

Abstract
Pathogenic variants in KMT2D, which encodes lysine specific methyltransferase 2D, cause autosomal dominant Kabuki syndrome, associated with distinctive dysmorphic features including arched eyebrows, long palpebral fissures with eversion of the lower lid, large protuberant ears, and fetal finger pads. Most disease-causing variants identified to date are putative loss-of-function alleles, although 15-20% of cases are attributed to missense variants. We describe here four patients (including one previously published patient) with de novo KMT2D missense variants and with shared but unusual clinical findings not typically seen in Kabuki syndrome, including athelia (absent nipples), choanal atresia, hypoparathyroidism, delayed or absent pubertal development, and extreme short stature. These individuals also lack the typical dysmorphic facial features found in Kabuki syndrome. Two of the four patients had severe interstitial lung disease. All of these variants cluster within a 40-amino-acid region of the protein that is located just N-terminal of an annotated coiled coil domain. These findings significantly expand the phenotypic spectrum of features associated with variants in KMT2D beyond those seen in Kabuki syndrome and suggest a possible new underlying disease mechanism for these patients.
Keywords: Kabuki syndrome, athelia, KMT2D
INTRODUCTION
The association between pathogenic variants in KMT2D (previously known as MLL2) and Kabuki syndrome 1 (MIM 147920) was first published in 2010 (Ng et al., 2010), and serves as an early example of the power of exome sequencing for gene discovery (Adam, Hudgins, & Hannibal, 1993-2019). Approximately 60-75% of patients with a clinical diagnosis of Kabuki syndrome are found to have disease-causing variants in KMT2D, which encodes histone-lysine N-methyltransferase 2D (Bogershausen et al., 2016). Somatic variants in KMT2D are also frequently observed in various cancers (Kandoth et al., 2013; Rao & Dou, 2015). An additional 3-8% of patients with features indistinguishable from Kabuki syndrome 1 have pathogenic variants in the X-linked gene KDM6A, which encodes lysine-specific demethylase 6A, and is associated with Kabuki syndrome 2 (MIM 300867). Consensus clinical diagnostic criteria for Kabuki syndrome were recently published and define the characteristic features of this disorder, including intellectual disability, developmental delay, infantile hypotonia, and “typical” dysmorphic features affecting the eyebrows, nose, ears, and fingertips, although multiple other organ systems can also be affected (Adam et al., 2019).
Variants in KMT2D that cause Kabuki syndrome are generally de novo and are presumed to result in loss of function, since >80% of reported variants are nonsense, splice site, or frameshift alleles. Missense variants are observed in approximately 15-20% of cases, and can occur along the entire length of the protein (Bogershausen et al., 2016). Recent reports have attempted to categorize these variants, which appear to cluster in specific functional domains, although their pathogenicity can be more difficult to interpret (Bogershausen et al., 2016; Cocciadiferro et al., 2018; Faundes, Malone, Newman, & Banka, 2019).
We report here the identification of a cohort of four patients with missense variants in KMT2D that cluster within a 40-amino-acid region of the protein, and who all have some features of Kabuki syndrome, but also have shared unusual clinical features that are not typically associated with Kabuki syndrome, including choanal atresia, absent nipples (athelia) or hypoplastic nipples, and hypoparathyroidism.
METHODS
Editorial Policies and Ethical Considerations
The parents of all patients provided informed consent for their participation, including consent for use of facial photographs. Institutional Review Boards at each institution reviewed and approved these studies.
Exome Sequencing
Clinical exome sequencing for Patient 1 was performed at GeneDx using Agilent Clinical Research Exome kit (Agilent Technologies, Santa Clara, CA) (Tanaka et al., 2015). Research exome sequencing was performed for Patients 2 and 3. For Patient 2, we used the Agilent SureSelect Human All Exon kit (38 Mb version) and ANNOVAR (Wang, Li, & Hakonarson, 2010) to annotate variants, which were filtered to select those that are novel or rare (minor allele frequency < 0.01 in the Exome Aggregation Consortium (ExAC) database (Lek et al., 2016)) and predicted to be damaging using outputs from dbNSFP (X. Liu, Wu, Li, & Boerwinkle, 2016). We manually reviewed candidate genes for possible clinical relevance. Patient 3 underwent research trio exome sequencing, and the methods for the sequencing and analyses have been previously published (Zhu et al., 2015). Expected familial relationships, including paternity, were observed in the analyses of the sequence data.
Protein Constraint and Domain Visualization
Evolutionary constraint of the patients’ missense variants was assessed using the Aminode tool (Chang, Guo, di Ronza, & Sardiello, 2018). Illustrator for Biological Sciences was used to draw the domains (W. Liu et al., 2015). Domains were identified by using InterPro (Mitchell et al., 2019). Regions of significant missense constraint were extracted from Supplemental Table 4 of a BioRxiv publication that used Exome Aggregation Consortium (ExAC) data to “identify sub-genic regions that are depleted of missense variation” (Samocha et al., 2017).
RESULTS
Patient 1
A female infant was born at 37 weeks’ estimated gestation to healthy, non-consanguineous, Caucasian parents with birth weight 2740g (42nd percentile), length 49cm (72nd percentile), and head circumference 32.5cm (38th percentile). She was delivered by normal spontaneous vaginal delivery, and had respiratory distress with associated oxygen desaturation, initially attributed to meconium aspiration. She was intubated and mechanically ventilated, and was found to have choanal atresia. A dopamine infusion was provided to treat hypotension. Initial physical exam revealed multiple dysmorphic features, including hypertelorism, thick and arched eyebrows, depressed nasal bridge, small nose and hypoplasia of the alae nasi, small dysplastic ears with unusual superior helical ear tags, right preauricular pit, right branchial cleft sinus tract, athelia (absent nipples), and an anteriorly placed anus (Figures 1A–C, 2A–B, 2F). She underwent repair of bilateral, mixed bony and membranous complete choanal atresia at one month of age. She was discharged at two months of life with supplemental oxygen due to persistent hypoxemia. Her fingers developed clubbing due to lung disease, and she did not have fetal fingertip pads (Figure 3C).
Figure 1. Facial features of three individuals with KMT2D missense variants.
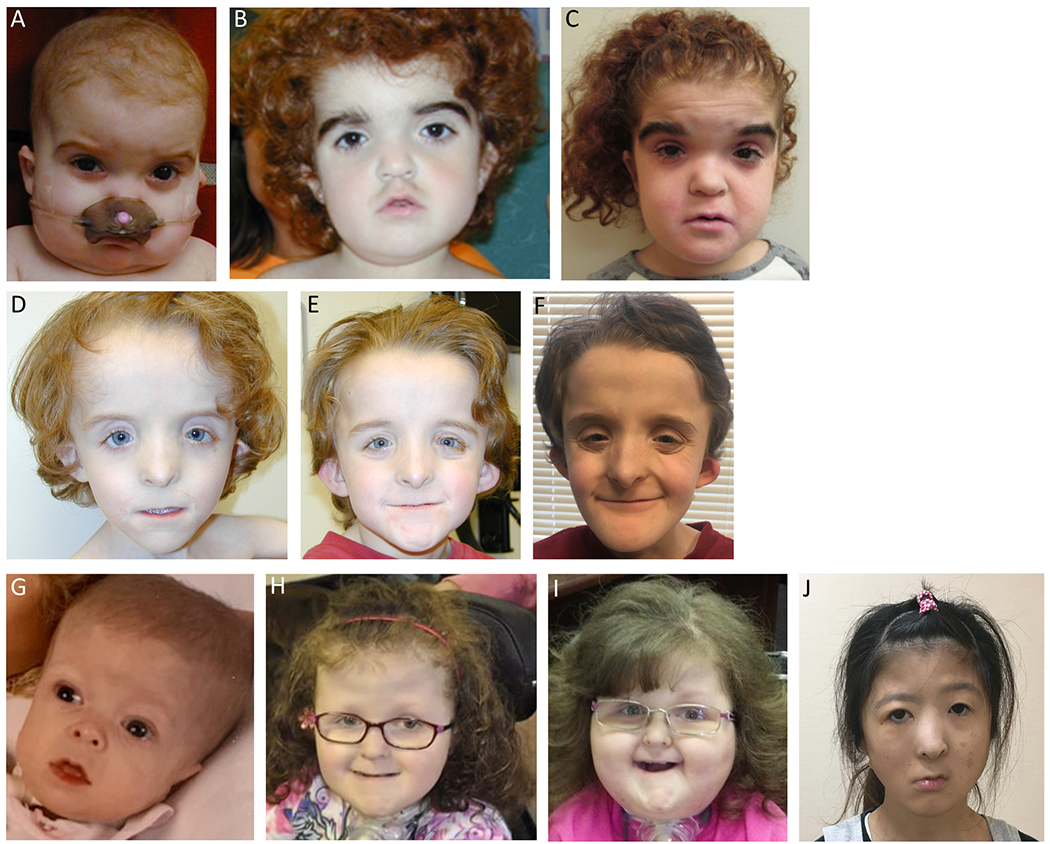
A-C: Patient 1, at age 12 months, 5 years, and 15 years. Arched eyebrows, depressed nasal bridge, small alae nasi, tented upper lip. D-F: Patient 2, at age 5 years, 8 years, and 15 years. Up-swept frontal hair line, widely spaced eyes, downslanting palpebral fissures, flat and wide nasal bridge, narrow and hypoplastic alae nasi, pointed nasal tip with low hanging columella, and a very thin upper lip with downturned corners. G-I: Patient 3, at age 8 months, 10 years, and 13 years. Tall, broad forehead with deep-set eyes, small nose, thin lips, dry, curly, and coarse hair. J: Patient 4, at age 24 years. Broad, depressed nasal tip and right facial nerve palsy. Reproduced, with permission, from Sakata et al., 2017.
