

Abstract
Targeted therapy with reduced side effects is a major goal in cancer research. We investigated the effects of JS-K, a nitric oxide (NO) prodrug designed to release high levels of NO when suitably activated, on human breast cancer cell lines, on non-transformed human MCF-10A mammary cells, and on normal human mammary epithelial cells (HMECs). Cell viability assay, flow cytometry, electron microscopy, and Western blot analysis were used to study the effects of JS-K on breast cancer and on mammary epithelial cells. After a 3-day incubation, the IC50s of JS-K against the breast cancer cells ranged from 0.8 to 3 μM. However, JS-K decreased the viability of the MCF-10A cells by only 20% at 10-μM concentration, and HMECs were unaffected by 10 μM JS-K. Flow cytometry indicated that JS-K increased the percentages of breast cancer cells under-going apoptosis. Interestingly, flow cytometry indicated that JS-K increased acidic vesicle organelle formation in breast cancer cells, suggesting that JS-K induced autophagy in breast cancer cells. Electron microscopy confirmed that JS-K-treated breast cancer cells underwent autophagic cell death. Western blot analysis showed that JS-K induced the expression of microtubule light chain 3-II, another autophagy marker, in breast cancer cells. However, JS-K did not induce apoptosis or autophagy in normal human mammary epithelial cells. These data indicate that JS-K selectively induces programmed cell death in breast cancer cells while sparing normal mammary epithelial cells under the same conditions. The selective anti-tumor activity of JS-K warrants its further investigation in breast tumors.
Keywords: JS-K, apoptosis, autophagy, breast cancer, nitric oxide
Introduction
Breast cancer is the second leading cause of cancer death in women. Great strides have been made in the diagnosis and treatment of breast cancer. Nonetheless, it was estimated that in 2010, about 39,840 women will die from breast cancer. Novel therapeutic strategies are urgently needed to decrease the mortality of breast cancer patients.
Nitric oxide (NO) is a free radical synthesized by nitric oxide synthases (NOS). NOSI and NOSIII are expressed constitutively and produce trace amounts of NO. In contrast, NOSII is the inducible isoform and can generate large amounts of NO. At high concentrations, such as those produced by NOSII, NO is cytotoxic, and can prevent tumors from developing or metastasizing (1,2). Transgenic studies indicated that lymphomas developed more rapidly in NOSII-null p53- knockout mice than in NOSII-wild-type p53-knockout mice (3,4). NOSII-null fibrosarcoma tumor cell lines grew faster and produced more lung metastases than NOSII-wild-type cell lines. EMT-6J murine breast carcinoma cells, which constitutively express NOSII, were less tumorigenic than NOSII-deficient EMT-6H cells (5). Human L3.3 pancreatic cancer cells transduced with the wild-type NOSII gene, which produced high levels of NO, did not form tumors or metastases in xenograft nude mouse models (6). On the other hand, L3.3 pancreatic cancer cells transduced with adenoviral vectors expressing mutant NOSII genes, which produced low levels of NO, formed tumors and metastases in nude mice (6).
We previously demonstrated that breast cancer cells possess intrinsic resistance mechanisms that can prevent the induction of NOSII (7,8); any therapeutic strategy designed to produce high NO levels in such cells should therefore not depend on NOSII induction. Given the suppressive effects of high levels of NO on tumorigenesis, drugs that supply NO exogenously could have potential in breast cancer therapy. The challenge is to deliver NO in a sustained and controlled manner so as to avoid NO-mediated systemic hypotension.
JS-K (O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl) piperazin-1-yl]diazen-1-ium-1,2-diolate) belongs to the diazeniumdiolate class of prodrug that is designed to release NO when metabolized by glutathione S-transferases (GSTs). GSTs are a superfamily of enzymes responsible for the detoxification of many xenobiotics by conjugating them to glutathione and increasing their cellular excretion. Among the major GST isoforms (α·, μ, π), GST-π is expressed at the highest concentration in breast tumors (9,10). High levels of GST-π have been associated with more aggressive phenotypes of breast cancer, increased risk of local relapse, and decreased disease-free survival in breast cancer patients (11–13). Since JS-K requires GST for its optimal activity, it can potentially turn GST overexpression to the tumor’s disadvantage by generating high intracellular concentrations of NO specifically within tumor cells. JS-K has been shown to have anti-tumor activity in both in vitro and in vivo models of prostate cancer, leukemia, liver cancer, and multiple myeloma (14–16). However, the effects of JS-K on the viability of breast cancer cells and normal breast epithelial cells have not been characterized.
In this study, we determined the effects of JS-K on the viability of breast cancer cells and normal mammary epithelial cells. Here we report that JS-K decreased the viability of breast cancer cell lines but hardly affected the viability of normal mammary epithelial cells. Furthermore, JS-K selectively induced programmed cell death in breast cancer cells but not in normal mammary epithelial cells.
Materials and methods
Cell lines and culture conditions.
