapy: An Evidence-Based Review. Oncolytic Virother. 2014, 3, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Poh AR; Ernst M Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol 2018, 8, No. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Takeuchi O; Akira S Innate Immunity to Virus Infection. Immunol. Rev 2009, 227, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hughes CE; Benson RA; Bedaj M; Maffia P Antigen-Presenting Cells and Antigen Presentation in Tertiary Lymphoid Organs. Front. Immunol 2016, 7, No. 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Denton NL; Chen C-Y; Scott TR; Cripe TP Tumor-Associated Macrophages in Oncolytic Virotherapy: Friend or Foe? Biomedicines 2016, 4, No. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bolyard C; Meisen WH; Banasavadi-Siddegowda Y; Hardcastle J; Yoo JY; Wohleb ES; Wojton J; Yu J-G; Dubin S; Khosla M; et al. BAI1 Orchestrates Macrophage Inflammatory Response to HSV Infection—Implications for Oncolytic Viral Therapy. Clin. Cancer Res 2017, 23, 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Masemann D; Kother K; Kuhlencord M; Varga G; Roth J; Lichty BD; Rapp UR; Wixler V; Ludwig S Oncolytic Influenza Virus Infection Restores Immunocompetence of Lung Tumor-Associated Alveolar Macrophages. Oncoimmunology 2018, 7, No. e1423171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ilett E; Kottke T; Donnelly O; Thompson J; Willmon C; Diaz R; Zaidi S; Coffey M; Selby P; Harrington K; et al. Cytokine Conditioning Enhances Systemic Delivery and Therapy of an Oncolytic Virus. Mol. Ther 2014, 22, 1851–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gordon S; Martinez FO Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [DOI] [PubMed] [Google Scholar]
- (12).Parihar A; Eubank TD; Doseff AI Monocytes and Macrophages Regulate Immunity through Dynamic Networks of Survival and Cell Death. J. Innate Immun 2010, 2, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gordon S Alternative Activation of Macrophages. Nat. Rev. Immunol 2003, 3, 23–35. [DOI] [PubMed] [Google Scholar]
- (14).Lehmann B; Biburger M; Bruckner C; Ipsen-Escobedo A; Gordan S; Lehmann C; Voehringer D; Winkler T; Schaft N; Dudziak D; et al. Tumor Location Determines Tissue-Specific Recruitment of Tumor-Associated Macrophages and Antibody-Dependent Immunotherapy Response. Sci. Immunol 2017, 2, No. eaah6413. [DOI] [PubMed] [Google Scholar]
- (15).Ciavarra RP.; Taylor L; Greene AR; Yousefieh N; Horeth D; van Rooijen N; Steel C; Gregory B; Birkenbach M; Sekellick M Impact of Macrophage and Dendritic Cell Subset Elimination on Antiviral Immunity, Viral Clearance and Production of Type 1 Interferon. Virology 2005, 342, 177–189. [DOI] [PubMed] [Google Scholar]
- (16).Martinez FO; Gordon S The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, No. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ushach I; Zlotnik A Biological Role of Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) and Macrophage Colony-Stimulating Factor (M-CSF) on Cells of the Myeloid Lineage. J. Leukocyte Biol 2016, 100, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lacey DC; Achuthan A; Fleetwood AJ; Dinh H; Roiniotis J; Scholz GM; Chang MW; Beckman SK; Cook AD; Hamilton JA Defining GM-CSF– and Macrophage-CSF– Dependent Macrophage Responses by In Vitro Models. J. Immunol 2012, 188, 5752–5765. [DOI] [PubMed] [Google Scholar]
- (19).Hamilton TA; Zhao C; Pavicic PG; Datta S Myeloid Colony-Stimulating Factors as Regulators of Macrophage Polarization. Front. Immunol 2014, 5, No. 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lisi L; Ciotti GMP; Braun D; Kalinin S; Currò D; Russo CD; Coli A; Mangiola A; Anile C; Feinstein DL; et al. Expression of INOS, CD163 and ARG-1 Taken as M1 and M2 Markers of Microglial Polarization in Human Glioblastoma and the Surrounding Normal Parenchyma. Neurosci. Lett 2017, 645, 106–112. [DOI] [PubMed] [Google Scholar]
