

Abstract
High-grade serous ovarian carcinoma (HGSOC) is the most lethal gynecological cancer with few effective, targeted therapies. HGSOC tumors exhibit genomic instability with frequent alterations in the protein kinome; however, only a small fraction of the kinome has been therapeutically targeted in HGSOC. Using multiplexed inhibitor beads and mass spectrometry (MIB-MS), we mapped the kinome landscape of HGSOC tumors from patients and patient-derived xenograft (PDX) models. The data revealed a prevalent signature consisting of established HGSOC-driver kinases, as well as several kinases previously unexplored in HGSOC. Loss-of-function analysis of these kinases in HGSOC cells indicated MRCKA (also known as CDC42BPA) as a putative therapeutic target. Characterization of the effects of MRCKA knockdown in established HGSOC cell lines demonstrated that MRCKA was integral to signaling that regulated the cell cycle checkpoint, focal adhesion and actin remodeling, as well as cell migration, proliferation, and survival. Moreover, inhibition of MRCKA using the small molecule BDP9066 decreased cell proliferation and spheroid formation and induced apoptosis in HGSOC cells, suggesting that MRCKA may be a promising therapeutic target for the treatment of HGSOC.
INTRODUCTION
High-grade serous ovarian carcinoma (HGSOC) is one of the most common and lethal forms of ovarian carcinoma, and current treatments have only modestly impacted survival (1). Frequent alterations in DNA repair proteins such as BRCA1/2, sensitize HGSOC tumors to DNA-damaging agents, such as carboplatin, and to drugs that target homologous recombination such as PARP inhibitors (2). Although front-line cytotoxic chemotherapy is initially effective for many patients, relapse and emergence of drug resistance are common with no subsequent therapies available (3). HGSOCs are characterized by near-universal mutation and/or loss of the TP53 tumor suppressor gene, and consequently, exhibit genome instability and aberrant cell signaling (1, 4). Signaling abnormalities in HGSOCs, particularly those involving aberrant protein kinases, represent potential therapeutic avenues for treatment (5–7).
Therapies targeting recognized, well understood, cancer-associated protein kinases such as EGFR, ERBB2, and SRC have shown limited clinical benefit in HGSOC, prompting the search for new anti-cancer kinase targets (8, 9). In the present study, we interrogated the HGSOC kinome for novel kinase vulnerabilities using Multiplexed Inhibitor Beads and Mass Spectrometry (MIB-MS) in combination with loss-of-function assays. MIB-MS, along with other kinase-enrichment techniques, such as Kinobeads™ and KiNativ™, provides a quantitative proteomics method to measure kinase levels for a substantial proportion of the kinome, including many largely unexplored kinases for which there are few existing references, reagents, or inhibitors (10–12). Functional interrogation of MIB-MS kinome signatures using siRNA knockdown strategies in cancer cells assigns growth and survival functions to unexplored kinases (11).
RESULTS
Mapping the kinome landscape of HGSOC tumors using MIB-MS uncovers a shared kinome signature
Purification of endogenous kinases from patient tumors was carried out using MIB-columns comprised of four immobilized ATP-competitive pan-kinase inhibitors (Purvalanol B, VI16832, PP58, and CTx-0249885) (13) (Fig. 1A). To quantify the MIB-bound kinome of HGSOC primary and PDX tumors, we used a super-stable isotope labeling by/with amino acids in cell culture (s-SILAC) strategy (14). High concordance of kinase levels of sections from a single HGSOC tumor demonstrates the robust reproducibility of combining MIB-MS and s-SILAC to kinome profile tumors (Fig. 1B and Data file S1). Using this workflow, we quantified the MIB-enriched kinomes of 25 stage III and IV primary HGSOC patient tumors, and 10 HGSOC patient-derived xenograft (PDX) models derived from patient tumor and ascites specimens (Data file S2).
Figure 1. Characterizing the HGSOC kinome in patient tumors using MIB-MS to identify previously unexplored therapeutic targets.
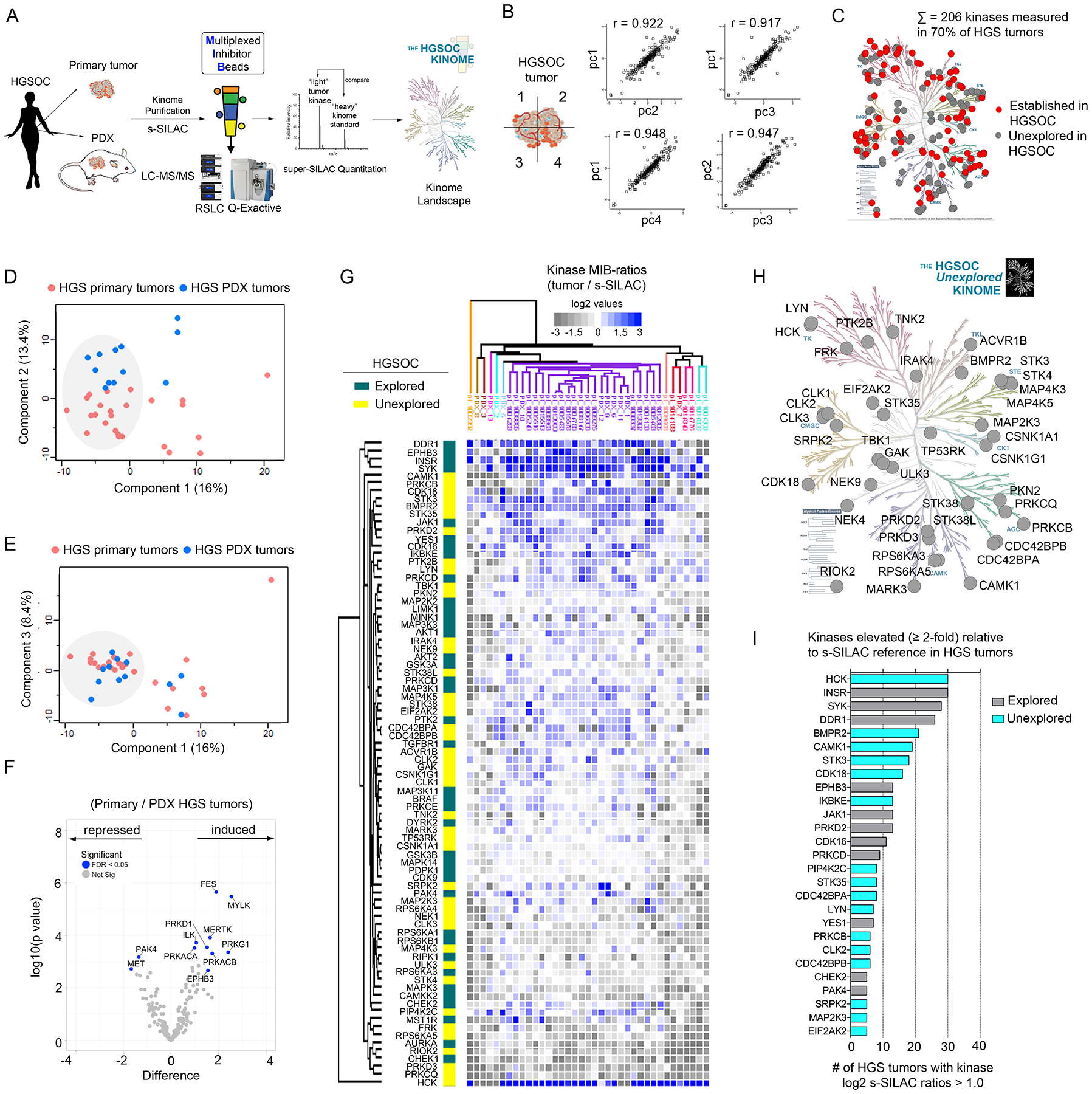
(A) Schematic of experimental approach. MIB-MS was used to quantify kinase abundance in HGSOC patient tumors to map the proteomic landscape of the kinome and identify prevalent kinases previously unexplored in HGSOC. (B) Measurement of kinase abundance in a patient HGSOC tumor sectioned into 4 pieces, each individually kinome-profiled by MIB-MS, and kinase abundance was determined by s-SILAC quantitation. Pearson’s correlation thereof was determined using Perseus. (C) Proportion of the kinome quantitated by MIB-MS in HGSOC (“HGS”) tumors that represent established (red) and unexplored (gray) targets in HGSOC. Data are from one independent assay per sample in 25 tumor tissues and 10 PDX tumor tissues. The kinome tree was generated in KinMap and was reproduced courtesy of Cell Signaling Technology. (D and E) PCA analysis, including PC1 vs PC2 (D) and PC1 vs PC3 (E), of MIB-MS determined kinome profiles of HGSOC primary and PDX tumors. Data are from one independent assay per sample in 25 tumor tissues and 10 PDX tumor tissues. (F) Volcano plot comparison of HGSOC primary and PDX tumor MIB-MS kinome profiles. Statistical differences in kinase log2 s-SILAC ratios comparing HGSOC primary vs PDX tumors was determined by paired t-test Benjamini-Hochberg adjusted p values at FDR of <0.05. Data are from one independent assay per sample in 25 tumor tissues and 10 PDX tumor tissues. (G) Predominant kinome signature identified by MIB-MS amongst HGSOC patient and PDX tumors, representing kinases detected at similar abundances amongst tumors. Statistical differences in kinase log2 s-SILAC ratios comparing HGSOC tumors from the main cluster relative to others was determined by paired t-test Benjamini-Hochberg adjusted p values at FDR of <0.05. Heat map depicts kinase log2 s-SILAC ratios, highlighting established HGSOC drivers, including those with drugs approved or in clinical trials, as well as those previously unexplored in HGOSC. Data are from one independent assay per sample in 25 tumor tissues and 10 PDX tumor tissues. (H) Kinases previously unexplored in HGSOC enriched in the MIB-MS kinome signature from (G). Data are from one independent assay per sample in 25 tumor tissues and 10 PDX tumor tissues. Kinome plot produced using KinMap. The kinome tree was reproduced courtesy of Cell Signaling Technology. (I) Bar graph depicting kinases that were increased ≥ 2-fold relative to the s-SILAC reference in HGSOC tumors. The number of HGSOC tumors with kinases exhibiting log2 s-SILAC values (≥ 1.0) was determined, and the bar graph was generated in Prism software. Related data (additional PCA, correlation, and hierarchical clustering of MIB-MS profiles of primary and PDX HGSOC tumors) can be found in figure S1.
In total, we measured MIB-binding s-SILAC values for 324 kinases, with 206 kinases quantitated in 70% of the tumors; the majority of these have not previously been explored as therapeutic targets in HGSOC (Fig. 1C and Data file S3). Principal component analysis (PCA) of kinase s-SILAC ratios revealed several of the HGSOC primary and PDX tumors grouped together exhibiting similar MIB-MS kinome profiles (Fig. 1D–E, fig. S1A–B). Volcano plot comparison of HGSOC primary and PDX tumor MIB-MS profiles showed some differences in kinase levels amongst tumor types, including elevated levels of myosin light chain kinase (MYLK), protein kinase D1 (PRKD1) and protein kinase G1 (PRKG1) in primary tumors, and increased p21-activated kinase 4 (PAK4) and hepatocyte growth factor receptor (MET) levels in PDX tumors (Fig. 1F). Notably, several of the kinases elevated in primary tumors had stromal-related functions, consistent with recent transcriptomic studies showing most of genes upregulated in primary vs matched HGSOC PDX tissues were stromal-specific genes (15). Unsupervised hierarchical clustering of kinase s-SILAC ratios identified a main cluster of HGSOC tumors (n=27) that exhibited a shared MIB-MS kinome signature - kinases that displayed a reproducible difference in expression relative to the s-SILAC reference across HGSOC tumors (Fig. 1G, fig. S1C, and Data file S4). The MIB-MS tumor kinome signature was comprised of established HGSOC driver kinases, as well as several kinases that had no established function in or previous association with HGSOC (Fig. 1H). Moreover, a number of these unexplored kinases were elevated ≥ 2-fold in HGSOC tumors relative to the s-SILAC cancer cell line reference, signifying these kinases were highly abundant in some HGSOC tumors (Fig. 1I, and fig. S1D).
Loss-of-function screen targeting the MIB-MS tumor kinome signature in HGSOC cells identifies MRCKA (CDC42BPA)
Next, we selected several kinases that were previously unexplored in HGSOC from the MIB-MS tumor signature and, using targeted RNA interference (RNAi), defined their functions in growth and survival in the HGSOC cell line OVCAR4 (Fig. 2A). A number of known kinase drivers of HGSOC from the tumor kinome signature, as well as others were used as controls. Notably, knockdown of Myotonic Dystrophy Kinase-Related CDC42-Binding Kinase Alpha (MRCKA, also known as CDC42BPA) inhibited cell growth to the greatest extent amongst unexplored kinases, similarly to established HGSOC kinases, Checkpoint kinase 1 (CHEK1), Protein tyrosine kinase 2 (PTK2 or FAK1), p21 activated kinase 4 (PAK4) or Aurora Kinase A (AURKA). Knockdown of Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2 (EIF2AK2) also inhibited viability of OVCAR4 cells as well as established HGSOC targets. Furthermore, knockdown of MRCKA reduced cell viability to varying degrees in 3 additional HGSOC cell lines, whereas, EIF2AK2 depletion showed no effect on these additional cell lines (Fig. 2B). Knockdown efficiency of MRCKA or EIF2AK2, and positive control CHEK1 in HGSOC cells was confirmed by Western blot (Fig. 2C). Of particular interest, knockdown of MRCKA induced apoptosis in OVCAR4 cells based on PARP cleavage (Fig. 2D), signifying MRCKA may be essential for HGSOC cell survival, nominating MRCKA as a plausible therapeutic kinase target in HGSOC. Knockdown of MRCKA in OVCAR4 or KURAMOCHI with siRNAs targeting distinct regions of the kinase gene (Fig. 2E) and siGENOME siRNA pools (containing individual siRNAs #4 and #8) confirmed on-target MRCKA ablation, growth inhibition and PARP cleavage (Fig. 2, F to I).
Figure 2. Knockdown screen targeting kinases from the MIB-MS HGSOC tumor signature identifies MRCKA as a candidate therapeutic target.