Figure 2. Chest, back, ear, scalp, and lung abnormalities in individuals with KMT2D missense variants.
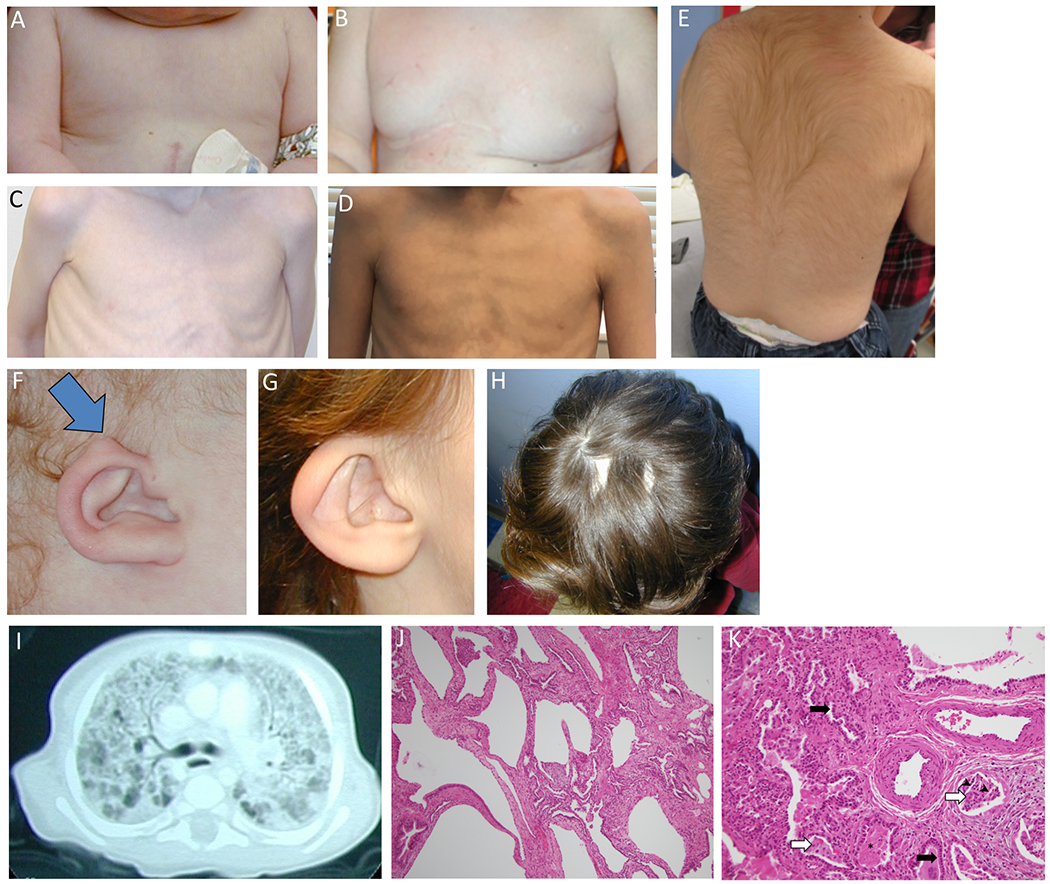
A-B: Patient 1, athelia at age 12 months and 5 years. C-D: Patient 2, hypoplastic, faintly pigmented nipples at age 5 years and 15 years. E: Patient 1, hypertrichosis at age 5 years. F: Patient 1, at age 12 months. Small ears with unusual superior helical ear tags (blue arrow). G: Patient 2, at age 8 years. Small, protruding, dysplastic, cup-shaped ears. H: Patient 2, at age 8 years. Scalp scars, after resolution of cutis aplasia at birth. I: Patient 1, at age 6 months. Computed tomography of lungs shows diffuse interstitial infiltrates and numerous, small, bilateral cysts. J: Patient 1. 10X hematoxylin and eosin (H&E) stained sections of explanted lung at age 19 months demonstrate an area of alveolar remodeling with enlarged airspaces; such areas were found throughout all lobes, interspersed with areas with smaller airspaces. K. Patient 1. 20X H&E. Areas with smaller airspaces show widened alveolar septa and prominent type II pneumocyte hyperplasia (black arrows). Airspaces contain macrophages (white arrows), degenerating cells, cholesterol clefts (black arrowheads) and dense eosinophilic and proteinaceous material (*). Pulmonary arterioles show mild medial and intimal thickening.
Figure 3. Teeth, hand, and feet abnormalities in individuals with KMT2D missense variants.
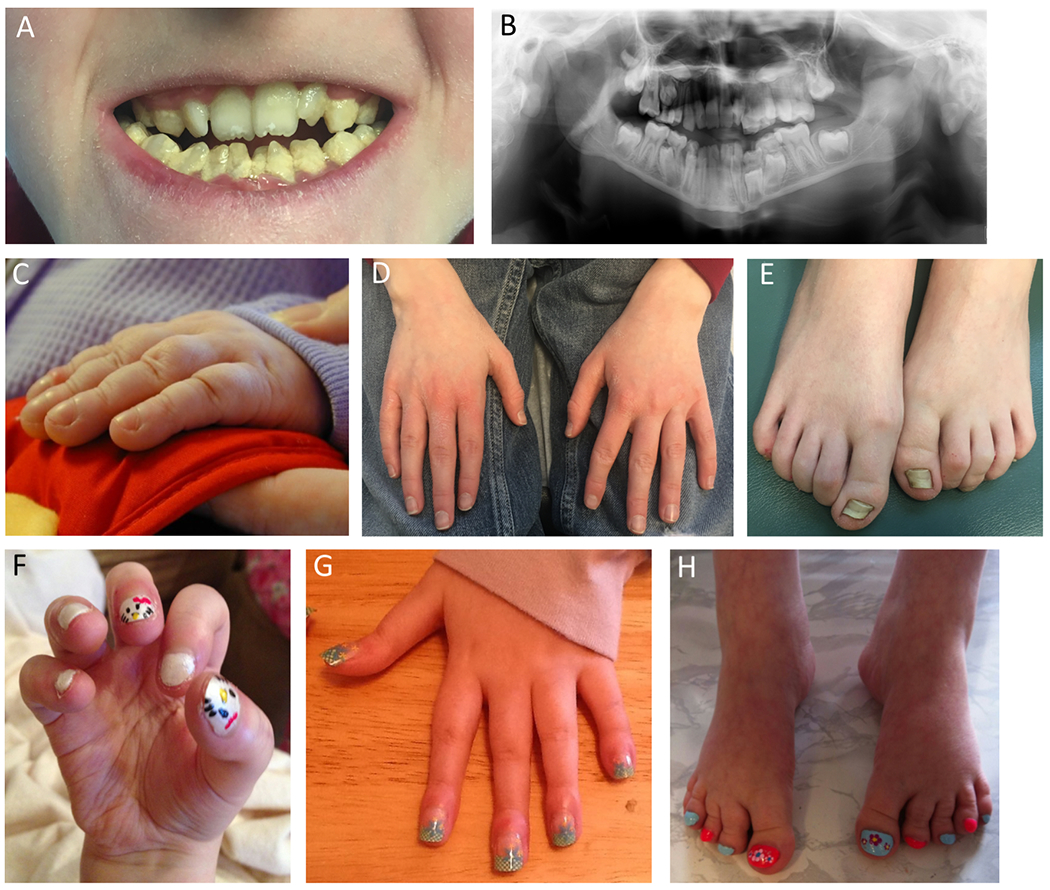
A-B: Patient 2, at age 15 years. Teeth are crowded, yellow, and covered with plaque, with abnormal conical shape of the lower central incisors. C: Patient 1, fingers at age 12 months prior to lung transplant showing clubbing due to lung disease, but absence of fetal fingertip pads. D-E: Patient 2, at age 15 years. Normal fingers with absence of fetal fingertip pads and dysplastic nails of great toes. F-H: Patient 3 at age 13 years, 14 years, and 13 years. Prominent clubbing of digits and absence of fetal fingertip pads.
