

Abstract
The microbiota influences host metabolism and obesity, yet organisms that protect from disease remain unknown. During studies interrogating an immune pathway that regulates microbiota composition, we observed the development of age-associated metabolic-syndrome driven by the microbiota. Expansion of Desulfovibrio and loss of Clostridia were key features associated with obesity in this model and replacement of Clostridia rescues obesity. T-cell dependent events jwere required to prevent loss of Clostridia and expansion of Desulfovibrio. Inappropriate IgA targeting of Clostridia and increased Desulfovibrio antagonized the colonization of beneficial Clostridia. Transcriptional and metabolic analysis revealed enhanced lipid absorption in the obese host. Colonization of germfree animals with Clostridia, but not Desulfovibrio, downregulated the expression of genes controlling lipid absorption and reduced adiposity. Moreover, supernatants from Clostridia suppressed the expression of lipid absorption genes within intestinal epithelia. Reduced Clostridia and increased Desulfovibrio were microbiota features found in humans with metabolic syndrome and obesity. Thus, immune control of the microbiota appears to maintain beneficial microbial populations that function to constrain lipid metabolism to prevent metabolic defects.
Introduction
Over the past century, obesity and metabolic syndrome have developed into a global epidemic. Currently, over 1.9 billion people are obese and at risk of developing metabolic dysfunctions such as type II diabetes, cardiovascular, and liver disease (1). Multiple studies have highlighted a role for immune-system regulation of metabolic disease. These reports have largely focused on the role of inflammatory responses during obesity. They reported increased macrophage infiltration and a reduction in regulatory T cells within the adipose tissue during weight gain (2, 3). However, a number of human studies suggest that suboptimal immune responses are also associated with metabolic syndrome and obesity. Indeed, obese adults show deficient immune responses to immunizations, increased incidence of infection and reduced mucosal IgA levels, suggesting that effective immunity cannot be mounted within these individuals (4–9). The mechanisms by which defective immune reactions influence metabolic disease remain unclear.
The microbiota has emerged as a key regulator of metabolism within the mammalian host, and the composition of the microbiota in obese individuals is sufficient to confer metabolic defects when transferred into animals (10). In particular, reductions in the gene richness of the microbiota have been reported during metabolic disease, including decreased butyrate and methane production. Conversely, some microbiota functions, such as hydrogen sulfide and mucus degradation, are enhanced in individuals with metabolic disease (11). We and others have recently shown that gut immune responses are critical in regulating the composition of the microbiota (12, 13). IgA, in particular, functions to constrain the outgrowth of certain microbes and diversify the microbiota; changes in IgA binding of microbes or, even slight reductions in gut IgA, can negatively affect diversity (12–14). Thus, defective immune control of the microbiota may contribute to metabolic disease.
Results
We recently identified a molecular pathway that instructs the appropriate development of T cell-dependent IgA targeting of the microbiota. Animals that possess a T cell specific ablation of the innate adaptor molecule, Myd88 (T-Myd88−/− mice) have defective T follicular helper (TFH) cell development and IgA production within the gut. This was associated with dysregulated IgA targeting of gut microbes and compositional differences within the microbiota between genotypes (12, 14). During these studies, we observed that older T-Myd88−/− mice were consistently obese compared to their wild-type controls (Fig. 1A). Despite being fed a standard chow diet, T-Myd88−/− mice exhibited significantly increased weight gain and fat accumulation as they aged (Fig. 1B and C and fig. S1A and B). By one year of age, male T-Myd88−/− mice weighed up to 60g and exhibited a 50% body fat composition based on NMR analysis (Fig.1D and E).
Fig. 1. Defective T cell signaling in the gut leads to age-associated obesity.
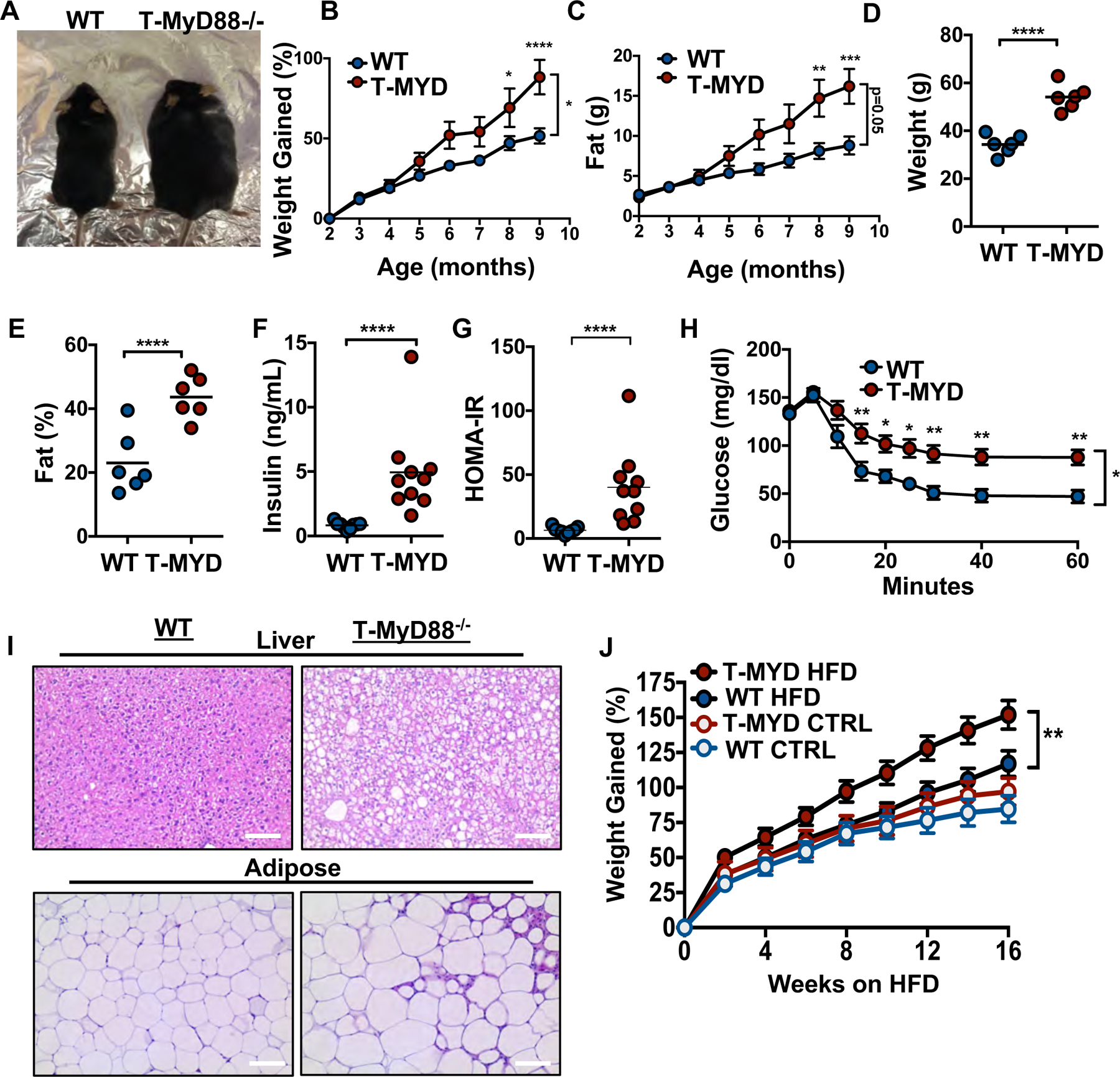
(A) Representative image of 6-month WT and T-Myd88−/− mice. (B) Percentage of weight gained as mice age, starting at 2 months of age (WT, n=8; T-Myd88−/−, n=7 plotted). Representative of three independent experiments. (C) Fat accumulation as mice age, starting at 2 months of age (WT, n=8; T-Myd88−/−, n=7 plotted.) Representative of three independent experiments. (D) Total weight of 1-year-old WT and T-Myd88−/− mice (n=6). Representative of three independent experiments. (E) Total fat percentage as measured by NMR of 1-year-old WT and T-Myd88−/− mice (n=6). Representative of three independent experiments. (F) Fasting serum insulin concentrations from 1-year-old WT and T-Myd88−/− mice (WT, n=9; T-Myd88−/−, n=10). Data pooled from three independent experiments. (G) Homeostatic model assessment (HOMA-IR) of 1-year-old WT and T-Myd88−/− mice. (WT, n=9; T-Myd88−/−, n=10). Data pooled from three independent experiments. (H) Blood glucose levels measured over time following i.p. insulin (0.75 U/kg) injection during insulin-resistance test (WT, n=9; T-Myd88−/−, n=10). Data pooled from three independent experiments. (I) Representative hematoxylin and eosin staining of liver and VAT tissue from WT and T-Myd88−/− mice, taken with 20X magnification. Scale bar indicates 100 μm. (J) Percentage of weight gained of WT and T-Myd88−/− mice fed a control or HFD (WT CTRL, n=8; WT HFD, n=15; T-Myd88−/− CTRL, n=9; T-Myd88−/− HFD, n=13). P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) using a two-tailed, unpaired t test (B-G) and a repeated measures ANOVA (H,J). Error bars indicate SD.