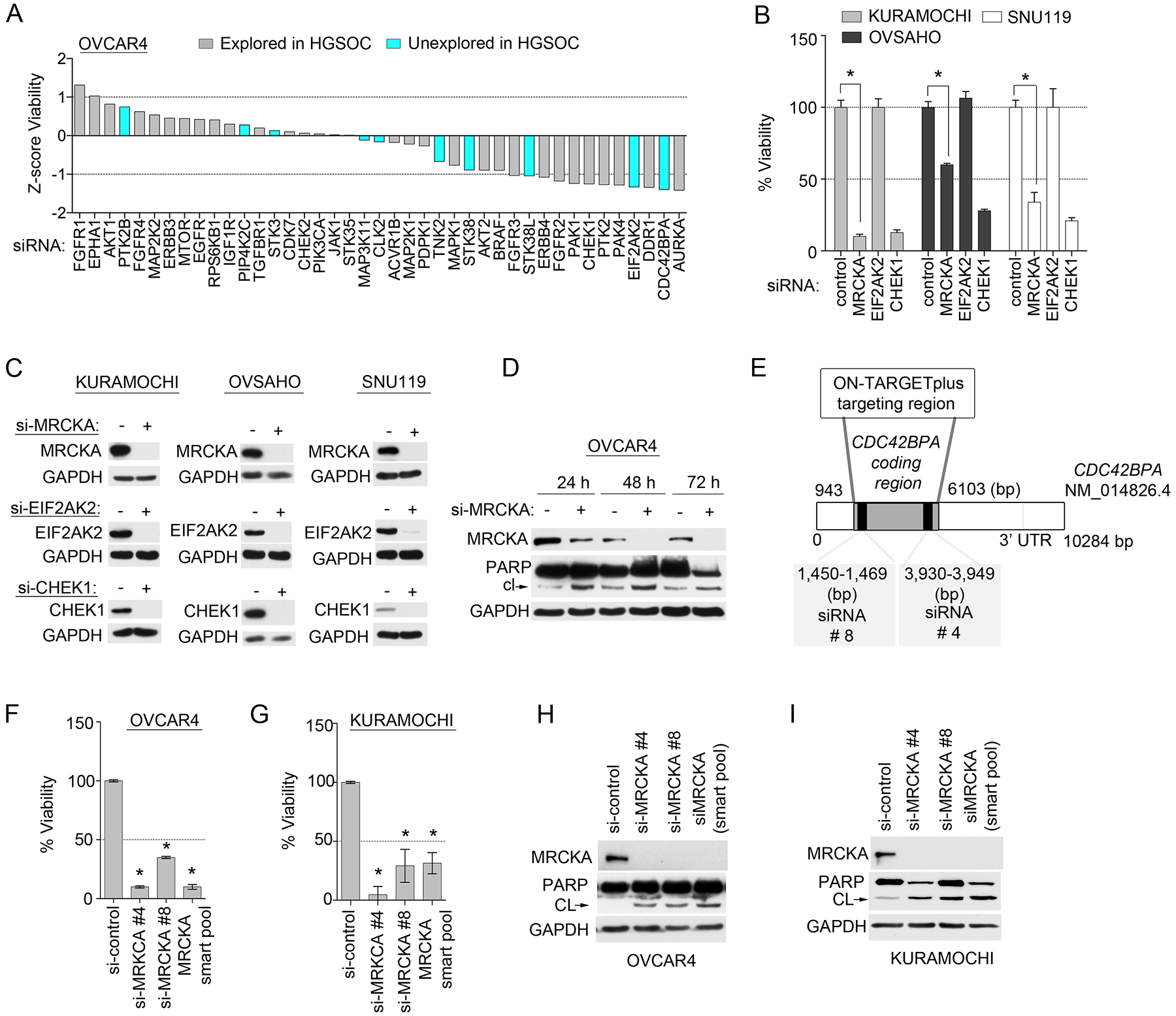
(A) Cell-Titer Glo assay for cell viability of established HGSOC cell line transfected with siRNAs targeting the indicated kinases or with control siRNA (NT2) and cultured for 120 hours. Data were analyzed as Z-scores; presented as means of 3 independent assays. (B) Cell viability in 3 additional established HGSOC cell lines to those presented in (A), cultured and assessed after knockdown of select kinases as described in (A). Data were analyzed as % cell viability; presented as means ± SD of 3 independent assays. *P≤0.05 by Student’s t-test. (C) Knockdown efficiency of MRCKA, EIF2AK2 and CHEK1 in HGSOC cell lines presented in (B). Cells were transfected with siRNAs targeting MRCKA, EIF2AK2, CHEK1/2 or with control siRNAs and cultured for 72 hours. MRCKA and cleaved PARP protein abundance was assessed by immunoblot every 24 hours. Blots are representative of 3 independent experiments. (D) Apoptosis assessed by immunoblotting for cleaved-PARP abundance in OVCAR4 cells. Cells were transfected with siRNAs targeting MRCKA or with control siRNAs and cultured for 72 hours. MRCKA and cleaved PARP protein abundance was assessed by immunoblot every 24 hours. Blots are representative of 3 independent experiments. (E) Targeted spectrum in CDC42BPA-coding gene of 2 distinct siRNAs used in study. CDC42BPA siRNAs 1 and 2 are present in the siGENOME pools. (F to I) Effect of MRCKA knockdown with 2 distinct siRNAs on cell viability by Cell-Titer Glo assay and apoptosis by cleaved-PARP immunoblotting in OVCAR4 (F and H) and KURAMOCHI (G and I) cells transfected with CDC42BPA-targeted siRNAs for 48 hours. Data are means ± SD of 3 independent experiments. *P≤0.05 by Student’s t-test.
MRCKA is frequently altered in HGSOC patient tumors and promotes proliferation, migration and survival of HGSOC cells
MRCKA is an established CDC42 downstream effector kinase that has recently emerged as a therapeutic target in breast, skin and lung cancers due to its role in actin cytoskeleton remodeling, cell migration, invasion and metastasis (16, 17). To characterize MRCKA expression across larger numbers of patient HGSOC tumors, we queried publicly available HGSOC genomics (TCGA) and proteomics databases (CPTAC) (1, 4). Frequent genomic alteration of CDC42BPA (44%, 259 of 594 HGSOC tumors) was observed in HGSOC tumors, with gene amplification (8%, 47 of 594), and altered gene expression (upregulation in 26%, 157 of 594; and downregulation in 11%, 64 of 594), but no homozygous deletions were detected (Fig. 3A). Only a single HGSOC tumor possessed a mutation in CDC42BPA (missense, L1719F). Additionally, global proteomics evaluation of HGSOC tumors by CPTAC revealed MRCKA was upregulated at the protein level (16%, 27 of 174) and downregulated (14%, 25 of 174), demonstrating correlation between CDC42BPA mRNA and protein levels (r = 0.53; fig. S2A). Levels of CDC42BPA mRNA expression were not statistically associated with survival, stage or molecular subtype (fig. S2, B to D); however, tumors with high levels of CDC42BPA did show notable pathway enrichment for CDC42 signaling, as well as focal adhesion, integrin, RHOA, RAC1, and inositol phosphate signaling (Fig. 3B and fig. S2E). Conversely, low CDC42BPA expressing tumors showed negative enrichment for these pathways, but were instead enriched for DNA repair signaling.
Figure 3. MRCKA is highly expressed amongst HGSOC tumors and promotes growth and survival of HGSOC cell lines.
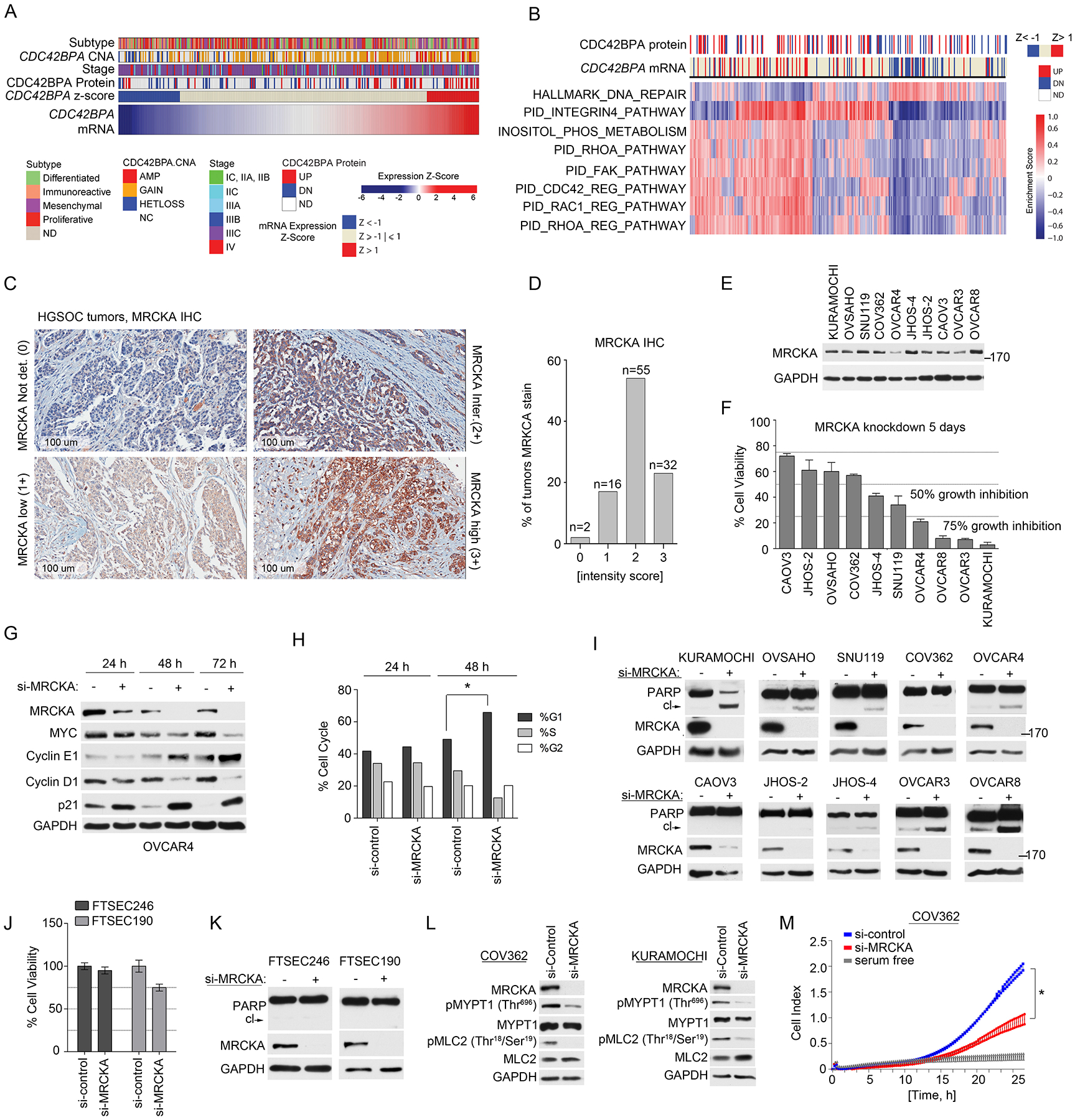
(A) Analysis of CDC42BPA copy number analysis (CNA), mRNA and protein expression changes in HGSOC tumors from TCGA and CPTAC studies (1, 18). mRNA levels determined by U133 microarray and protein level determined by mass spectrometry. Change in MRCKA abundance amongst tumors determined at z-score > 1 or < −1. Molecular subtype of HGSOC and disease stage are presented in relation to MRCKA mRNA levels. (B) Gene set enrichment analysis of HGSOC tumors expressing high or low CDC42BPA mRNA levels. Detailed description of bioinformatics methods described in Materials and Methods. (C and D) IHC analysis of MRCKA in HGSOC TMA’s. Immunoreactivity of MRCKA protein was evaluated by a pathologist and IHC score was given based on the intensity of protein stain. 0: Negative; 1+: mild(low)/weak; 2+: moderate/intermediate; 3+: Strong(high)/intensive. MRCKA IHC images captured at 100 um. Data are from duplicate analysis of 105 tumor tissues. (E) MRCKA protein abundance amongst HGSOC cell lines determined by immunoblot. Blots are representative of 3 independent experiments. (F) Cell-Titer Glo assay for cell viability of HGSOC cells transfected with siRNAs targeting MRCKA or control siRNAs and cultured for 120 hours. Data were analyzed as % cell viability of siRNA control treated cells; presented as means ± SD of 3 independent assays. (G) Immunoblot analysis for cell cycle markers in OVCAR4 cells transfected with MRCKA or control siRNAs for 24, 48 or 72 hours. Blots are representative of 2 independent experiments. (H) FACS analysis of OVCAR4 cells transfected with MRCKA or control siRNAs collected at the indicated time points. The cells were analyzed with a FACScan (BD) flow cytometer and the data were analyzed using FlowJo (BD). Data were analyzed as % phase of cell cycle; presented as means ± SD of 3 independent assays. *P≤0.05 by Student’s t-test. (I) Apoptosis assessed by immunoblotting for cleaved-PARP abundance in HGSOC cells transfected with siRNAs targeting MRCKA or control siRNA and cultured for 72 hrs. PARP cleavage and MRCKA protein levels were determined by western blot. Blots are representative of 3 independent experiments. (J) Cell-Titer Glo assay for cell viability of FTSEC cells transfected with siRNAs targeting MRCKA or control siRNAs and cultured for 120 hours. Data were analyzed as % cell viability of siRNA control treated cells; presented as means ± SD of 3 independent assays. (K) Apoptosis assessed by immunoblotting for cleaved-PARP abundance in FTSEC cells transfected with siRNAs targeting MRCKA or control siRNA and cultured for 72 hours. Blots are representative of 3 independent experiments. (L) Immunoblot analysis of MLC2 and MYPT1 phosphorylation in COV362 and KURAMOCHI cells transfected with MRCKA or control siRNAs for 72 hours. Blots are representative of 3 independent experiments. (M) Analysis of cell migration in COV362 cells transfected with MRCKA or control siRNAs and cultured for 72 hours. Migration monitored over a 24-hour period using the xCELLigence Real-Time Cell Analyzer (RTCA). Data were analyzed as cell index; presented as means ± SD of 3 independent assays. *P≤0.05 by Student’s t-test. Related data (impact of MRCKA expression on overall survival of HGSOC, gene set enrichment analysis, IHC of MRCKA in fallopian tube and ovarian surface epithelium, and immunoblot analysis of MRCKA protein abundance in HGSOC tumors and HGSOC cell lines) can be found in figure S2.
Widespread expression of MRCKA amongst HGSOCs was confirmed by immunohistochemical (IHC) analysis of tissue microarrays (TMAs) containing 105 HGSOC patient tumors (Fig. 3C and Data file S5). Nearly all HGSOC tumor cores stained positive for MRCKA with 30.5% of tumors exhibiting strong MRCKA staining (3+), 52.4% showing moderate staining (2+), and 15.2% showing weak/low MRCKA protein staining (1+), while only 1.9% of tumors showed no MRCKA staining (Fig. 3D). MRCKA protein was localized to the membrane and cytoplasmic regions of the tumor with predominant staining in the cytoplasm. MRCKA abundance was also detected in normal fallopian tube epithelium (FTE) and to a lesser extent in ovarian surface epithelium (OSE) (fig. S2, F to I).
To establish growth and survival functions of MRCKA in HGSOC, we performed loss-of-function siRNA studies targeting CDC42BPA (or MRCKA) in a panel of 10 ovarian cancer (OC) cell lines likely to be HGSOC based on genomic similarities with HGSOC tumors (18). Protein expression of MRCKA was detected across HGSOC cell lines, with most cells expressing MRCKA at similar protein levels, with the exception of OVCAR4, OVCAR3 and JHOS-2, which had lower levels (Fig. 3E and fig. S2J). Knockdown of MRCKA reduced cell viability across the majority of HGSOC cell lines, with SNU119, JHOS-4, OVCAR3, OVCAR4, OVCAR8, and KURMAOCHI cells exhibiting >50% growth inhibition (Fig. 3F). Notably, knockdown of MRCKA in OVCAR4 cells reduced MYC and cyclin D1 protein abundance and increased cyclin E1 and p21 protein abundance within 48 hours, suggesting a cell cycle arrest (Fig. 3G). FACS analysis of MRCKA knockdown in OVCAR4 cells confirmed an increase in percentage of cells in G1/S phase relative to cells transfected with control siRNA (Fig. 3H).
