

Abstract
The bromodomain (BrD) is a conserved protein modular domain found in many chromatin- and transcription-associated proteins that has the ability to recognize acetylated lysine residues. This activity allows bromodomains to play a vital role in many acetylation-mediated protein–protein interactions in the cell, ranging from substrate recruitment for histone acetyltransferases (HATs) to aiding in multiple-protein complex assembly for gene transcriptional activation or suppression in chromatin. In recent years, considerable efforts have been made to develop chemical inhibitors of these bromodomains in an effort to probe their cellular functions. Potent and selective inhibitors have been extensively developed for one group of the bromodomain family termed BET proteins that consist of tandem bromodomains followed by an extra terminal domain. Drug developers are actively designing inhibitors of other bromodomains and working to move the most successful inhibitors into the clinic.
Graphical Abstract
Acetylation of histone lysine residues is one of the many vital post-translational modifications that regulate chromatin structure and gene expression in the cell.1,2 Specifically, acetylation neutralizes the positive charge on the ε-amino group of a specific lysine residue, weakening the histone–DNA complex and creating an open chromatin state. This open state provides polymerases, transcription factors, and other components of transcriptional complexes with access to DNA, resulting in gene transcriptional activation.3,4 Acetylation levels are regulated by histone acetyltransferases (HATs; enzymes that “write” marks on lysine residues) and histone deacetylases (HDACs; enzymes that “erase” these marks). Acetylation marks are recognized by bromodomains (BrDs), which are typically found in chromatin- and transcription-associated proteins that partake in many protein–protein interactions, facilitating the formation of the protein complexes that drive active transcription.5–7
As the impact of epigenetic marks on gene expression has become increasingly recognized in recent years, the domains responsible for reading them have become popular drug targets, as modulating such proteins can help identify the major players in the progression of numerous diseases. While bromodomains are only one of the many reader domains encoded within the genome, they have certainly been the most frequently (and successfully) targeted by researchers over the past decade.8,9 Certain seminal small molecule discoveries have sparked a rich field of bromodomain biology that has yielded many high-affinity, highly selective inhibitors that have proven to be effective modulators both in vitro and in vivo. Many of these chemical inhibitors target the BET (bromodomain and extra-terminal) family of bromodomains and have led to a dramatic increase in both the biological knowledge of the BET (tandem bromodomains and an extra terminal domain) proteins and the desire to develop inhibitors of other bromodomains to deeply probe their functions in biology and disease pathways. As such, the advances in chemistry and biology are intimately related, and they drive one another forward.
In recent years, the desire to develop potent and selective inhibitors of both BET and non-BET bromodomains has led to considerable advances in the field of bromodomain biology, which have been reviewed extensively in the literature. These advances range from gaining a greater understanding of the molecular mechanisms underlying the BET bromodomain functions10 to an increase in structural knowledge of the bromodomain family of proteins11 to utilizing newly developed compounds to target cancer and other diseases.8,9,12 Importantly, a wide range of chemotypes of bromodomain inhibitors have been disclosed in both the scientific and patent literature.13,14 The increase in patented bromodomain inhibitors also reflects the increase in interest in moving top-performing compounds into clinical trials, where they are being tested against numerous types of cancer, inflammatory diseases, and autoimmune disorders.8,9
In this review, we reflect on the history of the study of bromodomains, providing an overview of the structural biology and medicinal chemistry discoveries that have helped this epigenome reader become such a popular and important drug target. We also look at recent progress in bromodomain inhibitor development, examining how the numerous, recently developed chemotypes have proven beneficial in the targeting of different bromodomains in different disease models. Finally, we take a look at what is on the horizon of this rapidly changing field and the questions that must be answered as bromodomain inhibitors enter and move forward through human clinical trials.
II. EARLY DISCOVERIES AND THE BIOLOGICAL FUNCTIONS OF BROMODOMAIN PROTEINS
The bromodomain was first discovered as an evolutionarily conserved sequence motif in the Drosophila brahma (brm) and female-sterile homeotic (fsh) proteins in 1992.15 Even though genetic evidence showed that it was a conserved domain found in multiple proteins, questions about its secondary structure, binding partners, and overall functional significance remained unanswered until later in the decade.16 In 1999, the structure of the bromodomain of the human transcriptional coactivator PCAF (p300/CBP-associated factor) was solved using nuclear magnetic resonance (NMR) spectroscopy, revealing its unique three-dimensional structure (Figure 1a).17 The structure showed that the four α helices of the bromodomain (αZ, αA, αB, and αC) form a left-handed four-helix bundle termed the “bromodomain fold” and that two interhelical loops, known as the ZA and BC loops, connect the αZ and αA and αB and αC helices, respectively, forming a hydrophobic binding pocket. This NMR study using 2D 1H–15N-heteronuclear single quantum coherence (HSQC) spectra demonstrated that s lysine-acetylated peptide perturbed residues within this binding pocket between the ZA and BC loops (Figure 1b), providing direct biochemical evidence that acetylated lysine bound at this location. Additionally, the 3D structure of the PCAF bromodomain showed that acetyl-histamine, a small molecule mimic of acetyl-lysine, bound within this pocket, providing further evidence that the bromodomain is an acetyl-lysine recognition domain.
Figure 1.

Bromodomain as the acetyl-lysine reader domain. (a) NMR structure of the PCAF bromodomain (PDB: 1N72). The four helices (αZ, αA, αB, αC) are noted, as are the N and C termini of the structure and the ZA and BC loops. The conserved asparagine residue, N803, is highlighted in yellow. (b) 2D 1H–15N-heteronuclear single quantum coherence (HSQC) spectra of the PCAF bromodomain (~0.5 mM) in its free form (red) and in complex with the acetylated H4 peptide (molar ratio 1:6; black). Figure adapted with permission from ref 17. Copyright 1999 Macmillan Publishers Limited. (c) Crystal structure of the GCN5 bromodomain (blue) in complex with the acetylated peptide H4K16ac (green; PDB: 1E6I). The five water molecules at the base of the bromodomain binding pocket are represented as red spheres, the conserved asparagine residue, N407, is highlighted in yellow, and the ZA and BC loops are labeled. The key hydrogen bond between the conserved asparagine residue and the acetyl-lysine is represented by a black dashed line. (d) Stick diagram of the network of interactions among the H4K16ac peptide and the residues and water molecules within the GCN5 binding pocket. Key residues are labeled. Water molecules are represented by red spheres, and the hydrogen bonding network is represented by black dashed lines.