T-Myd88−/− animals developed many of the metabolic disease co-morbidities found in humans (15). Although one-year-old T-Myd88−/− mice raised on a standard diet cleared glucose to similar levels as their WT counterparts (fig. S1C), they had higher levels of circulating insulin, resulting in a higher HOMA-IR index (Fig. 1F and G). Moreover, when challenged with additional insulin, T-Myd88−/− mice failed to clear glucose with similar kinetics as WT animals, indicating the development of insulin resistance (Fig. 1H). Food intake was decreased in T-Myd88−/− mice at two months of age compared to WT controls but was equivalent in one-year old animals (fig. S1D and E). Additionally, although energy expenditure was decreased in young mice, these changes did not persist over time (fig. S1D). Movement was also similar between WT and T-Myd88−/− mice at both ages and only a modest increase in heat production was measured in older T-MyD88−/− mice compared to WT controls suggesting that these are not the primary cause of increased weight gain as seen in other models (fig. S1F and G) (16). T-Myd88−/− mice also developed fatty liver disease and displayed inflammatory phenotypes within the adipose tissue that were marked by crown-like structures and dysregulated adipocyte size (Fig. 1I). Obesity on a standard mouse chow diet requires months to develop. In contrast, when animals were placed on a high-fat diet (HFD, 45% fat), T-Myd88−/− animals accumulated more weight and visceral adipose tissue (VAT) mass than WT mice as early as 8 weeks after initiation of the diet (Fig. 1J and fig. S2A and B). Thus, T-Myd88−/− animals are prone to developing metabolic syndrome and obesity, which can be accelerated by the increased intake of dietary fat.
We previously reported that the composition of the T-Myd88−/− microbiota is distinct from WT in young animals (12). The microbiota is a known contributor to metabolic function and has been linked with the development of human obesity (17, 18). To initially determine if the microbiota was involved in the metabolic syndrome seen in T-Myd88−/− mice, we placed WT and T-Myd88−/− mice on broad-spectrum antibiotics while feeding them a HFD. WT mice exhibited no difference in weight gain on antibiotics. In contrast, weight gain was completely rescued by antibiotic treatment in T-Myd88−/− animals (Fig. 2A and B). This was accompanied by a reduction in their body fat percentage and VAT mass to levels similar to the fat accumulation observed in lean animals (Fig. 2C and D).
Fig 2. The microbiota is required for weight gain associated with T-Myd88−/− mice.
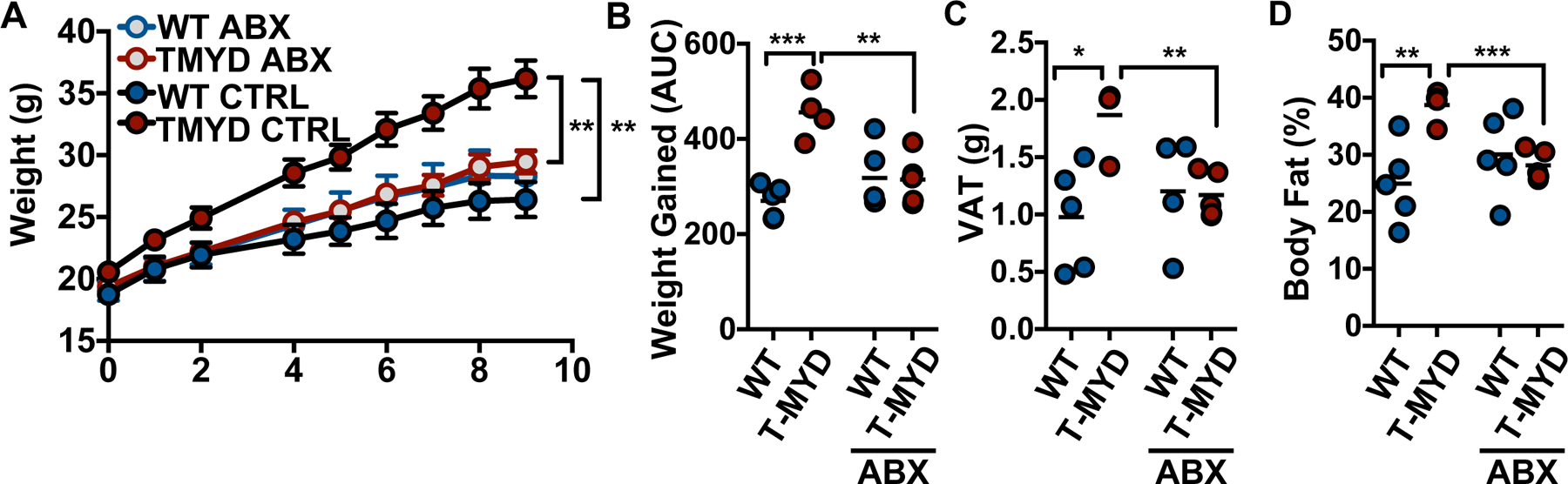
(A) Grams of weight gained measured over time (mean +/− SD), (B) total weight gained (AUC), (C) grams of VAT, and (D) final body fat percentage when WT and T-Myd88−/− mice were fed HFD with or without antibiotics (WT CTRL, n=5; TMYD CTRL, n=4; WT ABX, n=5, TMYD ABX, n=5). Representative of two independent experiments. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) using a repeated measures ANOVA (A) and a two-tailed, unpaired t test (B-D).
In order to determine the features of the microbiota that affect metabolic syndrome in T-Myd88−/− mice, we performed 16S rRNA gene sequencing on normal-chow-fed, aged animals to assess the taxonomic composition and diversity of the microbiota in obese T-Myd88−/− mice. There were significantly different communities in the ileum and fecal contents of aged WT and T-Myd88−/− mice (Fig 3A and fig. S3A). Additionally, there was a slightly reduced species richness in the feces of aged mice (Fig 3B). In order to identify organisms that could explain the major differences between WT and T-Myd88−/− microbiota communities, we performed a random forest analysis on the 16S rRNA data. The fecal microbiota was able to accurately classify genotype with 86% accuracy, whereas the ileal microbiota predicted genotype with 100% accuracy. Members of the microbiota that had the strongest influence on accuracy mostly belonged to the broad taxonomic class Clostridia and were enriched in WT mice compared to T-Myd88−/− mice (Fig 3C and fig. S3B). An additional random forest approach indicated that fecal and ileal microbiota could predict total weight with R2=0.5 and R2=0.76, respectively, with many members of Clostridia strongly influencing this prediction (Fig 3D and fig. S3B). Compared to WT mice, T-Myd88−/− mice showed broad reductions in diversity and overall abundance of multiple Clostridia taxa, including Dorea, SMB53, unclassified Peptostreptococcaceae, and Clostridium (fig S3C).
Fig. 3. Loss of diversity and Clostridia abundance are associated with weight gain in T-Myd88−/− mice.
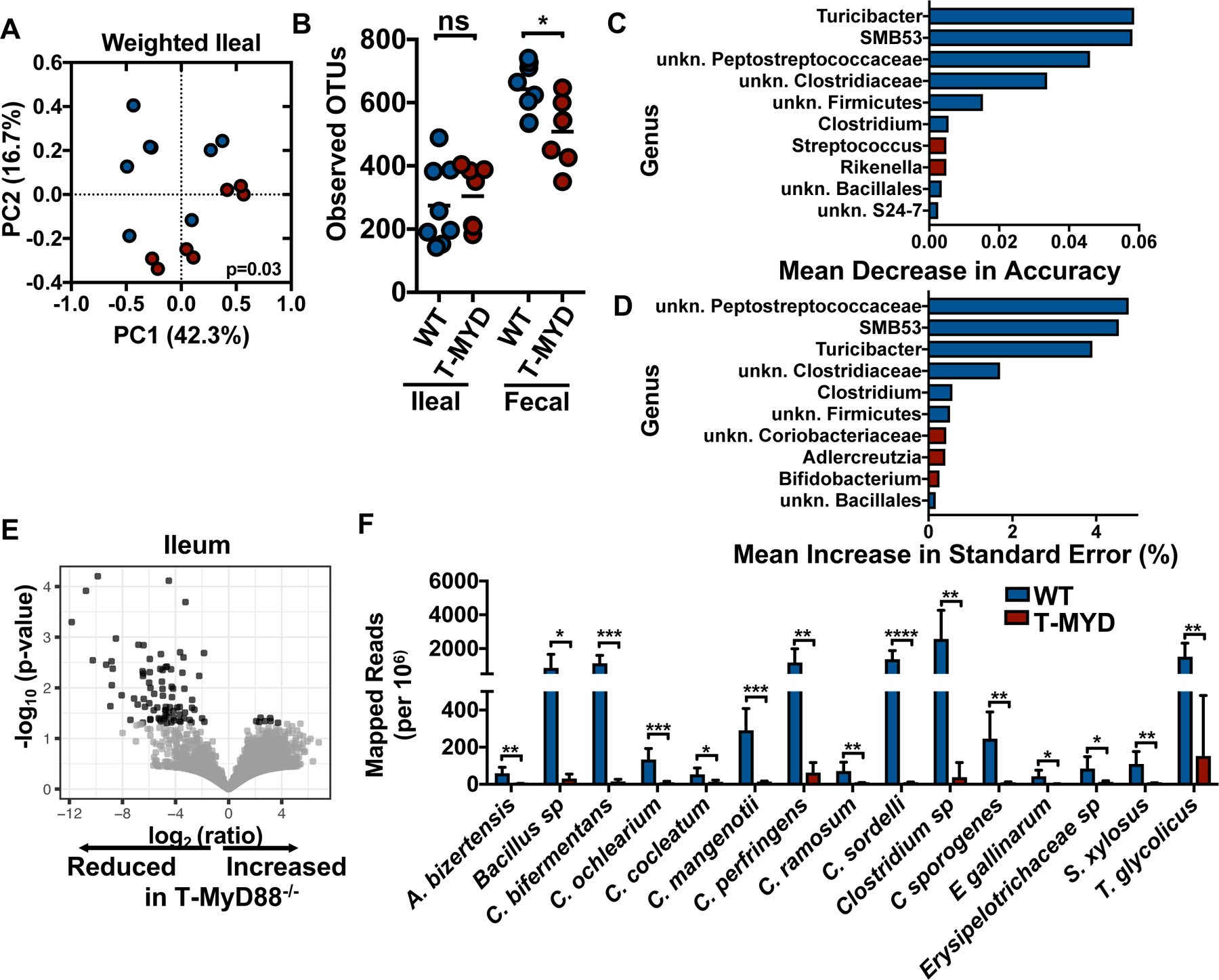
(A) PCoA plot and (B) number of observed OTUs from the ileal microbiota of indicated animals (WT, n=8; T-Myd88−/−, n=7). (C) Top ten bacterial genera influencing mean accuracy of random forest classification between WT and T-Myd88−/− ileal microbiota. Genera with enriched relative abundance in WT animals are shaded blue, genera with enriched relative abundance in T-Myd88−/− animals are shaded red (WT, n=8; T-Myd88−/−, n=7), (D) Top ten bacterial genera influencing standard error in random forest linearization of weight gain and ileal microbiota. Genera with enriched relative abundance in WT animals are shaded blue, genera with enriched relative abundance in T-Myd88−/− animals are shaded red (WT, n=8; T-Myd88−/−, n=7) (E) Volcano plot of ratio of bacterial UniRef90 gene family transcript abundances in ileal samples (n=6 per cohort). (F) Mapped reads per million of significantly different species from WT and T-Myd88−/− ileal microbiota transcripts (n=6 per genotype). Error bars indicate SD. Data in A, B, C and D are from one experiment and data from E, F are from one experiment. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) using permanova (A) and two-tailed unpaired t test (B, F).