MRCKA knockdown resulted in increased amounts of cleaved PARP in KURAMOCHI, OVSAHO, SNU119, OVCAR4, OVCAR3, JHOS-4 and OVCAR8 cells, but not in COV362, CAOV3, or JHOS-2 cells, demonstrating in 7 out of 10 OC cell lines MRCKA depletion induces apoptosis (Fig. 3I). Emerging findings suggest HGSOC arises from fallopian tube secretory epithelial cells (FTSEC) (19). To determine the effect of MRCKA disruption on putative HGSOC progenitor cells, siRNA-mediated depletion of MRCKA was evaluated in two independent immortalized human FTSEC lines (20) did not block cell viability > 50% or induce apoptosis, suggesting MRCKA is an emergent dependency in some HGSOC cell lines (Fig. 3, J and K).
MRCKA has been shown to regulate actin remodeling, migration and invasion through phosphorylation of MYPT1 and/or myosin light chain (MLC) in breast and lung cancer cells (21, 22). Consistent with these findings, MRCKA knockdown resulted in reduced phosphorylation of MLC2 (at Thr18/Ser19) and/or MYPT1 (at Thr696) in HGSOC cells (Fig. 3L). Next, we assayed cell migration in response to MRCKA knockdown in COV362 cells, which do not undergo apoptosis following MRCKA depletion in the timeframe migration was assessed. Depletion of MRCKA inhibited migration of COV362 cells (Fig. 3M), supporting MRCKA as an important mediating kinase in HGSOC cell migration. Notably, levels of phosphorylated MLC2 (at Thr18/Ser19) or MYPT1 (at Thr696) in HGSOC cell lines were not predictive of sensitivity towards MRCKA knockdown in HGSOC cells, suggesting MRCKA may have additional functions in HGSOC cells apart from its recognized role in actin remodeling (fig. S2, K to M).
Knockdown of MRCKA substantially rewires kinase signaling in HGSOC cells
To understand the molecular consequences of MRCKA inhibition in HGSOC cells, we carried out a detailed proteomic analysis following MRCKA knockdown in OVCAR4 cells using MIB-MS kinome profiling and global phosphoproteomics (TiO2) (23) (Fig. 4A). At 48 h post-knockdown, both induction and repression of kinase MIB-binding was observed, demonstrating MRCKA depletion dynamically reprograms kinase signaling in HGSOC cells (Fig. 4B, fig. S3A and Data file S6). Notably, MRCKA protein depletion following siRNA-mediated gene knockdown resulted in reduced MIB-binding of established kinase drivers known to promote HGSOC growth and survival [including CHEK1, PTK2 (FAK1), and AURKA] and induced MIB-binding of epidermal growth factor receptor (EGFR) and protein kinase B2 (AKT2). Similar to MIB-MS profiling, Kinome Substrate Enrichment Analysis (KSEA) (24) of control and MRCKA-knockdown phosphoproteomics datasets predicted inhibition of cell cycle kinases in MRCKA-depleted cells (Fig. 4C; fig. S3, B to D; and Data file S7). Activation of several kinases was predicted by KSEA following MRCKA knockdown, including p21 activated kinase 1 (PAK1).
Figure 4. MRCKA knockdown remodels the kinome of HGSOC cells.
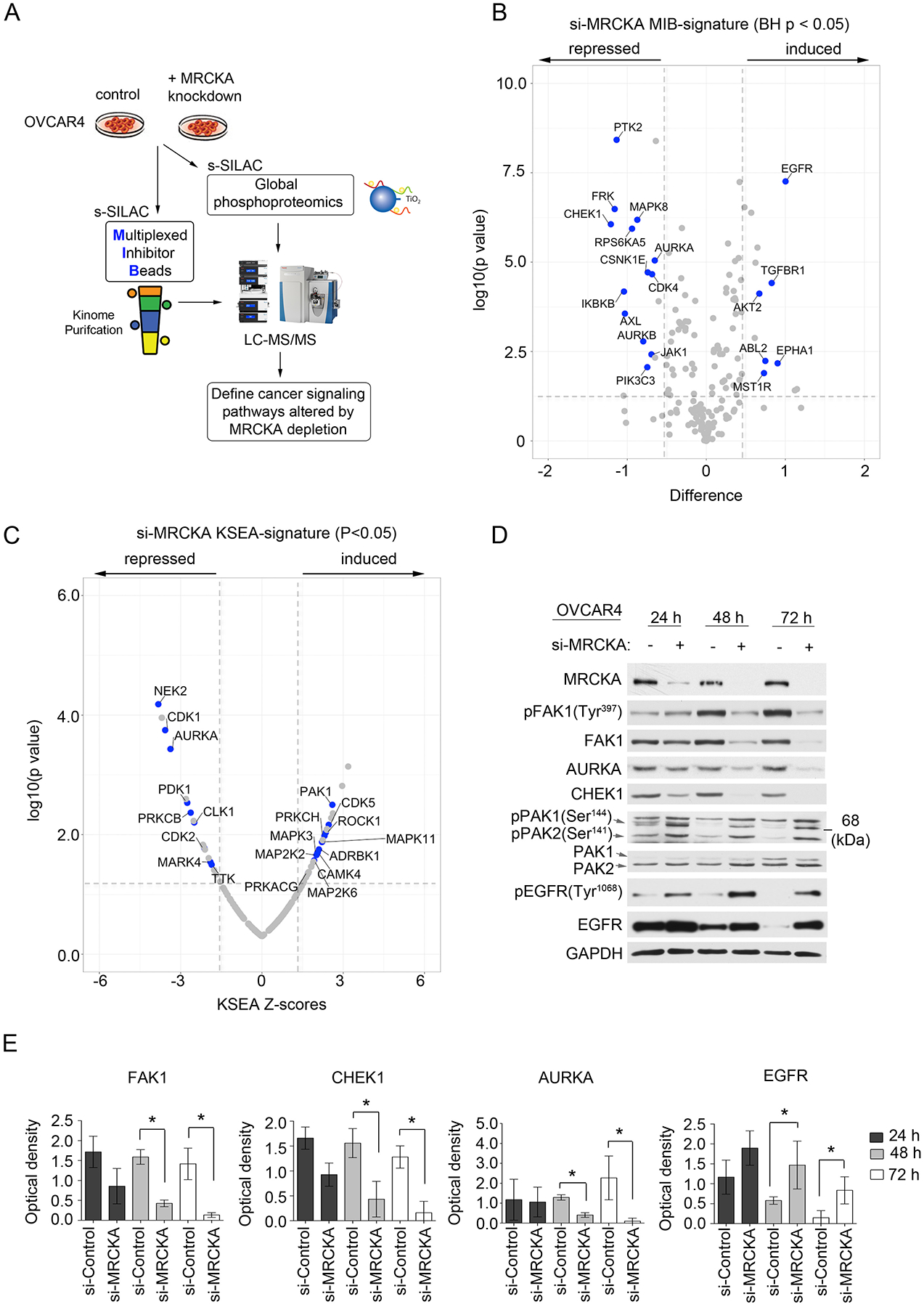
(A) Application of MIB-MS and global phosphoproteomics to explore the consequence of MRCKA knockdown on HGSOC signaling. (B) Volcano plot depicts kinases with induced or repressed MIB-binding after MRCKA knockdown. OVCAR4 cells were transfected with siRNAs targeting MRCKA or control siRNAs for 48 hours and subjected to MIB-MS analysis. Statistical differences in kinase log2 s-SILAC ratios comparing MRCKA relative to control siRNA were determined by ANOVA Benjamini-Hochberg adjusted p values at FDR of <0.05. Kinase log2 s-SILAC ratios were determined by comparing ratio of ratios (siMRCKA/s-SILAC relative siControl/s-SILAC). MIB-MS profiling was performed in biological duplicate. (C) Volcano plot depicts kinases predicted to be activated or inhibited in response to MRCKA knockdown using Kinase Substrate Enrichment Analysis (KSEA). Phosphoproteomics datasets were queried using PhosphoSitePlus at p value cutoff (0.05) and NetworkKIN score cutoff of 2 and a Set substrate count cutoff of 10. Statistical differences in log2 s-SILAC ratios of phosphosites comparing siMRCKA/s-SILAC relative to siControl/s-SILAC were determined by paired t-test, Benjamini-Hochberg adjusted p values at FDR of <0.05. Phosphoproteomics profiling was performed in biological duplicate. (D) Immunoblot analysis of kinase remodeling in OVCAR4 cells transfected with MRCKA or control siRNAs and cultured for 72 hours. Protein abundance and phosphorylation levels were assessed 24, 48 or 72 hours post MRCKA knockdown. Blots are representative of 3 independent experiments. (E) Densitometric analysis of immunoblots presented in (D). Values indicate the optical density of total protein levels from an immunoblot normalized to total protein content (loading control, GADPH). Quantitation of immunoblot bands was performed in ImageJ using 3 independent biological replicates. *P≤0.05 by Student’s t-test. Related data (hierarchical clustering of MIB-MS and phosphoproteomics profiles, a KSEA bar graph, and additional immunoblot and densitometry analysis) can be found in figure S3.
Immunoblot analysis confirmed a time-dependent reduction in protein abundance of FAK1, CHEK1 and AURKA following MRCKA knockdown, as well as an increase in total EGFR protein levels (Fig. 4, D and E). The decrease in phosphorylation of FAK1 (at Tyr397) and increase phosphorylation of EGFR (at Tyr1068) in response to MRCKA depletion was due to corresponding changes in total FAK1 or EGFR protein levels, respectively (fig. S3E). A visible increase in phosphorylation of PAK1 (at Ser144) was detected 24, 48 and 72 hours after MRCKA knockdown by immunoblot with minimal change in total PAK1 protein levels (Fig. 4D).
Together, the data suggest that MRCKA knockdown remodeled the kinome of HGSOC cells in a manner that impacted several established HGSOC signaling pathways, including the repression of FAK1 and CHEK1 with an induction of PAK1 and others. However, as OVCAR4 cells exhibited dependency on MRCKA for viability, subsequent validation studies will be required to parse out kinase changes directly due to MRCKA knockdown versus those associated with cellular stress or apoptosis.
Genetic depletion of MRCKA impairs focal adhesion signaling and sensitizes HGSOC cells to carboplatin and PAK inhibitors
Reduced abundance of FAK1 protein and phosphorylation of FAK1 (at Tyr397) was observed by immunoblot in all HGSOC cells following MRCKA ablation (Fig. 5A–B). This result was confirmed using two distinct MRCKA siRNAs (fig. S4A). Decreased levels of FAK1 phosphorylation in response to MRCKA knockdown were due to changes in total FAK1 protein levels for the majority of the HGSOC cell lines, with the exception of OVCAR8 and JHOS-4 cells, where MRCKA knockdown reduced phosphorylation of FAK1 (at Tyr397) in addition to FAK1 protein levels (fig. S4B). A minimal, insignificant reduction in PTK2 mRNA expression was observed in OVCAR4 cells after MRCKA knockdown (fig. S4C), indicating that MRCKA protein loss likely impacted FAK1 at the posttranslational, protein level. Additionally, we observed a time-dependent decrease in FAK1-mediated phosphorylation of its substrate paxillin at (Tyr118) following MRCKA knockdown (Fig. 5C). Furthermore, knockdown of MRCKA reduced filamentous actin fibers and the formation of focal contacts in HGSOC cells, supporting an integral role for MRCKA in facilitating focal adhesion signaling in HGSOC cells (Fig. 5D–E and fig. S4D). Clusters of viable tumor cells (spheroids) in HGSOC patients representing a major component of chemo-resistant and recurrent disease (25). Knockdown of MRCKA in HGSOC cells grown in three dimensional (3D) non-adherent cultures significantly reduced spheroid formation, supporting MRCKA as a potential therapeutic target for treatment of residual disease (Fig. 5, F and G).
Figure 5. MRCKA knockdown blocks focal adhesion signaling impairing spheroid formation and sensitizes HGSOC cells to carboplatin or PAK inhibitors.
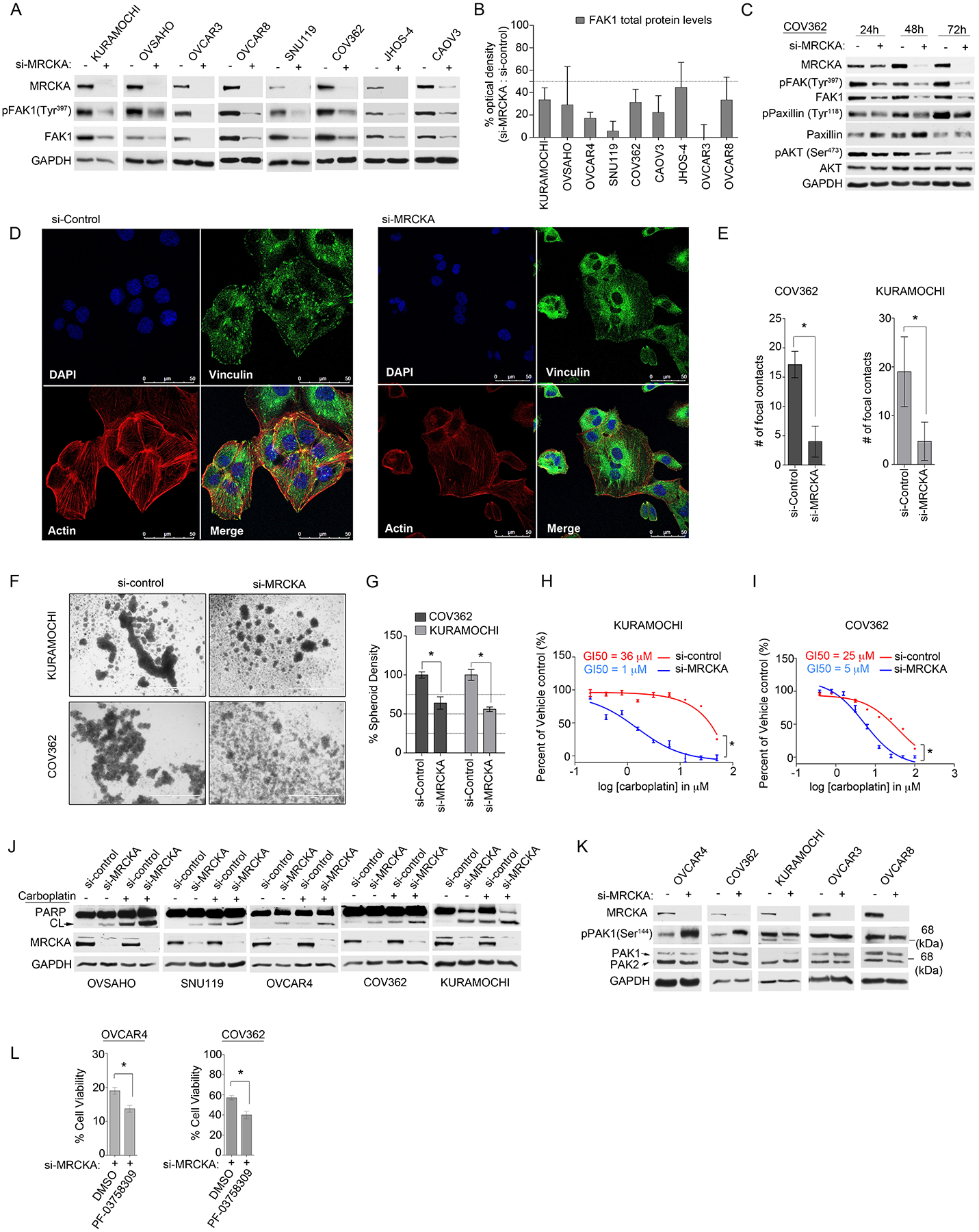
(A) Immunoblot analysis of FAK1 activating phosphorylation and total FAK1 protein abundance in HGSOC cells transfected with control or MRCKA siRNAs for 72 hours. Blots are representative of 3 independent experiments. (B) Densitometric analysis of immunoblots presented in (A). Values indicate the optical density of total protein levels from an immunoblot normalized to total protein content (loading control, GADPH) expressed as a percent change (si-MRCKA / si-control). Quantitation of immunoblot bands was performed in ImageJ using 3 independent biological replicates. (C) Immunoblot analysis of focal adhesion signaling markers in COV362 cells transfected with MRCKA or control siRNAs for 24, 48 or 72 hours. Blots are representative of 2 independent experiments. (D) Confocal fluorescence microscopy of focal adhesion and actin cytoskeleton in COV362 cells transfected with MRCKA or control siRNAs 72 hours. (DAPI) nuclear staining by DAPI, (Vinculin) focal contacts revealed by anti-vinculin antibody, (Actin) F-actin detected by TRITC-conjugated phalloidin and (Merge) merged stain of DAPI, phalloidin and vinculin. Images are representative of 3 independent experiments. Scale bar = 50 μm. (E) Quantitation of focal adhesion contacts in COV362 and KURAMOCHI cells transfected with MRCKA or control siRNAs 72 hrs. Data were analyzed as number of focal contracts; presented as means of 3 independent assays. *P≤0.05 by Student’s t-test. (F and G) Assessment of spheroid formation in COV362 and KURAMOCHI cells transfected with MRCKA or control siRNAs 72 hrs. Representative microscope images of KURAMOCHI or COV362 spheroids from 3 independent biological replicates. Scale bar = 1000 μm. (G) Quantitation of spheroid density determined by ImageJ. *P≤0.05 by Student’s t-test. (H and I) Cell-Titer Glo assay for cell viability of KURAMOCHI (H) or COV362 (I) cells transfected with siRNAs targeting MRCKA or control siRNAs, cultured for 72 hours (KURAMOCHI) or 120 hrs (COV362) and treated with increasing doses of carboplatin or DMSO. Data were analyzed as % of DMSO control; presented as means of 3 independent assays. GI50 were determined using PRISM. (J) Apoptosis assessed by immunoblotting for cleaved-PARP abundance in HGSOC cells transfected with siRNAs targeting MRCKA or with control siRNAs, cultured for 72 hours and treated with carboplatin (25 μM) or DMSO. Blots are representative of 2 independent experiments. (K) Immunoblot analysis of phosphorylated and total levels of PAK1 in HGSOC cells transfected with MRCKA or control siRNAs 72 hours. Blots are representative of 3 independent experiments. *P≤0.05 by Student’s t-test. (L) Cell-Titer Glo assay for cell viability of OVCAR4 or COV362 cells transfected with siRNAs targeting MRCKA or control siRNAs, cultured for 120 hrs and treated with PF-03758309 (3.5 nM, OVCAR4 or 50 nM, COV362) or DMSO. Data were analyzed as % of DMSO control; presented as means of 3 independent assays. Related data (immunoblot analysis of FAK1 protein levels using 2-distinct siRNAs, FAK1 mRNA levels following MRCKA-knockdown, confocal fluorescence microscopy of focal adhesion and actin cytoskeleton in KURAMOCHI cells, and Bliss synergy analysis of carboplatin and MRCKA siRNA drug synergy) can be found in figure S4.