- (21).Steidl C; Lee T; Shah SP; Farinha P; Han G; Nayar T; Delaney A; Jones SJ; Iqbal J; Weisenburger DD; et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N. Engl. J. Med 2010, 362, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).De Palma M; Lewis CE Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [DOI] [PubMed] [Google Scholar]
- (23).Coffey MC; Strong JE; Forsyth PA; Lee PW Reovirus Therapy of Tumors with Activated Ras Pathway. Science 1998, 282, 1332–1334. [DOI] [PubMed] [Google Scholar]
- (24).Gujar SA; Clements D; Dielschneider R; Helson E; Marcato P; Lee PWK Gemcitabine Enhances the Efficacy of Reovirus-Based Oncotherapy through Anti-Tumour Immunological Mechanisms. Br. J. Cancer 2014, 110, 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Marcato P; Shmulevitz M; Pan D; Stoltz D; Lee PW Ras Transformation Mediates Reovirus Oncolysis by Enhancing Virus Uncoating, Particle Infectivity, and Apoptosis-Dependent Release. Mol. Ther 2007, 15, 1522–1530. [DOI] [PubMed] [Google Scholar]
- (26).Fleetwood AJ; Lawrence T; Hamilton JA; Cook AD Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage CSF-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J. Immunol 2007, 178, 5245–5252. [DOI] [PubMed] [Google Scholar]
- (27).Livak KJ; Schmittgen TD Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- (28).Rappsilber J; Ishihama Y; Mann M Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem 2003, 75, 663–670. [DOI] [PubMed] [Google Scholar]
- (29).McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ting L; Rad R; Gygi SP; Haas W MS3 Eliminates Ratio Distortion in Isobaric Multiplexed Quantitative Proteomics. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Elias JE; Gygi SP Target-Decoy Search Strategy for Mass Spectrometry-Based Proteomics. Methods Mol. Biol 2010, 604, 5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Howe E; Holton K; Nair S; Schlauch D; Sinha R; Quackenbush J MeV: MultiExperiment Viewer. In Biomedical Informatics for Cancer Research;Ochs MF; Casagrande JT; Davuluri RV, Eds.; Springer US: Boston, MA, 2010; pp 267–277. [Google Scholar]
- (34).Huang DW; Sherman BT; Lempicki RA Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc 2009, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- (35).Clements DR; Murphy JP; Sterea A; Kennedy BE; Kim Y; Helson E; Almasi S; Holay N; Konda P; Paulo JA; et al. Quantitative Temporal in Vivo Proteomics Deciphers the Transition of Virus-Driven Myeloid Cells into M2 Macrophages. J. Proteome Res 2017, 16, 3391–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee M-C; Lacey DC; Fleetwood AJ; Achuthan A; Hamilton JA; Cook AD GM-CSF-and IRF4-Dependent Signaling Can Regulate Myeloid Cell Numbers and the Macrophage Phenotype during Inflammation. J. Immunol 2019, 3033. [DOI] [PubMed] [Google Scholar]
- (37).Xia C; Braunstein Z; Toomey AC; Zhong J; Rao X S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol 2018, 8, No. 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang Y; Zhang Z; Zhang L; Li X; Lu R; Xu P; Zhang X; Dai M; Dai X; Qu J; et al. S100A8 Promotes Migration and Infiltration of Inflammatory Cells in Acute Anterior Uveitis. Sci. Rep 2016, 6, No. 36140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rantakari P; Patten DA; Valtonen J; Karikoski M; Gerke H; Dawes H; Laurila J; Ohlmeier S; Elima K; Hubscher SG; et al. Stabilin-1 Expression Defines a Subset of Macrophages That Mediate Tissue Homeostasis and Prevent Fibrosis in Chronic Liver Injury. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 9298–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cai H; Zhu X-D; Ao J-Y; Ye B-G; Zhang Y-Y; Chai ZT; Wang C-H; Shi W-K; Cao M-Q; Li X-L; et al. Colony-Stimulating Factor-1-Induced AIF1 Expression in Tumor-Associated Macrophages Enhances the Progression of Hepatocellular Carcinoma. Oncoimmunology 2017, 6, No. e1333213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Na YR; Hong JH; Lee MY; Jung JH; Jung D; Kim YW; Son D; Choi M; Kim KP; Seok SH Proteomic Analysis Reveals Distinct Metabolic Differences Between Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) and Macrophage Colony Stimulating Factor (M-CSF) Grown Macrophages Derived from Murine Bone Marrow Cells. Mol. Cell. Proteomics 2015, 14, 2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lai CM; Boehme KW; Pruijssers AJ; Parekh VV; Van Kaer L; Parkos CA; Dermody TS Endothelial JAM-A Promotes Reovirus Viremia and Bloodstream Dissemination. J. Infect. Dis 2015, 211, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Thete D; Snyder AJ; Mainou BA; Danthi P Reovirus M1 Protein Affects Infectivity by Altering Virus-Receptor Interactions. J. Virol 2016, 90, 10951–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Maginnis MS; Forrest JC; Kopecky-Bromberg SA; Dickeson SK; Santoro SA; Zutter MM; Nemerow GR; Bergelson JM; Dermody TS B1 Integrin Mediates Internalization of Mammalian Reovirus. J. Virol 2006, 80, 2760–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Forrest JC; Campbell JA; Schelling P; Stehle T; Dermody TS Structure-Function Analysis of Reovirus Binding to Junctional Adhesion Molecule 1 IMPLICATIONS FOR THE MECHANISM OF REOVIRUS ATTACHMENT. J. Biol. Chem 2003, 278, 48434–48444. [DOI] [PubMed] [Google Scholar]
- (46).Mainou BA; Dermody TS Src Kinase Mediates Productive Endocytic Sorting of Reovirus during Cell Entry. J. Virol 2011, 85, 3203–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mainou BA; Dermody TS Transport to Late Endosomes Is Required for Efficient Reovirus Infection. J. Virol 2012, 86, 8346–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Danthi P; Guglielmi KM; Kirchner E; Mainou B; Stehle T; Dermody TS From Touchdown to Transcription: The Reovirus Cell Entry Pathway. Curr. Top. Microbiol. Immunol 2010, 343, 91–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mainou BA; Dermody TS In Search of Cathepsins: How Reovirus Enters Host Cells. DNA Cell Biol. 2012, 31, 1646–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ebert DH; Deussing J; Peters C; Dermody TS Cathepsin L and Cathepsin B Mediate Reovirus Disassembly in Murine Fibroblast Cells. J. Biol. Chem 2002, 277, 24609–24617. [DOI] [PubMed] [Google Scholar]
- (51).Terasawa Y; Hotani T; Katayama Y; Tachibana M; Mizuguchi H; Sakurai F Activity Levels of Cathepsins B and L in Tumor Cells Are a Biomarker for Efficacy of Reovirus-Mediated Tumor Cell Killing. Cancer Gene Ther. 2015, 22, 188–197. [DOI] [PubMed] [Google Scholar]
- (52).Alain T; Kim TS; Lun X; Liacini A; Schiff LA; Senger DL; Forsyth PA Proteolytic Disassembly Is a Critical Determinant for Reovirus Oncolysis. Mol. Ther 2007, 15, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Anafu AA; Bowen CH; Chin CR; Brass AL; Holm GH Interferon-Inducible Transmembrane Protein 3 (IFITM3) Restricts Reovirus Cell Entry. J. Biol. Chem 2013, 288, 17261–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Honda K; Takaoka A; Taniguchi T Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [DOI] [PubMed] [Google Scholar]