Compositional shifts in the microbiota, including reduced microbial diversity, can have negative effects on the functionality of the microbiota and have been correlated with a number of western-lifestyle-associated diseases including metabolic syndrome (16, 17). Additionally, individuals harboring a microbiota with lower gene richness are more likely to be obese (18). In fecal and ileal microbial transcriptomes, the representation of transcripts from a number of gene families within T-Myd88−/− animals was generally reduced. As there were the same number of organisms detected within the ileum by 16S rRNA gene sequencing, this supports the hypothesis that the microbiota at these sites has reduced metabolic functionality (Fig. 3E and fig. S4A). A comparable proportion of total reads uniquely mapped to reference genomes between the two genotypes, suggesting the same coverage of transcriptomes in all animals. However, the proportion of reads mapped to the Clostridiaceae reference genomes in ileal and fecal transcriptomes of T-Myd88−/− mice was, in particular, strikingly reduced (Fig. 3F and fig. S4B and C). Thus, Clostridia present in the obese animals have a reduced functional contribution to the microbiome. Furthermore, obesity is associated with a loss of microbial functional diversity within the Clostridia as has similarly been reported in humans with metabolic disease (11).
As loss of critical Clostridia organisms may play a role during disease, we performed a co-housing experiment to determine whether microbial transfer could rescue obesity (fig. S5A). As mice are coprophagic, co-housing allows for efficient and frequent transfer of microbes between genotypes and has known homogenizing effects on the microbiota. WT or T-Myd88−/− animals were either housed together with animals of the same genotype or co-housed with animals of the opposite genotype upon weaning. Prior to co-housing, T-Myd88−/− mice had a distinct microbiota composition, and one week of cohousing caused mixing of the two communities (fig. S6A). After 1 week, animals were placed on a HFD and monitored for signs of fat accumulation. Compared to separated WT mice, T-Myd88−/− mice and any animal cohoused with them gained significantly more weight, developed insulin resistance, and had increased VAT and total body fat (Fig. 4A and fig. S5B to E). Furthermore, after three months, the microbiota from cohoused WT animals became significantly distinct from separately housed WT mice and showed greater similarity to the microbiota of separately housed T-Myd88−/− (fig. S6B). Thus, a transferable component of the microbiota formed in a T-Myd88−/− animal that can cause metabolic syndrome in an otherwise healthy WT animal.
Fig. 4. Manipulation of gut microbiota influences T-Myd88−/− associated weight gain.
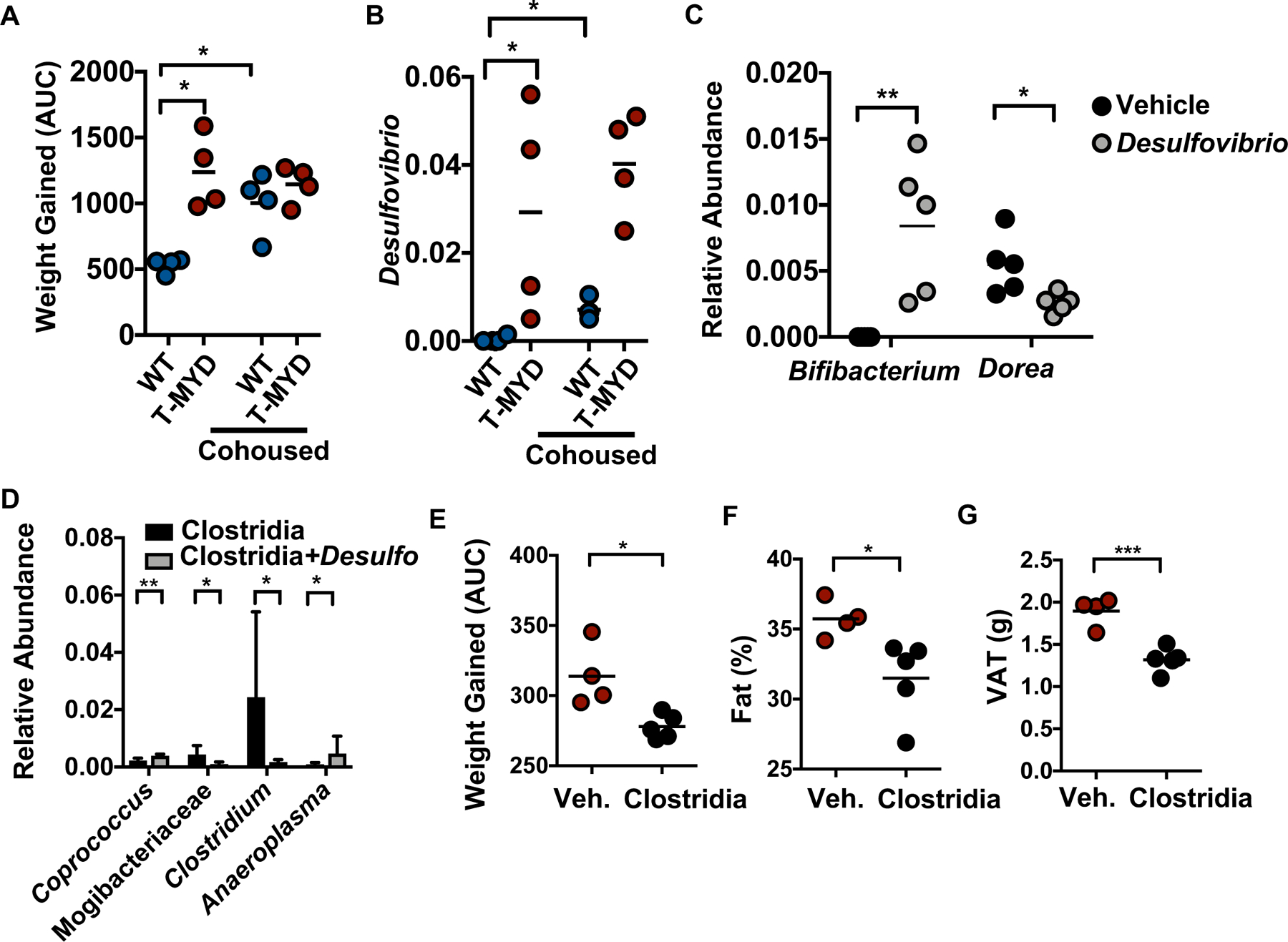
(A) Area under the curve (AUC) of weight gained and (B) relative abundance of Desulfovibrio in WT and T-Myd88−/− mice maintained in separate cages or cohoused and fed a HFD (n=4 per genotype). Representative of two independent experiments. (C) Relative abundance of indicated bacteria within fecal samples from SPF mice colonized with or without D. desulfuricans (n=5 per genotype). (D) Relative abundance of indicated bacteria from 16S sequencing in germfree mice colonized with the Clostridia consortium alone or together with D. desulfuricans (n=5 per cohort). Error bars indicate SD. (E-G), T-Myd88−/− mice were gavaged with vehicle control or spore-forming Clostridia consortium (Vehicle (CTRL), n=4; Clostridia consortium, n=5). Representative of two independent experiments. (E) AUC of weight gained. (F) Total fat percentage as measured by NMR. (G) Grams of VAT. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) using a two-tailed, unpaired t test (A,E-G) and a Mann–Whitney U test (B-D).
As differences in weight gain of cohoused animals were detected within the first 3 weeks, we focused on differences in microbial composition that were detectable at both the early and final time points. After three months of cohousing, Desulfovibrio, Lactobacillales, and Bifidobacterium pseudolongum were all present at greater abundances within cohoused WT mice (Fig. 4B and fig. S6C and D). However, only the Desulfovibrio genus showed significantly greater abundance in separately housed T-Myd88−/− animals and co-housed animals after just one week of co-housing (fig. S6E). Desulfovibrio are mucolytic δ-proteobacteria that produce hydrogen sulfide as a byproduct of disulfide-bond degradation within mucin (19–22). In addition to its association with inflammatory bowel disease (IBD), increased colonization of Desulfovibrio and genes associated with hydrogen sulfide production are detected in patients with type II diabetes and obesity (11). Thus, the community changes in obese mice mimics much of what is seen in humans and suggests that loss of Clostridia and increases in Desulfovibrio is highly relevant to metabolic disease (11, 23, 24).