Oncogenic FAK signaling has been shown to promote chemotherapy resistance in HGSOC and inhibition of FAK1 sensitizes HGSOC cells to DNA damaging agents such as platinum (26). Based on our observations that MRCKA knockdown reduced FAK1 activity in HGSOC cells, we hypothesized MRCKA-depletion may enhance sensitivity towards chemotherapy agents. Indeed, co-treatment of KURAMOCHI or COV362 cells with MRCKA siRNAs and carboplatin was synergistic at blocking cell viability (Fig. 5H–I and fig S4, E to F). Additionally, the combination of MRCKA siRNAs and carboplatin induced a greater amount of PARP cleavage in HGSOC cell lines than did knockdown or drug alone, supporting MRCKA as a candidate therapeutic target to be combined with chemotherapy agents (Fig. 5J).
Kinome reprogramming or re-wiring in response to kinase inhibition represents an established resistance mechanism in cancer cells (11). Consistent with this idea, MIB-MS profiling and phosphoproteomics analysis of MRCKA inhibition in OVCAR4 cells had revealed that MRCKA depletion resulted in enhanced activity of several kinases, including PAK1/2 (described above). Notably, MRCKA knockdown increased phosphorylation of PAK1 (at Ser144) in OVCAR4 and COV362 but not in OVCAR3, KURAMOCHI or OVCAR8 cells (Fig. 5K), suggesting MRCKA inhibition may induce a compensatory response involving Rac1-PAK1/2 signaling in some HGSOC cells. As both MRCKA and PAK1 compete for CDC42 binding in cells (27), knockdown of MRCKA may enable more binding of PAK1 to CDC42, thereby resulting in PAK1 activation. Co-treatment of OVCAR4 or COV362 cells with MRCKA siRNAs and pan-PAK inhibitor PF-03758309 (28) inhibited growth beyond MRCKA knockdown alone, suggesting that co-targeting MRCKA and PAKs may be more effective at inhibiting viability than targeting MRCKA alone in some HGSOC cells (Fig. 5L).
BDP9066 represents a highly selective MRCKA and MRCKB inhibitor for the treatment of HGSOC cells
Our RNAi-based studies revealed a role for MRCKA in HGSOC cell growth, migration and survival through promoting a variety of oncogenic signaling, highlighting MRCKA as a promising kinase drug target in HGSOC. Recently, selective kinase inhibitors, BDP5290 and BDP9066, targeting MRCKA and related kinase Myotonic Dystrophy Kinase-Related CDC42-Binding Kinase Beta (MRCKB or CDC42BPB) respectively, were developed and shown to block migration and invasion of cancer cells (16, 21). Kinase profiling of selectivity of BDP5290 revealed little activity against MRCKA or MRCKB but some presumably off-targets effects on several kinases, including AURKB (fig. S5A, and Data file S8). Conversely, in vitro kinase assays confirmed BDP9066 as a potent inhibitor of MRCKA and MRCKB, with EC50 values of 65 nM and 72 nM, respectively (Fig. 6, A and B). Moreover, MIB-MS kinome profiling of OVSAHO cells treated with 2 μM of BDP9066 for 4 hours revealed selective inhibition of MRCKA and MRCKB amongst the MIB-bound kinome (Fig. 6C, and Data file S9). In comparison, treatment of OVSAHO cells with A-674563, a pan-kinase inhibitor that targets MRCKA (29), reduced MIB-binding of MRCKA and MRCKB, as well as several other kinases (fig. S5B, and Data file S9). Treatment of HGSOC cells with increasing doses of BDP9066 for 4 hours reduced phosphorylation of MRCKA-substrate MLC2 at (Thr18/Ser19), confirming inhibition of MRCKA kinase activity (Fig. 6D). BDP9066-treatment reduced filamentous actin staining in HGSOC cells and significantly reduced migration of COV362 and KURAMOCHI cells (Fig. 6, E and F, and fig. S5, C and D), demonstrating small-molecule targeting of MRCKA using BDP9066 inhibits actin remodeling in HGSOC cells.
Figure 6. Characterizing MRCKA small molecule inhibitors in HGSOC cells.
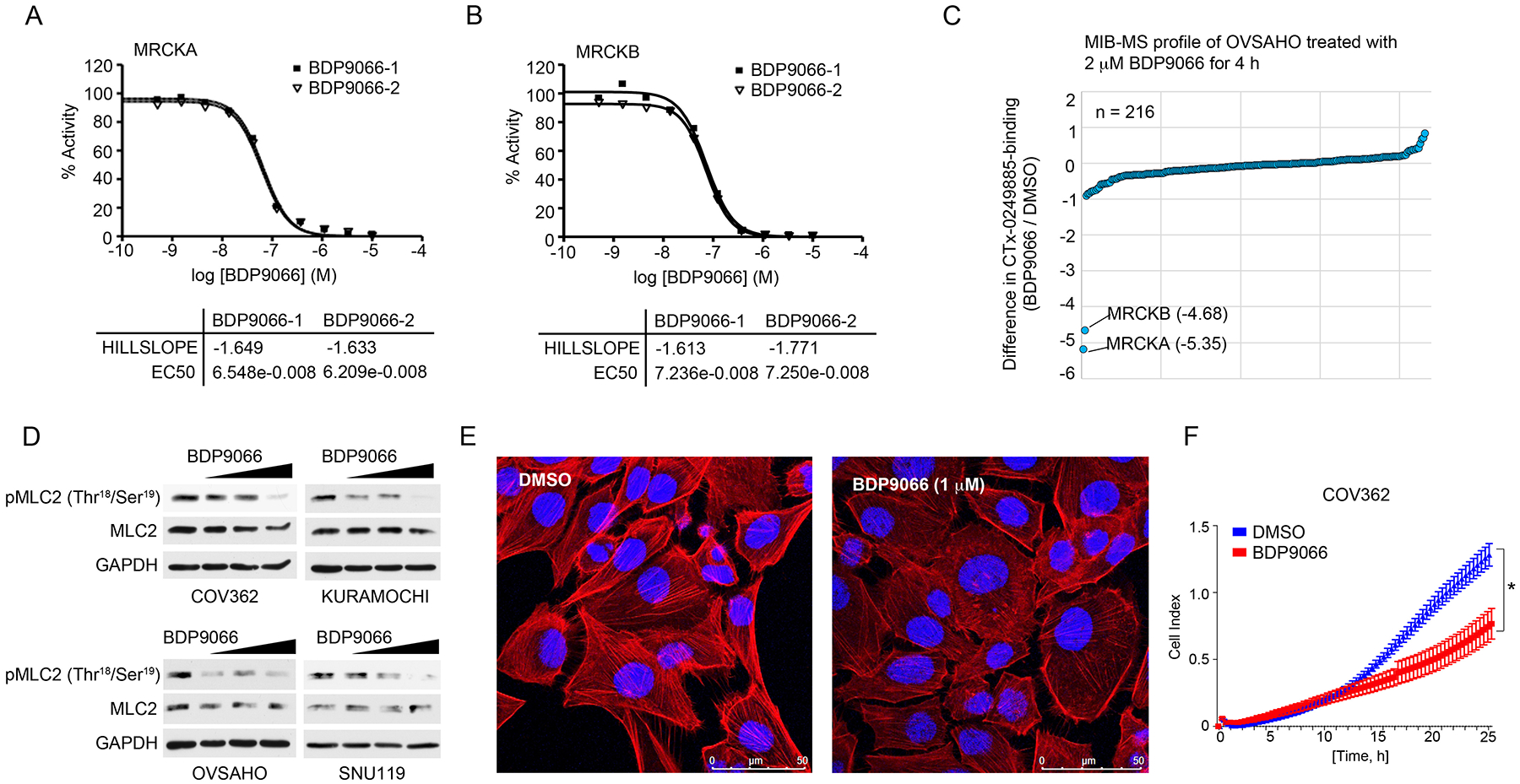
(A and B) In vitro radioisotope-based 33P assays testing the effect of BDP9066 on MRCKA and MRCKB activity. BDP9066 was tested in a 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM. Reactions were carried out at 10 μM ATP. Curve fits were performed where MRCKA or MRCKB activities at the highest concentration of BDP9066 were less than 65%. Hillslope and EC50 values were determined in mM. (C) MIB-MS kinome profile of OVSAHO cells treated with BDP9066 for 4 hours. SILAC-labeled OVSAHO cells were treated with 2 μM BDP9066 or DMSO for 4 hours and lysates incubated with CTx-0249885 beads. Line graph depicts average SILAC-determined log2fold changes in kinase MIB binding as a ratio of BDP9066/DMSO. Biological duplicates of SILAC Heavy-BDP9066/Light-DMSO or Light-BDP9066/Heavy-DMSO were performed. (D) Immunoblot analysis of MLC2 phosphorylation in HGSOC cells treated with increasing concentrations of BDP9066 (0, 0.5, 1 and 3 μM) for 4 hours. Blots are representative of 3 independent experiments. (E) Confocal fluorescence microscopy of actin cytoskeleton in COV362 treated with DMSO or 1 μM BDP9066 for 48 hours. F-actin was detected by TRITC-conjugated phalloidin and nuclear staining by DAPI. Images are representative of 3 independent experiments. Scale bar = 50 μm. (F) Analysis of cell migration in COV362 cells treated with DMSO or 1 μM of BDP9066. Migration monitored over a 24-hour period using the xCELLigence Real-Time Cell Analyzer (RTCA). Data were analyzed as cell index; presented as means ± SD of 3 independent assays. *P≤0.05 by Student’s t-test. Related data (in vitro kinase assays testing BDP5290 selectivity, a comparison of BDP9066 and A-674563 MIB-MS profiles, and the consequence of BDP9066-treatment on actin cytoskeleton and migration of KURAMOCHI cells) can be found in figure S5.
MRCKA inhibition induces apoptosis and blocks spheroid formation, establishing MRCKA as a therapeutic target in HGSOC
Next, we explored the impact of small molecule inhibition of MRCKA kinase activity on cell growth and survival. BDP9066-treatment inhibited cell viability to varying degrees across the HGSOC cell lines, with KURAMOCHI, OVCAR8, SNU119, and OVCAR3 among the most affected by MRCKA inhibition, exhibiting GI50 values < 0.5 μM (Fig. 7A). Moreover, BDP9066 reduced colony formation of HGSOC cells with nearly complete inhibition of colonies at a dose of 0.312 μM in SNU119, OVCAR8, OVCAR3, KURAMOCHI and OVCAR4 (Fig. 7B). These cell lines were among the most sensitive to MRCKA knockdown, demonstrating the overlap between small molecule inhibition and genetic depletion of MRCKA. BDP9066-treatment increased levels of cleaved PARP in SNU119, OVCAR8, OVCAR3, and KURAMOCHI cells (Fig. 7C), indicating that small-molecule inhibition of MRCKA kinase activity can trigger apoptosis in HGSOC cells. Notably, COV362 cells showed no increase in cleaved PARP levels, consistent with MRCKA knockdown findings. Because MRCKB is also targeted by BDP9066, we depleted MRCKB using siRNAs in HGSOC cells to determine whether MRCKB inhibition contributed to the observed response. Knockdown of MRCKB marginally inhibited cell viability (~40% growth inhibition) of OVCAR4, JHOS-4 and OVCAR3, but not other HGSOC cell lines (fig. S6, A and B), suggesting MRCKA inhibition is likely the major contributing factor to the observed anti-growth effects of BDP9066.
Figure 7. Treatment of HGSOC cells with BDP9066 blocks cell growth, induces apoptosis and inhibits focal adhesion signaling impairing spheroid formation of HGSOC cells.