In the years that followed, additional studies revealed more about the interaction between acetylated lysine and the bromodomain. The year after the initial NMR structure was published, a crystal structure showed that the double bromodomain module of TAFII250, the largest subunit of TFIID (RNA polymerase II transcription factor D), could bind selectively to multiply acetylated H4 peptides.18 That same year, the crystal structure of the bromodomain of the HAT GCN 5p in complex with acetylated histone H4 was solved, providing a more detailed view of the interactions between the bromodomain and an acetylated ligand (Figure 1c,d).19 Within the hydrophobic pocket formed by the ZA and BC loops, the carbonyl oxygen of the acetyl group on the lysine residue forms a hydrogen bond with the amide nitrogen of an asparagine residue that is highly conserved throughout the bromodomain family (Asn407 in GCN 5p). This crystal structure also revealed that a network of water molecules is found at the base of the acetyl-lysine binding pocket, where they form a network of hydrogen bonds among themselves, the acetyl-lysine, and multiple protein backbone residues.19
Additional research also provided a greater scope into just how important bromodomains and lysine-mediated interactions are to a wide variety of cellular processes. The human genome contains 61 unique bromodomains encoded in 46 proteins,20 referred to in this text as bromodomain proteins. Each bromodomain is approximately 110 amino acids in length. The members of the human bromodomain family can be clustered into eight groups—the members of each group share similar sequence lengths and have at least 30% sequence identity.7,11,21 Branches of the tree in Figure 2 are numbered such that group 1 (shaded in blue) and group 2 (shaded in light orange) consist of the histone lysine methyltransferases, the subfamily from which the conserved bromodomain sequence and structure were first reported.17,19 Group 3 contains the BET bromodomains, and the remaining groups of the tree are numbered clockwise.
Figure 2.

Phylogenetic tree of the human bromodomain proteins. Sequence similarity-based phylogenetic tree of the 61 human bromodomains divided into eight groups. Generated using the web knowledgebase Chepimod; modified with permission from ref 11. Copyright 2014 Elsevier.
Bromodomain proteins, chromatin remodelers, and histone acetyl- and methyl-transferases are among the many transcription-associated proteins that work together to regulate the complex process of gene transcription. The majority of bromodomain proteins are comprised of multiple modular domains that work either alone or in concert with one another to convey a series of epigenetic signals within the cell. While the ability to bind acetyl-lysine is necessary for the proper function of a bromodomain protein, it is important to note that this singular quality does not necessarily define the overarching function of a given bromodomain protein.22 In fact, the idea that a conserved fold can have the same function in so many differently functioning proteins is part of the appeal of studying the entire bromodomain family. Among the multidomain bromodomain proteins are transcriptional coactivators, such as the HATs PCAF, GCN5, and p300/CBP. Possessing a bromodomain helps these proteins anchor themselves to an acetylated residue on chromatin, driving additional acetylation and active transcription. The ability of a bromodomain to anchor a protein to chromatin also highlights the roles played by bromodomain proteins in substrate recruitment and complex formation, two very important lysine-mediated protein–protein interactions.7 Some bromodomain proteins, such as SMARC2 and SMARC4, utilize their ability to anchor to acetylated lysine residues on chromatin to drive the chromatin remodeling process.20 Members of the BET family, such as BRD4, are associated with the complexes responsible for transcriptional activation and elongation.5 Growing evidence indicates that the bromodomain structural fold can confer functions other than acetyl-lysine binding. For instance, the bromodomain in the PHD finger-bromodomain tandem module of human transcriptional corepressor KAP1 (also known as TIF-1β) does not bind acetyl-lysine, rather the tandem PHD finger-BrD module functions as a SUMO E3 ligase for transcriptional silencing.23,24 Other proteins such as the histone methyltransferases (HMTs) ASH1L and MLL possess atypical bromodomains whose functions are still under investigation.
III. BREAKTHROUGHS SHIFT THE SPOTLIGHT TO THE BET FAMILY
The identification of the bromodomain as the acetyl-lysine binding motif created a tremendous opportunity for researchers interested in developing inhibitors of protein–protein interactions. With a definitive binding partner in place, the knowledge that bromodomains were a part of many transcriptional-associated proteins, and an increasing appreciation of the importance of lysine acetylation-mediated interactions to many cellular processes, interest in bromodomains as druggable targets grew rapidly. This interest clearly has not waned, as evidenced by the vast number of bromodomain inhibitors that have been published and patented over the past decade. Chemical structures of selected inhibitors can be seen in Tables 1 and 3, and the processes behind their discovery and importance are both described here (see below). A typical bromodomain inhibitor is a small molecule that binds deep within the hydrophobic pocket between the ZA and BC loops and displaces an acetylated peptide, preventing a protein–protein interaction from taking place. To secure itself within the pocket, these inhibitors form the same hydrogen bonds with the conserved asparagine residue and the network of water molecules that a lysine-acetylated peptide would.
Table 1.
Chemical Structures of Selected BET Family Bromodomain Inhibitors
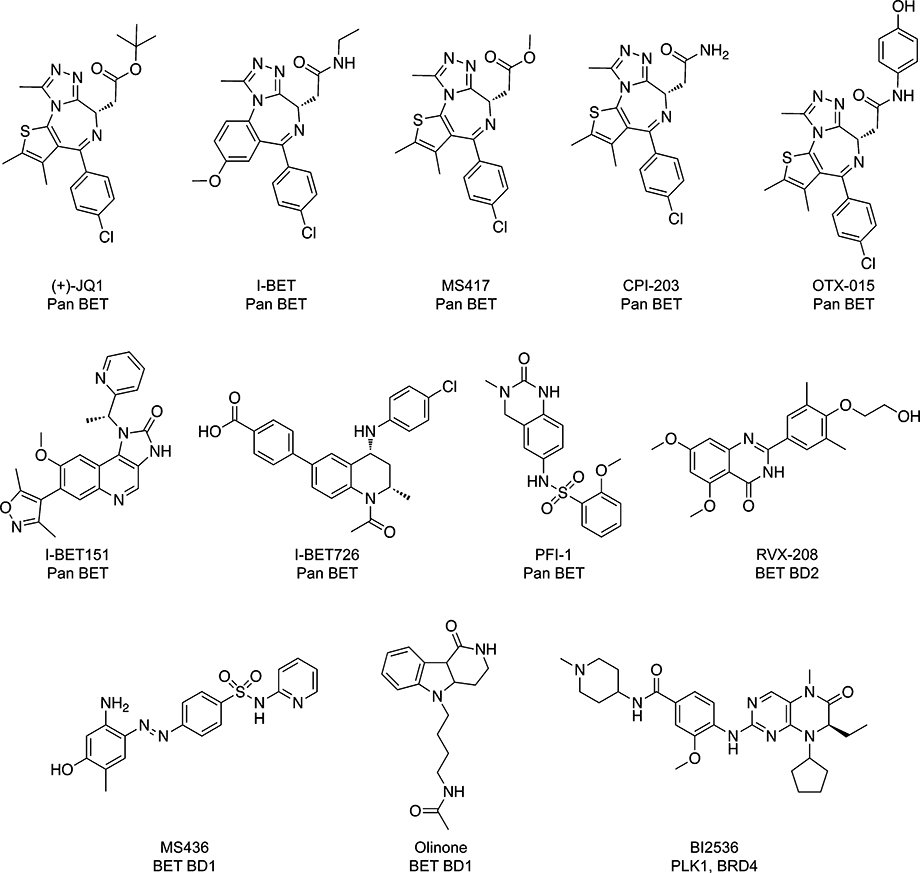 |
Table 3.