Cohousing of WT mice with T-Myd88−/− animals that leads to obesity is also associated with reduced colonization of members of Clostridia in WT animals (fig. S6F). Therefore, we hypothesized that Desulfovibrio colonization may reduce the abundance of these organisms. Specific-pathogen-free (SPF) mice were colonized for one week with Desulfovibrio desulfuricans subsp. desulfuricans, a strain that has a 16S rRNA gene sequence similarity of greater than 97% to the Desulfovibrio identified in our mice. Consistent with our hypothesis, WT SPF animals had significant reductions in the Clostridiales family Lachnospiraceae and genus Dorea (Fig. 4C and fig. S7A). Colonization with Desulfovibrio did not result in an overall reduction to all organisms as there was a significant increase in Bifidobacterium (Fig. 4C). As these changes to the community could be an indirect effect of Desulfovibrio colonization, we tested whether Desulfovibrio could influence the colonization of Clostridia members in a germfree system. Germfree animals colonized with chloroform-treated fecal slurries were enriched for Clostridiaceae and Lachnospiraceae (fig. S8). We then analyzed this community in the presence or absence of D. desulfuricans. Desulfovibrio colonization lead to a significant reduction in Clostridium, a genus which strongly influenced the predictive accuracy of both genotype and weight (Fig. 4D). Thus, an expansion of Desulfovibrio species, as seen in T-Myd88−/− mice and humans with type II diabetes, can antagonize the colonization of microbes associated with leanness.
We sought to identify whether reintroducing these lean-associated microbes could protect against obesity within T-Myd88−/− mice. Treatment of obesity-prone T-Myd88−/− animals every other day with a cocktail of spore-forming bacteria significantly reduced weight gain and fat accumulation (Fig. 4E and F). At the end of 3 months, T-Myd88−/− mice treated with spore-forming microbes had a lower body fat percentage and a reduced VAT mass when compared to untreated T-Myd88−/− mice (Fig. 4F and G). Thus, loss of Clostridia is causally associated with obesity and metabolic syndrome in T-Myd88−/− mice.
Microbiota formed during defective gut immunity appears to result in metabolic syndrome. Although co-housing of animals for 12 weeks led to the transmission of obesity into WT hosts, fecal transplants from T-Myd88−/− into WT germfree recipients was insufficient to transfer obesity (fig. S9A). Additionally, when either SPF WT or T-Myd88−/− pregnant dams were co-housed with germfree WT pregnant dams, the resulting colonized pups separated at weaning did not transmit the obesity phenotype (fig. S9A). We hypothesized that immune defects in T-Myd88−/− mice were necessary to allow the persistence of the obesogenic microbiota. In contrast, the microbiota was appropriately controlled in the presence of a fully intact immune system. Tcrb−/− mice, which lack endogenous T cells, were depleted of endogenous microbiota with broad spectrum antibiotic treatment and then colonized with a 1:1 mixture of WT and T-Myd88−/− microbiota prior to adoptive transfer of either WT or T-Myd88−/−CD4+ T cells (fig. S9B). Mice were separated into individually housed cages so that microbiota formation would not be influenced by the presence of other animals within the cage and each microbial community would be shaped independently. Despite the fact that these mice were initially colonized with the same microbiota, Tcrb−/− mice given T-Myd88−/− CD4+ T cells gained significantly more weight when compared to Tcrb−/− mice given WT CD4+ T cells (Fig. 5A). Thus, defects in Myd88 signaling within T cells drives the metabolic defects in animals. Only 10% of bacteria were coated by IgA within Tcrb−/−mice, demonstrating the importance of T cells for IgA targeting of the microbiota (Fig. 3B). However, 1 week post-T cell transfer, mice given WT T cells showed a threefold increase in IgA-bound microbes (fig. S9C). IgG1 or IgG3 responses against the microbiota took longer to develop but were detectable 8 weeks post-T cell transfer (fig. S9D to F). Although, total IgA levels were similar in all animals within this experimental setting, IgA and IgG1 binding to the microbiota was defective in animals receiving knockout T cells (Fig. 5B and fig. S9E and G). Targeting of the microbiota by IgG3, which is believed to be governed by T-cell-independent mechanisms, was not defective in Tcrb−/− mice receiving T-Myd88−/− T cells (fig. S9H) (25). The microbiota composition increasingly differed between genotypes over time (Fig. 5C). Moreover, community changes in animals receiving T cells from obesogenic mice are similar those observed in T-Myd88−/− animals. Indeed, there was a significant negative correlation between the abundance of Desulfovibrionaceae and Clostridiaceae in both genotypes. Animals receiving T-Myd88−/− T cells were ultimately colonized with significantly fewer Clostridiaceae despite starting with the same microbiota admixture (Fig. 5D and E). Three taxa at the genus level were differentially targeted by IgA including the Oscillospira genus of Clostridia, whereas most Clostridia genera were highly variable at this level of taxonomic resolution (Fig S10A). We assessed the IgA-binding index at the finer OTU-level (97% similarity) and found an enrichment of Clostridia-classified OTUs differentially targeted by IgA in animals receiving T-Myd88−/− T cells (Fig 5F). We observed only trending increases in IgA targeting of Desulfovibrio (fig. S10A and B). Thus, reductions in Clostridia and their functional contributions may arise from a combination of inappropriate targeting by IgA and the expansion of Desulfovibrio.
Fig. 5. TFH cell regulation of the microbiota prevents obesity.
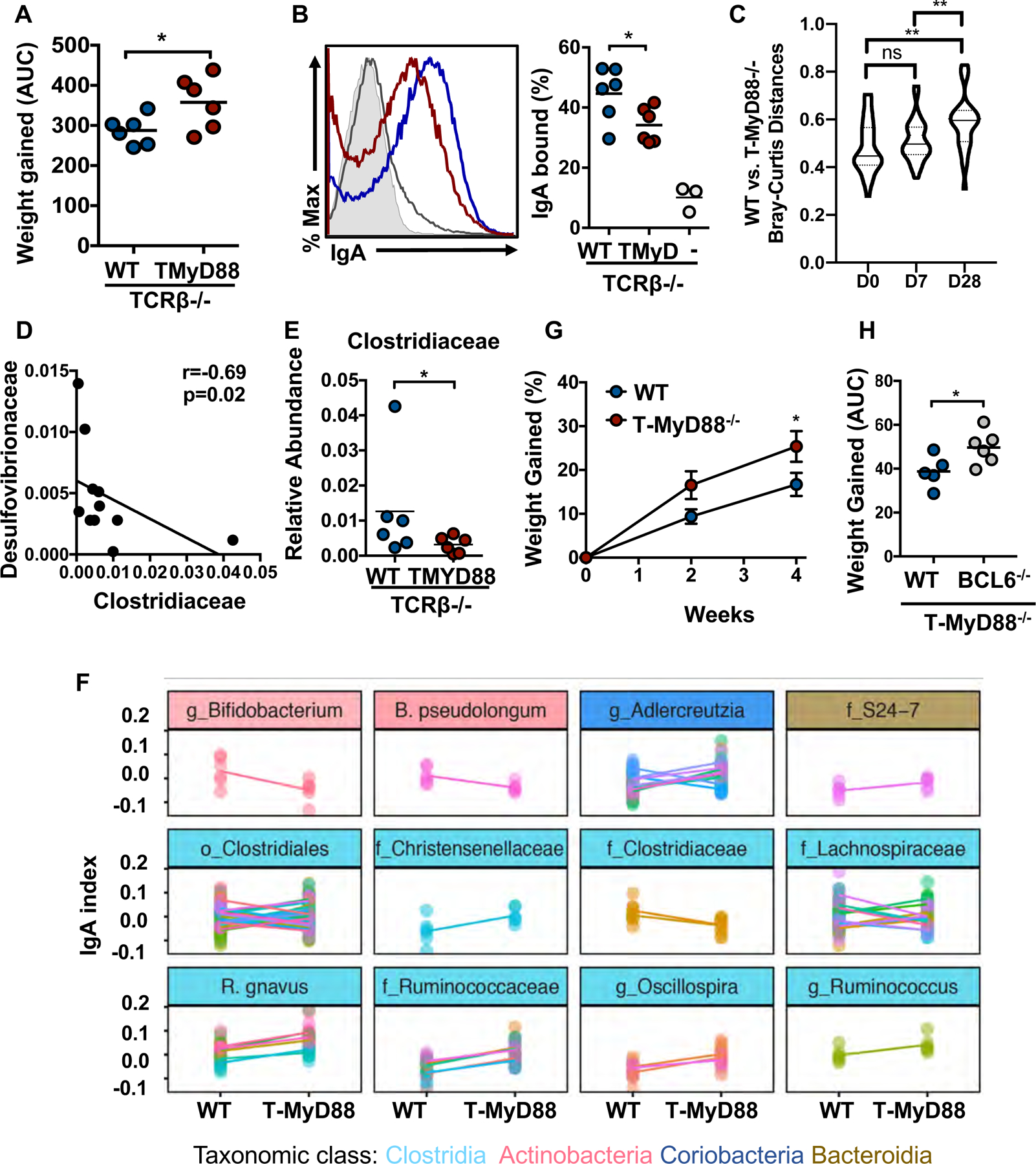
(A-H) Tcrb−/− mice were given a mixture of WT and T-Myd88−/− microbiota one week before being given either WT or T-Myd88−/− T cells. Mice were then individually housed for 8 weeks and measured for weight gain and microbiota composition while being fed a normal chow (n=6 per cohort). (A) Area under the curve (AUC) analysis of weight gained. (B) Representative flow cytometry plot was previously gated on SYBR Green+ cells in order to quantify the percentage of antibody bound bacteria at 8 weeks. Rag1−/− feces control (grey shaded area); Tcrb−/− feces (gray line); WT (blue line); T-Myd88−/− (red line). Quantitation of multiple animals to the right. (C) Violin plot of Bray–Curtis distances between microbiota of TCRb−/− mice given WT or T-Myd88−/− T cells at days 0, 7 and 28. (D) Correlation between Desulfovibrionaceae abundance and Clostrideaceae abundance in Tcrb−/− mice given WT or T-Myd88−/− CD4+ T cells (n=12). (E) Relative abundance of Clostridiaceae (4 weeks). Error bars indicate SD. (F) IgA-bound and IgA-unbound bacteria were analyzed from cecal contents of Tcrb−/− mice given WT or T-Myd88−/− CD4+ T cells. An IgA index was calculated for each OTU to show differences in binding. Positive values indicate enrichment in the bound fraction and negative values enrichment in the unbound fraction. All OTUs with statistically significant differences are shown (p<0.05, Wilcoxon rank sum test). Each panel groups OTUs with the same taxonomic call according to their finest classification level (genus (g), family (f), or order (o)). Each dot represents an individual animal, while different colors within a panel distinguish OTUs within a taxa, and each line connects the means from each OTU. Data from one experiment. (G) Percent weight gained in Rag1−/− mice colonized with WT or T-Myd88−/− fecal microbiota (n= 7 per cohort). Representative of two independent experiments. (H) AUC of weight gained in T-Myd88−/− mice receiving donor WT or Bcl6−/− T cells and fed a normal chow (WT donor, n=5; T-Myd88−/− donor, n=6). Representative of two independent experiments. P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) using a two-tailed, unpaired t test (A, B, H), repeated measures ANOVA with Tukey’s multiple comparison (C), Spearman’s rank-order correlation (D), a repeated measures ANOVA Sidak’s correction for multiple comparisons (G) and a Mann–Whitney U test (E). Error bars indicate SD (D,G).