MCF-7, MDA-MB-231, SKBr3, MDA-MB-453, and MDA-MB-468 human breast cancer cell lines were obtained from American Type Cell Culture (ATCC, Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (Invitrogen Corp., Carlsbad, CA) supplemented with 5% heat-inactivated fetal bovine serum (FBS) (Invitrogen). MCF-10A human mammary epithelial cells, originated from spontaneous immortalization of breast epithelial cells obtained from a patient with fibrocystic disease, were obtained from ATCC and cultured in DMEM/F12 medium supplemented with 10% heat-inactivated FBS, 20 ng/ml epidermal growth factor, 20 ng/ml insulin-like growth factor-I, and 500 ng/ml hydrocortisone. MCF-10AT and MCF-10CA1a cells, which are MCF-10A sublines, were obtained from Dr Fred Miller (Karmanos Cancer Institute, Detroit, MI) and cultured in DMEM/F12 medium supplemented with 10% heat-inactivated FBS. MCF-10AT cells were created by stable transfection of MCF-10A cells with the activated c-Ha-ras oncogene (17). MCF-10A cells do not survive in vivo in immunodeficient mice. However, MCF-10AT cells form small nodules in nude/beige mice that persist for at least 1 year and sporadically progress to carcinomas (17). MCF-10CA1a cells, derived from MCF-10AT1 xenograft, form carcinomas in nude/beige mice with 100% efficiency (18). Normal human mammary epithelial cells (HMECs), which are primary cells isolated from a healthy woman donor, were obtained from Cambrex (Rockland, MA) and cultured in mammary epithelial growth medium supplemented with bovine pituitary extract (Cambrex). To ensure that these primary cells are mammary epithelial cells, the company uses immunofluorescent staining technique to confirm that the mammary epithelial cells stain positive for cytokeratins 14 and 18 and negative for cytokeratin 19.
Cell viability assay.
Mammary epithelial cells and breast cancer cells (with the exception of MDA-MB-468 cells) were plated in 96-well plates in 0.1 ml of their respective media. MDA-MB-468 cells were plated in the HMEC medium (Cambrex). Cells were plated in different cell densities because the cells have different size and growth rate. MCF-7 and MDA-MB-231 cells were plated at 1,000 cells per well. SKBr3 and MDA-MB-453 cells were plated at 2,500 cells per well. MDA-MB-468, MCF-10A, MCF-10AT, MCF-10CA1a, and HMEC cells were plated at 7,500 cells per well. The next day, JS-K, its metabolites (DNP-SG, DNP, JS-9-29, or piperazine, structures in Fig. 1) or its non-NO-releasing, non-arylating control (JS-43-126) were diluted from their DMSO stock solutions, and added to cells (within 30 min of its dilution) at final concentrations of 1, 5, and 10 μM. After 3 days of incubation, cell viability was determined by the Promega (Madison, WI) Celltiter 96® AQueous non-radioactive cell proliferation assay. The CellTiter 96 AQueous Assay is composed of tetrazolium 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and the electron coupling reagent phenazine methosulfate (PMS). MTS is reduced by live cells into a formazan product, whose absorbance can be read at 490 nm. The quantity of formazan product is directly proportional to the number of living cells in culture. The absorbance of the formazan product is typically read within 1–2 h after the MTS/PMS dye addition. Cell viability was expressed as percentage of untreated cells.