At 6 months of age, chest computed tomography and open lung biopsy were performed due to increasing oxygen requirement and failure to gain appropriate weight. These studies revealed tiny cysts scattered throughout both lungs and diffuse interstitial pneumonitis without features to suggest a definitive etiology (Figure 2I–K). An upper GI study showed an aberrant right subclavian artery, and a gastrostomy tube was placed for nutritional supplementation. At age 14 months, she had worsening respiratory failure requiring prolonged mechanical ventilation. She received bilateral lung transplantation at age 19 months. Hematoxylin and eosin stained sections of explanted lung demonstrated extensive alveolar remodeling with septal widening, areas of enlarged airspaces, extensive fibrosis with peripheral honeycombing, and prominent type II pneumocyte hyperplasia (Figure 2J–K). Airspaces contained macrophages, degenerating cells, cholesterol clefts and dense eosinophilic and proteinaceous material. Pulmonary arterioles showed mild medial and intimal thickening. Electron microscopy of type II pneumocytes demonstrated normal appearing lamellar bodies (not shown).
Her initial hearing screen showed profound bilateral sensorineural hearing loss. Subsequently, an MRI showed bilateral absence of the posterior semicircular canals, and she began wearing hearing aids. A left cochlear implant was placed at age 3 years, although she has not worn the external device due to behavioral issues.
She was diagnosed with hypothyroidism (thyroid stimulating hormone (TSH) elevated at 14.06 mcIUnits/ml [normal range 0.40 - 6.20 mcIUnits/ml], free T4 inappropriately low-normal at 0.9 ng/dl [normal range 0.7-1.8 ng/dl]) at 22 months and has been treated with levothyroxine since then. Hypoparathyroidism was confirmed at age 3 years (low ionized calcium at 3.52 mg/dl [normal range 3.90 - 5.20 mg/dl], low total calcium at 7.3 mg/dl [normal range 8.6 - 10.3 mg/dl], elevated phosphorus at 10.4 mg/dl [normal range 3.0 - 6.0 mg/dl], intact PTH inappropriately normal at 35 pg/ml [normal range 14 - 72 pg/ml]), and has been treated with calcium and calcitriol. She developed osteopenia and acquired stable thoracic vertebral compression fractures. Estradiol therapy was initiated at age 16 years to treat her osteopenia.
At age 6 years, she developed acute renal insufficiency during an illness. This progressed to chronic kidney disease with a peak creatinine of 5 mg/dl (normal range 0.40 - 1.00 mg/dl). She continues to have a persistently elevated but stable creatinine (0.9 to 1.5 mg/dl). Her chronic renal disease is attributed to toxicity from her immunosuppressive medications. She developed anemia, treated with erythropoietin, and chronic hypertension treated with amlodipine, both attributed to her kidney disease.
She has severe growth failure with extreme short stature (at age 16 years, height is 106 cm [9 standard deviations (SD) below the mean] and weight is 16.9 kg [−20 SD]), as well as absent pubertal development by physical exam and biochemical evaluation. She had a delayed bone age of 7 years when measured at chronological age of 14 years (−11 SD). Of note, she was not a candidate for growth hormone therapy due to the risk of inducing bronchiolitis obliterans in her transplanted lungs (Sweet, de la Morena, Schuler, Huddleston, & Mendeloff, 2004).
With regard to her development at her current age of 17 years, she smiles, laughs, and uses about 40 signs to communicate. Her fine motor skills are intact. She had delayed walking at age 3.5 years. She is not toilet trained. She is unable to read, but enjoys watching movies. She will not eat anything by mouth and receives all of her nutrition through a gastrostomy tube. She has repetitive behaviors, and is periodically violent and self-destructive, for which she is treated with risperidone. She has excess body hair on her back and extremities (Figure 2E). She has dental overcrowding with retained deciduous teeth. She underwent removal of squamous papilloma on hard palate at age 10 years.
In summary, this patient’s features include choanal atresia, athelia, profound sensorineural hearing loss, progressive lung disease with interstitial fibrosis requiring lung transplantation, hypothyroidism, hypoparathyroidism, chronic renal disease, feeding difficulties, developmental disabilities, extreme short stature, and lack of pubertal development (Table 1). Previous genetic testing included a normal karyotype and chromosomal microarray analysis in 2008. Family history is non-contributory, with no similarly affected individuals.
Table 1. Genotype and clinical features of individuals with KMT2D missense variants.
ID: intellectual disability, GH: growth hormone, SNHL: sensorineural hearing loss, IDDM: insulin-dependent diabetes mellitus.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 (Sakata et al., 2017) | |
|---|---|---|---|---|
| Genetic information | ||||
| Genomic coordinates (hg19) | chr12:49428016A>G | chr12:49428008G>C | chr12:49427969C>G | chr12:49427900G>C |
| cDNA change | c.10574T>C (NM_003482.3) | c.10582C>G (NM_003482.3) | c.10621G>C (NM_003482.3) | c.10690C>G (NM_003482.3) |
| Protein change | p.(Leu3525Pro) | p.(Leu3528Val) | p.(Ala3541Pro) | p.(Leu3564Val) |
| Mutation type | Missense | Missense | Missense | Missense |
| Inheritance | De novo | De novo | De novo | De novo |
| Method of detection | Clinical exome | Research exome | Research exome | Clinical testing at Illumina, TruSight One sequencing panel |
| Clinical information | ||||
| Age at last evaluation | 17 years | 14 years | Deceased at 14 years | 24 years |
| Sex | Female | Male | Female | Female |
| Features Observed In Kabuki Syndrome | ||||
| Neurodevelopment | Developmental delay; ID | Development delay; mild ID | Developmental delay; ID | Developmental delay |
| Growth | Growth failure; low IGF1 | Growth failure; low IGF1 | Growth failure | Growth retardation; GH deficient |
| Hearing | Profound SNHL | Profound SNHL | Hearing loss | SNHL |
| Ear abnormalities | Small ears with superior helical tags and right preauricular pit | Small cup-shaped dysplastic ears | Preauricular pit | Dysplastic cupped ears |
| Dental | Retained deciduous teeth | Retained deciduous teeth and conical adult teeth | Small wide spaced teeth | Hypoplasia of teeth and malalignment |
| Thyroid function | Hypothyroidism | Hypothyroidism | Central hypothyroidism due to pituitary hypoplasia | Mild hypothyroidism |
| Features Not Typically Observed in Kabuki Syndrome | ||||
| Airway | Choanal atresia | Choanal atresia | Choanal atresia | Choanal atresia |
| Nipples | Bilateral athelia | Bilateral hypoplastic nipples | Bilateral athelia | Bilateral hypoplastic nipples |
| Inner ear | Bilateral absence of posterior semicircular canals | Imaging not available | Vestibular and cochlear abnormalities | Bilateral absence of posterior semicircular canals |
| Parathyroid function | Hypoparathyroidism | Transient hypoparathyroidism | Hypoparathyroidism | Hypoparathyroidism |
| Pubertal development | Absent pubertal development | Absent pubertal development | Pre-pubertal at 14 years | Uterine hypoplasia and hypogonadotropic hypogonadism |
| Pancreas | Normal | Enzyme insufficiency | Normal | Hypoplastic pancreas, IDDM |
| Lung disease | Progressive interstitial lung disease; lung transplant | None | Bronchopulmonary dysplasia; interstitial lung disease; tracheostomy and ventilator | None |
| Neck | Branchial cleft fistula | Branchial cleft fistula | Normal | Normal |
| Facial dysmorphisms | Hypoplastic alae nasi | Hypoplastic alae nasi; Lacrimal duct abnormalities | Lacrimal duct abnormalities | Right facial nerve palsy; broad and depressed nasal tip |
Clinical trio exome sequencing in 2017 revealed a de novo missense variant in the KMT2D gene, NM_003482.3:c.10574T>C p.(Leu3525Pro). The variant is not present in ExAC or gnomAD (Karczewski et al., 2019; Lek et al., 2016), and the CADD score (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019) is 23.5. The diagnostic laboratory interpreted this variant as Likely Pathogenic.