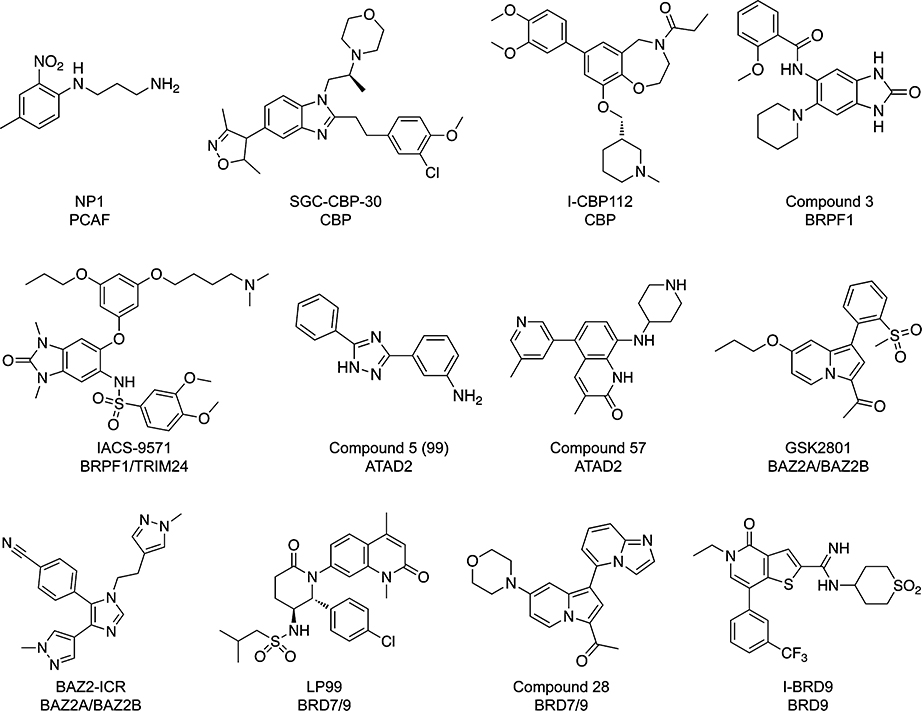
Chemical Structures of Selected Inhibitors of Bromodomains Outside of the BET Family
 |
The first bromodomain inhibitor, which targets the transcriptional coactivator PCAF, was discovered via an NMR-based screen in 2005.25 Interestingly, this inhibitor, an N1-arylpropane-1,3-diamine named NP1, differed slightly from the “typical” bromodomain inhibitors that would follow, as it did not bind deep within the acetyl-lysine binding pocket nor did it interact with the conserved asparagine residue of that bromodomain (Asn798 in PCAF). Yet still, the compound, which was shown to disrupt the interaction between HIV-1 Tat and the PCAF bromodomain, provided a very strong proof of concept that bromodomain-mediated protein–protein interactions could be targeted by small molecular compounds. A similar compound published the following year, a tetrahydrocarbazolone inhibitor of the CBP bromodomain, provided additional evidence that these interactions could be targeted and further provided the impetus for researchers to develop such inhibitors.26
Following these early studies, the Mitsubishi Tanabe Pharmaceutical Corporation reported a series of highly potent thienotriazolodiazepines that targeted the bromodomains of the BET family proteins in two landmark patents filed in 2006 and 2008.27,28 These compounds showed efficacy against multiple tumor cell lines as well as selected autoimmune disorders. Notably, these compounds belong to the diazepine family of compounds and are analogs of benzodiazepines, which have been used in the clinic for many years for anxiolytic and sedative purposes.29 The fact that this scaffold had been proven to be safe, bioavailable, and effective in humans combined with the potential for structure-based drug design research to be conducted to tailor these compounds directly to the BET family bromodomains made the publication of these compounds a seminal moment in the timeline of bromodomain drug discovery and development.
As a result of the findings of the Mitsubishi Tanabe Corporation, researchers at academic institutions and other companies soon developed numerous diazepine-based compounds with high affinity and selectivity for the BET family. These compounds, the specific structural features of which can be viewed in Table 1, make use of two different ring structures to confer their specificity for the BET bromodomains. Each of the compounds highlighted contains a triazole ring, which serves as the ligand’s acetyl-lysine mimic, sitting deep within the hydrophobic binding pocket and forming hydrogen bonds with the conserved water molecules and conserved asparagine residue. Some diazepine-based compounds in the literature have experimented with replacing the triazole ring with a similar hydrogen-bond forming ring, but on the whole, the triazole-based compounds have been the most successful to date. Second, these compounds have either a thienodiazepine or a benzodiazepine core, which allows for different groups to be presented to different residues within the binding pocket. Medicinal chemists have taken advantage of this versatile core region to improve the affinity and/or specificity of a given ligand.29 Importantly, these triazolodiazepine compounds have been widely used in the research community over the past five years and have shown the true breadth of biological pathways in which the BET family bromodomains play an integral role.