Figure 1.
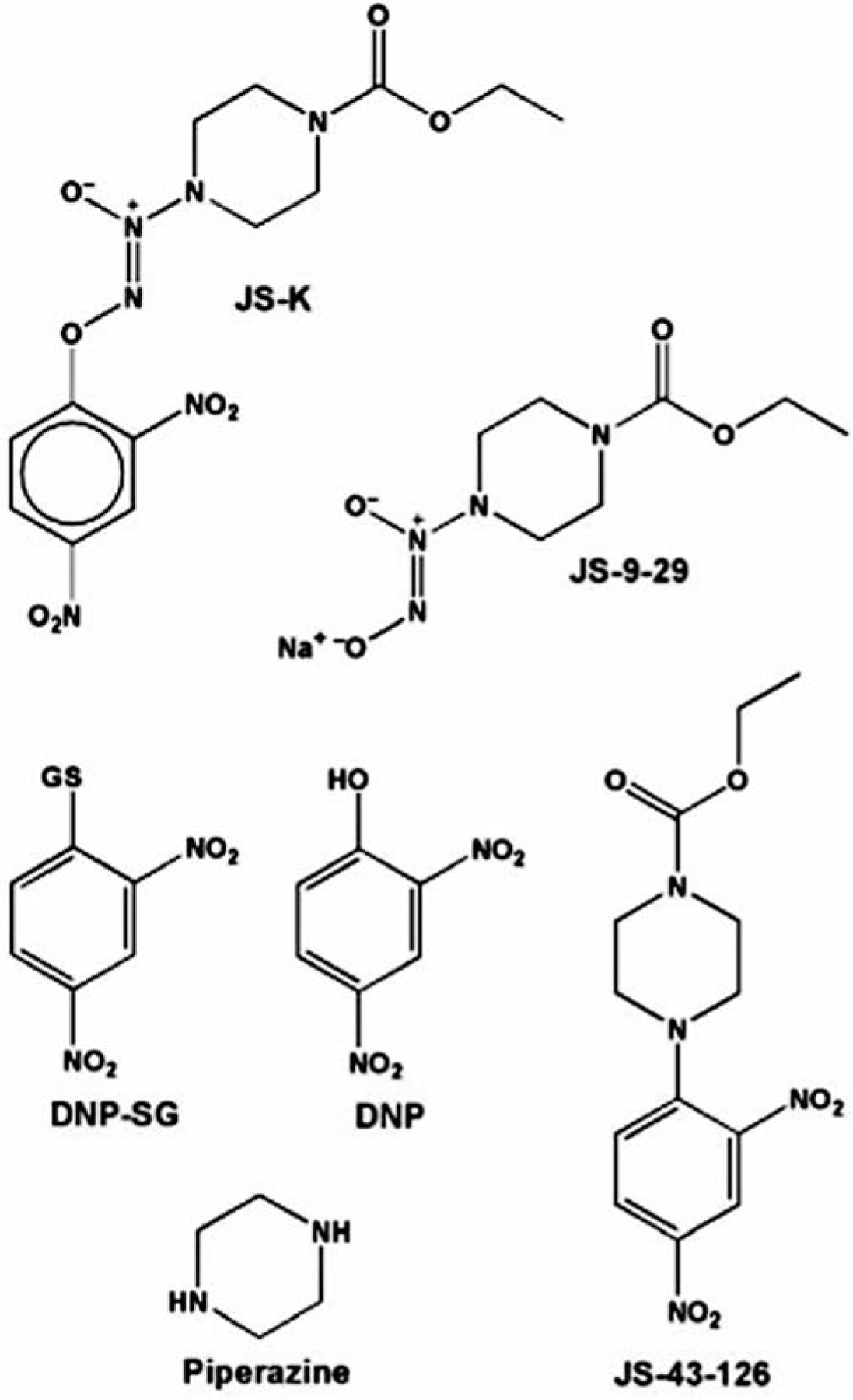
Structures of JS-K, four of its presumed metabolites (JS-9-29, DNP-SG, DNP, and piperazine), and a non-NO-releasing, non-arylating control (JS-43-126). SG and GS represent the S-substituted glutathionyl residue.
Apoptosis analysis.
The effect of JS-K on apoptosis was analyzed by flow cytometry as described (8,19). Breast cancer cells were plated at 1×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 5% FBS. MCF-10A cells were plated at 4×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 10% FBS, 20 ng/ml epidermal growth factor, 20 ng/ml insulin-like growth factor-I, and 500 ng/ml hydrocortisone. Cells were untreated or treated with 5 μM JS-K for 3 days. Floating cells and adherent cells were collected, washed, and resuspended in 1 ml of PBS. The cell suspension was added to 1 ml of cold 70% ethanol and incubated overnight at −20°C. Cells were centrifuged at 1500 rpm for 10 min at 4°C and then washed twice in PBS, and the pellet was left loose. Approximately 0.5–1 ml of PBS containing RNase (20 μg/ml) and propidium iodide (50 μg/ml) was added to each cell pellet, followed by 20 min of incubation at room temperature. Flow cytometric analysis was performed using a Coulter Epics Profile 488 laser.
Electron microscopy.
To morphologically demonstrate the induction of autophagy in JS-K-treated cancer cells, ultrastructural analysis was performed as described previously (20). MDA-MB-231 breast cancer cells, plated at 1×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 5% FBS, were untreated or treated with 5-μM JS-K for 3 days. Cells were washed with serum-free medium before being fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), post-fixed in 1% OsO4 in the same buffer, and then analyzed by electron microscopy. Representative areas were chosen for ultrathin sectioning and viewed with a Hitachi electron microscope.
Electrophoresis and immunoblotting.
Fifty μg of protein lysates from untreated, JS-K-treated, or JS-43-126-treated cells were loaded on polyacrylamide gels (Bio-Rad, Hercules, CA). Proteins were electrophoresed and electrotransferred to nitrocellulose membranes (Bio-Rad) with the use of a wet transfer apparatus (Bio-Rad) for ~24 h at 22 V. The membranes were incubated in blocking buffer (1X Tris-buffered saline, 0.1% Tween-20, and 5% non-fat dry milk) for 30 min at room temperature and then incubated overnight with polyclonal antibodies specific for microtubule-associated light chain 3 (LC3) protein (1:1000 dilution). The LC-3 antibody was raised against a synthetic peptide corresponding to the N-terminus of LC3B (20,21). After washing in 1X Tris-buffered saline containing 0.1% Tween-20, membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (1:1000 dilution) (Cell Signaling Technology) for 2 h at room temperature. Monoclonal antibodies specific for ß-actin (1:10000 dilution) (Sigma Chemical Co., St. Louis, MO) were used to ensure equal loading. Protein bands were visualized by enhanced chemiluminescence (Kirkgaard & Perry Laboratories, Gaithersburg, MD). Images were scanned using an Alpha Imager and its application program (Alpha Innotech, San Leandro, CA).