Patient 2
A Caucasian male infant was born at 38 weeks’ estimated gestation by spontaneous vaginal delivery to healthy parents with birth weight 2959g (33rd percentile), length 50cm (62nd percentile), and head circumference 36.5cm (96th percentile). The mother was treated for hypothyroidism throughout gestation, but the pregnancy was otherwise uncomplicated. Shortly after birth, the infant became cyanotic despite supplemental oxygen and CPAP. Passage of a catheter through either nostril was not possible, and the infant improved after placement of an oral airway. He was diagnosed with choanal atresia by CT scan, which showed no bony component to the posterior membranous soft tissue density, and was surgically corrected at three days of age. Mild micrognathia was also observed, as well as low-set, posteriorly rotated, and malformed ears. Due to a diagnosis of severe gastroesophageal reflux combined with poor feeding, he underwent Nissen fundoplication and placement of a gastrostomy tube at one month of age. He developed oral aversion which has continued through childhood, and he has had poor growth.
He was treated with calcitriol for transient hypoparathyroidism, diagnosed shortly after birth, with inappropriately normal intact PTH (34 pg/ml [normal range 14 - 72 pg/ml]), during a period of significant hypocalcemia (6.6 mg/dl [normal range 8.6 - 10.3 mg/dl]). Treatment was discontinued at 2 months of age due to normalization of calcium and PTH levels. He was diagnosed with hypothyroidism (TSH elevated at 7.77 mcIUnits/ml [normal range 0.40 - 6.20 mcIUnits/ml], free T4 low at 0.57 ng/dl [normal range 0.7-1.8 ng/dl]) at 7 months and has been treated with levothyroxine since then. At age 12 months, brain MRI showed a relatively enlarged sella with an enlarged and abnormal pituitary gland, most consistent with a Rathke cleft cyst.
Bilateral profound sensorineural hearing loss was diagnosed at age 4 months. He has periodically used hearing aids, but now communicates primarily by signing. Severe bilateral nasolacrimal duct abnormalities were noted at age 22 months, and subsequently surgically repaired with multiple procedures. He also has a right branchial cleft fistula.
He has had global developmental delays and received occupational and physical therapy. At 13 months, he was unable to sit unassisted and only started to roll over. He began to walk at 4 years. At 5 years, he had 8 words and 25 signs with no sentences. At 13 years, he was in the seventh grade, and able to read at the second-grade level and communicate via signs.
He was initially evaluated by genetics at 5 years, and suspicion for Johanson-Blizzard syndrome was raised. His physical exam was notable for an up-swept frontal hair line, widely spaced eyes, downslanting palpebral fissures, flat and wide nasal bridge, narrow and hypoplastic alae nasi, and a convex nasal ridge (Figure 1D–F). He has two scars on either side of the midline of the vertex of the scalp, which appeared like cutis aplasia at birth (Figure 2H). He has a very thin upper lip with downturned corners. His teeth are abnormally shaped (the lower central incisors are conical), crowded, yellow, and covered with plaque (Figure 3A–B). His ears are small, dysplastic, cup-shaped, and protrude from the sides of the head (Figure 2G). He has very small, faintly pigmented nipples (Figure 2C–D). His fingers are normal with absence of fetal fingertip pads, and the nails of his great toes are dysplastic (Figure 3D–E). He has mild scoliosis. Because of the concern for Johanson-Blizzard syndrome, UBR1 sequencing was sent and was negative. Karyotype was normal, and chromosomal microarray analysis was non-diagnostic.
Stool elastase was low on several measurements consistent with mild to moderate exocrine pancreatic dysfunction, which is seen in Johanson-Blizzard syndrome. Pancreatic enzyme treatment was initiated, and he began to gain weight from 5 years to 8 years. Then, the pancreatic enzyme preparation he was taking was discontinued, and his growth plateaued. At 12 years, he was initiated on pancrelipase, 12,000 units four times per day, and began to gain weight again. He had a delayed bone age of 11 years when measured at chronological age of 13 years (−2 SD). At age 16, he had delayed puberty and extreme short stature (height −4.5 SD and weight −9 SD). He has had significant constipation, requiring hospitalization twice with fecal impaction, which has improved with polyethylene glycol 3350.
Research exome sequencing revealed a de novo missense variant in KMT2D, c.10582C>G p.(Leu3528Val), which was confirmed in a CLIA lab. The variant is not present in ExAC or gnomAD, and the CADD score is 17.5.
Patient 3
A 37 weeks’ estimated gestation Caucasian female was born via spontaneous vaginal delivery with birth weight 2098g (4th percentile) and length 48cm (57th percentile). Pregnancy was complicated by gestational diabetes, and intrauterine growth retardation noted during the 3rd trimester. Multiple congenital anomalies were present, including a preauricular pit, bilateral athelia, anterior placement of the anus, and bilateral 5th finger clinodactyly with tapering of the remaining fingers. She was diagnosed with choanal atresia soon after birth, confirmed by CT and underwent surgical correction. Her postnatal course was notable for sepsis, meningitis and recurrent lung infections, and she was eventually discharged home at 4.5 months of age.
Due to persistent respiratory insufficiency and infections, a wedge lung biopsy was performed at 3 months, and pathology was consistent with bronchopulmonary dysplasia. When she was 13 years, a review of the lung biopsy results was suggestive of surfactant dysfunction, although no further details are available. Overall, her clinical course suggested evolving interstitial lung disease, and she had persistent clubbing, without fetal fingertip pads (Figure 3F–H). At 1 month of age, she required placement of a tracheostomy for ventilation at night. She typically did not require ventilator support or oxygen supplementation during the day.
She had a history of gastroesophageal reflux, poor feeding, and failure to thrive for which she underwent Nissen fundoplication and placement of a gastrostomy tube. She continued to have oral aversion and poor somatic growth.
She was found to have significant hypocalcemia (6.1mg/dl) as a neonate and was diagnosed with hypoparathyroidism which was treated with calcitriol and calcium supplementation. She was diagnosed with hypothyroidism at 1 year (believed to be caused by a small anterior pituitary seen on a brain MRI), and treated with levothyroxine. At 10 years, she was diagnosed with insulin-dependent, non-autoimmune diabetes mellitus (negative antibodies). At 13 years, she had not yet shown signs of puberty. A pelvic ultrasound indicated that both ovaries were present, but were smaller than expected for age. Hormonal testing indicated she had low gonadotropins (LH 0.010 mIU/ml [normal range <0.02 - 11.7 mIU/ml], FSH <0.017 mIU/ml [normal range 0.9 - 8.9 mIU/ml]) and estradiol (<1.0 pg/ml [normal range 0 - 20 pg/ml]), and she was diagnosed with hypogonadotropic hypogonadism.
She failed the newborn hearing screen and was subsequently diagnosed with bilateral profound sensorineural hearing loss. A CT scan confirmed vestibular and cochlear abnormalities, and cochlear implants were placed at 2 years with replacement implants at 9 years. She was able to hear conversations and communicated through sign language and facial expressions. She was noted to have bilateral nasolacrimal duct abnormalities and had surgical repair at 2 years although she continued to have difficulties with frequent tearing.
Initial abdominal ultrasound at birth was normal; however, at 6 months, a repeat renal ultrasound showed hyperechoic renal parenchyma. Additional renal ultrasounds confirmed her kidneys were small raising continual concern for renal dysplasia. Most recently at 13 years of age, the kidneys measured lengths of 5.4cm on the right and 5.2cm on the left. She was diagnosed with chronic kidney disease, stage 2, and her estimated creatinine clearance (75ml/min/1.73m2) and creatinine were stable (0.97mg.dl).
She was hospitalized at 13 years due to chest pain and fatigue. An echocardiogram showed systemic right ventricular systolic pressure and moderate tricuspid insufficiency. An ECG revealed right ventricular enlargement with a strain pattern. A prior echocardiogram at 11 years was normal.