To support the hypothesis that antibody responses influence metabolic defects, we transferred the obesogenic microbiota into either antibiotic-treated WT or Rag1−/− animals. Indeed, the transfer of the obesogenic microbiota to WT mice did not confer the phenotype, whereas transfer into Rag1−/− animals, which lack antibodies, resulted in significantly greater weight gain compared to animals receiving WT microbiota (Fig. 5G and fig S9I). TFH are T cells that function to instruct antibody class switching and mutation within B cells in germinal centers. We previously established that the only T cell developmental defect in T-Myd88−/− mice was within TFH cells. T-Myd88−/− animals receiving Bcl6−/− T cells, which cannot differentiate into TFH cells, weighed significantly more compared to animals receiving WT T cells (Fig. 5H) (26). Thus, T cells that do not have the capacity to develop into TFH cells fail to rescue the obesity phenotype. Appropriate TFH cell function is therefore required to regulate the microbiota to prevent obesity.
Short-chain fatty acids (SCFAs) are a well-studied microbiota-dependent mechanism that influences host metabolism. However, SCFA production did not differ between WT and T-Myd88−/− animals (fig. S11A). Increased intestinal permeability and leakage of bacterial products that induce low-grade inflammation within adipose tissue has also been proposed (21, 27). However, we failed to detect differences in bacterial ligands within the serum of T-Myd88−/− animals. Furthermore, placement of T-Myd88−/− on a diet infused with an anti-inflammatory, 5-ASA, (28) also failed to rescue weight gain (fig. S11B). Liver RNA-seq and gene set enrichment analysis (GSEA) revealed that, despite animals being fed a standard mouse chow, pathways involved in lipid metabolism, including glycerolphospholipid and glycerolipid metabolism, were the most significantly enriched pathways within T-Myd88−/− animals compared to WT controls (Fig. 6A). Particularly, expression of genes required for the synthesis of lipids, including Fasn, Dgat2, and Srebpf1, and genes involved in lipid absorption including Slc27a4 and Cd36, were all highly upregulated within the liver of T-Myd88−/− animals (Fig. 6B). Although CD36 was upregulated in T-Myd88−/− animals, antibiotic treatment significantly downregulated CD36 expression (Fig. 6C). Moreover, Clostridia treatment of obese T-Myd88−/− animals produced a significant downregulation of CD36, suggesting that Clostridia function to reduce lipid uptake (Fig. 6D). Indeed, gnotobiotic animals colonized with only the Clostridia consortia had significant reductions within hepatic CD36 expression when compared to germfree mice (Fig. 6E). Thus lipid uptake in T-Myd88−/− appears to be in a microbiota-dependent manner.
Fig. 6. Clostridia inhibit lipid absorption within the intestine.
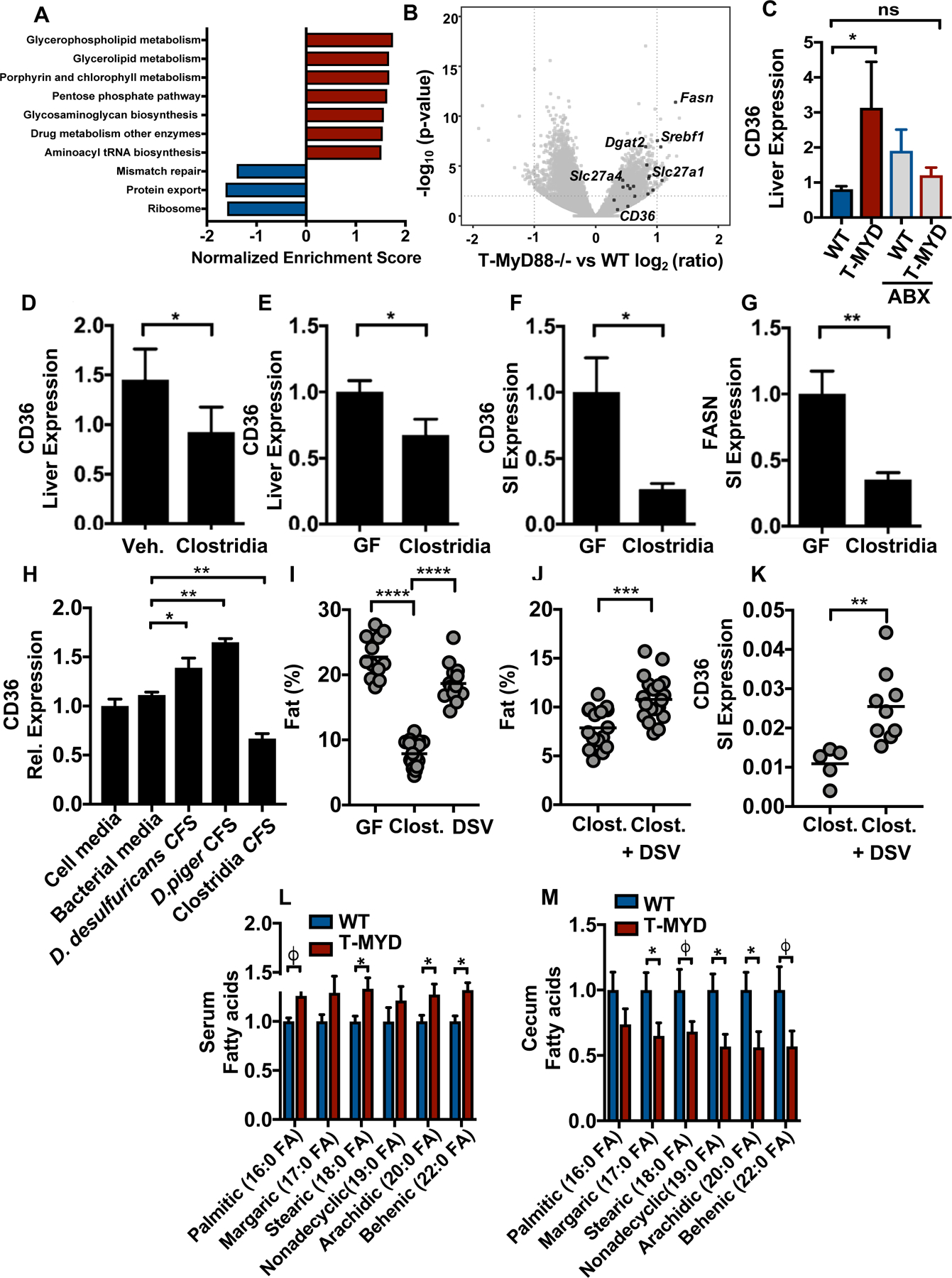
(A) GSEA analysis from RNA expression in livers from 1-year-old WT and T-Myd88−/− mice, pathways that had a significant FDR of .25 or smaller were included. (B) Volcano plot of ratio of liver transcripts. Highlighted genes are involved in lipid metabolism. (C) Cd36 RNA expression within livers of WT and T-Myd88−/− mice fed HFD with or without antibiotics (ABX) (WT, n=5; T-Myd88−/−, n=4; WT ABX, n=5, T-Myd88−/− ABX, n=5). Representative of two independent experiments. (D) Cd36 RNA expression in livers of T-Myd88−/− mice gavaged with vehicle control or spore-forming Clostridia consortium (control n=4; Clostridia consortium, n=5). Representative of two independent experiments. (E-G) Germfree mice with or without colonization of a Clostridia consortium (GF, n= 8; Clostridia, n= 10). (E) Cd36 RNA expression in the liver. (F) Cd36 RNA expression in the small intestines (SI). (G) Fasn RNA expression in the SI. (H) Cd36 RNA expression in MODE-K cells incubated for 4 hours with media or bacterial cell-free-supernatant (CFS). Representative of three independent experiments. (I) Germfree mice were associated with the Clostridia consortia or two Desulfovibrio species (D. piger and D. desulfuricans). Body fat percentage was measured by NMR analysis. (Germfree mice n=12; Clostridia, n=16, Desulfovibrio, n=14). (J,K) Germfree mice were associated with the Clostridia consortia with our without D. piger and D. desulfuricans (DSV). Body fat percentage was measured by NMR (Clostridia alone n=16; Clostridia+DSV n=21) (J) and Cd36 within the small intestine by q-PCR (K). (L,M) GC–MS-detected metabolites within serum and cecum contents of WT and T-Myd88−/− mice fed HFD (n=6 per cohort). P-value of <0.06 (j), P-value<0.05 (*); P-value<0.01 (**); P-value<0.001 (***); P-value<0.0001 (****) a two-tailed, unpaired t test (C-G, I-K), and one-way ANOVA Sidak’s correction for multiple comparisons (H). Data are presented as mean +/− SD.