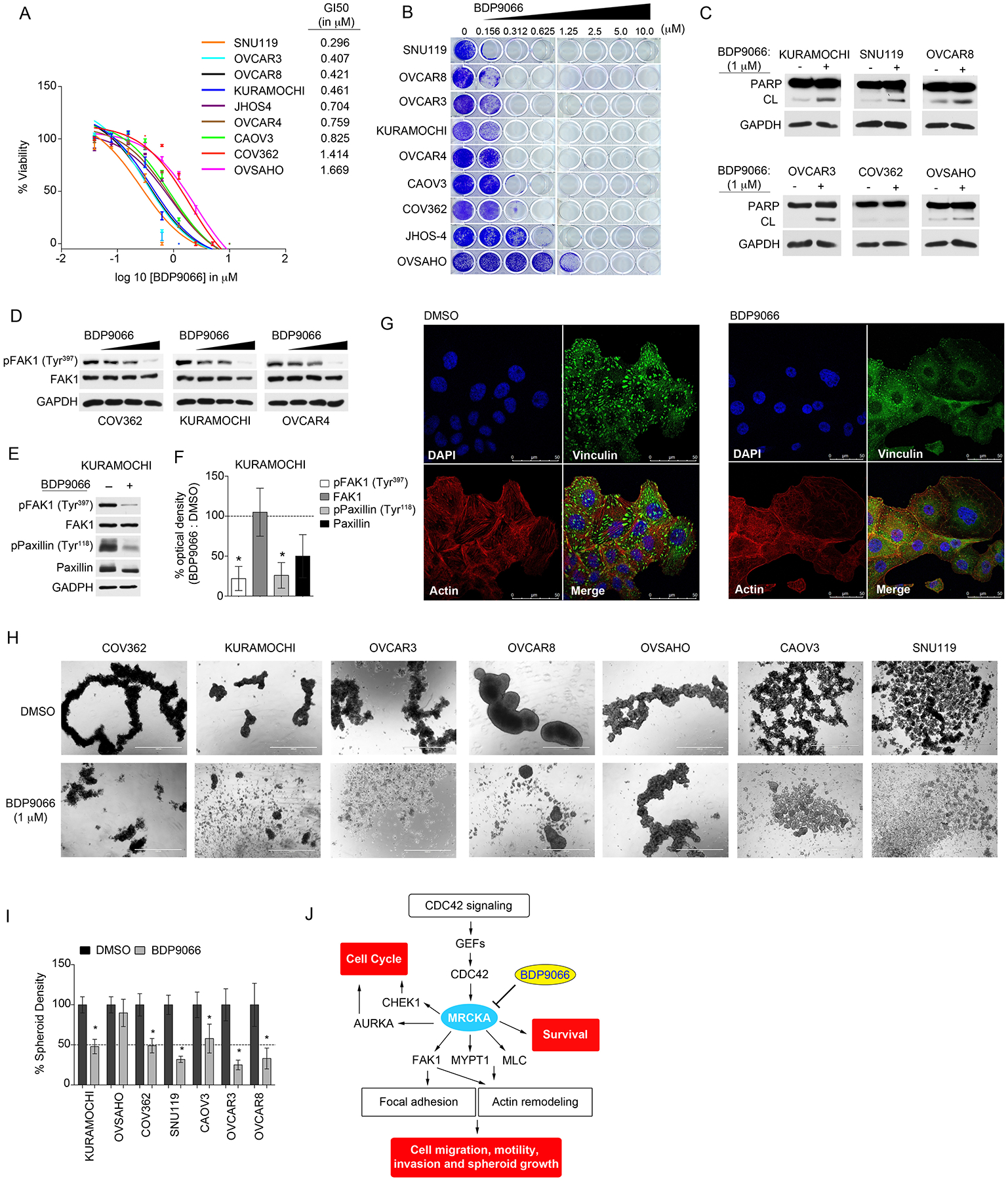
(A) Cell-Titer Glo assay for cell viability of HGSOC cell lines treated with increasing concentrations of BDP9066 or DMSO and cultured for 120 hours. Data were analyzed as % cell viability of DMSO control; presented as means of 3 independent assays. GI50 values for BDP9066 were generated in Prism. (B) Long-term 14-day colony formation assay of HGSOC cells treated with BDP9066 or DMSO. Colony formation was assessed by crystal violet staining. Representative images of 3 biological replicates. (C) Apoptosis assessed by immunoblotting for cleaved-PARP abundance in HGSOC cells treated with DMSO or 1 μM of BDP9066 for 72 hours. Blots are representative of 3 independent experiments. (D) Immunoblot analysis of FAK1 phosphorylation and total FAK1 protein abundance in HGSOC cells treated with escalating doses of BDP9066 (0, 0.5, 1 and 3 μM) for 48 hours. Blots are representative of 3 independent experiments. (E) Immunoblot analysis of phosphorylation of FAK1 and Paxillin in HGSOC cells treated with 2 μM BDP9066 for 48 hours. Blots are representative of 3 independent experiments. (F) Densitometric analysis of immunoblots presented in (E). Values indicate the optical density of phosphorylated or total protein levels from an immunoblot normalized to total protein content expressed as a percent change (BDP9066/DMSO). Phosphorylated proteins were normalized to loading control, then normalized to total abundance of the respective protein. Quantitation of immunoblot bands was performed in ImageJ using 3 independent biological replicates. *P≤0.05 by Student’s t-test. (G) Confocal fluorescence microscopy of focal adhesion and actin cytoskeleton in KURAMOCHI cells treated with DMSO or 1 μM BDP9066 for 24 hours. (DAPI) nuclear staining by DAPI, (Vinculin) focal contacts revealed by anti-vinculin antibody, (Actin) F-actin detected by TRITC-conjugated phalloidin and (Merge) merged stain of DAPI, phalloidin and vinculin. Images are representative of 3 independent experiments. Scale bar = 50 μm. (H and I) Assessment of spheroid formation in HGSOC cells treated with DMSO or 1 μM BDP9066 for 72 hrs. Representative images of HGSOC cell spheroids (H) from 3 independent biological replicates. Scale bar = 1000 μm. (I) Quantitation of spheroid density determined by ImageJ. *P≤0.05 by Student’s t-test. (J) Model of the mechanism, depicting the integral role of MRCKA in promoting proliferation, migration and survival of HGSOC cells, thus highlighting MRCKA as new kinase inhibitor target for the treatment of HGSOC. Related data (knockdown of MRCKB’s impact on HGSOC cell viability, drug synergy studies involving BDP9066 and carboplatin or PAK1 inhibitors, as well as quantitation of number of focal contacts in KURAMOCHI cells in response to BDP9066) can be found in figure S6.
In our loss-of-function studies (described above), we showed MRCKA knockdown sensitized HGSOC cells to carboplatin or PAK inhibitors. In agreement, co-treatment of HGSOC cells with BDP9066 and carboplatin improved growth inhibition relative to single agents in HGSOC cell lines trending towards drug synergy (fig. S6, C to F). Similar to MRCKA knockdown, increased activating phosphorylation of PAK1 (at Ser144) was observed in response to BDP9066-treatment in COV362 cells, and combination therapies involving BDP9066 and PAK1 inhibitor FRAX1036 (30) further inhibited growth compared to single agent drugs, with a Bliss synergy score of 6.4 (fig. S6, G and H). Thus, PAK1 induction may represent a potential compensatory resistance mechanism to BDP9066 therapy in some HGSOC cells, requiring combination therapies that co-target MRCKA and PAK1.
Treatment of HGSOC cells with escalating doses of BDP9066 reduced activating phosphorylation of FAK1 (at Tyr397), but did not lower total protein abundance of FAK1 that was observed with MRCKA-depletion, signifying MRCKA may have both kinase-dependent and independent functions in focal adhesion signaling (Fig. 7D). Treatment of KURAMOCHI cells with BDP9066, decreased phosphorylation of FAK1 at (Tyr 397) and reduced both total Paxillin levels and phosphorylation of Paxillin at (Tyr118) (Fig. 7E and F). Moreover, BDP9066-treatement reduced the number of focal contacts relative to control treated cells, demonstrating small-molecule inhibition of MRCKA kinase activity blocks focal adhesion signaling similarly to MRCKA knockdown (Fig. 7G and fig. S6I). Furthermore, treatment of HGSOC cell lines grown in 3D cultures with BDP9066 blocked spheroid formation (Fig. 7, H and I), supporting BDP9066 as a promising drug for the treatment of HGSOC, including metastatic ascites. Notably, BDP9066-treatment had no impact on spheroid formation of OVSAHO cells, indicating that some HGSOC cells are intrinsically resistant to small-molecule inhibition of MRCKA.
DISCUSSION
Protein kinases are arguably one of the most important regulatory enzymes in cells, and drugs targeting these enzymes have led to great improvements in treating cancer. However, the vast majority of kinase publications focus on a comparatively small group of well-understood kinases, yet synthetic lethality screens repeatedly identify various understudied kinases as playing a vital role in cancer cell proliferation and survival (31). This is particularly evident in ovarian cancer, where only a small percentage of the kinome has been therapeutically explored. Here, we profiled the kinome in HGSOC tumors using MIB-MS and identified MRCKA, a CDC42 effector kinase not previously linked to HGSOC to be highly abundant in HGSOC patient and PDX tumors. Loss-of-function studies targeting MRCKA in HGSOC cell lines, revealed an essential role for MRCKA in HGSOC cell proliferation, survival, and migration, establishing MRCKA as a plausible drug target in this disease (Fig. 7J).
In addition to MRCKA’s established role in actin remodeling (32), here, we provide evidence that MRCKA protein is essential for FAK1 activity and protein stability, as knockdown of MRCKA reduced both activating FAK1 phosphorylation and total protein levels independent of RNA changes. Moreover, focal adhesions were reduced in HGSOC cells following MRCKA inhibition blocking spheroid formation, supporting a previously unknown role for MRCKA in focal adhesion signaling. Future experiments assessing focal adhesion formation following forced expression of FAK1 or constitutively active FAK1 mutants in MRCKA-depleted cells will be required to establish a causal link of MRCKA with FAK1 in focal adhesion signaling. We have extensively characterized the loss of FAK1 protein in response to MRCKA knockdown, but the mechanism by which MRCKA protein depletion influences FAK1 protein levels in HGSOC cells has not yet been defined. FAK1 protein stability has been shown to be controlled by E3 ligase activity regulating focal adhesion signaling in cells (33). Evaluation of FAK1 ubiquitylation and E3 ligase activity following MRCKA depletion could provide insight into the mechanism linking MRCKA activity and FAK1 protein stability. One could envisage MRCKA directly or indirectly regulating a critical E3 ligase that controls FAK1 protein levels, where blockade of MRCKA induces E3 ligase activity promoting FAK1 degradation.
Intriguingly, we observed inhibition of MRCKA kinase activity using BDP9066 reduced phosphorylation of FAK1 but did not reduce FAK1 protein abundance as observed with MRCKA knockdown. These findings suggest MRCKA may have kinase-independent functions that promote FAK1 protein stability and focal adhesion signaling in HGSOC cells. Rescue experiments expressing kinase-dead MRCKA in cells depleted of MRCKA protein could help parse kinase-dependent vs independent functions of MRCKA in HGSOC cells. Additional proteomics studies investigating whether MRCKA influences FAK1 activity and/or stability directly through phosphorylation, or indirectly through protein interaction will be required. Notably, MRCKA is a large protein that has several binding domains, (34), where MRCKA may function as a scaffold facilitating FAK1 activity in focal adhesion signaling. Recently, PROTACs have been employed to selectively degrade proteins providing a therapeutic strategy to block both the kinase-dependent and independent functions of a specific kinase (35). This was recently applied to FAK1, which showed degradation of FAK1 was superior to kinase inhibition alone, owing to the blockade of both FAK1 kinase signaling and scaffolding capabilities (36). Development of PROTACs that degrade MRCKA may provide a promising technique to gain insight into MRCKA’s kinase-dependent and independent functions, as well as offer a therapeutic strategy to block both MRCKA kinase and non-kinase functions in HGSOC.
Tumors often bypass targeted kinase inhibition by activating alternative kinase-mediated survival pathways, a process termed kinome reprogramming (37). Proteomic characterization of MRCKA depleted HGSOC cells showed activation of several kinases including PAK1, suggesting a potential kinome reprogramming mechanism linked to MRCKA protein loss in HGSOC cells. Previous studies have shown that both MRCKA and PAK1 compete for CDC42 binding in cells (27); therefore, knockdown of MRCKA protein may facilitate binding of PAK1 to CDC42 resulting in PAK1 activation. Because PAK1 is an established oncogenic kinase in ovarian cancer (38), combination therapies blocking PAK1 feedback activation may be required to achieve durable responses when targeting MRCKA in some HGSOC cells. Small molecule inhibitors of CDC42/RAC1 have emerged that can block cell migration and metastasis in cancer models (39). Targeting upstream of MRCKA using a dual CDC42/RAC1 inhibitor such as MBQ-167 (39) may represent an effective strategy to shut down both MRCKA and PAK signaling in HGSOC cells.
Our evaluation of current MRCK inhibitors revealed BDP5290 showed minimal activity against MRCK kinases, whereas BDP9066 was a highly selective and potent inhibitor of MRCKA and MRCKB in HGSOC cells. Of particular interest, MIB-MS analysis of HGSOC cells treated with 2 μM of BDP9066 showed selective reduction in MRCKA and MRCKB MIB-binding (out of 216 kinases) but not the closely related kinase ROCK1, which possesses similar protein structure overlap (21). The MRCK family of kinases consists of 3 members: MRCKA, MRCKB and Myotonic Dystrophy kinase-related CDC42-binding kinase gamma (CDC42BPG or MRCKG), with the latter exhibiting a more tissue-specific expression pattern (34). We did not detect MRCKG in our proteomics analysis of BDP9066 selectivity in OVSAHO cells; however, further studies will be required to determine if MRCKG is expressed and/or inhibited by BDP9066 in other HGSOC cells. Notably, knockdown of MRCKB reduced the proliferation of some HGSOC cell lines, suggesting inhibition of MRCKB may contribute to the overall efficacy of BDP9066. Future studies exploring the overlapping or distinct functions of these related MRCK kinases in HGSOC signaling will be of particular interest.
Malignant ascites, a buildup of fluid containing cancer cells in the abdomen, often occurs in late-stage HGSOC patients with residual chemoresistant disease, causing substantial morbidity and death (25). Tumor cells within ascites of ovarian cancer patients often grow as spheroids, or aggregates of tumors cells which are typically difficult to treat with conventional therapies (40). Our findings showed BDP9066-treatment impaired focal adhesion signaling, resulting in reduced spheroid formation of HGSOC cells grown in 3D cultures, suggesting MRCKA inhibition may have therapeutic potential in late-stage patients with residual disease. Of particular interest, our proteogenomics analysis of HGSOC patient tumors revealed MRCKA was overexpressed in ~25% of tumors, and these tumors were enriched for CDC42 and FAK1 signaling. Preclinical studies in HGSOC tumor xenograft models, including those overexpressing MRCKA, will be required to test both the efficacy of BDP9066 therapy to inhibit tumor growth and cause tumor regression, as well to define potential toxicities associated with targeted MRCKA inhibition.