One such compound, the thienotriazolodiazepine JQ1, inhibits the BRD4-NUT fusion oncoprotein that causes NUT midline carcinoma (NMC), a rare and aggressive form of cancer.30–32 By blocking the acetyl-lysine binding capabilities of the bromodomains of BRD4, JQ1 displaces BRD4-NUT from chromatin, causing tumor regression, squamous differentiation, and growth arrest in patient-derived xenograft models of NMC.32 The benzodiazepine I-BET was disclosed at the same time as JQ1 and was shown to be an effective disruptor of the chromatin complexes that controlled gene expression in activated macrophages.33 More recently, three additional thienotriazolodiazepines have been published, providing further evidence that this scaffold is robust in its ability to target the BET bromodomains. One thienotriazolodiazepine, MS417, was shown to down-regulate HIV-driven NF-κB transcriptional activity in HIV-associated nephropathy (HIVAN).34 This compound showed improved potency over JQ1 due to the fact that the former possesses a methyl ester moiety at the chiral carbon on the diazepine ring that engages additional interactions with the protein residues, which are absent in JQ1 owing to its bulky t-butyl ester group. A second, CPI-203, has been shown to have a synergistic effect against both mantle cell lymphoma and pancreatic neuroendocrine tumors.35–37 Finally, another recently published thienotriazolodiazepine, OTX015, has shown efficacy against multiple human cancer cell lines.38
Even as the triazolodiazepines have become increasingly visible as a successful class of BET bromodomain inhibitors, researchers are still actively searching for alternate chemotypes that could also serve as potent and selective inhibitors of these bromodomains. Compounds with different chemotypes may present a different selectivity/affinity profile than the diazepine-based compounds and may also have superior pharmacokinetic/pharmacodynamics properties that make them more suitable for in vivo studies. One widely used chemotype has been the 3,5-dimethylisoxazole, which serves as a replacement for the triazole ring found in the thienotriazolodiazepine and benzodiazepine classes of inhibitors.39 An isoxazole ring is a main feature of the potent and selective BET inhibitor I-BET151, which also features a quinoline core in place of a diazepine-based core.40 In separate studies, this compound has been shown to block the growth of MLL-fusion leukemia cells by displacing BET bromodomains from chromatin, thus altering the expression of a series of key genes,40 and to inhibit the progression of JAK2 V617F-driven myeloproliferative neoplasms.41 Aside from I-BET151, many bromodomain inhibitors with a quinoline or quinoline-like core have recently been disclosed. These nondiazepine scaffolds have been reviewed extensively in the recent literature8,9,13 and are represented by the tetrahydroquinoline I-BET726,42 the dihydroquinazolinone PFI-1,43 and the quinazolinone RVX-208 in Table 1.44
While the diazepine-based compounds show impressive selectivity for the BET bromodomains over other bromodomains, questions remain about selectivity within the BET family. The functional differences between members of the BET family (such as BRD2 vs BRD4), different bromodomains of the same protein (such as BRD4-BD1 vs BRD4-BD2), or even the analogous bromodomains of different BET family members (such as BRD4-BD1 vs BRD2-BD1) are not fully understood, but newly developed compounds with alternate chemotypes may prove to be helpful tools in answering these questions. The promising quinazolinone RVX-208 shows 15–30-fold selectivity for the BD2s of the BET family over the BD1s,44 and three other compounds, the diazobenzenes MS43645 and MS61146 as well as the tetrahydro-pyrido indole compound Olinone,46 show a preference for the BD1s of the BET family over the BD2s. Olinone appears to have the best selectivity profile of this series of compounds, with a dissociation constant of 3.4 μM toward BRD4-BD1, as opposed to a dissociation constant of over 300 μM toward BRD4-BD2.46 The developers of Olinone found that BD1-specific inhibition promoted the differentiation of oligodendrocyte progenitors, but treatment of these cells with a nonspecific inhibitor of both domains, MS417, did not have the same effect. As more inhibitors that are selective within the BET family are developed, similar discoveries of the precise functions of individual bromodomains in different model systems are on the horizon.
The concept of dual kinase-bromodomain inhibitors is also one that has been explored in recent years, especially as BRD4 itself has been shown to be an atypical kinase that phosphorylates Ser2 of the RNA Pol II carboxy-terminal domain.35 The CDK inhibitor dinaciclib47 and the PI3K inhibitor LY29400248 show modest affinity for the BET bromodomains, but the PLK1 inhibitor BI2536 and the JAK2 inhibitor TG10120949 are very potent inhibitors of the BET bromodomains, with IC50 values of 25 nM and 130 nM, respectively, against BRD4. Structural analyses have shown that several kinase inhibitors (including those listed here) have excellent shape complementarity with the bromodomain binding pocket and the ability to form hydrogen bonds with the conserved water molecules and residues found within the ZA channel of the bromodomains and, importantly, interact with the conserved asparagine residue within the binding pocket.47,49,50 Some kinase inhibitors utilize their “hinge-binding” groups to interact with the acetyl-lysine recognition site, while others interact independently of these groups, indicating the wide variety of ways by which these inhibitors can exert their polypharmacology.47,49 These early findings provide the impetus for the rational design of inhibitors with the ability to target a selected kinase and a selected bromodomain that are both known to play a role in the progression of a given disease, as well as inhibitors that would only target one of these domains, possibly designed in an effort to limit off-target effects.50 On the whole, these findings are noteworthy because kinase inhibitors are often included in combination therapies in the clinic, and being able to accomplish this aim with a single compound with polypharmacology may make a given therapeutic avenue easier to test.50 This could prove to be an effective treatment strategy for a disease that possesses multiple dysregulated pathways, specifically one driven by a kinase, and another driven by an epigenetic reader domain.
IV. INCREASED KNOWLEDGE OF BET BIOLOGY AND POTENTIAL DISEASE TREATMENT
After the publication of the earliest diazepine-based bromodomain inhibitors, it became quite clear that these chemical tools would spark an incredible interest in the many functions of bromodomain proteins. Because bromodomains, especially the BET family members, are so intimately involved in transcription, there existed the potential for many disease pathways to be elucidated through the use of the inhibitors. Sure enough, as the number of inhibitors developed and available for public research use grew, so too did the number of studies initiated to look more closely at these bromodomain-impacted pathways. Many of these studies have centered around the transcriptional regulation of Myc by the BET bromodomains. Myc has been an especially interesting target for many years, as it is a major player in the development of many forms of cancer that has eluded direct modulation via small molecule inhibitors.51,52 Bromodomain inhibition provides a mechanism by which Myc levels can be indirectly modulated, and the multitude of pathways that it takes part in can be more closely examined. Furthermore, researchers have found the chemical inhibitors of the BET bromodomains described in section III to be effective against cell growth in numerous cancer cell lines, including acute myeloid leukemia,51,52 Burkitt’s lymphoma,51 multiple myeloma,52 lung adenocarcinoma,53 neuroblastoma,54 a genetically diverse glioblastoma,55 castration-resistant prostate cancer,56 and basal-like breast cancer.57 BET inhibitors have also been used to study the activation of HIV from latency,58 autoimmune indications and TH17 pathology,59 and the role of transcriptional pause release in heart failure,60 among many other viral, autoimmune, and inflammatory indications. Unquestionably, the success of these chemical inhibitors in the laboratory will lead to many more studies testing the roles played by the BET proteins in disease pathways in the near future.
The success of the BET inhibitors in the laboratory setting has prompted multiple companies to enter these compounds into clinical trials. One of the diazepine compounds, I-BET (also known as I-BET762 and GSK525762), has been entered into two separate phase I clinical trials by GlaxoSmithKline—one for NUT midline carcinoma and multiple other types of cancer and another for relapsed, refractory hematologic malignancies. The company OncoEthix, which was recently acquired by Merck, has entered a thienotriazolodiazepine it had in-licensed from Mitsubishi Tanabe Pharmaceuticals, OTX015, into four clinical trials for a variety of cancers. Two other companies, Constellation Pharmaceuticals (CPI-0610) and Tensha Therapeutics (TEN-010), have entered their compounds into clinical trials for different types of cancer, as well. Full details of these trials, including disease indications and ClinicalTrials.gov identifiers, can be found in Table 2.
Table 2.