Colonization of germfree animals with Clostridia significantly downregulates both CD36 and FASN within the small intestine (Fig. 6F and G), suggesting that Clostridia influence lipid absorption and metabolism within the gut. Moreover, cell-free supernatant (CFS) collected from the cultured Clostridia consortia significantly downregulated CD36 in cultured intestinal epithelial cells (IECs) (Fig. 6H). In contrast, CFS collected from cultured Desulfovibrio species directly elevated the expression of CD36 on IECs (Fig. 6H). Furthermore, germfree animals mono-associated with the Clostridia consortia showed a significant decrease in body fat percentage compared to animals mono-associated with Desulfovibrio or germfree animals (Fig. 6I). Notably, the addition of Desulfovibrio to germfree mice colonized with the Clostridia consortia alone led to an increase in body fat percentage and CD36 expression in the small intestine (Fig. 6J and K). Thus, the microbiota can directly regulate lipid metabolism within gut epithelia.
Supporting increased lipid absorption, HFD-fed T-Myd88−/− had significant decreases in several long-chain fatty acids (LCFAs) within the cecum and concomitant increases in the serum (Fig. 6L and M). Comparison of lumenal lipid profiles and 16S sequencing revealed opposing correlations between Desulfovibrio and members of Clostridia and the abundance of LCFAs and other lipids. The depletion of LCFAs within the cecal content was significantly correlated with the presence of Desulfovibrio. In contrast, multiple members of Clostridia, including SMB53 and Dorea, were associated with LCFA accumulation (fig. S11C), further supporting the hypothesis that microbial composition can regulate lipid absorption. Thus, the loss of particular Clostridia species seen in individuals with obesity and T2D may lead to increased intestinal absorption and metabolism of fats, highlighting the importance of an appropriate microbiota composition to health.
Discussion
The microbiota has been implicated in a wide variety of autoimmune and metabolic conditions. However, these diseases are not always associated with the acquisition of a pathogenic organism, and instead the loss of beneficial species has been proposed to be a causative factor (29). Mechanisms leading to the loss of beneficial bacteria can include antibiotic use, increased sanitation and a low-fiber diet (30). Our studies indicate that another mechanism to maintain healthy microbial communities is through appropriate immune control of these populations within the intestine. The microbiota formed within T-Myd88−/− animals mirrors the dysbiosis seen in individuals with type II diabetes and obesity, including an expansion of Desulfovibrio and a loss of Clostridia (11). Although comprehensive human studies are lacking, individuals with obesity and type II diabetes have also been reported to have lower mucosal IgA and decreased responses to immunizations. This suggests that these individuals have a sub-optimal, but not completely deficient, immune response to gut microbiota that, coupled with dietary deficiencies, leads to metabolic disease. Our data suggest that T cell-dependent targeting of the microbiota is important for the maintenance of a healthy community. Although IgA binding of bacteria is typically thought to lead to its eradication, IgA can regulate the functional gene expression of certain bacteria and even aid in mucosal association of certain commensals (31–33). Indeed, we find that despite lower levels of IgA in T-Myd88−/− animals, Desulfovibrio and several Clostridia species display increased IgA coating. Thus, inappropriate targeting of Clostridia by IgA may either reduce their colonization or change their metabolic functions to influence development of obesity. Additionally, several Clostridia are targeted less by IgA. Interestingly, a recent evaluation of the microbiota within individuals with IgA deficiency showed a significant reduction in colonization by several Clostridia (34). Therefore, IgA may also function to enhance colonization of some Clostridia species as has been shown for Bacteroides fragilis (31). The mechanism by which Desulfovibrio expands in this model and in individuals with metabolic syndrome is still unclear.
Our studies, however, indicate that this expansion can directly influence the colonization of specific Clostridia members, although how this occurs remains enigmatic. Understanding how IgA targeting of gut microbes influences their colonization and function in a germfree setting may provide insight into how the immune system influences this microbial relationship. As members of Clostridia are increasingly recognized in several settings (24), it will be important to determine how colonization by other micro-organisms and the immune system together influence the function of Clostridia.
CD36 is a critical regulator of lipid absorption within the intestine and its deficiency results in resistance to the development of obesity and metabolic syndrome upon HFD feeding (35) (36). Increased expression of CD36 within the human liver is associated with fatty liver disease. Furthermore, individuals with polymorphisms in CD36, which produce just a twofold decrease in its expression within the gut, are resistant to metabolic disease (37). Thus, relative expression levels of CD36 are important for lipid absorption and homeostasis within mammals. Recent studies have demonstrated that the microbiota can upregulate host absorption of lipids within the intestine through enhanced CD36 expression (38). However, we find that bacteria may also be able to restrain host lipid absorption.
Thus, gut bacteria can differentially regulate lipid metabolism. Indeed, products secreted by Desulfovibrio upregulate CD36 expression, whereas products produced by Clostridia can downregulate CD36 expression. Therefore, the loss of organisms that function to temper CD36 expression may lead to the inappropriate absorption of lipids, which can accumulate over time, leading to obesity and metabolic syndrome. Further characterization of the interaction of organisms such as Desulfovibrio and Clostridia as well as identification of the molecules secreted that influence CD36 expression may inform future targeted therapies.
Materials and Methods
Mice
C57Bl/6 Myd88LoxP/LoxP mice (Jackson Laboratories) were crossed to C57Bl/6 CD4-Cre animals (Taconic) to produce Myd88+/+; CD4-Cre+ mice (WT) and Myd88LoxP/LoxP; CD4-Cre+ (T-MyD88−/−) animals. Age-matched male mice were used to compare the spontaneous weight phenotype, including immune and microbiota responses, on a standard diet. Age-matched male and female mice were used to compare the weight phenotype, including immune and microbiota responses, on a high-fat diet (HFD). To measure T cell-dependent shaping of the microbiota, 4-week old Tcrb−/− mice (Jackson Laboratories) were used. To investigate Desulfovibrio desulfuricans-dependent shaping of the microbiota, 6-week-old WT C57Bl/6 mice (Jackson Laboratories) were used or age-matched CD4-Cre+ (WT) mice from our facility were used. To measure microbiota effects on weight gain in immunodeficient mice, 4-week old Rag1−/− mice (Jackson Laboratories) were used. GF mice were maintained in sterile isolators and verified monthly for GF status by plating and PCR of feces. GF C57Bl/6 animals were used in this study. The use of animals in all experiments was in strict adherence to federal regulations as well as the guidelines for animal use set forth by the University of Utah Institutional Animal Care and Use Committee.
Colonization of mice with spore-forming microbes
Fecal pellets were taken from WT mice and incubated in reduced PBS containing 3% chloroform (v/v) for 1 hour at 37°C in an anaerobic chamber. A control tube containing only reduced PBS and 3% chloroform was also incubated for 1 hour at 37°C in an anaerobic chamber. After ncubation, tubes were gently mixed and fecal material was allowed to settle for 10 seconds. Supernatant was transferred to a fresh tube and chloroform was removed by forcing CO2 into the tube. For spore-forming (SF) experiments in conventional conditions, mice within the SF cohort were orally gavaged with 100 μL of spore forming fecal fraction, and mice within the CTRL cohort were orally gavaged with 100 μL of PBS control that also had chloroform removed every third day. For spore-forming associations with germ-free animals, tubes containing gavage material were sterilized in the port of a germfree isolator for 1 hour before pulling them into the isolator for gavage. Breeder pairs were then orally gavaged with 100 μL of the spore-forming cocktail. Their offspring were sacrificed at 8 weeks of age for analysis of the small intestine and liver.
T cell transfer into T-Myd88−/−mice
T-Myd88−/− mice were sublethally irradiated with 500 rads the day before T cell transfer. Spleens from WT(CD4-cre+) and BCL6KO (Bcl6LoxP/LoxP CD4-cre+) mice were provided by Shane Crotty from La Jolla Institute, and T cells were isolated using the CD4+ T Cell Isolation Kit (Miltenyi). T-Myd88−/− were retro-orbitally injected with 5×106 of either the WT or Bcl6−/− MACS-enriched T cells and weighed weekly for 5 weeks.
Diet treatment
Animals housed within the SPF facility were fed a standard chow of irradiated 2920x (Envigo). Mice were fed a high fat diet of 45 kcal% fat DIO mouse feed (Research Diets) or a diet of 10 kcal% fat DIO mouse feed (Research Diets) as a control during HFD experiments. Mice were also fed a custom diet containing irradiated standard 2020 chow containing 1% 5-ASA (Envigo) or a control diet lacking the 5-ASA (Envigo) during 5-ASA inflammation experiments.
Antibiotics treatment
WT and T-Myd88−/− mice were maintained on 0.5 mg/mL of ampicillin (Fisher Scientific), neomycin (Fisher Scientific), erythromycin (Fisher Scientific), and gentamicin (GoldBio) within their drinking water for 14 weeks while being fed a HFD in order to determine the relative contribution of the microbiota to the weight gain phenotype. TCRb−/− and Rag1−/− mice were placed on 0.5 mg/mL of ampicillin (Fisher Scientific), neomycin (Fisher Scientific), erythromycin (Fisher Scientific), and gentamicin (GoldBio) within their drinking water for 1 week to reduce the endogenous microbiota before being recolonized by fecal transfers.
T cell shaping of the microbiota within Tcrb−/− mice
Three separate cages of four Tcrb−/− mice were placed on an antibiotic cocktail within their drinking water for one week. Antibiotics was removed for 24 hours before any further treatment. One fecal pellet from a WT donor and one fecal pellet from a T-Myd88−/− donor were mashed in reduced PBS containing 0.1% cysteine and immediately orally gavaged into the Tcrb−/− mice. This oral gavage was repeated every other day for one week. Forty-eight hours following the final gavage, mice were placed into individually housed cages and retro-orbitally injected with 5×106 CD4+ MACS-enriched WT or T-Myd88−/− cells. This was labeled as D0.
Glucose tolerance test
Mice were fasted for 6 hours prior to being challenged with glucose. Fasting levels of glucose were detected using a Contour Glucose Meter (Bayer) and Contour Glucose Strips (Bayer). One milligram of glucose per gram of body weight was injected intraperitoneally into animals at timepoint zero. Blood levels of glucose were measured at 5-,15-,30-,60-, and 120-min time points using the glucose meter and strips.