MATERIALS AND METHODS
Cell lines
Cell lines were verified by IDEXX laboratories and were verified Mycoplasma-negative (1/7/19) using the Hoechst DNA stain method. OVCAR4, KURAMOCHI, OVSAHO and OVCAR8 cell lines were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin and 2mM GlutaMAX. OVCAR3 cells were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin, 2mM GlutaMAX, and 5 μg/mL insulin. SNU-119 cells were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin, 2mM GlutaMAX and 25mM HEPES. COV362 cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin and 2mM GlutaMAX. CAOV3 cells were maintained in DMEM supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin, 2mM GlutaMAX, and 100mM Sodium Pyruvate. JHOS-2 and JHOS4 cells were maintained in DMEM/F12 supplemented with 10% FBS, 100 U/ml Penicillin-Streptomycin, 2mM GlutaMAX and Non-Essential Amino Acids. FTSEC cells were maintained in DMEM supplemented with 4% FBS, 100 U/ml Penicillin-Streptomycin, 2mM GlutaMAX, and 1x Insulin-Transferrin-Selenium (Gibco). Cell lines used for the super-SILAC reference sample (MOLT4, UACC257, ACHN, Colo205, and PC-3) were grown for seven doublings in arginine- and lysine-depleted media supplemented with heavy isotope labeled [13C6,15N4]arginine (Arg10) (84 mg/L) and [13C6]lysine (Lys8) (48 mg/L) (Sigma), and unlabeled leucine (50 mg/L) (ThermoFisher Scientific) as described previously (41). OVSAHO SILAC cells were grown for seven doublings in arginine- and lysine-depleted RPMI supplemented with either unlabeled L-arginine (84 mg/L), L-lysine (48 mg/L) and leucine (50 mg/L) (ThermoFisher Scientific) or equimolar amounts of heavy isotope labeled [13C6,15N4] arginine (Arg10) and [13C6] lysine (Lys6) (Sigma). All cells were kept at 37°C in a 5% CO2 incubator.
Patient Samples
All patient samples used in this study were obtained from the Fox Chase Cancer Center (FCCC) Biosample Repository Facility (BRF). The FCCC BRF maintains a longstanding Institutional Review Board-approved protocol for collection, banking and distribution of de-identified biospecimens and associated clinical data. Consent and authorization for the use of de-identified specimens and associated clinical data for unrestricted research was obtained from all BRF participants prior to specimen collection.
Frozen Tumor Samples
Ovarian tumors from 28 chemo-naïve patients with stage III or IV high grade serous ovarian carcinoma (HGSOC) were obtained from the FCCC BRF for this IRB-approved research (16–9031 and 14–809). Tumor tissue was snap frozen at the time of collection and stored at −80°C in the FCCC BRF. Tumor histology and cellularity was confirmed by the FCCC BRF pathologist at the time of banking. Tumors isolated from HGSOC patient-derived xenografts (PDX) models were snap frozen and stored at −80°C until sample processing for MIB-MS analysis. A description of patient tumors and PDX tumor tissue details can be found in (Data file S2).
Compounds
A-674563, Carboplatin, FRAX1036, and PF-03758309 were purchased from Selleckchem. BDP5290 and BDP9066 were purchased from Glixx Labs and ProbeChem respectively (Data file S10). For the compounds utilized in MIB synthesis, Purvalanol B was purchased from Abcam. PP58 (42) and VI16832 (43) were custom synthesized according to previously described methods by The Center for Combinatorial Chemistry and Drug Discovery, Jilin University, P.R. China. CTx-0294885 (44) was purchased from MedKoo Biosciences, Inc (406457). Conjugation of inhibitors to beads was performed by carbodiimide coupling to ECH Sepharose 4B (CTx-0294885, VI16832 and PP58) or EAH Sepharose 4B (purvalanol B) (GE Healthcare).
In vitro kinase profiling
In vitro kinase assays for BDP5290 and BDP9066 were performed by Reaction Biology Corp. (Malvern, PA). BDP5290 compound was tested in a single dose duplicate mode at a concentration of 10 μM against 371 kinases. BDP9066 was tested against MRCKA/CDC42BPA or MRCKB/CDC42BPB. BDP9066 was tested in 10-dose mode with 3-fold serial dilution starting at 10 μM. Reactions were carried out at 1 μM ATP control. Staurosporine was tested in 10-dose IC50 mode with 4-fold serial dilution starting at 20 or 100 μM. Alternative control compounds were tested in 10-dose IC50 mode with 3 or 4-fold serial dilution starting at 10, 20, or 50 μM. Data pages presented in Data file S8, include raw data % enzyme activity (relative to DMSO controls), and curve fits. List of kinases sources used in kinase profiling can be found at: (http://www.reactionbiology.com/webapps/site/KinaseDetail.aspx?page=Kinases&id=1&filter=512).
Immunoblotting
Samples were harvested in MIB lysis buffer, subjected to SDS-PAGE chromatography and transferred to PVDF membranes before western blotting with primary antibodies. For pMYPT and pMLC2 blots, cells were harvested in a buffer containing 1% (w/v) SDS, 50 mM Tris pH 7.5 supplemented with protease and phosphatase inhibitors buffer and were passed through QIAshredder columns (Qiagen, 79654) (21). For the list of primary antibodies used, see (Data file S10). Secondary HRP-anti-rabbit and HRP-anti-mouse were obtained from ThermoFisher Scientific. SuperSignal West Pico and Femto Chemiluminescent Substrates (Thermo Scientific) were used to visualize blots. Western blot images were quantified using the Analyze>Gels function in Image J open source software (National Institutes of Health). Optical density values were normalized by sum (fig. S2, G–J) or by a fixed point (Fig. 4E, Fig. 5B and Fig. 7F) (45). Phosphorylated proteins were normalized to loading control, then normalized to total abundance of the respective protein.
Actin cytoskeleton and focal adhesion staining
KURAMOCHI and COV362 cells were plated at a density of 2×105 cells per well in a 6 well plate with an 18 mm2 glass coverslip inside each well. Following treatment, cells were fixed and stained using the Actin Cytoskeleton / Focal Adhesion Staining Kit (Millipore Sigma) following manufacturer’s instructions. Anti-vinculin (1:500), TRITC-conjugated phalloidin (1:1000) and donkey anti-mouse Alexa Fluor 488-conjugated secondary antibodies (Thermo Scientific, 1:1000) were applied to the coverslips for 1 hour at room temperature. Following antibody incubation, coverslips were mounted on slides using ProLong Gold antifade reagent with DAPI (Thermo Scientific) and allowed to set overnight. Images were taken with a Leica SP8 confocal microscope at 40x magnification. Focal contacts were counted using the 3D Objects Counter tool in Image J.
Spheroid formation
Cells were plated at a density of 50,000 per well in 24 well ultra-low attachment plates (Corning 3473). For drug treatment studies, drug was added to the cells at time of plating and images were taken 72 hours later on an EVOS digital inverted microscope. For siRNA studies, cells were transfected with siRNA in 2D culture plates 48 hours prior to addition to the ultra-low attachment plates, and images were taken 72 hours later (5 days post-transfection). Spheroid area was quantified in ImageJ using the Analyze Particles tool after the color threshold was set to identify larger clusters of cells.
Drug synergy analysis
Drug synergy was determined using SynergyFinder (https://synergyfinder.fimm.fi) using the Bliss model and viability as the readout (46). Each drug combination was tested in triplicate.
Ovarian Tissues and Tumor Specimens for IHC
Cancerous and benign ovarian specimens were obtained from patients who underwent surgical resection at Fox Chase Cancer Center. Seven ovarian tumor tissue microarrays (TMAs) containing a total of 129 ovarian tumor cases in duplicate and one normal ovary and fallopian tube TMA containing a total of 24 normal ovaries and 11 fallopian tube cores in duplicate were provided by the Biosample Repository of Fox Chase Cancer Center (Data file S5). All these samples were used with informed patient consent, and the study was approved by the Institutional Review Boards of Fox Chase Cancer Center.
Typically, tissue specimens were fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. All tumors were histologically classified according to the World Health Organization (WHO) classification, and the surgical stages were determined according to the classification of International Federation of Gynecology and Obstetrics (FIGO). Two-Tiered histological grading system was also used for grading the serous carcinoma.
Immunohistochemistry (IHC) and IHC Evaluation
Immunohistochemical staining was carried out according to standard methods by the FCCC Histopathology Facility. Briefly, 5μm formalin-fixed, paraffin-embedded TMA sections were deparaffinized and hydrated. Sections were then subjected to heat-induced epitope retrieval with 0.01 M citrate buffer (pH 6.0). Endogenous peroxidases were quenched by the immersion of slides in 3% hydrogen peroxide solution. The sections were incubated overnight with primary antibodies to MRCKA (Abcam, ab96659) (16) (Rabbit, 1:200, Abcam) at 4 °C in a humidified slide chamber. Immunodetection was performed using the Dako Envision+ polymer system and immunostaining was visualized with the chromogen 3, 3’-diaminobenzidine. The sections were then washed, counterstained with hematoxylin, dehydrated with ethanol series, cleared in xylene, and mounted. Known positive cancer tissues were used as positive controls. As a negative control, the primary antibody was replaced with normal rabbit IgG to confirm absence of specific staining. All slides were viewed with a Nikon Eclipse 50i microscope and photomicrographs were taken with an attached Nikon DS-Fi1 camera (Melville, NY, USA). Immunoreactivity of MRCKA protein was evaluated by a pathologist and IHC score was given based on the intensity of the staining. 0: Negative; 1: mild/weak; 2: moderate/intermediate; 3: Strong/intensive.
Cell proliferation Assays
For short-term growth assays, 1500–5000 cells were plated per well in 96-well plates and allowed to adhere and equilibrate overnight. Drug was added the following morning and after 120 hours of drug treatment, cell viability was assessed using the CellTiter-Glo Luminescent cell viability assay according to manufacturer (Promega). Student’s t-tests were performed for statistical analyses and p values ≤ 0.05 were considered significant.
qRT-PCR
GeneJET RNA purification kit (Thermo Scientific) was used to isolate RNA from cells according to manufacturer’s instructions. qRT-PCR on diluted cDNA was performed with inventoried TaqMan® Gene Expression Assays on the Applied Biosystems 7500 Fast Real-Time PCR System. The TaqMan Gene Expression Assay probes (ThermoFisher Scientific) used to assess changes in gene expression include MRCKA (Assay ID: Hs00177522_m1), and PTK2 (Assay ID: Hs00178587_m1).
RNAi Knockdown Studies
siRNA transfections were performed using 25 nM siRNA duplex and the reverse transfection protocol. 1500–5000 cells per well were added to 96 well plates with media containing the siRNA and transfection reagent (Lipofectamine RNAiMax) according to the manufacturer’s instructions. In experiments where inhibitors were used, the inhibitor was added at the time of transfection. Cells were allowed to grow for 72 to 120 hours post-transfection prior to CellTiter Glo (Promega) analysis. Two-to-three independent experiments were performed with each cell line and siRNA. Student’s t-tests were performed for statistical analyses and p values ≤0.05 were considered significant. For western blot studies, the same procedure was performed with volumes and cell numbers proportionally scaled to a 60mm or 10 cm dish, and cells were collected 24 to 72 hours post-transfection. siRNA product numbers and manufacturers are listed in (Data file S10).
FACS Analysis of MRCKA knockdown
OVCAR4 cells were transfected with 25 nM siRNA and collected at the indicated time points. Cells were trypsinized, washed twice with PBS and fixed in ice cold 70% ethanol for at least 24 hours. Prior to analysis, the fixed cells were washed once again with PBS, resuspended in 300 μl FxCycle PI/RNAse (Thermo Scientific F10797) and incubated for 30 min at room temperature. The cells were analyzed with a FACScan (BD) flow cytometer and the data were analyzed using FlowJo (BD) at the Flow Cytometry Facility, FCCC.
Migration Assay
Cell migration assays were performed using the xCELLigence RTCA DP Instrument (ACEA Biosciences) in a CIM-Plate 16 (ACEA Biosciences) and the Cell Culture Facility, FCCC. Complete medium was added to the lower chambers of the CIM-Plate (or serum free medium for negative control). In the upper chambers, 30 μL serum free medium was added to each well and the CIM-Plate was placed in the instrument to equilibrate for 1 hour at 37°C. Following this equilibration step, a background measurement was taken. The CIM-Plate was removed from the instrument, and cells were added to the upper chambers at 50,000 cells per well in serum free medium. The CIM-Plate was left at 25°C for 30 minutes to allow the cells to settle at the bottom of the well. The plate was then returned to the instrument, and cell index reads were taken once every 15 minutes for 24 hours. For the siRNA experiment, the cells were transfected 24 hours prior to the start of the assay. For the BDP9066 experiments, cells were pre-treated with 1 μM inhibitor or DMSO 24 hours prior, and both upper and lower chambers contained 1 μM inhibitor or DMSO.
MIBs preparation and chromatography
Experiments using MIB/MS were performed as previously described (13). Briefly, cells or tumors were lysed on ice in buffer containing 50 mM HEPES (pH 7.5), 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 2.5 mM sodium orthovanadate, 1X protease inhibitor cocktail (Roche), and 1% each of phosphatase inhibitor cocktails 2 and 3 (Sigma). Particulate was removed by centrifugation of lysates at 21,000 g for 15 minutes at 4°C and filtration through 0.45 μm syringe filters. Protein concentrations were determined by BCA analysis (Thermo Scientific). To compare kinome signatures within and HGSOC tumors, we designed a super-SILAC method consisting of a cocktail of cancer cell lines that encompasses the activity of the “cancer kinome” to be used as a control for individual samples. Five cancer cell lines were selected from the NCI-60 panel (UACC257, MOLT4, COLO205, ACHN and PC3) that differed in their origin, gene expression patterns and mutation status. The cancer cell lines were labeled using SILAC and mixed equally providing a diverse SILAC ([13C6, 15N4] arginine (Arg 10) and [13C6] lysine (Lys 8)) heavy reference standard (s-SILAC) to be used repeatedly in kinome profiling assays. An equal amount of the s-SILAC reference (5 mg) lysate was added to our non-labeled (5 mg) sample (cell, or tumor tissue) and analyzed on MIB-beads. Endogenous kinases were isolated by flowing lysates over kinase inhibitor-conjugated Sepharose beads (purvalanol B, VI16832, PP58 and CTx-0294885 beads) in 10 ml gravity-flow columns. After 2×10 ml column washes in high-salt buffer and 1×10 ml wash in low-salt buffer (containing 50 mM HEPES (pH 7.5), 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, and 10 mM sodium fluoride, and 1M NaCl or 150 mM NaCl, respectively), retained kinases were eluted from the column by boiling in 2 × 500 μl of 0.5% SDS, 0.1 M TrisHCl (pH 6.8), and 1% 2-mercaptoethanol. Eluted peptides were reduced by incubation with 5 mM DTT at 65°C for 25 minutes, alkylated with 20 mM iodoacetamide at room temperature for 30 minutes in the dark, and alkylation was quenched with DTT for 10 minutes. For MIBs competition experiments related to Fig. 6C and Fig. S5B, heavy-labeled (drug treated or control) or light-labeled (drug-treated or control) lysates were run on MIB-columns separately, eluted, then mixed prior to reduction and alkylation. Samples were concentrated to approximately 100 μl with Millipore 10kD cutoff spin concentrators. Detergent was removed by chloroform/methanol extraction, and the protein pellet was resuspended in 50 mM ammonium bicarbonate and digested with sequencing-grade modified trypsin (Promega) overnight at 37°C. Peptides were cleaned with PepClean C18 spin columns (ThermoFisher Scientific) dried in a speed-vac, resuspended in 50 μl 0.1% formic acid, and extracted with ethyl acetate (10:1 ethyl acetate:H2O). Briefly, 1 mL ethyl acetate was added to each sample, vortexed and centrifuged at max speed for 5 minutes, then removed. This process is repeated 4 more times. After removal of ethyl acetate following the 5th centrifugation, samples were placed at 60°C for 10 minutes to evaporate residual ethyl acetate. The peptides were dried in a speed vac, and subsequent LC-/MS/MS analysis was performed.