Detection and quantification of acidic vesicular organelles.
The acridine orange dye was used as described previously (20) to detect and quantify acidic vesicular organelles in JS-K-treated cells. Breast cancer cells (with the exception of MDA-MB-468 cells) were plated at 1×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 5% FBS. MDA-MB-468 cells and normal HMECs were plated at 4×105 cells per well in 6-well plates in 2 ml of HMEC medium. MCF-10A cells were plated at 4×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 10% FBS, 20 ng/ml epidermal growth factor, 20 ng/ml insulin-like growth factor-I, and 500 ng/ml hydrocortisone. Cells were treated with JS-K (0, 1.0, 1.5, 2.5, or 5.0 μM) or JS-43-126 (0, 1, 5 μM) for 3 days. Breast cancer cells were stained with acridine orange at a final concentration of 1 μg/ml for 15 min at 37°C, removed from the plate with trypsin-EDTA, and collected in their culture medium. Green (510–530 nm) and red (>650 nm) fluorescence emission from 1×104 cells illuminated with blue (488 nm) excitation light was measured with a FACSCalibur from Becton-Dickinson using CellQuest software. FL1-H (x-axis) indicates green color intensity, while FL3-H (y-axis) indicates red color intensity. Percentage of acridine orange-stained positive cells is the sum of percentage of cells in the upper left and the upper right quadrants.
Breast cancer cells were plated in triplicates in 2-well, 4.0-cm2 chamber slides (Fisher Scientific Inc., Hampton, NH) at 3×104 cells per well in 2 ml of DMEM/F12 medium containing 5% FBS. Transfection with the LC3 expression vector which was tagged with green fluorescent protein (GFP) (20) was performed with 1 μg plasmid per chamber with 3 μl FuGene 6 Transfection Reagent (Roche, Indianapolis, IN) according to the manufacturer’s instructions. The next day, cells were treated without or with JS-K (5 μM). After 3 days of treatment, cells were fixed with 4% paraformaldehyde and counted in five fields for each chamber slide under a fluorescence microscope at magnification x10.
Statistical analysis.
The cell viability data are expressed as the means ± standard deviation. For statistical analysis of the cell viability experiments, the Shapiro-Wilk test was first performed to assess the normality assumption of the data. Given that the data were normally distributed, two-tailed Student’s t-test was performed to compare the viability of untreated cells and JS-K-treated cells. Criterion for statistical significance was taken as P<0.05. All analyses were performed using SAS®.
Results
JS-K is more cytotoxic to breast cancer cells than to normal mammary epithelial cells.
Breast cancer cells and normal HMECs were incubated with JS-K. JS-K decreased the viability of all breast cancer cell lines tested (Fig. 2 top panel). At 1-μM concentration, JS-K decreased the viability of MCF-7 cells by 67%, MDA-MB-231 cells by 57%, SKBr3 cells by 73%, and MDA-MB-453 cells by 74% (Fig. 2 top panel). On the contrary, JS-K, at 10-μM concentration, did not affect the viability of the HMECs (Fig. 2 top panel). To ensure that this selective cytotoxic effect is not due to different cell density or different medium used, MDA-MB-468 breast cancer cells were plated at the same cell density in the same medium as the HMECs. Under these conditions, JS-K remained to be effective in decreasing the viability of MDA-MB-468 cells (Fig. 2 top panel). At 1- and 5-μM concentrations, JS-K decreased the viability of MDA-MB-468 cells by 39 and 72%, respectively (Fig. 2 top panel).
Figure 2.