Insulin ELISA
Serum was collected from 6-hour fasted mice, and insulin was measured using a mouse insulin ELISA kit (Crystal Chem). Serum samples were run in duplicate according to the manufacturer instructions.
Insulin resistance test
Mice were fasted for 6 hours prior to being challenged with glucose. Fasting levels of glucose were detected using a Contour Glucose Meter (Bayer) and Contour Glucose Strips (Bayer). Insulin (0.75U/kg of body weight) was injected intraperitoneally into animals at timepoint zero. Blood levels of glucose were measured at 5-, 10-, 15-, 20-, 25-, 30-, 40-, and 60-min time points using the glucose meter and strips. Animals were removed from the experiment following an 150 μL i.p. injection of 25% glucose if blood glucose levels dropped to 30 mg/dL.
In vitro experiments using mouse intestinal epithelial cells (MODE-K cells)
Mouse intestinal epithelial cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), with L-glutamine and sodium pyruvate. DMEM was supplemented with 10% FBS, 1% (v/v) glutamine, penicillin–streptomycin, and 1% HEPES. To determine if bacteria regulated gene expression, a confluent monolayer of cells was incubated with (1:1) DMEM w/o penicillin–streptomycin:CFS collected from either cultured Clostridia consortia or Desulfovibrio species for 4 hours. Media was then aspirated and cells were placed in 600 μL RiboZol (VWR) for later analysis.
RNA isolation from small intestine, cell culture and liver tissue for qPCR and RNA-seq
Tissue sections 0.5 cm in length or 1×105 cells were stored at −70°C in 700 μL of RiboZol (VWR). RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymoresearch). cDNA was synthesized using qScript cDNA synthesis kit (Quanta Biosciences). qPCR was conducted using LightCycler 480 SYBR Green I Master (Roche). qPCR experiments were conducted on a Lightcycler LC480 instrument (Roche). For liver RNA sequencing, RNA was prepped following QC via an Illumina TruSeq Stranded RNA Sample Prep with RiboZero treatment (human, mouse, rat, etc.) and analyzed using Illumina HiSeq Sequencing.
Quantification of fecal immunoglobulins
To quantify luminal IgA, fecal pellets were collected in 1.5 mL microcentrifuge tubes and weighed. Luminal contents were resuspended in 10 μL of sterile 1X HBSS per milligram of fecal weight and spun at 100 × g for 5 minutes to remove course material. Supernatants were then placed in a new 1.5 mL microcentrifuge tube and spun at 8000 × g for 5 min to pellet bacteria.
Supernatants (containing IgA) were then placed in a new 1.5 mL microcentrifuge tube and used as samples (1/10 and 1/100 (v/v) dilutions) for an IgA-specific ELISA kit (eBioscience; performed according to manufacturer instructions). Absorbance was read at 450 nm and concentrations of IgA were calculated using a standard curve. Concentrations were normalized to fecal weight.
Bacterial pellets were resuspended in 500 μL of sterile PBS and washed twice by spinning at 8000× g for 5 min. The washed bacterial pellet was then resuspended in 10 μL of sterile PBS per mg of feces. Five microliters of each sample was plated on to a 96-well round-bottom plate. Bacteria were blocked for 15 min at room temperature with 100 μL of sterile HBSS containing 10% (v/v) FBS. Without washing cells, 100 μL of anti-IgA (ebioscience clone mA-6E1 PE), anti-IgG1(Santacruz CruzFluor555), or anti-IgG3 (Santacruz CruzFluor555) diluted at 1:500 in sterile HBSS containing 10% (v/v) FBS was added to the wells. Wells were incubated at 4°C for 30 min. The plate was washed twice by spinning at 2500× g for 5 min before removing the supernatant and resuspending cells in sterile HBSS. After final wash, bacterial wells were resuspended in 250 μL of HBSS containing 5 μL of 1x SYBR green stain (Invitrogen cat #S7563). Wells were incubated for 20 min at 4°C before immediate enumeration on a flow cytometer. Rag1−/− fecal pellets were included in all experiments as negative controls.
Growth of Desulfovibrio desulfuricans ATCC 27774 and Desulfovibrio piger ATCC 29098
The bacterial species Desulfovibrio desulfuricans was purchased from ATCC (#27774). The bacterial species Desulfovibrio piger was purchased from ATCC (#29098). The vial was handled and opened per ATCC instructions for anaerobic bacteria and cells were grown in Desulfovibrio media described previously (21). Media was composed of NH4Cl (1 g/L) (Fisher Chemical), Na2SO4 (2 g/L) (Fisher Chemical), Na2S2O3•5H2O (1 g/L) (Sigma), MgSO4•7H2O (1 g/L) (Fisher Chemical), CaCl2•2H2O (0.1 g/L) (Fisher Chemical), KH2PO4 (0.5 g/L) (Fisher Bioreagents), Yeast Extract (1 g/L) (Amresco), Resazurin (0.5 mL/L) (Sigma), cysteine (0.6 g/L) (Sigma), DTT (0.6 g/L) (Sigma), NaHCO3 (1 g/L) (Fisher Chemical), pyruvic acid (3 g/L) (Acros Organics), malic acid (3 g/L) (Acros Organics), ATCC Trace Mineral Mix (10 mL/L), ATCC Vitamin Mix (10 mL/L) and adjusted to pH of 7.2. Bacteria were grown for 48 hrs in an anaerobic chamber (Coy Labs) and stored in growth media containing 25% glycerol at 70°C. 2.5×108 bacterial CFUs were added to 250 μL of drinking water of mice for 1 week.
Isolation and 16S sequencing of fecal, ileal and IgA bound microbial DNA
Animals were sacrificed and their entire lower digestive tract (from duodenum to rectum) was removed and longitudinally sectioned. One fecal pellet and luminal content from lower 10cm of small intestine were collected from each animal to characterize the fecal and ileal microbiota communities, respectively. Fecal and ileal samples were immediately frozen at −70°C in 2 mL screw cap tubes containing ~250 mg of 0.15 mm garnet beads (MoBio, cat# 13122–500). DNA was extracted using the Power Fecal DNA Isolation Kit (MoBio), according to manufacturer instructions. IgA-bound and -unbound bacteria from T cell transfer experiments were isolated from cecal contents and frozen at −70 C before processing. IgA bound bacteria eparation, 16S rDNA amplification, sequencing and sequence processing was done as previously described (12), using paired-end 300 cycle MiSeq reads. The IgA index was calculated as previously described (39).
Metatranscriptomics
Fecal pellets or lumenal ileal contents were placed directly into Trizol and stored at −20 C until RNA extraction. Total RNA was extracted from samples using Direct-zol (Zymo Research, #R2052), then prepared for Illumina sequencing by the University of Utah high-throughput genomics core facility using the Ribo-Zero Gold rRNA (epidemiology) removal kit (Illumina, #MRZE724). Illumina libraries were multiplexed and sequenced on a HiSeq 2500 with single-end 50 cycle sequencing. The humann2(v 0.9.9) analysis framework was used for all subsequent sequencing processing and data analysis (40). First, using the knead data script implemented in Humann2, raw sequences were quality trimmed and filtered using Trimmomatic (41), then filtered to remove host reads against the Mus musculus genome build GRCm38 using bowtie2 (42). No significant difference in quality-filtered reads was observed among genotypes, although across all samples many more reads from ileal samples mapped to the mouse genome, providing less bacterial transcript coverage. Then, to improve mapping of these short reads, we restricted mapping of the quality-filtered reads to a custom database of mouse isolated bacterial reference genomes with UniRef90 gene annotations. This custom database consisted of 53 organisms isolated and sequenced recently as part of the mouse intestinal bacterial collection (miBC) (43), as well as nine reference genomes included in humman2’s chocophlan database, representing species we detected in 16S sequencing but that were not included in the miBC collection already. These nine genomes were: Bifidobacterium pseudolongum, Bifidobacterium animalis, Bifidobacterium longum, Bacteroides fragilis, Mucispirillum schaedleri, Lactobacillus reuteri, Clostridium perfringens, Desulfovibrio_desulfuricans, and Candidatus Arthromitus. To create the custom database with Uniref90 annotations, we aligned the amino acid sequences from the miBC genomes to the Uniref90 database using the Diamond aligner(44) and requiring 50% query coverage and 90% identity. Then, these uniref90-annotated miBC amino acid sequences were used to annotate each corresponding gene’s nucleotide sequences and combined with the nine genomes already annotated to create our custom nucleotide mapping reference containing mouse-specific bacterial genomes. For mapping filtered sequence reads to the custom reference using Humann2, we used only the nucleotide alignments (no translated alignments) due to the short read length. The counts of aligned reads per kilobase for uniref90 gene families output from humann2 were then normalized to counts per million (within a sample), or regrouped to Gene Ontology (GO) terms then normalized, for all subsequent analyses.
Metabolic Phenotyping
Total body fat composition was measured on an NMR Bruker Minispec. CLAMS Metabolic Cages were used to measure indirect calorimetry. Both services were provided by the Metabolic Phenotyping Core, a part of the Health Sciences Cores at the University of Utah. Energy expenditure (EE) was calculated using the following formulas. Calorific Value (CV) = 3.815+(1.232 * RER). EE = CV * VO2
Liver and adipose tissue microscopy
Liver and adipose tissue were fixed in formalin, embedded in wax, and Hematoxylin and eosin stained. Microscopy images were collected using an EVOS core XL imaging system from Thermofisher.
Serum and cecal content metabolomics (Excluding SCFA measurements)
Sample Extraction and Preparation
Cecal contents were stored at −70°C prior to analysis. Five mililiters of 75% ethanol solution containing internal standards (1 μg of d4-succinic acid and 5 μg of labeled amino acids (13C, 15N-labeled) mixture per sample) was added to each sample. Samples were vigorously vortexed and then incubated in boiling water for 10 min. Cooled samples were spun down at 5,000× g for 5 min. Supernatants were transferred to fresh tubes and then speed-vacuumed overnight to dry.