Nano-LC-MS/MS
Proteolytic peptides were resuspended in 0.1% formic acid and separated with a Thermo Scientific RSLC Ultimate 3000 on a Thermo Scientific Easy-Spray C18 PepMap 75μm × 50cm C-18 2 μm column with a 240 min gradient of 4–25% acetonitrile with 0.1% formic acid at 300 nL/min at 50°C. Eluted peptides were analyzed by a Thermo Scientific Q Exactive plus mass spectrometer utilizing a top 15 methodology in which the 15 most intense peptide precursor ions were subjected to fragmentation. The AGC for MS1 was set to 3×106 with a max injection time of 120 ms, the AGC for MS2 ions was set to 1×105 with a max injection time of 150 ms, and the dynamic exclusion was set to 90 s.
Data processing and analysis
Raw data analysis of SILAC experiments was performed using Maxquant software 1.6.1.0 and searched using andromeda 1.5.6.0 against the swiss-prot human protein database (downloaded on July 26, 2018). The search was set up for full tryptic peptides with a maximum of two missed cleavage sites. All settings were default and searched using acetylation of protein N-terminus and oxidized methionine as variable modifications. Carbamidomethylation of cysteine was set as fixed modification. The precursor mass tolerance threshold was set at 10 ppm and maximum fragment mass error was 0.02 Da. SILAC quantification was performed using MaxQuant by choosing multiplicity as 2 in group-specific parameters and Arg10 and Lys8 as heavy labels. The match between runs was employed and the significance threshold of the ion score was calculated based on a false discovery rate of < 1%. For HGSOC tumor MIB-MS data analysis, MaxQuant normalized ratios were imported into Perseus software (1.6.2.3) for quantitation (Data file S2). HGSOC tumor MIB-MS profiles were processed in the following manner: normalized MIB-MS s-SILAC ratios were transformed 1/(x) to generate light / heavy ratios, followed by log2/(1/x) transformed. Columns were then filtered based on a valid value of 150 and rows filtered for present in 70% of samples leaving 206 kinases. Imputation of missing values was performed as previously described (47), where in the super-SILAC data, a width of 0.3 and the downshift of 0.5, was employed. Principal component analysis (PC1 vs PC2, PC2 vs PC3 and PC1 vs PC3) was then performed to visualize kinome profiles amongst HGSOC tumors (Fig. 1, D and E, and fig S1, A and B). Hierarchical clustering (Euclidean) of s-SILAC ratios was then performed and column clusters annotated selecting 8 clusters at a distance threshold of 21.59 (fig. S1D). No imputation of missing MIB-MS s-SILAC values was used for clustering analysis. The prominent cluster (Cluster-27) of HGSOC tumors was then compared to other HGSOC tumors using a two-sample Student’s t-test with the following parameters, (S0 0.1, and Side, Both) using p-value 0.05 (Data file S4). Kinases statistically enriched in the main cluster were isolated (n=82), the s-SILAC ratios hierarchical clustered and depicted in a heat map (Fig. 1G). For MRCKA knockdown MIB-MS data analysis, MaxQuant normalized ratios were imported into Perseus software (1.6.2.3) for quantitation (Data file S6). MIB-MS profiles were processed in the following manner: normalized MIB-MS s-SILAC ratios were transformed 1/(x) to generate light / heavy ratios, followed by log2/(1/x) transformed. Rows were filtered for presence in 100% of samples leaving 168 kinases. No imputation of missing values was performed. Differences in s-SILAC ratios amongst cells treated with control siRNAs or MRCKA siRNAs was determined using a two-sample Student’s t-test with the following parameters, (S0 0.1, and Side, Both) using Benjamini-Hochberg FDR, FDR < 0.05 (Data file S6). A Volcano plot was then generated in RStudio 1.1.423 using ggplot2 and ggrepel libraries (Fig 4B) to visualize differences in MIB-binding amongst treatments. For BDP9066 and A-674563 MIB-MS data analysis, MaxQuant normalized ratios were imported into Perseus software (1.6.2.3) for quantitation (Data file S9). MIB-MS profiles were processed in the following manner: normalized MIB-MS s-SILAC ratios were transformed 1/(x) to generate light / heavy ratios, followed by log2/(1/x) transformed. Rows were filtered for presence in 50% of samples leaving 216 kinases No imputation of missing values was performed. Average SILAC ratio for kinases was calculated using Perseus software (1.6.2.3) and a line graph was generated to visualize the primary targets of BDP9066 (Fig. 6B). The MIB-MS mass spectrometry data were deposited to the ProteomeXchange Consortium via PRIDE partner repository with the dataset identifier PDX015849.
Titanium dioxide enrichment and phosphoproteomics data analysis
To define kinase and kinase substrate phosphorylation at baseline or in response to kinase knockdown, we performed global phosphoproteomics analysis, as previously described (23). Using methanol/chloroform protein precipitation methods, 4 mg lysate (2 mg OVCAR4 si-NT2 or si-CDC42BPA and 2 mg of s-SILAC reference sample) was mixed and precipitated, resuspended in 360 μl 50 mM Tris-HCl pH 8.5 and digested with 20 μL of 1% Protease Max (Promega) plus 20 μg of Promega Trypsin/LysC mix for 4 hours at 37°C with vigorous shaking. Samples were reduced with 5 mM DTT at room temperature for 25 minutes, alkylated with 10 mM iodoacetamide at room temperature for 25 minutes in the dark, and alkylation was quenched with 5 mM DTT for 15 minutes. Samples were then digested with sequencing-grade modified trypsin (Promega) overnight at 37°C in an incubator shaker. Peptides were cleaned by subsequent elution from C-18 (Phenominex) and Hypercarb/Hypersep PGC columns (Thermo Scientific) and dried via speed-vac. Peptides were resuspended in 100 μl 0.1% formic acid and quantified using a Nanodrop spectrophotometer at A280 absorbance. TiO2 beads (GL Sciences) were resuspended in binding buffer (2M lactic acid in 50% ACN) at a concentration of 100 μg/μl and added to the peptides at a ratio of 4 mg beads per 1 mg peptide. Prior to the addition of the TiO2 beads, lactic acid and acetonitrile were added to the peptides for a final concentration of 2M lactic acid and 50% ACN. Samples were vortexed and incubated for 30 min with end over end rotation. Following incubation, samples were centrifuged for 1 minute at 8000g and the supernatant was added to fresh TiO2 beads for a second 30 minute incubation. The beads from both incubations were combined and washed three times with 1 mL binding buffer, then three times with 1 mL 50% ACN. Peptides were eluted from the TiO2 beads with 600 μL 5% ammonium hydroxide in 50% ACN by vortexing and passed through C-18 stage tips. 100 μl 80% ACN was passed through the stage tip and peptides were dried in a speed-vac and subsequent LC-/MS/MS analysis was performed using the Q Exactive.
Raw data analysis of s-SILAC phosphoproteomics experiments was performed as described above with phosphorylation of Ser, Thr and Tyr being included as variable modifications, with a maximum number of modifications per peptide being 5. The MaxQuant normalized ratios “Phospho(STY).txt” were used for quantitation data analysis in Perseus software (1.6.2.3) (Data file S7). s-SILAC phosphosite ratios were processed in the following manner: MaxQuant normalized s-SILAC ratios were transformed 1/(x) to generate light / heavy ratios, followed by log2/(1/x) transformed. Phosphorylated serine, threonine, and tyrosine (pSTY) sites were filtered for only those that were confidently localized (class I, localization probability ≥ 0.75) followed by filtering for phosphosites identified in at least 70% of runs leaving 2906 phosphosites. Log2- transformed s-SILAC values were normalized to each column by subtracting its median. Imputation of missing values was performed the same as in MIB-MS analysis, where in the s-SILAC data, a width of 0.3 and the downshift of 0.5, was employed (Data file S7). Quantitative differences in s-SILAC ratios amongst control siRNAs or MRCKA siRNA treatments was determined using a two-sample Student’s t-test with the following parameters, (S0 0.1, and Side, Both) using p value < 0.05 (Data file S7). Kinase Substrate Enrichment Analysis (KSEA) (24) software (https://casecpb.shinyapps.io/ksea/) that uses pre-existing phosphoproteomics databases to determine candidate active kinases was used to predict changes in kinase activity following MRCKA knockdown. A .csv file was generated from the two-sample Student’s t-test comparing control siRNA vs MRCKA siRNA treated cells Perseus export matrix (Data file S7) that included Protein ID, Gene ID, Peptide, Residue.Both, p value and fold change. Parameters used in KSEA analysis were as follows; PhosphoSitePlus + NetworkKIN, Set NetworkKIN score cutoff = 2, [for plot] Set p-value cutoff = 0.05, [for plot] Set substrate count cutoff = 10. A Volcano plot was then generated in RStudio 1.1.423 using ggplot2 and ggrepel libraries (Fig 4C) using KSEA Kinase Scores (Data file S7) to visualize kinases predicted to activated or inhibited. The phosphoproteomic mass spectrometry data were deposited to the ProteomeXchange Consortium via PRIDE partner repository with the dataset identifier PXD015849.
Bioinformatics analyses of TCGA and CPTAC datasets
Analysis of MRCKA (CDC42BPA) CNA, protein and mRNA alterations in HGSOC from TCGA and CPTAC studies was performed at the Biostatistics and Bioinformatics Facility, FCCC. Heat map presented in (Fig. 3A) was generated using R packages and shows the relationship between MRCKA (CDC42BPA) gene and protein expression with stage of tumors, or subtype. MRCKA expression is not associated with stage of tumors (Fisher’s two-sided p=0.9) or subtype (Fisher’s two-sided p = 0.17), however, a positive correlation was observed between mRNA and protein expression (Pearson correlation = 0.5, p < 0.001). Stage and subtype data were obtained from TCGA data repository. Overall survival was based on vital status and “days to death” from initial pathologic diagnosis. Individuals who were still alive at the time of the last follow-up were censored. Survival curves were compared with log-rank tests, and these calculations were done using the R ‘survival’ package (Therneau, T. M. & Grambsch, P. M. Modeling Survival Data: Extending the Cox Model (Springer-Verlag, 2010). Survival data was obtained from TCGA data repository.
To assess the pathway profiles enriched in low and high MRCKA expressing tumors, we applied single sample gene set variation analysis using RSEM gene expression measures for 307 HGSOC obtained from Broad Institute’s GDAC Firehose portal (https://gdac.broadinstitute.org/) and 12,915 gene sets obtained from MSigDB (48) as input. To identify enriched gene sets and pathways for low and high MRCKA expressing HGSOC cases, we split the 307 cases into high and low MRCKA expressors (Z-score > 1.28 and Z-score < 1.28) and Wilcoxon-rank sum test was applied. Significant pathways (C2 pathway sets of MSigDB) and gene sets (Benjamini-Hochberg False Discovery Rate (FDR) < 0.05%) enriched for low and high MRCKA cases (enrichment score > 0.3 or < −0.3) were selected. Resulting enrichment scores were plotted as heat map and expression of MRCKA gene and protein were plotted as bars (Z-score). Selected pathways were plotted as bar plots (p-values were transformed into –log p-values). Similarly, we also evaluated differentially expressed genes between low and high MRCKA expressors using DESeq2 algorithm with absolute gene counts for the same data as input. Selected differentially expressed genes (FDR < 0.05) were used as input to Gene Set Enrichment Analysis (49).
Supplementary Material
Data file S1: Raw and processed s-SILAC ratios from MIB-MS profiling of sections from a single HGSOC tumor tissue.
Data file S2: Characteristics of HGSOC primary and PDX tumors used in MIB-MS studies.
Data file S3: Raw and processed s-SILAC ratios from MIB-MS profiling of HGSOC primary and PDX tumors.
Data file S4: Prevalent MIB-MS kinome signature amongst HGSOC primary and PDX tumors.
Data file S5: Immunohistochemical analysis of MRCKA protein levels in HGSOC tumor sections, normal fallopian tube and ovarian surface epithelial tissues.
Data file S6: Raw and processed s-SILAC ratios from MIB-MS profiling of MRCKA knockdown in HGSOC cells.
Data file S7: Raw and processed s-SILAC ratios from phosphoproteomics analysis of MRCKA knockdown in HGSOC cells.
Data file S8: In vitro kinase inhibitor profiling report for BDP5290 and BDP9066.
Data file S9: Raw and processed SILAC ratios MIB-MS profiling of OVSAHO cells treated with BDP9066 or A-674563.
Data file S10: Reagents used in the study, including small molecules, siRNAs and antibodies.
Figure S1: Quantitation of kinase abundance in HGSOC tumors using MIB-MS and s-SILAC.
Figure S2: Characterization of MRCKA in HGSOC tumors and HGSOC cell lines.
Figure S3. Exploring MRCKA knockdown in HGSOC cells using MIB-MS and phosphoproteomics.
Figure S4: MRCKA promotes focal adhesion signaling in HGSOC cells.
Figure S5: Evaluation of MRCKA kinase inhibitors in HGSOC cells.
Figure S6. Characterization of proliferation, migration, apoptosis, and spheroid formation in BDP9066-treated HGSOC cells.
Acknowledgements:
We thank Ronny Drapkin (Penn Ovarian Cancer Research Center, Department of Obstetrics and Gynecology, Perelman School of Medicine, University of Pennsylvania) for providing the FTSEC cell lines.
Funding: This work was supported by grants from NIH CORE Grant CA06927 (Fox Chase Cancer Center), R01 CA211670 (J.S.D.), R01 CA142928 (J.C.), a grant from the Pennsylvania Department of Health (SAP#4100068716, D.C.C.), NIH T32 CA009035 (A.M.K), FCCC Interdisciplinary Translational Cancer Research Grants (J.S.D., A.J.), ACS IRG-92-027-20 (J.S.D.) and generous donations from the Teal Tea Foundation (J.B., G.M.S., J.S.D.), Goodis Fund (J.B., J.S.D.) Sandy Rollman Ovarian Cancer Foundation (J.S.D), Dubrow Fund (D.C.C.), Bucks County Board of Associates (D.C.C.) and Mainline Board of Associates (D.C.C.). Kinome profiling studies were funded by the Cancer Kinome Initiative (CKI) at FCCC, which was established by a donation from Don Morel.
Footnotes
Competing interests: J.S.D. is an inventor on International patent application PCT/US2019/048053 for using multiplexed inhibitor beads for measuring kinase levels in patient tumors. The other authors declare that they have no competing interests.