She had global developmental delays. She was able to sit unsupported at 2 years and crawled at 4 years. She was unable to walk unsupported. She started to write her name at 5 years and achieved a 3rd-4th grade level in homebound school. She was able to communicate via sign language, facial expressions, and writing, although she was never able to speak. She was described as being very social and enjoyed being around other people.
She died at 14 years from a hypoglycemic coma in the context of a brief but severe gastrointestinal illness, despite efforts from her parents and emergency personnel to increase her blood glucose.
She was most recently evaluated by genetics at 14 years. At that time, her height was 124.5 cm (−5.6 SD), weight was 24.4 kg (−5.9 SD), and her head circumference was 52 cm (at the 3rd percentile). On physical exam, she had a tall, broad forehead with deep-set eyes (Figure 1G–I). Her hair was dry, curly and coarse. Her ears were low-set with a simplified pinna and a preauricular pit of the left ear. She had a small nose and thin lips. She had wide-spaced and peg shaped teeth with several adult teeth missing. She was Tanner stage 1 with absent nipples. She had significantly reduced tone in her lower extremities with brisk reflexes. Family history was non-contributory.
Research exome sequencing found a de novo missense variant in KMT2D, c.10621G>C p.(Ala3541Pro). The variant is not present in ExAC or gnomAD, and the CADD score is 24.4.
Patient 4 (Published as Sakata et al., 2017)
A search of the published literature revealed an additional patient with overlapping clinical features and a nearby missense variant in KMT2D (Sakata et al., 2017) (Figure 1J). In brief, this adult female had choanal atresia, bilateral hypoplastic nipples, hypoparathyroidism, hypothyroidism, and sensorineural hearing loss with abnormal external and internal ear development associated with bilateral agenesis of the posterior semicircular canals, all of which are present in the other patients reported here (Table 1). She also had developmental delay, growth failure, abnormal teeth, and genitourinary abnormalities, which can be seen in patients with Kabuki syndrome. However, because of the presence of additional features, including right facial nerve palsy and cleft palate, which are not seen in the other patients above, she satisfied both the Blake and Verloes criteria that define CHARGE syndrome, and received this clinical diagnosis (Blake et al., 1998; Verloes, 2005). However, no pathogenic variants in CHD7 were found. A comprehensive sequencing panel revealed a de novo missense variant in KMT2D, c.10690 C>G p.(Leu3564Val). The variant is not present in ExAC or gnomAD, and the CADD score is 25.1.
DISCUSSION
The strong phenotypic overlap and the clustering of missense KMT2D variants in these four patients support the conclusion that these variants are the cause of the observed clinical findings. The 2015 ACMG-AMP variant interpretation guidelines caution against using those classification rules until a gene’s association with disease has been validated, which may require multiple published observations of the same gene-disease relationship (Richards et al., 2015). Regardless, when these criteria are applied, we determine that each of the three KMT2D variants found in Patients 1-3 is Likely Pathogenic, by applying both one strong (PS2: de novo) and one moderate (PM2: absent from controls) criteria to each variant. Applying the recently published consensus clinical diagnostic criteria for Kabuki syndrome demonstrates the challenge of having diverse phenotypes associated with variants in the same gene (Adam et al., 2019). Technically, because all four of these patients have a history of developmental delay and a likely pathogenic variant in KMT2D, they satisfy the criteria for a definitive diagnosis of Kabuki syndrome. However, these patients do not have craniofacial features typical of Kabuki syndrome, nor persistent fetal fingertip pads, arguing against a diagnosis of Kabuki syndrome.
The available crystal structures for KMT2D-encoded histone-lysine N-methyltransferase 2D and closely related proteins only include small portions of this relatively large, 5,537 amino acid-long protein, and do not include the region where these missense variants are located (residues 3,525 to 3,564) (Bienert et al., 2017). However, UniProt displays an apparent coiled-coil domain at the overlapping and nearby residues of 3,562 to 3,614, as predicted by the COILS program (Lupas, Van Dyke, & Stock, 1991) (Figure 5A–B). This is one of five predicted coiled-coil domains in this protein, and these missense variants may disrupt the appropriate protein folding in this region. Further, we observe that our patients’ variants fall in regions of evolutionary constraint, as identified by the Aminode program, affirming their biological importance (Chang et al., 2018) (Figure 5A).
Figure 5. KMT2D missense variants, evolutionary conservation, protein domains, and population variation.
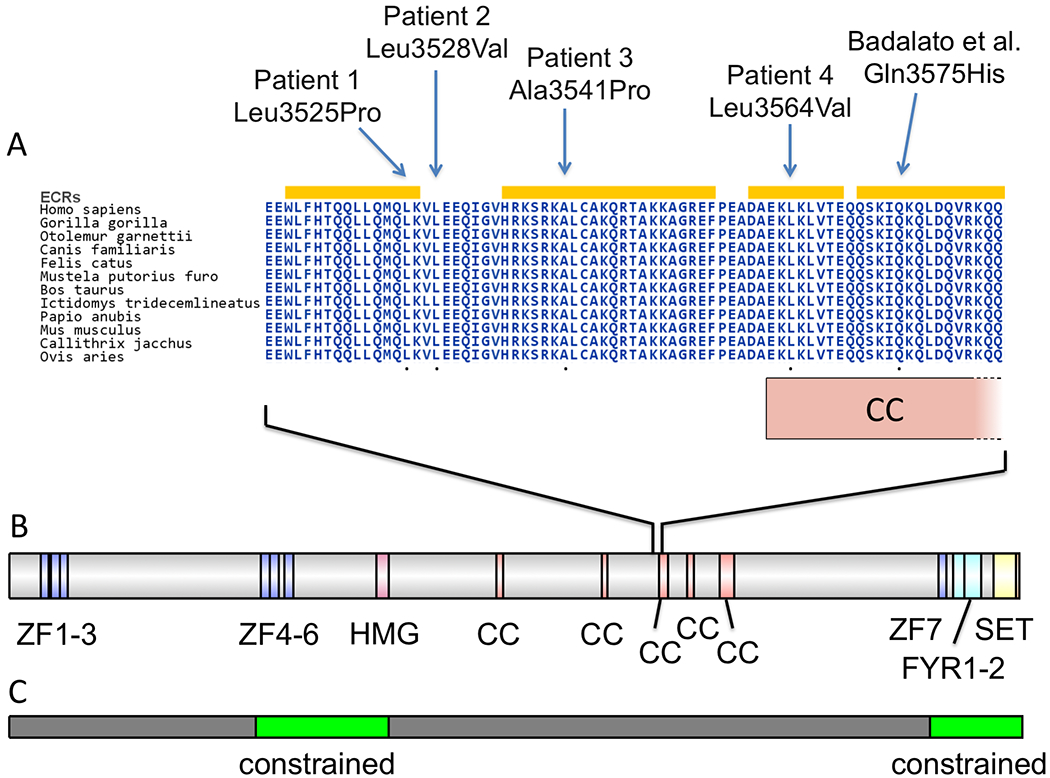
A: Location of patients’ KMT2D missense variants on a highly conserved multi-species alignment of KMT2D protein sequence. ECRs: evolutionarily constrained regions as identified by Aminode (Chang et al., 2018). CC: coiled-coil domain, which extends from residues 3562-3614. B: Schematic of domains of KMT2D protein from InterPro (Mitchell et al., 2019). ZF: zinc-finger. HMG: high mobility group. CC: coiled-coil. FYR: FY-rich. SET: SET methyltransferase domain. C: In green are shown two regions of missense constraint from human population data (Samocha et al., 2017).