Bromodomain Inhibitors in Human Clinical Trials
| compound | company | indication | stage/status | ClinicalTrials.gov identifier |
|---|---|---|---|---|
| GSK525762 | GlaxoSmithKline | NUT midline carcinoma and other cancers | phase I (recruiting) | NCT01587703 |
| relapsed, refractory hematologic malignancies | phase I (recruiting) | NCT01943851 | ||
| OTX015 | OncoEthix | acute myeloid leukemia and diffuse large B-cell lymphoma | phase I (recruiting) | NCT01713582 |
| advanced solid tumors | phase IB (recruiting) | NCT02259114 | ||
| acute myeloid leukemia | phase IB/II (withdrawn) | NCT02303782 | ||
| glioblastoma multiforme | phase IIA (ongoing, not recruiting) | NCT02296476 | ||
| CPI-0610 | Constellation Pharmaceuticals | progressive lymphoma | phase I (recruiting) | NCT02296476 |
| multiple myeloma | phase I (recruiting) | NCT02157636 | ||
| acute leukemia, myelodysplastic syndrome, myelodysplastic/myeloproliferative neoplasms | phase I (recruiting) | NCT02158858 | ||
| TEN-010 | Tensha | acute myeloid leukemia and myelodysplastic therapeutics syndrome | phase I (recruiting) | NCT02308761 |
| solid tumors | phase I (recruiting) | NCT01987362 | ||
| RVX-208 | Resverlogix Corp. | dyslipidemia, atherosclerosis, acute coronary syndrome, cardiovascular disease | phase II (completed) | NCT00768274 |
| atherosclerosis and coronary artery disease | phase II (completed) | NCT01058018 | ||
| coronary artery disease | phase II (completed) | NCT01067820 | ||
| coronary artery disease and dyslipidemia | phase II (completed) | NCT01423188 | ||
| diabetes | phase II (completed) | NCT01728467 | ||
| dyslipidemia and coronary artery disease | phase II (terminated) | NCT01863225 |
Resverlogix Corp. has been testing their BET family inhibitor, RVX-208, in clinical trials against indications other than cancer. This compound has completed phase II clinical trials for diabetes, coronary artery disease, atherosclerosis, dyslipidemia, and cardiovascular diseases (Table 2). The data from these trials have suggested that RVX-208 usage could lead to a reduction in Major Adverse Cardiac Events (MACE) in high-risk cardiovascular and diabetes mellitus patients, prompting the planning of a phase III clinical trial set to begin in fall 2015.61
V. NON-BET BROMODOMAIN INHIBITORS
The success of the BET family inhibitors in displaying the critical roles played by BET bromodomains in numerous biological pathways has inspired researchers to develop potent and selective inhibitors of non-BET bromodomains in an effort to study their biological functions to the same degree. The bromodomains outside of the BET family appear to present a greater challenge from a drug discovery/design perspective, as they are considered less druggable targets than their BET counterparts, due to factors such as a shallower binding pocket or suboptimal hydrophobicity within the pocket.62 However, a number of challenging non-BET bromodomains previously considered to have druggabilities in the low-to-medium ranges are among those that have been targeted within the past three years by novel, high-affinity, high-selectivity compounds (Table 3). Researchers conducting high-throughput screens and structure-based drug design have applied the lessons learned from BET ligand development to other bromodomains and are now reaping the benefits.
One of the non-BET bromodomains that has recently been successfully targeted is the CBP bromodomain, which had been categorized as a bromodomain with a medium druggability.62 While the earliest CBP inhibitors were not highly potent,26,63 two selective inhibitors recently developed by the SGC, SGC-CBP-30 and I-CBP112, have low nanomolar potencies for the CBP/EP300 bromodomains.64,65 The literature has linked CBP/EP300 to a number of disorders, including (but not limited to) acute myeloid leukemia and a number of neurodegenerative diseases.64,65 These newly developed probes may provide more insight into the pathways that cause these diseases and if bromodomain inhibition is a reasonable long-term treatment strategy in patients.
Two separate laboratories have developed inhibitors of the bromodomain of BRPF1 (Bromodomain-PHD finger protein 1), a protein with multiple reader domains that interacts with multiple acetylated residues on different histones66 and is a part of complexes that also include the MOZ/MORF transcriptional coactivators.67 Inhibitors specific for the BRPF1 bromodomain would help isolate its function from the other reader domains of the protein and of the other members of the multiprotein complexes it takes part in.68 One inhibitor, the benzimidazolone compound 3, binds BRPF1 with a pIC40 of 7.1 and shows selectivity for BRPF1 over the other BRPF family members (BRPF2 and BRPF3), as well as the members of the BET family.68 The other compound published as a BRPF1 inhibitor, IACS-9571, binds BRPF1 and TRIM24 with very low nanomolar potency in vitro (BRFP1, Kd = 14 nM; TRIM, Kd = 31 nM) and has very promising cellular activity (EC50 = 50 nM), as well.69 This inhibitor does show selectivity over a panel of other bromodomains, but is not very selective within the BRPF bromodomain family.69
The bromodomain of ATAD2 (ATPase family, AAA domain containing 2) is seen as a very challenging target, even relative to the other bromodomains of the family.62 However, this bromodomain has been linked to a wide variety of cancers in the literature, as it plays a role in the transcription of a number of genes that play a role in cancer cell proliferation and survival.70 Within the past two years, two small molecule fragment screens were conducted in an effort to find inhibitors of the ATAD2 bromodomain that could help determine if it is a viable therapeutic target. The first study, an NMR-based fragment screen, yielded a number of low-affinity core chemotypes that could potentially be built upon in the development of a high quality ATAD2 probe.71 The second study yielded more potent quinolinone- and napthyridone-based inhibitors, with the top performing compounds having IC50 values in the low micromolar range.70 Further improvements upon these first-generation compounds could lead to a greater understanding of the specific role of the ATAD2 bromodomain in numerous disease pathways.
Importantly, inhibitors are currently being developed for bromodomains that had previously unknown functions. One such bromodomain, BAZ2B (bromodomain adjacent to zinc finger domain protein 2B), has been successfully targeted by multiple research groups over the past three years, despite its shallow binding pocket and predicted low druggability.72 The first BAZ2B bromodomain inhibitor to be discovered was a tetrahydro-γ-carboline (THγC) with a Kd value of 9 μM against BAZ2B.73 A second compound, GSK2801, has shown high affinity and selectivity for both BAZ2B and the closely related BAZ2A (Kd of 136 nM and 257 nM, respectively),74 and a third, BAZ2-ICR, is also selective for both BAZ2A/B (IC50 of 130 nM and 180 nM, respectively).72 Another previously understudied bromodomain, BRD9 (bromodomain-containing protein 9), has also had multiple inhibitors developed to probe its function within the past year. Two of these compounds, the quinolone-fused lactam LP9975 and the imidazopyridine compound 28,76 are inhibitors of both BRD9 and the structurally similar BRD7 bromodomain. Both LP99 and compound 28 have high affinities for BRD9 (Kd values of 99 nM and 68 nM, respectively), have some degree of activity against BRD7, and show little-to-no activity against bromodomains outside of those two. An additional series of compounds with 9H-purine scaffolds also possesses nanomolar activity against BRD9 and shows moderate selectivity over BRD4-BD1.77 However, the most potent and selective BRD9 inhibitor developed to date is the thienopyridine I-BRD9, which shows low nanomolar potency against BRD9, >700-fold selectivity over the BET family, and, in contrast to the other BRD7/9 inhibitors, >200-fold selectivity over BRD7.78 As is the aim in the development of inhibitors of little-studied bromodomains such as BAZ2B and BRD9, the use of I-BRD9 in a cell-based assay uncovered a number of genes specifically regulated by BRD9 in cancer and immunological pathways.78 With these inhibitors and other comparable inhibitors of other little-studied bromodomains likely forthcoming, additional discoveries are on the horizon.