Data and materials availability: Data have been deposited via the consortium through the PRIDE partner repository with the dataset identifier PXD015849. All other data needed to evaluate the conclusion in the paper are present in the paper or the Supplementary Materials. An MTA is available for the HGSOC PDX tissues used in the manuscript upon request
REFERENCES AND NOTES
- 1.N. The Cancer Genome Atlas Research, Integrated Genomic Analyses of Ovarian Carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musella A, Bardhi E, Marchetti C, Vertechy L, Santangelo G, Sassu C, Tomao F, Rech F, D’Amelio R, Monti M, Palaia I, Muzii L, Benedetti Panici P, Rucaparib: An emerging parp inhibitor for treatment of recurrent ovarian cancer. Cancer Treat. Rev 66, 7–14 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, Frankum J, Ranes M, Pemberton HN, Rafiq R, Fenwick K, Swain A, Guettler S, Lee J-M, Swisher EM, Stoynov S, Yusa K, Ashworth A, Lord CJ, Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature Communications 9, 1849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Liu T, Zhang Z, Payne Samuel H., Zhang B, McDermott Jason E., Zhou J-Y, Petyuk Vladislav A., Chen L, Ray D, Sun S, Yang F, Chen L, Wang J, Shah P, Cha Seong W., Aiyetan P, Woo S, Tian Y, Gritsenko Marina A., Clauss Therese R., Choi C, Monroe Matthew E., Thomas S, Nie S, Wu C, Moore Ronald J., Yu K-H, Tabb David L., Fenyö D, Bafna V, Wang Y, Rodriguez H, Boja Emily S., Hiltke T, Rivers Robert C., Sokoll L, Zhu H, Shih I-M, Cope L, Pandey A, Zhang B, Snyder Michael P., Levine Douglas A., Smith Richard D., Chan Daniel W., Rodland Karin D., Carr Steven A., Gillette Michael A., Klauser Karl R., Kuhn E, Mani DR, Mertins P, Ketchum Karen A., Thangudu R, Cai S, Oberti M, Paulovich Amanda G., Whiteaker Jeffrey R., Edwards Nathan J., McGarvey Peter B., Madhavan S, Wang P, Chan Daniel W., Pandey A, Shih I-M, Zhang H, Zhang Z, Zhu H, Cope L, Whiteley Gordon A., Skates Steven J., White Forest M., Levine Douglas A., Boja Emily S., Kinsinger Christopher R., Hiltke T, Mesri M, Rivers Robert C., Rodriguez H, Shaw Kenna M., Stein Stephen E., Fenyo D, Liu T, McDermott Jason E., Payne Samuel H., Rodland Karin D., Smith Richard D., Rudnick P, Snyder M, Zhao Y, Chen X, Ransohoff David F., Hoofnagle Andrew N., Liebler Daniel C., Sanders Melinda E., Shi Z, Slebos Robbert J. C., Tabb David L., Zhang B, Zimmerman Lisa J., Wang Y, Davies Sherri R., Ding L, Ellis Matthew J. C., Townsend RR, Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhaak RGW, Tamayo P, Yang J-Y, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, Mermel CH, Kostic AD, Etemadmoghadam D, Saksena G, Cibulskis K, Duraisamy S, Levanon K, Sougnez C, Tsherniak A, Gomez S, Onofrio R, Gabriel S, Chin L, Zhang N, Spellman PT, Zhang Y, Akbani R, Hoadley KA, Kahn A, M. bel xF, Huntsman D, Soslow RA, Defazio A, Birrer MJ, Gray JW, Weinstein JN, Bowtell DD, Drapkin R, Mesirov JP, Getz G, Levine DA, Meyerson M, Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. The Journal of Clinical Investigation 123, 517–525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, Gaillard S, Phillip Jude M., Huang T-C, Pinto Sneha M., Tessarollo Nayara G., Zhang Z, Pandey A, Wirtz D, Ayhan A, Davidson B, Wang T-L, Shih I-M, Inhibition of Spleen Tyrosine Kinase Potentiates Paclitaxel-Induced Cytotoxicity in Ovarian Cancer Cells by Stabilizing Microtubules. Cancer Cell 28, 82–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Q, Yu Y, Upregulated CDK16 Expression in Serous Epithelial Ovarian Cancer Cells. Medical Science Monitor : International Medical Journal of Experimental and Clinical Research 21, 3409–3414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Secord AA, Teoh DK, Barry WT, Yu M, Broadwater G, Havrilesky LJ, Lee PS, Berchuck A, Lancaster J, Wenham RM, A phase I trial of dasatinib, a Src-family kinase Inhibitor, in combination with paclitaxel and carboplatin in patients with advanced or recurrent ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 18, 5489–5498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilken JA, Badri T, Cross S, Raji R, Santin AD, Schwartz P, Branscum AJ, Baron AT, Sakhitab AI, Maihle NJ, EGFR/HER-targeted therapeutics in ovarian cancer. Future Med. Chem 4, 447–469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G, Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol 25, 1035 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Duncan James S., Whittle Martin C., Nakamura K, Abell Amy N., Midland Alicia A., Zawistowski Jon S., Johnson Nancy L., Granger Deborah A., Jordan Nicole V., Darr David B., Usary J, Kuan P-F, Smalley David M., Major B, He X, Hoadley Katherine A., Zhou B, Sharpless Norman E., Perou Charles M., Kim William Y., Gomez Shawn M., Chen X, Jin J, Frye Stephen V., Earp HS, Graves Lee M., Johnson Gary L., Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 149, 307–321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee J-D, Gray NS, Kozarich JW, In Situ Kinase Profiling Reveals Functionally Relevant Properties of Native Kinases. Chem. Biol 18, 699–710 (2011) 10.1016/j.chembiol.2011.04.011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurimchak AM, Shelton C, Duncan KE, Johnson KJ, Brown J, O’Brien S, Gabbasov R, Fink LS, Li Y, Lounsbury N, Abou-Gharbia M, Childers WE, Connolly DC, Chernoff J, Peterson JR, Duncan JS, Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell reports 16, 1273–1286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neubert TA, Tempst P, Super-SILAC for tumors and tissues. Nat Meth 7, 361–362 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Chanana P, Davila JI, Hou X, Zanfagnin V, McGehee CD, Goode EL, Polley EC, Haluska P, Weroha SJ, Wang C, Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep 9, 6314–6314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Unbekandt M, Belshaw S, Bower J, Clarke M, Cordes J, Crighton D, Croft DR, Drysdale MJ, Garnett MJ, Gill K, Gray C, Greenhalgh DA, Hall JAM, Konczal J, Lilla S, McArthur D, McConnell P, McDonald L, McGarry L, McKinnon H, McMenemy C, Mezna M, Morrice NA, Munro J, Naylor G, Rath N, Schüttelkopf AW, Sime M, Olson MF, Discovery of Potent and Selective MRCK Inhibitors with Therapeutic Effect on Skin Cancer. Cancer Res. 78, 2096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kale VP, Hengst JA, Desai DH, Dick TE, Choe KN, Colledge AL, Takahashi Y, Sung S-S, Amin SG, Yun JK, A novel selective multikinase inhibitor of ROCK and MRCK effectively blocks cancer cell migration and invasion. Cancer Lett. 354, 299–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, Montag A, Lastra RR, Lengyel E, Mann M, Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nature Communications 7, 12645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, Hruban CA, Hirsch MS, Lin DI, Schwartz L, Maire CL, Tille J-C, Bowden M, Ayhan A, Wood LD, Scharpf RB, Kurman R, Wang T-L, Shih I-M, Karchin R, Drapkin R, Velculescu VE, High grade serous ovarian carcinomas originate in the fallopian tube. Nature communications 8, 1093–1093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karst AM, Drapkin R, Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat. Protoc 7, 1755–1764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unbekandt M, Croft DR, Crighton D, Mezna M, McArthur D, McConnell P, Schüttelkopf AW, Belshaw S, Pannifer A, Sime M, Bower J, Drysdale M, Olson MF, A novel small-molecule MRCK inhibitor blocks cancer cell invasion. Cell Communication and Signaling : CCS 12, 54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Z, Manser E, Myotonic dystrophy kinase-related Cdc42-binding kinases (MRCK), the ROCK-like effectors of Cdc42 and Rac1. Small GTPases 6, 81–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thingholm TE, Jørgensen TJD, Jensen ON, Larsen MR, Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc 1, 1929 (2006); published online Epub11/22/online ( 10.1038/nprot.2006.185). [DOI] [PubMed] [Google Scholar]
- 24.Wiredja DD, Koyutürk M, Chance MR, The KSEA App: a web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 33, 3489–3491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed N, Stenvers KL, Getting to know ovarian cancer Ascites: opportunities for targeted therapy-based translational research. Front. Oncol 3, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S, Bean LM, Sulzmaier FJ, Jean C, Tancioni I, Anderson K, Uryu S, Cordasco EA, Li J, Chen XL, Fu G, Ojalill M, Rappu P, Heino J, Mark AM, Xu G, Fisch KM, Kolev VN, Weaver DT, Pachter JA, Győrffy B, McHale MT, Connolly DC, Molinolo A, Stupack DG, Schlaepfer DD, FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. eLife 8, e47327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeder MK, Serebriiskii IG, Golemis EA, Chernoff J, Analysis of Small GTPase Signaling Pathways Using p21-activated Kinase Mutants That Selectively Couple to Cdc42. J. Biol. Chem 276, 40606–40613 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, Dagostino E, Knighton D, Loi C-M, Zager M, Kraynov E, Popoff I, Christensen JG, Martinez R, Kephart SE, Marakovits J, Karlicek S, Bergqvist S, Smeal T, Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proceedings of the National Academy of Sciences 107, 9446–9451 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP, Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol 29, 1046 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Ong CC, Gierke S, Pitt C, Sagolla M, Cheng CK, Zhou W, Jubb AM, Strickland L, Schmidt M, Duron SG, Campbell DA, Zheng W, Dehdashti S, Shen M, Yang N, Behnke ML, Huang W, McKew JC, Chernoff J, Forrest WF, Haverty PM, Chin S-F, Rakha EA, Green AR, Ellis IO, Caldas C, O’Brien T, Friedman LS, Koeppen H, Rudolph J, Hoeflich KP, Small molecule inhibition of group I p21-activated kinases in breast cancer induces apoptosis and potentiates the activity of microtubule stabilizing agents. Breast Cancer Res. 17, 59 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell J, Ryan Colm J., Brough R, Bajrami I, Pemberton Helen N., Chong Irene Y., Costa-Cabral S, Frankum J, Gulati A, Holme H, Miller R, Postel-Vinay S, Rafiq R, Wei W, Williamson Chris T., Quigley David A., Tym J, Al-Lazikani B, Fenton T, Natrajan R, Strauss Sandra J., Ashworth A, Lord Christopher J., Large-Scale Profiling of Kinase Dependencies in Cancer Cell Lines. Cell Reports 14, 2490–2501 (2016); published online Epub2016/03/15/ ( 10.1016/j.celrep.2016.02.023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kale VP, Hengst JA, Desai DH, Amin SG, Yun JK, The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett. 361, 185–196 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Rafiq K, Guo J, Vlasenko L, Guo X, Kolpakov MA, Sanjay A, Houser SR, Sabri A, c-Cbl ubiquitin ligase regulates focal adhesion protein turnover and myofibril degeneration induced by neutrophil protease cathepsin G. The Journal of biological chemistry 287, 5327–5339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unbekandt M, Olson MF, The actin-myosin regulatory MRCK kinases: regulation, biological functions and associations with human cancer. J. Mol. Med. (Berl.) 92, 217–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J, Crews CM, The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell chemical biology 25, 67–77.e63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cromm PM, Samarasinghe KTG, Hines J, Crews CM, Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc 140, 17019–17026 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Graves LM, Duncan JS, Whittle MC, Johnson GL, The dynamic nature of the kinome. Biochem. J 450, 1–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prudnikova TY, Villamar-Cruz O, Rawat SJ, Cai KQ, Chernoff J, Effects of p21-activated kinase 1 inhibition on 11q13 amplified ovarian cancer cells. Oncogene 35, 2178–2185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphries-Bickley T, Castillo-Pichardo L, Hernandez-O’Farrill E, Borrero-Garcia LD, Forestier-Roman I, Gerena Y, Blanco M, Rivera-Robles MJ, Rodriguez-Medina JR, Cubano LA, Vlaar CP, Dharmawardhane S, Characterization of a Dual Rac/Cdc42 Inhibitor MBQ-167 in Metastatic Cancer. Mol. Cancer Ther 16, 805–818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kipps E, Tan DS, Kaye SB, Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat. Rev. Cancer 13, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ong SE BB, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M, Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Klutchko SR, Hamby JM, Boschelli DH, Wu Z, Kraker AJ, Amar AM, Hartl BG, Shen C, Klohs WD, and Steinkampf RW, Driscoll DL, Nelson JM, Elliott WL, Roberts BJ, Stoner CL, Vincent PW, Dykes DJ, Panek RL, Lu GH, Major TC, Dahring TK, Hallak H, Bradford LA, H. D. Hollis Showalter HDH, and Doherty AM, 2-Substituted Aminopyrido[2,3-d]pyrimidin-7(8H)-ones. Structure-Activity Relationships Against Selected Tyrosine Kinases and in Vitro and in Vivo Anticancer Activity. J. Med. Chem 41, 3276–3292 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Oppermann FS, Gnad F, Olsen JV, Hornberger R, Greff Z, Kéri G, Mann M, Daub H, Large-scale Proteomics Analysis of the Human Kinome. Mol. Cell. Proteomics 8, 1751–1764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Holmes IP, Hochgräfe F, Walker SR, Ali NA, Humphrey ES, Wu J, de Silva M, Kersten WJA, Connor T, Falk H, Allan L, Street IP, Bentley JD, Pilling PA, Monahan BJ, Peat TS, Daly RJ, Characterization of the Novel Broad-Spectrum Kinase Inhibitor CTx-0294885 As an Affinity Reagent for Mass Spectrometry-Based Kinome Profiling. J. Proteome Res 12, 3104–3116 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN, Evaluating strategies to normalise biological replicates of Western blot data. PLoS One 9, e87293–e87293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ianevski A, He L, Aittokallio T, Tang J, SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics 33, 2413–2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deeb SJ, D’Souza RCJ, Cox J, Schmidt-Supprian M, Mann M, Super-SILAC Allows Classification of Diffuse Large B-cell Lymphoma Subtypes by Their Protein Expression Profiles. Molecular & Cellular Proteomics : MCP 11, 77–89 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics (Oxford, England) 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP, GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 23, 3251–3253 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1: Raw and processed s-SILAC ratios from MIB-MS profiling of sections from a single HGSOC tumor tissue.
Data file S2: Characteristics of HGSOC primary and PDX tumors used in MIB-MS studies.
Data file S3: Raw and processed s-SILAC ratios from MIB-MS profiling of HGSOC primary and PDX tumors.
Data file S4: Prevalent MIB-MS kinome signature amongst HGSOC primary and PDX tumors.
Data file S5: Immunohistochemical analysis of MRCKA protein levels in HGSOC tumor sections, normal fallopian tube and ovarian surface epithelial tissues.
Data file S6: Raw and processed s-SILAC ratios from MIB-MS profiling of MRCKA knockdown in HGSOC cells.
Data file S7: Raw and processed s-SILAC ratios from phosphoproteomics analysis of MRCKA knockdown in HGSOC cells.
Data file S8: In vitro kinase inhibitor profiling report for BDP5290 and BDP9066.
Data file S9: Raw and processed SILAC ratios MIB-MS profiling of OVSAHO cells treated with BDP9066 or A-674563.
Data file S10: Reagents used in the study, including small molecules, siRNAs and antibodies.
Figure S1: Quantitation of kinase abundance in HGSOC tumors using MIB-MS and s-SILAC.
Figure S2: Characterization of MRCKA in HGSOC tumors and HGSOC cell lines.
Figure S3. Exploring MRCKA knockdown in HGSOC cells using MIB-MS and phosphoproteomics.
Figure S4: MRCKA promotes focal adhesion signaling in HGSOC cells.
Figure S5: Evaluation of MRCKA kinase inhibitors in HGSOC cells.
Figure S6. Characterization of proliferation, migration, apoptosis, and spheroid formation in BDP9066-treated HGSOC cells.