- (55).Tähtinen S; Kaikkonen S; Merisalo-Soikkeli M; Grönberg-Vähä-Koskela S; Kanerva A; Parviainen S; Vähä-Koskela M; Hemminki A Favorable Alteration of Tumor Microenvironment by Immunomodulatory Cytokines for Efficient T-Cell Therapy in Solid Tumors. PLoS One 2015, 10, No. e0131242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Brown JM; Recht L; Strober S The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res 2017, 3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ahn G-O; Tseng D; Liao C-H; Dorie MJ; Czechowicz AA. ; Brown JM Inhibition of Mac-1 (CD11b/CD18) Enhances Tumor Response to Radiation by Reducing Myeloid Cell Recruitment. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 8363–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Noy R; Pollard JW Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bagby GC; Dinarello CA; Wallace P; Wagner C; Hefeneider S; McCall E Interleukin 1 Stimulates Granulocyte Macrophage Colony-Stimulating Activity Release by Vascular Endothelial Cells. J. Clin. Invest 1986, 78, 1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zucali JR; Dinarello CA; Oblon DJ; Gross MA; Anderson L; Weiner RS Interleukin 1 Stimulates Fibroblasts to Produce Granulocyte-Macrophage Colony-Stimulating Activity and Prostaglandin E2. J. Clin. Invest 1986, 77, 1857–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Nebiker CA; Han J; Eppenberger-Castori S; Iezzi G; Hirt C; Amicarella F; Cremonesi E; Huber X; Padovan E; Angrisani B ; et al. GM-CSF Production by Tumor Cells Is Associated with Improved Survival in Colorectal Cancer. Clin. Cancer Res 2014, 20, 3094–3106. [DOI] [PubMed] [Google Scholar]
- (62).Heo J; Reid T; Ruo L; Breitbach CJ; Rose S; Bloomston M ; Cho M; Lim HY; Chung HC; Kim CW; et al. Randomized Dose-Finding Clinical Trial of Oncolytic Immunotherapeutic Vaccinia JX-594 in Liver Cancer. Nat. Med 2013, 19, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kaufman H; Wagner VJ; Goldsweig H; Yao B; Coffin R OPTiM: A Randomized Phase III Trial to Evaluate the Efficacy and Safety of Talimogene Laherparepvec (T-VEC) Compared with Subcutaneously (Sc) Administered GM-CSF for the Treatment (Tx) of Unresectable Stage IIIb, IIIc, and IV Melanoma. J. Clin. Oncol 2012, 30, No. TPS8604. [Google Scholar]
- (64).Kaufman HL; Bines SD OPTIM Trial: A Phase III Trial of an Oncolytic Herpes Virus Encoding GM-CSF for Unresectable Stage III or IV Melanoma. Future Oncol. 2010, 6, 941–949. [DOI] [PubMed] [Google Scholar]
- (65).Andtbacka RHI; Collichio FA; Amatruda T; Senzer NN ; Chesney J; Delman KA; Spitler LE; Puzanov I; Doleman S; Ye Y; et al. OPTiM: A Randomized Phase III Trial of Talimogene Laherparepvec (T-VEC) versus Subcutaneous (SC) Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) for the Treatment (Tx) of Unresected Stage IIIB/C and IV Melanoma. J. Clin. Oncol 2013, 31, No. LBA9008. [Google Scholar]
- (66).Kim S-G; Ha HK; Lim S; De Silva NS; Pelusio A; Mun JH; Patt RH; Breitbach CJ; Burke JM Phase II Trial of Pexa-Vec (Pexastimogene Devacirepvec; JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus, in Patients with Metastatic, Refractory Renal Cell Carcinoma (RCC). J. Clin. Oncol 2018, 36, No. 671. [Google Scholar]
- (67).Samson A; Scott KJ; Taggart D; West EJ; Wilson E; Nuovo GJ; Thomson S; Corns R; Mathew RK; Fuller MJ; et al. Intravenous Delivery of Oncolytic Reovirus to Brain Tumor Patients Immunologically Primes for Subsequent Checkpoint Blockade. Sci. Transl Med 2018, 10, No. eaam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Temporal reovirus infection in GM-BMM and M-BMM cells (Figure S1); characterization of the GM-BMM and M-BMM proteomic data set (Figure S2); fluorescence microscopy of GM-BMM and M-BMM cells infected with reovirus (Figure S3); IRF3 expression in GM-BMM compared to M-BMM cells (Figure S4) (PDF)