Focused analyses of missense variants in KMT2D have recently been published. Cocciadiferro et al. (2018) identified missense variants associated with Kabuki syndrome along the entire length of the protein and used functional studies to demonstrate that some missense variants abrogate histone methylation activity, confirming these are loss-of-function variants. Faundes et al. (2019) analyzed three groups of missense variants in KMT2D: 1) germline variants associated with Kabuki syndrome, 2) somatic variants found in various cancers, and 3) a compilation of “control” variants observed in ExAC (Lek et al., 2016) and similar databases. This study found statistically significant clustering of Kabuki-associated variants in the several protein domains, some of which also had significant clustering for variants found in cancers. There was no significant clustering or presence of any Kabuki-associated variants in the region of the gene that harbors the variants found in the four patients described in this report. However, there were thirteen non-Kabuki KMT2D variants identified in this region (9 from cancers and 4 in controls), including one observation of the same variant (c.10574T>C p.(Leu3525Pro)) that was found in Patient 1 above. This observation is found in COSMIC (Tate et al., 2019) and was reported as a homozygous variant in tumor sequence from a patient with head and neck squamous cell carcinoma (Seiwert et al., 2015). We also note that this same variant is annotated in ClinVar (Landrum et al., 2018) as Variation ID 449909, and we confirmed that this submission is from Patient 1 who was sequenced at GeneDx. Variants in many genes cause both developmental disorders when germline and cancer when somatic; the significance of the observation of this single variant in a tumor is unclear with regard to the pathogenesis of our patients. We observe that our patients’ variants do not fall in either of the two regions of the KMT2D protein that are significantly depleted from missense variation, according to an analysis of the distribution of missense variants in ExAC (Samocha et al., 2017) (Figure 5C). We speculate that result could be due to the specific effects of our patients’ particular missense variants as opposed to variants found in unaffected controls, or due to the limited sensitivity of this method to identify a signal of constraint within a very small 50 amino acid region.
In 2016, Badalato et al. reported a mother, and her son and daughter with choanal atresia, hearing loss, and dental abnormalities, and who all harbor a missense variant (c.10725G>C p.(Gln3575His)) in KMT2D, which was de novo in the mother (Badalato et al., 2017). While their facial appearances were not typical for Kabuki syndrome, all three did have fetal fingertip pads, and the authors characterized these individuals as cases of “atypical Kabuki syndrome.” As noted by Sakata et al. (2017), this variant is only eleven amino acids away from the variant found in Patient 4. Of note, the son had absent right and small left posterior semicircular canals and hypoplastic alae nasi with anteverted nares, and the daughter had short stature (<3%), features which are present in some of our patients. However, unlike all four of the patients presented in our report, there is no description of whether these individuals have absent or hypoplastic nipples or endocrine abnormalities. Given the close proximity of the variant to our patients and the overlap of the unusual finding of choanal atresia, the same underlying molecular mechanism may be present in these “atypical” cases and our patients.
Choanal atresia (present in all four patients and the Badalato patients) and abnormal semicircular canals (present in Patient 1 and 4 and one of the Badalato patients) are two of the major diagnostic criteria for CHARGE syndrome (Verloes, 2005). Further, the ear shape of Patients 1, 2, and 4 could be characterized as similar to that observed in CHARGE syndrome. No clinically significant variants in CHD7 were identified in our patients. The overlap between Kabuki syndrome and CHARGE syndrome has been reported previously, including an assessment of the presence of coloboma in cases with clinically diagnosed Kabuki syndrome prior to the discovery of the molecular basis of either syndrome (Ming, Russell, Bason, McDonald-McGinn, & Zackai, 2003). Several cases with Kabuki syndrome and pathogenic variants in KMT2D also have features of CHARGE syndrome including: 1) a case with choanal atresia and anal atresia (p.(Gln1755*)) (Schulz et al., 2014), 2) a case with coloboma and anal atresia (p.(Gln4329Leufs*47)) (Verhagen, Oostdijk, Terwisscha van Scheltinga, Schalij-Delfos, & van Bever, 2014), and 3) a case with coloboma and absent right inner ear (p.(Arg2771*)) (Patel & Alkuraya, 2015). It has been established that KMT2D and CHD7 proteins both interact with members of the WAR complex (Schulz et al., 2014) and that patterns of aberrant DNA methylation are in common between Kabuki and CHARGE syndromes (Butcher et al., 2017), suggesting that a single common molecular pathway involving chromatin modification may be responsible for both conditions. The added observation of the CHARGE-like findings in these four patients, which have missense variants within a restricted 40-amino-acid region of the protein, further suggests that this region might be the particular portion of the KMT2D protein that is involved in CHD7 interaction or interaction with WAR complex members.
It is possible that the complex medical issues affecting these patients may have been causative of their growth failure and delayed puberty. However, particularly in Patient 1 (Figure 4A–B), the near complete absence of growth may indeed be primary.
Figure 4. Growth curves for individuals with KMT2D missense variants.
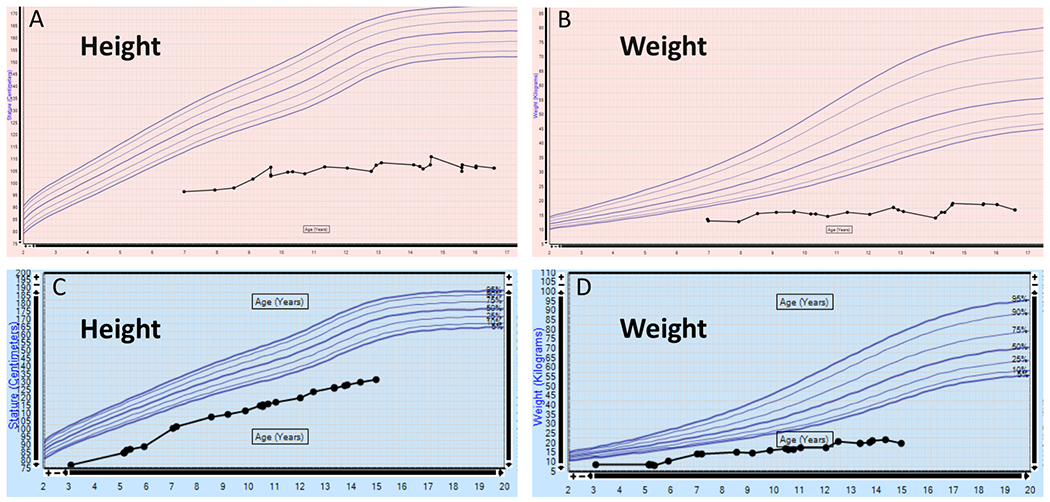
A-B: Patient 1. C-D: Patient 2.
Athelia is an unusual clinical feature (Ishida et al., 2005), and we report it here in association with missense variants in KMT2D. Seven other genes have been associated with athelia in humans, and no clinically significant variants in these genes were identified in our patients. Scalp-Ear-Nipple (SEN) syndrome (MIM 181270) is caused by pathogenic variants in KCTD1 and manifests as cutis aplasia, minor malformations of the external ears, and athelia or amastia (Marneros et al., 2013). The observation of the scalp abnormality in Patient 2 suggests a potential mechanistic connection between KMT2D and KCTD1, which both code for proteins involved in transcriptional regulation. Additional genes that are associated with abnormal breast development include: A) TBX3, which causes Ulnar-Mammary syndrome (MIM 181450) (M. Bamshad et al., 1997), B) TP63 which causes multiple developmental disorders (M. J. Bamshad, 2016; Rinne, Hamel, van Bokhoven, & Brunner, 2006), C) EDA, which causes X-Linked Hypohidrotic Ectodermal Dysplasia (XLHED) (MIM 305100) (Wahlbuhl-Becker, Faschingbauer, Beckmann, & Schneider, 2017), D) EDAR, which causes autosomal recessive hypohidrotic ectodermal dysplasia (MIM 224900) (Haghighi et al., 2013), and E) FIG4, which causes Yunis-Varon syndrome (MIM 216340) (Campeau et al., 2013). Two families with variants that disrupt PTPRF have been reported to have athelia (MIM 616001) (Ausavarat et al., 2011; Borck et al., 2014). Ausavarat et al., 2011 is supplemented with a summary of 62 other cases with absent breast tissue / nipples, most of which are molecularly undefined. We identified an additional seven reports of cases with phenotypic similarity to our patients, but without a molecular diagnosis (Al-Gazali et al., 2002; Dumic, Cvitanovic, Saric, Spehar, & Batinica, 2002; Hisama, Reyes-Mugica, Wargowski, Thompson, & Mahoney, 1998; Horvath & Armstrong, 2007; Papadimitriou, Karapanou, Papadopoulou, Nicolaidou, & Fretzayas, 2009; Qazi, Kanchanapoomi, Beller, & Collins, 1982; Uchida, Konishi, Inoue, Otake, & Kusunoki, 2006).