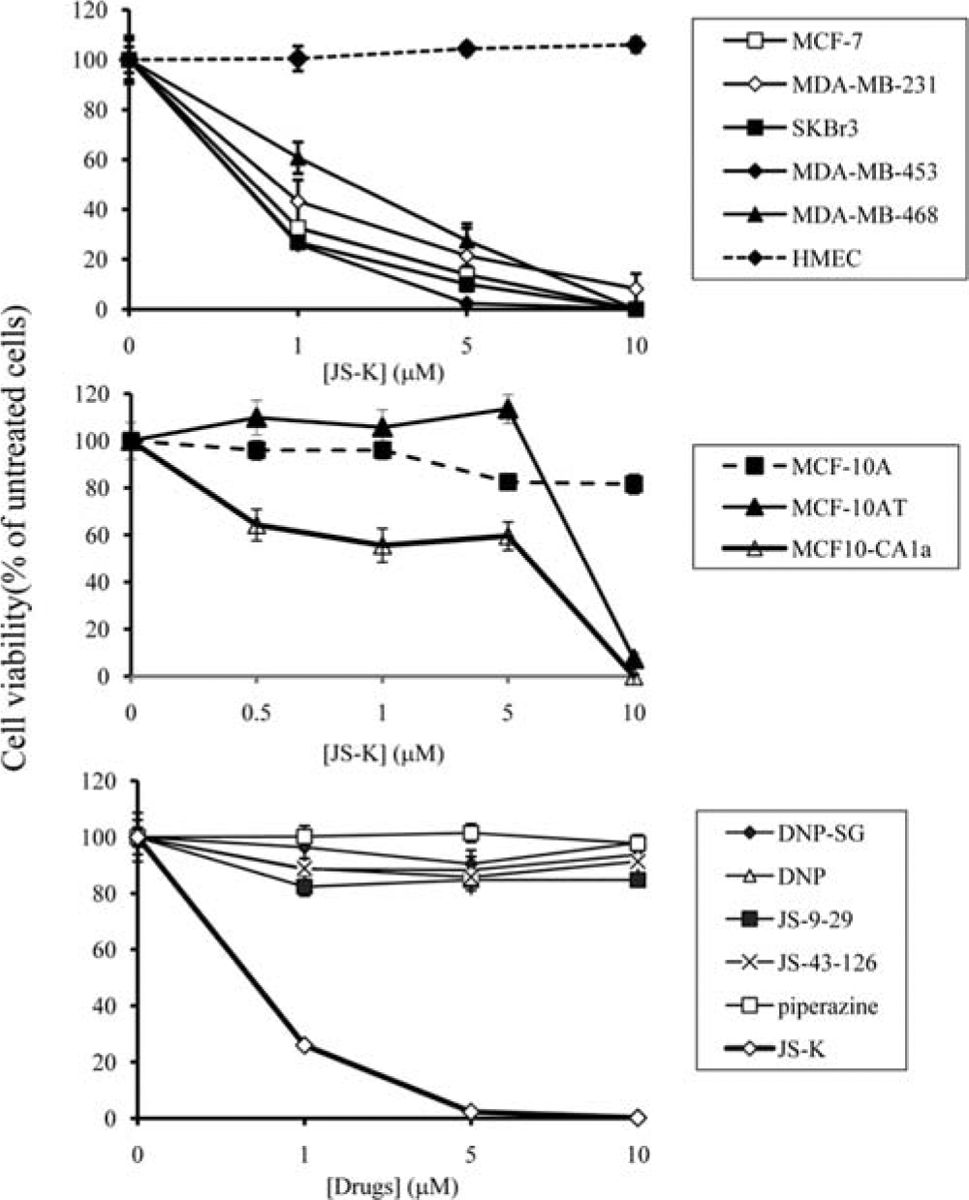
JS-K decreases the viability of breast cancer cells but not that of normal mammary epithelial cells. Breast cancer cells and normal HMECs were incubated with increasing concentrations (1, 5, 10 μM) of JS-K for 3 days (top panel). Cell viability was determined by the Promega Celltiter 96 AQueous non-radioactive proliferation assay. Data points represent the mean of five replicates. Data are expressed as mean percentage of untreated cells ± standard deviation. Non-transformed MCF-10A, premalignant MCF-10AT and fully malignant MCF-10CA1a mammary epithelial cells were incubated with increasing concentrations (1, 5, 10 μM) of JS-K for 3 days (middle panel). Cell viability was determined by the Promega Celltiter 96 AQueous non-radioactive proliferation assay. Data points represent the mean of five replicates. Data are expressed as mean percentage of untreated cells ± standard deviation. MDA-MB-453 breast cancer cells were treated with JS-K, its presumed metabolites (DNP-SG, DNP, JS-9-29, and piperazine), and a non-NO-releasing, non-arylating control (JS-43-126) for 3 days (bottom panel). Cell viability was determined by the Promega Celltiter 96 AQueous non-radioactive proliferation assay. Data points represent the mean of five replicates. Data are expressed as mean percentage of untreated cells ± standard deviation.
Furthermore, the cytotoxic activity of JS-K was assessed in the MCF-10A series of cell lines, which includes cell lines representing non-malignant immortalized (MCF-10A), premalignant (MCF-10AT), and fully malignant (MCF-10CA1a) mammary epithelial cells. All these cell lines were derived from a single patient, thus all are on a common genetic background. These cell lines are useful tools to investigate different stages in the breast cancer progression process (18). The non-malignant MCF-10A mammary epithelial cells were essentially resistant to JS-K; at 10-μM concentration, JS-K decreased the viability of MCF-10A cells by only 18% (Fig. 2 middle panel). However, at 1-μM concentration, JS-K decreased the viability of the fully malignant MCF-10CA1a cells by ~50% (Fig. 2 middle panel). JS-K did not decrease the viability of the premalignant MCF-10AT cells until >5-μM concentration was used (Fig. 2 middle panel). These data indicate that JS-K had potent cytotoxic effects against malignant breast cancer cells, with moderate effects against premalignant breast cells and minimal effects on non-malignant mammary epithelial cells.