VI. AN EYE ON THE FUTURE
As interest has grown in the development of novel epigenetic therapeutics to treat a variety of different diseases, the members of the human bromodomain family have become extensively studied drug targets. With their ability to recognize and bind to acetyl-lysine, bromodomain proteins are at the heart of lysine acetylation-mediated processes within the cell, including the opening of condensed chromatin to facilitate transcription and the recruitment of transcription factors to open chromatin, where they can form the complexes that drive active transcription. The inhibitors that have been developed over the past five years, largely the ones targeting the BET family, have provided tremendous insight into the functions of these bromodomains as well as the disease pathways they take part in. The successes of these inhibitors will guide the paths taken in the near future by researchers studying bromodomain biology in many ways.
Of primary interest is the fate of the existing BET family inhibitors that are currently undergoing clinical trials. As these trials begin to yield results, we will be able to see either how impactful these compounds can be or how future generations of BET inhibitors will need to be modified to yield the desired level of therapeutic success. If the trials prove unsuccessful, different chemotypes, different selectivity profiles, and the potential for using these inhibitors in combinatory treatments for different types of cancer are all potential avenues to take that could lead to an improved chance at success in the clinic in the long term. These clinical trials will also show the side effects of using bromodomain inhibitors in humans, which researchers and doctors will need to combat, possibly through altered doses or, again, using these inhibitors in conjunction with other therapeutics. On the other hand, successful trials would certainly lead to more entries into the bromodomain inhibitor marketplace and likely an even deeper commitment to laboratory research into other therapeutic areas that could be affected by bromodomain inhibition.
An additional trend to watch in the coming years will be the development of inhibitors of the less druggable non-BET bromodomains and how targets of interest will be selected. For the bromodomains of which little is known about their function, the development of a tight-binding, selective inhibitor will likely come first and be used to elucidate the bromodomain’s biological function and potential role in disease pathways. For other bromodomains with known functions, researchers targeting specific diseases or biological complexes may initiate drug discovery programs to help improve their model systems, test additional hypotheses, or even target a protein they believe has long-term clinical potential. With improvements in structure-based drug design of bromodomain inhibitors coming from the rich collection of structure–activity relationship data that has been generated by all bromodomain inhibitors to date, advances in this arena seem very near.
Moving forward, it is quite clear that the future of bromodomain inhibitors is filled with considerable potential. This emerging class of compounds will continue to drive an increase in knowledge of transcriptional regulation in the context of numerous diseases and potentially serve as new epigenetic therapeutics to treat diseases such as cancer, autoimmune disorders, and inflammatory conditions in the clinic.
ACKNOWLEDGMENTS
We would like to thank the past and current members of the Zhou Group for helpful discussion. This work was supported in part by research grants from the National Institutes of Health (to M.-M.Z.).
Glossary
- Acetylation
a type of post-translational protein modification that affects lysine residues, associated with active transcription in the context of chromatin
- Bromodomain
conserved structural module responsible for “reading,” or recognizing, acetylation marks on lysine residues on proteins
- Chromatin
the complex within the nuclei of eukaryotic cells that contains DNA tightly wrapped around histone proteins
- Epigenetics
the study of how changes within the cellular environment regulate the expression of genes without altering the actual DNA sequence
- Inhibitors
chemical ligands developed to inhibit the functions of a specific protein or protein domain
- Lysine
basic amino acid involved in many protein–protein interactions in the cell, largely due to the numerous post-translational modifications to which it is subject
- Reader Domain
domain of a protein that recognizes and binds to certain post-translational modifications, such as an acetyl group or methyl group
- Transcription
the process by which the DNA of a cell is read and converted to an RNA message
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Kouzarides T (2007) Chromatin Modifications and Their Function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- (2).Strahl BD, and Allis CD (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (3).Thomas MC, and Chiang C-M (2006) The General Transcription Machinery and General Cofactors. Crit. Rev. Biochem. Mol. Biol. 41, 105–178. [DOI] [PubMed] [Google Scholar]
- (4).Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412. [DOI] [PubMed] [Google Scholar]
- (5).Chiang C-M (2009) Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4, F1000 Biol. Rep., 1, DOI: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Josling GA, Selvarajah SA, Petter M, and Duffy MF (2012) The Role of Bromodomain Proteins in Regulating Gene Expression. Genes 3, 320–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sanchez R, and Zhou M-M (2009) The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discovery Devel. 12, 659–665. [PMC free article] [PubMed] [Google Scholar]
- (8).Brand M, Measures AM, Wilson BG, Cortopassi WA, Alexander R, Hoss M, Hewings DS, Rooney TPC, Paton RS, and Conway SJ (2015) Small Molecule Inhibitors of Bromodomain-Acetyl-lysine Interactions. ACS Chem. Biol. 10, 22–39. [DOI] [PubMed] [Google Scholar]
- (9).Filippakopoulos P, and Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 13, 337–356. [DOI] [PubMed] [Google Scholar]
- (10).Shi J, and Vakoc CR (2014) The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 54, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sanchez R, Meslamani J, and Zhou M-M (2014) The bromodomain: From epigenome reader to druggable target. Biochim. Biophys. Acta, Gene Regul. Mech. 1839, 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, and Haendler B (2015) Targeting BET bromodomains for cancer treatment. Epigenomics 7, 487–501. [DOI] [PubMed] [Google Scholar]
- (13).Gallenkamp D, Gelato KA, Haendler B, and Weinmann H (2014) Bromodomains and Their Pharmacological Inhibitors. ChemMedChem 9, 438–464. [DOI] [PubMed] [Google Scholar]
- (14).Garnier J-M, Sharp PP, and Burns CJ (2014) BET bromodomain inhibitors: a patent review. Expert Opin. Ther. Pat. 24, 185–199. [DOI] [PubMed] [Google Scholar]
- (15).Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, and Kennison JA (1992) brahma: A Regulator of Drosophila Homeotic Genes Structurally Related to the Yeast Transcriptional Activator SNF2/SWI2. Cell 68, 561–572. [DOI] [PubMed] [Google Scholar]
- (16).Haynes SR, Dollard C, Winston F, Beck S, Trowsdale J, and Dawid IB (1992) The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 20, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, and Zhou M-M (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. [DOI] [PubMed] [Google Scholar]
- (18).Jacobson RH, Ladurner AG, King DS, and Tijan R (2000) Structure and Function of a Human TAF(II)250 Double Bromodomain Module. Science 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- (19).Owen DJ, Ornaghi P, Yang J-C, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, and Travers AA (2000) The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J. 19, 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras A-C, Arrowsmith CH, and Knapp S (2012) Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sanchez R, Pieper U, Melo F, Eswar N, Marti-Renom MA, Madhusudhan MS, Mirkovic N, and Sali A (2000) Protein structure modeling for structural genomics. Nat. Struct. Biol. 7, 986–990. [DOI] [PubMed] [Google Scholar]