The characteristics of SWI/SNF-related intellectual disability disorders, also known as Coffin-Siris spectrum disorders, include some overlap with our patients, including hypertrichosis, thick eyebrows, coarse facial features, and feeding problems (Bogershausen & Wollnik, 2018), although other facial characteristics of our patients are distinct. Some cases also have bronchopulmonary disease, like our patients. This observation raises the possibility of a potential molecular interaction between KMT2D and the SWI/SNF chromatin remodeling complex. There is also overlap between the abnormalities in our patients and the features reportedly associated with methimazole and carbimazole embryopathy. Specifically, children born to mothers taking these hyperthyroidism treatments during the first trimester of pregnancy sometimes have choanal atresia, athelia, hypothyroidism, hearing loss, and cutis aplasia of the scalp (Foulds, Walpole, Elmslie, & Mansour, 2005; Greenberg, 1987; Wilson, Kerr, Wilkinson, Fossard, & Donnai, 1998), suggesting a potential overlap in the underlying disease mechanisms affecting these patients.
Johanson-Blizzard syndrome (JBS, MIM 243800) is a rare autosomal recessive disorder caused by pathogenic variants in UBR1 (Zenker et al., 2005). This condition has some features in common with our patients suggesting possible causal mechanistic commonality, including hypoplastic or notched alae nasi, exocrine pancreatic insufficiency, cutis aplasia of the scalp, hearing loss, abnormal hair and teeth, hypothyroidism, and short stature. Additional features of JBS that are not observed in our patients include urogenital, anorectal, and cardiac defects. Before the KMT2D variants were found, this condition was strongly considered in the differential diagnosis of both Patients 1 and 2, but no abnormalities in the UBR1 gene were identified.
Finally, while this manuscript was under review, we became aware of a study which describes the same genotype-phenotype relationship that we report here, but in a separate cohort of 10 affected individuals (Cuvertino et al., 2020). We note that two of these patients share the same de novo missense variant as in our Patient 2 (p.(Leu3528Val)), and another patient’s variant (p.(Leu3542Pro)) is only one amino acid away from our Patient 3’s variant (p.(Ala3541Pro)). This other report also includes functional evidence of pathogenicity, including the observation of a distinct signature of DNA methylation compared to patients with Kabuki syndrome. Among the variants profiled in this assay was our Patient 2’s variant (p.(Leu3528Val)). These data, together with the close phenotypic overlap we observe among the patients, provide compelling evidence for the pathogenicity of the identified KMT2D de novo missense variants.
In conclusion, comparing our patients to those with Kabuki syndrome, we speculate that a distinct biological mechanism and disease process may be affecting the development of multiple tissues with abnormalities in our patients, including the ear, nipple, nasal passages, lung, parathyroid gland, and pancreas. While it has been established that Kabuki syndrome results from haploinsufficiency caused by loss of function variants in KMT2D, we propose that the missense variants reported here may instead cause a gain of function, possibly altering affinity for enzymatic substrates or binding partners. Further experimental studies, such as assessment of H3K4 tri-methyltransferase activity (Bjornsson et al., 2014; Shilatifard, 2012) or detailed examination of additional patterns of DNA methylation (Butcher et al., 2017; Sobreira et al., 2017), are needed to verify this proposition.
ACKNOWLEDGMENTS
We are grateful to the patients and their families for participating in this publication. We acknowledge Alexander Paul for his expert exome sequence analysis. We appreciate the helpful comments provided by the reviewers of this manuscript. Research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG010215 (FSC, JAW, DB), by the National Heart, Lung, and Blood Institute under Award Number K12HL120002 (DB), and by the National Human Genome Research Institute under Award Number K08HG010154 (DB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for this project was provided by the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (FSC, JAW). Patient 3 in this publication was enrolled in Protocol #32301: Genomic Study of Medical, Developmental, or Congenital Problems of Unknown Etiology, which was supported by the Duke University Health system and partially funded by UCB Celltech (VS).
Grant Numbers: National Institutes of Health U01HG010215, K12HL120002, and K08HG010154; Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital; Protocol #32301: Genomic Study of Medical, Developmental, or Congenital Problems of Unknown Etiology, supported by the Duke University Health system and partially funded by UCB Celltech.
Footnotes
CONFLICT OF INTEREST
The authors do not have any conflict of interest to declare.
REFERENCES
- Adam MP, Banka S, Bjornsson HT, Bodamer O, Chudley AE, Harris J, … Kabuki Syndrome Medical Advisory, B. (2019). Kabuki syndrome: international consensus diagnostic criteria. J Med Genet, 56(2), 89–95. doi: 10.1136/jmedgenet-2018-105625 [DOI] [PubMed] [Google Scholar]
- Adam MP, Hudgins L, & Hannibal M (1993-2019). Kabuki Syndrome. 2011 Sep 1 [Updated 2019 Oct 21]. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, & Amemiya A (Eds.), GeneReviews((R)) Seattle (WA). [PubMed] [Google Scholar]
- Al-Gazali LI, Hamid Z, Hertecant J, Bakir M, Nath D, & Kakadekar A (2002). An autosomal recessive syndrome of choanal atresia, hypothelia/athelia and thyroid gland anomalies overlapping bamforth syndrome, ANOTHER syndrome and methimazole embryopathy. Clin Dysmorphol, 11(2), 79–85. [DOI] [PubMed] [Google Scholar]
- Ausavarat S, Tongkobpetch S, Praphanphoj V, Mahatumarat C, Rojvachiranonda N, Snabboon T, … Shotelersuk V (2011). PTPRF is disrupted in a patient with syndromic amastia. BMC Med Genet, 12, 46. doi: 10.1186/1471-2350-12-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badalato L, Farhan SM, Dilliott AA, Care4Rare Canada C, Bulman DE, Hegele RA, & Goobie SL (2017). KMT2D p.Gln3575His segregating in a family with autosomal dominant choanal atresia strengthens the Kabuki/CHARGE connection. Am J Med Genet A, 173(1), 183–189. doi: 10.1002/ajmg.a.38010 [DOI] [PubMed] [Google Scholar]
- Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, … Jorde LB (1997). Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet, 16(3), 311–315. doi: 10.1038/ng0797-311 [DOI] [PubMed] [Google Scholar]
- Bamshad MJ (2016). Epstein’s Inborn Errors of DevelopmentThe Molecular Basis of Clinical Disorders of Morphogenesis TP63 and the Ectodermal Dysplasia, Ectrodactyly, and Cleft Lip and/or Palate (EEC), Limb-Mammary (LMS), Ankyloblepharon, Ectrodactyly, and Cleft Lip/Palate (AEC, Hay-Wells), Acro-Dermato-Ungual-Lacrimal-Digit (ADULT), and Rapp-Hodgkin Syndromes and Ectrodactyly (Split Hand/Foot Malformation): Oxford University Press. [Google Scholar]
- Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, & Schwede T (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res, 45(D1), D313–D319. doi: 10.1093/nar/gkw1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, … Dietz HC (2014). Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med, 6(256), 256ra135. doi: 10.1126/scitranslmed.3009278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KD, Davenport SL, Hall BD, Hefner MA, Pagon RA, Williams MS, … Graham JM Jr. (1998). CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila), 37(3), 159–173. doi: 10.1177/000992289803700302 [DOI] [PubMed] [Google Scholar]
- Bogershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, … Wollnik B (2016). Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum Mutat, 37(9), 847–864. doi: 10.1002/humu.23026 [DOI] [PubMed] [Google Scholar]
- Bogershausen N, & Wollnik B (2018). Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Front Mol Neurosci, 11, 252. doi: 10.3389/fnmol.2018.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, de Vries L, Wu HJ, Smirin-Yosef P, Nurnberg G, Lagovsky I, … Basel-Vanagaite L (2014). Homozygous truncating PTPRF mutation causes athelia. Hum Genet, 133(8), 1041–1047. doi: 10.1007/s00439-014-1445-1 [DOI] [PubMed] [Google Scholar]
- Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, … Weksberg R (2017). CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am J Hum Genet, 100(5), 773–788. doi: 10.1016/j.ajhg.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, Turnpenny P, … Lee BH (2013). Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet, 92(5), 781–791. doi: 10.1016/j.ajhg.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Guo J, di Ronza A, & Sardiello M (2018). Aminode: Identification of Evolutionary Constraints in the Human Proteome. Sci Rep, 8(1), 1357. doi: 10.1038/s41598-018-19744-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocciadiferro D, Augello B, De Nittis P, Zhang J, Mandriani B, Malerba N, … Merla G (2018). Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum Mol Genet, 27(21), 3651–3668. doi: 10.1093/hmg/ddy241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvertino S, Hartill V, Colyer A, Garner T, Nair N, Al-Gazali L, … Banka S (2020). A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med. doi: 10.1038/s41436-019-0743-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumic M, Cvitanovic M, Saric B, Spehar A, & Batinica S (2002). Choanal stenosis, hypothelia, deafness, recurrent dacryocystitis, neck fistulas, short stature, and microcephaly: report of a case. Am J Med Genet, 113(3), 295–297. doi: 10.1002/ajmg.10783 [DOI] [PubMed] [Google Scholar]