The presumed metabolites of JS-K, such as DNP-SG, DNP, JS-9-29, and piperazine, and the non-NO-releasing control JS-43-126 (22) did not have any effect on the viability of MDA-MB-453 cells (Fig. 2 bottom panel) and MDA-MB-231 cells (not shown). Thus, the cytotoxic effects of JS-K are attributed mainly to its ability to produce NO and/or its arylating ability.
JS-K induces programmed cell death in breast cancer cells.
Apoptosis was reported to be the main mechanism by which JS-K exerts its toxic effects in leukemia, hepatoma and multiple myeloma cells (14,16,23,24). Flow cytometric analysis of propidium iodide-stained sub-G1 cells was used to quantify the extent of apoptosis induced by JS-K in breast cancer cells and in the non-malignant, immortalized MCF-10A mammary epithelial cells. Cells were untreated or treated with 5-μM concentration of JS-K. JS-K increased apoptosis from 1.4 to 13.6% in MCF-7 cells and from 0.5 to 7.6% in MDA-MB-231 cells. JS-K increased the percentages of SKBr3 apoptotic cells from 0.4% to 3.0%, and the percentages of MDA-MB-453 apoptotic cells from 0.5 to 5.4%. But JS-K did not affect the percentages of apoptotic MCF-10A cells; 0.8 and 1.9% for untreated and JS-K-treated MCF-10A cells, respectively. These data indicate JS-K selectively induced apoptosis in breast cancer cells.
Previously, using the same apoptosis quantification assay, Shami et al (14) and Kiziltepe et al (16), showed that a 1-μM concentration of JS-K increased HL-60 and MM.1S apoptotic cells by 43 and 60%, respectively. It appears that JS-K-induced apoptosis was more potent in leukemia and myeloid cells than in breast cancer cells. We speculate that other mechanisms, such as autophagy, may be involved in the cytotoxic effects of JS-K in breast cancer cells. Autophagic cell death is the process of sequestrating cytoplasmic proteins into the lytic compartment and characterized by the formation and promotion of acidic vesicular organelles. To determine if JS-K increased the formation of acidic vesicular organelles in breast cancer cells, cells were stained with the acridine orange dye as described (20). In acridine orange-stained cells, the cytoplasm and nucleolus fluoresce bright green and dim red, whereas acidic compartments fluoresce bright red. The intensity of the red fluorescence is proportional to the degree of acidity and/or the volume of the cellular acidic compartment. Therefore, we could measure a change in the degree of acidity and/or the fractional volume of their cellular acidic compartment. At 5-μM concentration, JS-K increased the percentage of acridine orange-stained SKBr-3 cells from 4.8 to 45.8% (Fig. 3). JS-K increased the percentages of acridine orange-stained cells in all