- (22).Basu MK, Carmel L, Rogozin IB, and Koonin EV (2008) Evolution of protein domain promiscuity in eukaryotes. Genome Res. 18, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zeng L, Yap KL, Ivanov AV, Wang X, Mujtaba S, Plotnikova O, Rauscher FJ III, and Zhou M-M (2008) Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 15, 626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou M-M, and Rauscher FJ III (2007) PHD Domain-Mediated E3 Ligase Activity Directs Intramolecular Sumoylation of an Adjacent Bromodomain Required for Gene Silencing. Mol. Cell 28, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, Wang Z, and Zhou M-M (2005) Selective Small Molecules Blocking HIV-1 Tat and Coactivator PCAF Association. J. Am. Chem. Soc. 127, 2376–2377. [DOI] [PubMed] [Google Scholar]
- (26).Sachchidanand Resnick-Silverman L, Yan S, Mujtaba S, Liu W.-j., Zeng L, Manfredi JJ, and Zhou M-M (2006) Target Structure-Based Discovery of Small Molecules that Block Human p53 and CREB Binding Protein Association. Chem. Biol. 13, 81–90. [DOI] [PubMed] [Google Scholar]
- (27).Adachi K, Hikawa H, Hamada M, Endoh J.-i., Ishibuchi S, Fujie N, Tanaka M, Sugahara K, Oshita K, and Murata M (2006) Thienotriazolodiazepine Compound and Medicinal Use Thereof, Mitsubishi Tanabe Pharma Corporation, Japan. [Google Scholar]
- (28).Miyoshi S, Ooike S, Iwata K, Hikawa H, and Sugahara K (2008) Antitumor Agent, citMitsubishi Tanabe Pharma Corporation, Japan. [Google Scholar]
- (29).Smith SG, Sanchez R, and Zhou M-M (2014) Privileged Diazepine Compounds and Their Emergence as Bromodomain Inhibitors. Chem. Biol. 21, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, and Fletcher JA (2003) BRD4-NUT Fusion Oncogene: A Novel Mechanism in Aggressive Carcinoma. Cancer Res. 63, 304–307. [PubMed] [Google Scholar]
- (31).French C, Ramirez C, Kolmakova J, Hickman T, Cameron M, Thyne M, Kutok J, Toretsky J, Tadavarthy A, Kees U, Fletcher J, and Aster J (2008) BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 27, 2237–2242. [DOI] [PubMed] [Google Scholar]
- (32).Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C. w., Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, and Tarahovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, Rusinova E, Gerona-Navarro G, Moshkina N, Joshua J, Chuang PY, Ohlmeyer M, He JC, and Zhou M-M (2012) Down-regulation of NF-κB Transcriptional Activity in HIV-associated Kidney Disease by BRD4 Inhibition. J. Biol. Chem. 287, 28840–28851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, Ozato K, Sims RJ, and Singer DS (2012) BRD4 is an atypical kinase that phosphorylates Serine2 of the RNA Polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 109, 6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Moros A, Rodriguez V, Saborit-Villarroya I, Montraveta A, Balsas P, Sandy P, Martinez A, Wiestner A, Normant E, Campo E, Perez-Galan P, Colomer D, and Roue G (2014) Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 28, 2049–2059. [DOI] [PubMed] [Google Scholar]
- (37).Wong C, Laddha S, Tang L, Vosburgh E, Levine A, Normant E, Sandy P, Harris C, Chan C, and Xu E (2014) The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 5, No. e1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Noel JK, Iwata K, Ooike S, Sugahara K, Nakamura H, and Daibata M (2013) Development of the BET bromodomain inhibitor OTX015. Mol. Cancer Ther. 12, C244. [Google Scholar]
- (39).Hewings DS, Wang M, Philpott M, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Vuppusetty C, Marsden B, Knapp S, Conway SJ, and Heightman TD (2011) 3,5-Dimethylisoxazoles Act As Acetyl-lysine-mimetic Bromodomain Ligands. J. Med. Chem. 54, 6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, Robson SC, Chung C. w., Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJP, and Kouzarides T (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wyspianska B, Bannister A, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto F, Robson S, Rioja I, Li J, Wiese M, Cannizzaro E, Dawson M, Huntly B, Prinjha R, Green A, Gottgens B, and Kouzarides T (2014) BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia 28, 88–97. [DOI] [PubMed] [Google Scholar]
- (42).Gosmini R, Nguyen VL, Toum J, Simon C, Brusq J-MG, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung C. w., Cutler L, Demont EH, Kaur R, Lewis AJ, Schilling MB, Soden PE, Taylor S, Walker AL, Walker MD, Prinjha RK, and Nicodeme E (2014) The Discovery of I-BET726 (GSK1324726A), a Potent Tetrahydroquinoline ApoA1 Up-Regulator and Selective BET Bromodomain Inhibitor. J. Med. Chem. 57, 8111–8131. [DOI] [PubMed] [Google Scholar]
- (43).Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, Owen DR, Bradner JE, Taniere P, O’Sullivan B, Muller S, Schwaller J, Stankovic T, and Knapp S (2013) PFI-1, a Highly Selective Protein Interaction Inhibitor, Targeting BET Bromodomains. Cancer Res. 73, 3336–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Muller S, Brennan PE, Knapp S, and Filippakopoulos P (2013) RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A. 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, and Zhou M-M (2013) Structure-Guided Design of Potent Diazobenzene Inhibitors for the BET Bromodomains. J. Med. Chem. 56, 9251–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gacias M, Gerona-Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B, Vincek A, Joshua J, Casaccia P, and Zhou M-M (2014) Selective Chemical Modulation of Gene Transcription Favors Oligodendrocyte Lineage Progression. Chem. Biol. 21, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Martin MP, Olesen SH, Georg GI, and Schonbrunn E (2013) Cyclin-Dependent Kinase Inhibitor Dinaciclib Interacts with the Acetyl-Lysine Recognition Site of Bromodomains. ACS Chem. Biol. 8, 2360–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dittmann A, Werner T, Chung C-W, Savitski MM, Fälth Savitski M, Grandi P, Hopf C, Lindon M, Neubauer G, Prinjha RK, Bantscheff M, and Drewes G (2014) The Commonly Used PI3-Kinase Probe LY294002 Is an Inhibitor of BET Bromodomains. ACS Chem. Biol. 9, 495–502. [DOI] [PubMed] [Google Scholar]
- (49).Ember SWJ, Zhu J-Y, Olesen SH, Martin MP, Becker A, Berndt N, Georg GI, and Schonbrunn E (2014) Acetyl-lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 9, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, Martin S, Wodicka LM, Shah NP, Treiber DK, and Knapp S (2014) Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 10, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, and Sims RJ III (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U. S. A. 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, and Mitsiades CS (2011) BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lockwood WW, Zejnullahu K, Bradner JE, and Varmus H (2012) Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. U. S. A. 109, 19408–19413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, and Stegmaier K (2013) Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discovery 3, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, Thompson RC, Muller S, Knapp S, and Wang J (2013) Inhibition of BET Bromodomain Targets Genetically Diverse Glioblastoma. Clin. Cancer Res. 19, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu Y-M, Cao X, Qin ZS, Wang S, Feng FY, and Chinnaiyan AM (2014) Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng Y-X, Evers BM, Zhou M-M, and Zhou BP (2014) Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell 25, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li P-C, Planelles V, Bradner JE, Zhou MM, Siliciano RF, Weinberger L, Verdin E, and Ott M (2013) BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 12, 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Mele DA, Salmeron A, Ghosh S, Huang H-R, Bryant BM, and Lora JM (2013) BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 210, 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, and Haldar SM (2013) BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell 154, 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Resverlogix. (2015) News Release - Resverlogix Officially Attains Phase 3 Status with a European Regulatory Authority.