- Faundes V, Malone G, Newman WG, & Banka S (2019). A comparative analysis of KMT2D missense variants in Kabuki syndrome, cancers and the general population. J Hum Genet, 64(2), 161–170. doi: 10.1038/s10038-018-0536-6 [DOI] [PubMed] [Google Scholar]
- Foulds N, Walpole I, Elmslie F, & Mansour S (2005). Carbimazole embryopathy: an emerging phenotype. Am J Med Genet A, 132A(2), 130–135. doi: 10.1002/ajmg.a.30418 [DOI] [PubMed] [Google Scholar]
- Greenberg F (1987). Choanal atresia and athelia: methimazole teratogenicity or a new syndrome? Am J Med Genet, 28(4), 931–934. doi: 10.1002/ajmg.1320280419 [DOI] [PubMed] [Google Scholar]
- Haghighi A, Nikuei P, Haghighi-Kakhki H, Saleh-Gohari N, Baghestani S, Krawitz PM, … Mundlos S (2013). Whole-exome sequencing identifies a novel missense mutation in EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia with bilateral amastia and palmoplantar hyperkeratosis. Br J Dermatol, 168(6), 1353–1356. doi: 10.1111/bjd.12151 [DOI] [PubMed] [Google Scholar]
- Hisama FM, Reyes-Mugica M, Wargowski DS, Thompson KJ, & Mahoney MJ (1998). Renal tubular dysgenesis, absent nipples, and multiple malformations in three brothers: a new, lethal syndrome. Am J Med Genet, 80(4), 335–342. [PubMed] [Google Scholar]
- Horvath GA, & Armstrong L (2007). Report of a fourth individual with a lethal syndrome of choanal atresia, athelia, evidence of renal tubulopathy, and family history of neck cysts. Am J Med Genet A, 143A(11), 1231–1235. doi: 10.1002/ajmg.a.31734 [DOI] [PubMed] [Google Scholar]
- Ishida LH, Alves HR, Munhoz AM, Kaimoto C, Ishida LC, Saito FL, … Ferreira MC (2005). Athelia: case report and review of the literature. Br J Plast Surg, 58(6), 833–837. doi: 10.1016/j.bjps.2005.01.018 [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, … Ding L (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502(7471), 333–339. doi: 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, … MacArthur DG (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv, 531210. doi: 10.1101/531210 [DOI] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, … Maglott DR (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 46(D1), D1062–D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation C (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, … Ren J (2015). IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics, 31(20), 3359–3361. doi: 10.1093/bioinformatics/btv362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, & Boerwinkle E (2016). dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat, 37(3), 235–241. doi: 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, & Stock J (1991). Predicting coiled coils from protein sequences. Science, 252(5009), 1162–1164. doi: 10.1126/science.252.5009.1162 [DOI] [PubMed] [Google Scholar]
- Marneros AG, Beck AE, Turner EH, McMillin MJ, Edwards MJ, Field M, … University of Washington Center for Mendelian, G. (2013). Mutations in KCTD1 cause scalp-ear-nipple syndrome Am J Hum Genet, 92(4), 621–626. doi: 10.1016/j.ajhg.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming JE, Russell KL, Bason L, McDonald-McGinn DM, & Zackai EH (2003). Coloboma and other ophthalmologic anomalies in Kabuki syndrome: distinction from charge association. Am J Med Genet A, 123A(3), 249–252. doi: 10.1002/ajmg.a.20277 [DOI] [PubMed] [Google Scholar]
- Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, … Finn RD (2019). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res, 47(D1), D351–D360. doi: 10.1093/nar/gky1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, … Shendure J (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet, 42(9), 790–793. doi: 10.1038/ng.646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitriou A, Karapanou O, Papadopoulou A, Nicolaidou P, & Fretzayas A (2009). Congenital bilateral amazia associated with bilateral choanal atresia. Am J Med Genet A, 149A(7), 1529–1531. doi: 10.1002/ajmg.a.32935 [DOI] [PubMed] [Google Scholar]
- Patel N, & Alkuraya FS (2015). Overlap between CHARGE and Kabuki syndromes: more than an interesting clinical observation? Am J Med Genet A, 167A(1), 259–260. doi: 10.1002/ajmg.a.36804 [DOI] [PubMed] [Google Scholar]
- Qazi QH, Kanchanapoomi R, Beller E, & Collins R (1982). Inheritance of posterior choanal atresia. Am J Med Genet, 13(4), 413–416. doi: 10.1002/ajmg.1320130409 [DOI] [PubMed] [Google Scholar]
- Rao RC, & Dou Y (2015). Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer, 15(6), 334–346. doi: 10.1038/nrc3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res, 47(D1), D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne T, Hamel B, van Bokhoven H, & Brunner HG (2006). Pattern of p63 mutations and their phenotypes--update. Am J Med Genet A, 140(13), 1396–1406. doi: 10.1002/ajmg.a.31271 [DOI] [PubMed] [Google Scholar]
- Sakata S, Okada S, Aoyama K, Hara K, Tani C, Kagawa R, … Kobayashi M (2017). Individual Clinically Diagnosed with CHARGE Syndrome but with a Mutation in KMT2D, a Gene Associated with Kabuki Syndrome: A Case Report. Front Genet, 8, 210. doi: 10.3389/fgene.2017.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, … Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 148353. [Google Scholar]
- Schulz Y, Freese L, Manz J, Zoll B, Volter C, Brockmann K, … Pauli S (2014). CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet, 23(16), 4396–4405. doi: 10.1093/hmg/ddu156 [DOI] [PubMed] [Google Scholar]
- Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, … Hammerman PS (2015). Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res, 21(3), 632–641. doi: 10.1158/1078-0432.CCR-13-3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A (2012). The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem, 81, 65–95. doi: 10.1146/annurev-biochem-051710-134100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Brucato M, Zhang L, Ladd-Acosta C, Ongaco C, Romm J, … Bjornsson HT (2017). Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur J Hum Genet, 25(12), 1335–1344. doi: 10.1038/s41431-017-0023-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet S, de la Morena M, Schuler P, Huddleston C, & Mendeloff E (2004). Association of growth hormone therapy with the development of bronchiolitis obliterans syndrome in pediatric lung transplant recipients. The Journal of Heart and Lung Transplantation, 23(2), S127. [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, … Chung WK (2015). Mutations in SPATA5 Are Associated with Microcephaly, Intellectual Disability, Seizures, and Hearing Loss. Am J Hum Genet, 97(3), 457–464. doi: 10.1016/j.ajhg.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, … Forbes SA (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res, 47(D1), D941–D947. doi: 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Konishi N, Inoue M, Otake K, & Kusunoki M (2006). A case of congenital jejunal atresia associated with bilateral athelia and choanal atresia: new syndrome spectrum. Clin Dysmorphol, 15(1), 37–38. [DOI] [PubMed] [Google Scholar]
- Verhagen JM, Oostdijk W, Terwisscha van Scheltinga CE, Schalij-Delfos NE, & van Bever Y (2014). An unusual presentation of Kabuki syndrome: clinical overlap with CHARGE syndrome. Eur J Med Genet, 57(9), 510–512. doi: 10.1016/j.ejmg.2014.05.005 [DOI] [PubMed] [Google Scholar]
- Verloes A (2005). Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A, 133A(3), 306–308. doi: 10.1002/ajmg.a.30559 [DOI] [PubMed] [Google Scholar]
- Wahlbuhl-Becker M, Faschingbauer F, Beckmann MW, & Schneider H (2017). Hypohidrotic Ectodermal Dysplasia: Breastfeeding Complications Due to Impaired Breast Development. Geburtshilfe Frauenheilkd, 77(4), 377–382. doi: 10.1055/s-0043-100106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, & Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 38(16), e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LC, Kerr BA, Wilkinson R, Fossard C, & Donnai D (1998). Choanal atresia and hypothelia following methimazole exposure in utero: a second report. Am J Med Genet, 75(2), 220–222. [DOI] [PubMed] [Google Scholar]
- Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, … Reis A (2005). Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat Genet, 37(12), 1345–1350. doi: 10.1038/ng1681 [DOI] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, … Goldstein DB (2015). Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med, 17(10), 774–781. doi: 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]