GC–MS analysis
All GC–MS analysis was performed with a Waters GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler. Dried samples were suspended in 40 μL of a 40 mg/mL O-methoxylamine hydrochloride (MOX) in pyridine and incubated for 1 hour at 30°C. To autosampler vials was added 25 μL of this solution. Forty microliters 40 μL of N-methyl-N-trimethylsilyltrifluoracetamide (MSTFA) was added automatically via the autosampler and incubated for 60 minutes at 37°C with shaking. After incubation 3 μL of a fatty acid methyl ester standard (FAMES) solution was added via the autosampler then 1 uL of the prepared sample was injected to the gas chromatograph inlet in the split mode with the inlet temperature held at 250°C. A 10:1 split ratio was used for analysis. The gas chromatograph had an initial temperature of 95°C for 1 minute followed by a 40°C/min ramp to 110°C and a hold time of 2 minutes. This was followed by a second 5°C/min ramp to 250°C, a third ramp to 350°C, then a final hold time of 3 minutes. A 30-m Phenomex ZB5–5 MSi column with a 5-m long guard column was employed for chromatographic separation. Helium was used as the carrier gas at 1 mL/min. Due to the high amounts of several metabolites the samples were analyzed once more at a tenfold dilution.
Analysis of GC–MS data
Data were collected using MassLynx 4.1 software (Waters). Metabolites were identified and their peak area was recorded using QuanLynx. This data was transferred to an Excel spread sheet (Microsoft, Redmond WA). Metabolite identity was established using a combination of an in house metabolite library developed using pure purchased standards and the commercially available NIST library. Not all metabolites are observed using GC–MS. This was due to several reasons. For example, some metabolites were present at very low concentrations. Second, metabolites may not be amenable to GC-MS due to either being too large to volatilize, are a quaternary amine such as carnitine, or just do not ionize well. Metabolites that do not ionize well include oxaloacetate, histidine, and arginine. Cysteine is observed depending upon cellular conditions, often forms disulfide bonds with proteins, and is at a low intracellular concentration.
Short chain fatty acid detection of cecal contents
Sample extraction and preparation
Samples were removed from freezer and allowed to thaw at RT for 5 min. To these samples were added 400 μL of dd-H2O, 10 μL of 5-sulfosalicylic acid (1 mg/μL), and 2 μL of internal standard (1 M pivalic acid). Samples were vortexed for 30 sec and rested on ice for 30 min. Samples were then centrifuged at 2000 × g for 10 min at 4°C. The supernatants were then transferred to glass vials with PTFE lined caps containing 10 μL of concentrated HCl. Next, 3 mL of ether was added and the samples were vortexed for 30 sec, then centrifuged at 1,200 × g for 10 min at 4°C. The supernatants were then transferred to new glass vials with PTFE-lined caps and derivatized with 50 μL of N-Methyl-N-(tert-butyldimethylsilyl) trifluoroacetamide, tertbutyldimetheylchlorosilane (MTBSTFA; Thermo Scientific). Samples were vortexed and placed in a 60°C sand bath for 30 min. Samples were allowed to cool to RT and partially evaporated under a gentle stream of nitrogen to a volume of approximately 250 μL and transferred to glass GC–MS vials.
GC–MS analyses
GC/MS analyses were conducted on an Agilent 6890 gas chromatograph coupled to an Agilent 5793 mass spectrometer and an Agilent 7683 (Santa Clara, CA, USA) auto-injector equipped with a DB-1 column (15 m × 0.25-mm internal diameter × 0.25-μm film thickness; J&W Scientific, Folsom, CA, USA). Helium carrier gas was used with a flow rate of 1.0 mL/min. Split ratio 10:1 with injections of 1-μL samples were made into an inlet held at 250°C. The GC oven ramp used was 40°C (hold 1 min); ramp at 5°C/min to 70°C (hold 3 min); ramp at 20°C/min to 160°C (hold 0 min); ramp at 40°C/min to 330°C (hold 6 min). Data were acquired in scan mode with a mass range of 44–200 m/z, targets were quantitated using m/z 117.0 for acetic acid, m/z 131.0 for butanoic acid, m/z 145.0 for propanoic acid and m/z 159.0 pivalic acid.
Primers used in this study:
| Primers | Sequence |
|---|---|
| L32 | For: 5′-AAGCGAAACTGGCGGAAAC-3′ |
| CD36 | For: 5′-TCCTCTGACATTTGCAGGTCTATC-3′ |
| FASN | For: 5′-GGAGGTGGTGATAGCCGGTAT-3′ |
Supplementary Material
Acknowledgments:
We would like to thank members of the Round and O’Connell labs for their critical reviews of the manuscript. Some of the GF mice used in this publication were provided from the UNC Gnotobiotic Facility which is supported by grants 5-P39-DK034987 AND 5-P40-OD010995. We thank Dr. S.Crotty for the CD4-cre BCL6fl/fl animals. B. Dalley and T. Mosbruger in the High-Throughput Genomics and Bioinformatics Analysis core facility performed the liver RNA-seq and analysis respectively. R.M.O. is supported by the NIH New Innovator Award DP2GM111099-01, the NHLBI R00HL102228-05, an American Cancer Society Research Grant, Kimmel Scholar Award and R01AG047956. S.R.C. is supported by NIGMS RO1 grant GM114817. Support for this project came from the Pews Scholar Program and NIH R56AI107090. Other support for the lab came from the Edward Mallinckrodt Jr. Foundation, NSF CAREER award (IOS-1253278), Packard Fellowship in Science and Engineering, NIAID K22 (AI95375), Burrough’s Wellcome Investigator in Pathogenesis Award, the American Asthma Foundation, Margolis Foundation, the MS Society Center grant, and the Crohn’s and Colitis foundation Senior research award to J.L.R. All sequence data are accessible under NCBI archives under the BioProject accession number PRJNA542126.
References
- 1.Mozaffarian D et al. , Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 131, e29–322 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Gregor MF, Hotamisligil GS, Inflammatory mechanisms in obesity. Annu Rev Immunol 29, 415–445 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Emanuela F et al. , Inflammation as a Link between Obesity and Metabolic Syndrome. Journal of nutrition and metabolism 2012, 476380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pallaro A et al. , Total salivary IgA, serum C3c and IgA in obese school children. J Nutr Biochem 13, 539 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Must A et al. , The disease burden associated with overweight and obesity. JAMA 282, 1523–1529 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Nieman DC et al. , Influence of obesity on immune function. J Am Diet Assoc 99, 294–299 (1999). [DOI] [PubMed] [Google Scholar]
- 7.McMurray DN, Beskitt PA, Newmark SR, Immunologic status in severe obesity. Int J Obes 6, 61–68 (1982). [PubMed] [Google Scholar]
- 8.Hirokawa J et al. , Identification of macrophage migration inhibitory factor in adipose tissue and its induction by tumor necrosis factor-alpha. Biochem Biophys Res Commun 235, 94–98 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Tanaka S et al. , Impaired immunity in obesity: suppressed but reversible lymphocyte responsiveness. Int J Obes Relat Metab Disord 17, 631–636 (1993). [PubMed] [Google Scholar]
- 10.Turnbaugh PJ et al. , An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Qin J et al. , A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Kubinak JL et al. , MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawamoto S et al. , Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Wang S et al. , MyD88 Adaptor-Dependent Microbial Sensing by Regulatory T Cells Promotes Mucosal Tolerance and Enforces Commensalism. Immunity 43, 289–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pi-Sunyer FX, Comorbidities of overweight and obesity: current evidence and research issues. Med Sci Sports Exerc 31, S602–608 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Vijay-Kumar M et al. , Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ussar S et al. , Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab 22, 516–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Chatelier E et al. , Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Gibson GR, Macfarlane GT, Cummings JH, Occurrence of sulphate-reducing bacteria in human faeces and the relationship of dissimilatory sulphate reduction to methanogenesis in the large gut. J Appl Bacteriol 65, 103–111 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Pitcher MC, Cummings JH, Hydrogen sulphide: a bacterial toxin in ulcerative colitis? Gut 39, 1–4 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rey FE et al. , Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci U S A 110, 13582–13587 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desai MS et al. , A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353 e1321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zierer J et al. , The fecal metabolome as a functional readout of the gut microbiome. Nat Genet 50, 790–795 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harakeh SM et al. , Gut Microbiota: A Contributing Factor to Obesity. Front Cell Infect Microbiol 6, 95 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch MA et al. , Maternal IgG and IgA Antibodies Dampen Mucosal T Helper Cell Responses in Early Life. Cell 165, 827–841 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crotty S, Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29, 621–663 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Cani PD et al. , Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Luck H et al. , Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 21, 527–542 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Cho I et al. , Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koropatkin NM, Cameron EA, Martens EC, How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol 10, 323–335 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donaldson GP et al. , Gut microbiota utilize immunoglobulin A for mucosal colonization. Science 360, 795–800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cullender TC et al. , Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14, 571–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson DA, McNulty NP, Guruge JL, Gordon JI, IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2, 328–339 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Fadlallah J et al. , Microbial ecology perturbation in human IgA deficiency. Sci Transl Med 10, (2018). [DOI] [PubMed] [Google Scholar]
- 35.Buttet M et al. , Deregulated Lipid Sensing by Intestinal CD36 in Diet-Induced Hyperinsulinemic Obese Mouse Model. PLoS One 11, e0145626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buttet M et al. , From fatty-acid sensing to chylomicron synthesis: role of intestinal lipid-binding proteins. Biochimie 96, 37–47 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Love-Gregory L, Abumrad NA, CD36 genetics and the metabolic complications of obesity. Curr Opin Clin Nutr Metab Care 14, 527–534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y et al. , The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 357, 912–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kau AL et al. , Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med 7, 276ra224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abubucker S et al. , Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8, e1002358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagkouvardos I et al. , The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat Microbiol 1, 16131 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Buchfink B, Xie C, Huson DH, Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.