breast cancer cell lines tested (Table I). On the other hand, JS-43-126, the non-NO-releasing control (22), did not increase the percentages of acridine orange-stained cells (Table I). Furthermore, JS-K hardly affected the formation of acidic vesicular organelles in HMECs or MCF-10A cells; at 5-μM concentration, JS-K increased the percentages of acridine orange-stained HMECs and MCF-10A cells only by 1.3% and 2.2%, respectively (Fig. 3 and Table I).
Figure 3.
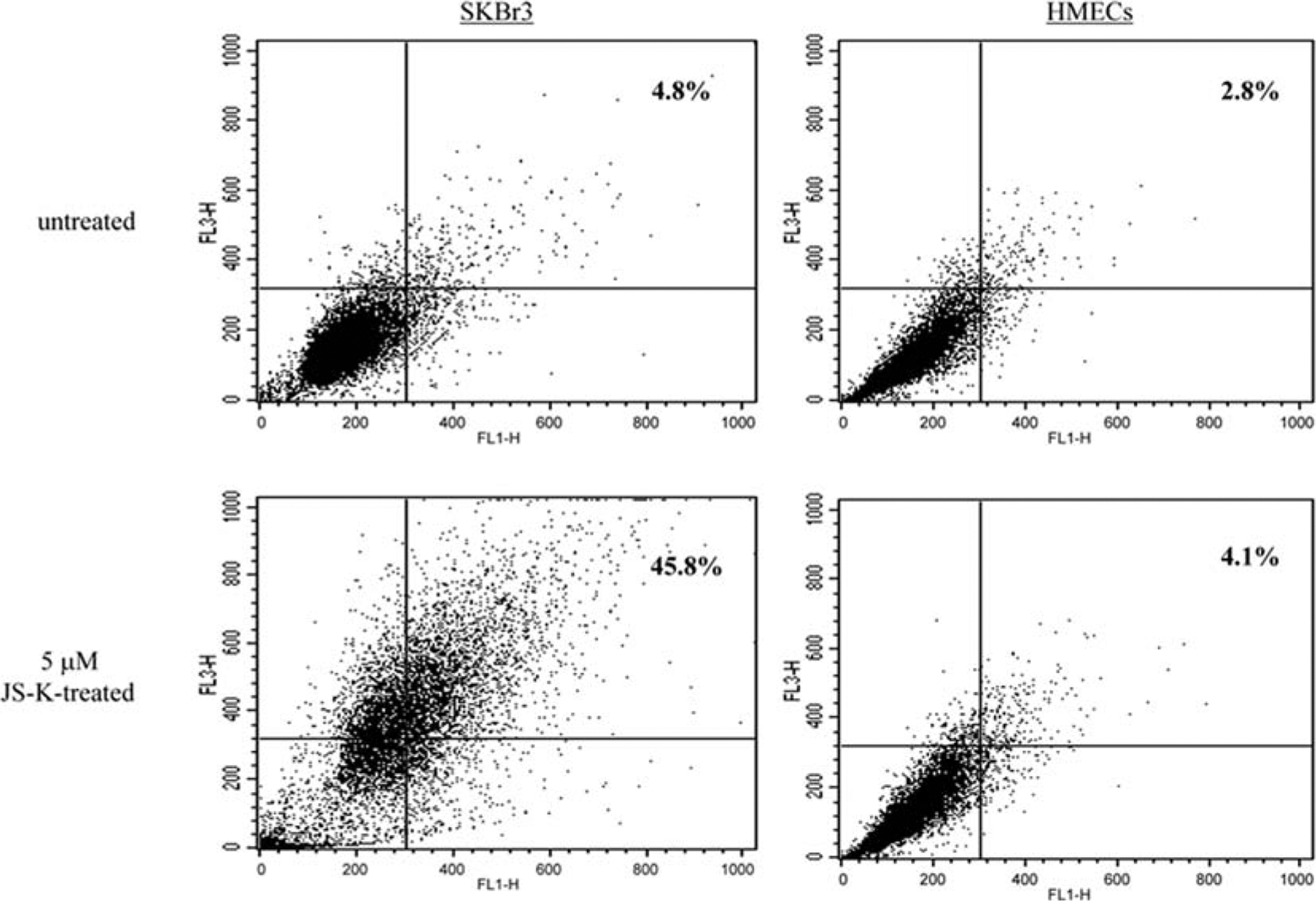
JS-K increases the formation of acidic vesicular organelles in breast cancer cells but not in normal mammary epithelial cells. SKBr-3 breast cancer cells and normal HMECs were incubated with JS-K (0, 5 μM) for 3 days, and stained with the acridine orange dye. FACS analysis was used to detect the green and the red fluorescence in the acridine orange-stained cells. FL1-H (x-axis) indicates green color intensity, while FL3-H (y-axis) shows red color intensity. Percentage of acridine orange-stained positive cells is the sum of percentage of cells in the upper left and the upper right quadrants.
Table I.
JS-K increases the formation of acidic vesicular organelles in breast cancer cells but not in non-transformed or normal mammary epithelial cells.
| Cells | JS-K (μM) | Acridine orange-stained positive cells (%) |
|---|---|---|
| MCF-7 | 0 | 4.7 |
| 1 | 19.3 | |
| 5 | 29.2 | |
| MDA-MB-231a | 0 | 3.1 |
| 1 | 13.6 | |
| 5 | 39.6 | |
| SKBr-3a | 0 | 4.8 |
| 1 | 29.7 | |
| 5 | 45.8 | |
| MDA-MB-453a | 0 | 4.4 |
| 1 | 17.9 | |
| 5 | 41.1 | |
| MDA-MB-468b | 0 | 2.6 |
| 5 | 18.4 | |
| HMECb | 0 | 2.8 |
| 5 | 4.1 | |
| MCF-10Ac | 0 | 7.1 |
| 5 | 9.3 | |
| Cells | JS-43-126 (μM) | Acridine orange-stained positive cells (%) |
| MDA-MB-231a | 0 | 3.8 |
| 5 | 3.5 | |
| MDA-MB-453a | 0 | 2.2 |
| 5 | 4.5 |
Breast cancer cells, plated at 1×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 5% FBS, were treated with JS-K for 3 days and stained with acridine orange. The red fluorescence in the acridine orange-stained cells was detected by flow cytometry.
MDA-MB-468 and HMEC cells, plated at 4×105 cells per well in 6-well plates in 2 ml of HMEC medium containing bovine pituitary extract, were treated with JS-K for 3 days and stained with acridine orange. The red fluorescence in the acridine orange-stained cells was detected by flow cytometry.
MCF-10A cells, plated at 4×105 cells per well in 6-well plates in 2 ml of DMEM/F12 medium containing 10% FBS, 20 ng/ml epidermal growth factor, 20 ng/ml insulin-like growth factor-I, and 500 ng/ml hydrocortisone, were treated with JS-K for 3 days and stained with acridine orange. The red fluorescence in the acridine orange-stained cells was detected by flow cytometry.