- (62).Vidler LR, Brown N, Knapp S, and Hoelder S (2012) Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 55, 7346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, Moshkina N, Morohashi K, Zhang W, Gerona-Navarro G, Hajjar RJ, and Zhou M-M (2011) A Small Molecule Binding to the Coactivator CREB-Binding Protein Blocks Apoptosis in Cardiomyocytes. Chem. Biol. 18, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).SGC. (2015) SGC-CBP30 - a CREBBP/EP300-selective chemical probe.
- (65).SGC. (2015) I-CBP112 - a CREBBP/EP300-selective chemical probe.
- (66).Poplawski A, Hu K, Lee W, Natesan S, Peng D, Carlson S, Shi X, Balaz S, Markley JL, and Glass KC (2014) Molecular Insights into the Recognition of N-Terminal Histone Modifications by the BRPF1 Bromodomain. J. Mol. Biol. 426, 1661–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh S-L, Champagne N, Cote J, and Yang X-J (2008) Molecular Architecture of Quartet MOZ/MORF Histone Acetyltransferase Complexes. Mol. Cell. Biol. 28, 6828–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Demont EH, Bamborough P, Chung C. w., Craggs PD, Fallon D, Gordon LJ, Grandi P, Hobbs CI, Hussain J, Jones EJ, Le Gall A, Michon A-M, Mitchell DJ, Prinjha RK, Roberts AD, Sheppard RJ, and Watson RJ (2014) 1,3-Dimethyl Benzimidazolones Are Potent, Selective Inhibitors of the BRPF1 Bromodomain. ACS Med. Chem. Lett. 5, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP, Leo E, Shepard HE, Tieu TN, Shi X, Zhan Y, Zhao S, Barton MC, Draetta G, Toniatti C, Jones P, Geck Do M, and Andersen JN (2015) Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J. Med. Chem, 150706120257007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Demont EH, Chung C. w., Furze RC, Grandi P, Michon A-M, Wellaway C, Barrett N, Bridges AM, Craggs PD, Diallo H, Dixon DP, Douault C, Emmons AJ, Jones EJ, Karamshi BV, Locke K, Mitchell DJ, Mouzon BH, Prinjha RK, Roberts AD, Sheppard RJ, Watson RJ, and Bamborough P (2015) Fragment-Based Discovery of Low-Micromolar ATAD2 Bromodomain Inhibitors. J. Med. Chem. 58, 5649–5673. [DOI] [PubMed] [Google Scholar]
- (71).Harner MJ, Chauder BA, Phan J, and Fesik SW (2014) Fragment-Based Screening of the Bromodomain of ATAD2. J. Med. Chem. 57, 9687–9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Drouin L, McGrath S, Vidler LR, Chaikuad A, Monteiro O, Tallant C, Philpott M, Rogers C, Fedorov O, Liu M, Akhtar W, Hayes A, Raynaud F, Muller S, Knapp S, and Hoelder S (2015) Structure Enabled Design of BAZ2-ICR, A Chemical Probe Targeting the Bromodomains of BAZ2A and BAZ2B. J. Med. Chem. 58, 2553–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Ferguson FM, Fedorov O, Chaikuad A, Philpott M, Muniz JRC, Felletar I, von Delft F, Heightman T, Knapp S, Abell C, and Ciulli A (2013) Targeting Low-Druggability Bromodomains: Fragment Based Screening and Inhibitor Design against the BAZ2B Bromodomain. J. Med. Chem. 56, 10183–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Chen P, Chaikuad A, Bamborough P, Bantscheff M, Bountra C, Chung C. w., Fedorov O, Grandi P, Jung D, Lesniak R, Lindon M, Muller S, Philpott M, Prinjha R, Rogers C, Selenski C, Tallant C, Werner T, Willson TM, Knapp S, and Drewry DH (2015) Discovery and Characterization of GSK2801, a Selective Chemical Probe for the Bromodomains BAZ2A and BAZ2B. J. Med. Chem, 150418091209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Clark PGK, Vieira LCC, Tallant C, Fedorov O, Singleton DC, Rogers CM, Monteiro OP, Bennett JM, Baronio R, Muller S, Daniels DL, Mendez J, Knapp S, Brennan PE, and Dixon DJ (2015) LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed. 54, 6217–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hay DA, Rogers CM, Fedorov O, Tallant C, Martin S, Monteiro OP, Muller S, Knapp S, Schofield CJ, and Brennan PE (2015) Design and synthesis of potent and selective inhibitors of BRD7 and BRD9 bromodomains. MedChemComm 6, 1381–1386. [Google Scholar]
- (77).Picaud S, Strocchia M, Terracciano S, Lauro G, Mendez J, Daniels DL, Riccio R, Bifulco G, Bruno I, and Filippakopoulos P (2015) 9H-Purine Scaffold Reveals Induced-Fit Pocket Plasticity of the BRD9 Bromodomain. J. Med. Chem. 58, 2718–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung C. w., Dittmann A, Drewes G, Drewry DH, Gordon L, Grandi P, Leveridge M, Lindon M, Michon A-M, Molnar J, Robson SC, Tomkinson NCO, Kouzarides T, Prinjha RK, and Humphreys PG (2015) Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem, 150430080108003. [DOI] [PMC free article] [PubMed] [Google Scholar]