To confirm that JS-K induces autophagy in breast cancer cells, electron microscopy was used. Untreated MDA-MB-231 breast cancer cells exhibited few autophagic features (Fig. 4). However, numerous empty vacuoles and autophagic vacuoles were observed in MDA-MB-231 cells treated with 5-μM JS-K (Fig. 4). Most of the autophagic vacuoles contained intact cytoplasmic structure, lamellar structure, or residual digested materials. Together, these data indicate that JS-K induces autophagy in breast cancer cells.
Figure 4.
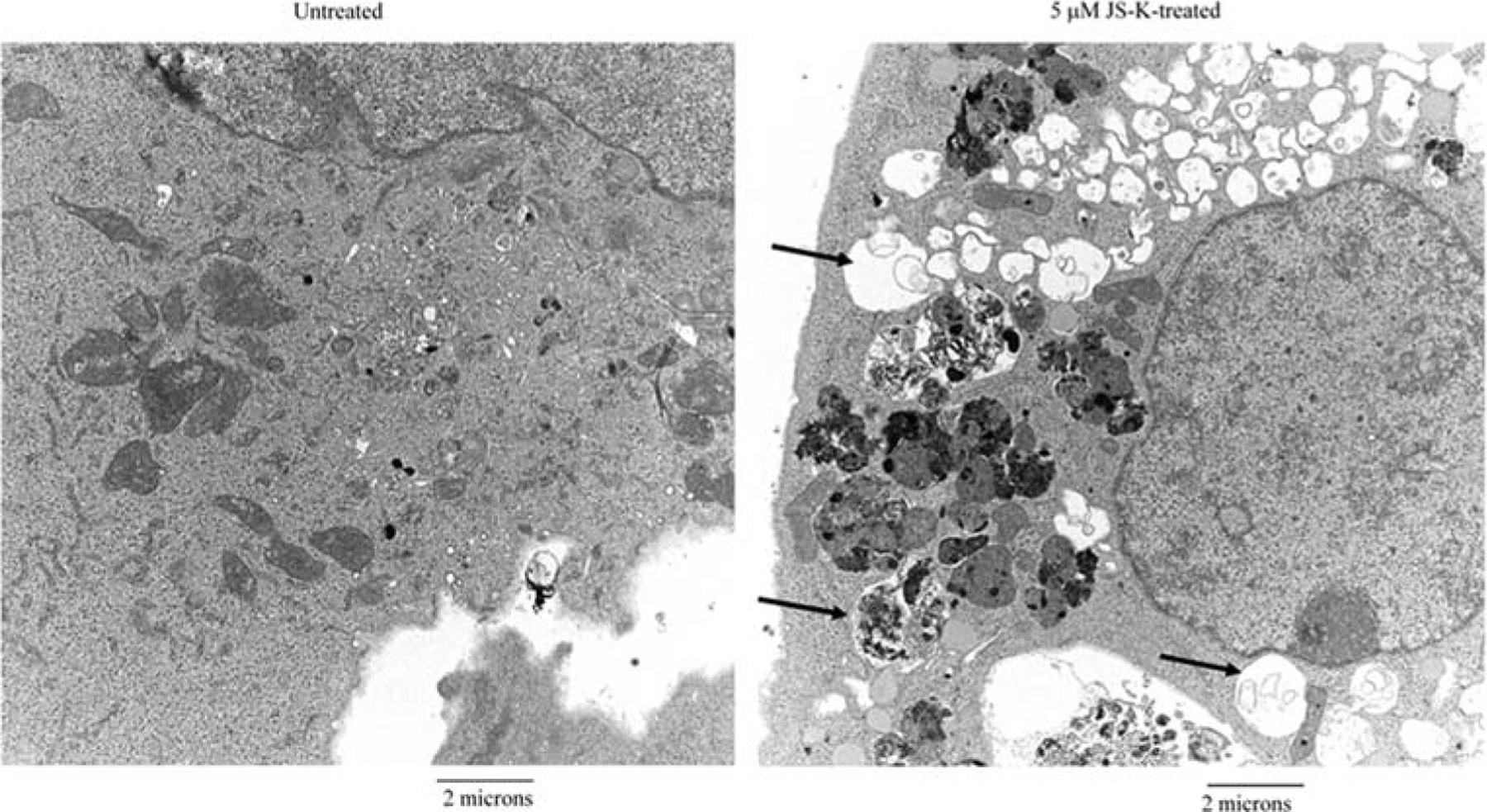
Ultrastructural features of JS-K-treated breast cancer cells. MDA-MB-231 cells treated with 0 or 5 μM JS-K for 3 days were harvested and fixed. Representative areas were chosen for ultrathin sectioning and viewed with a Hitachi electron microscope. Arrows indicate autophagic vesicles in JS-K-treated breast cancer cells.
There are two forms of LC3: LC3-I and LC3-II (25). LC3-I (18 kDa) is cytoplasmic, whereas LC3-II (16 kDa) is membrane-bound and essential for autophagic vesicle formation. To further confirm that JS-K induced autophagy in breast cancer cells, the effect of JS-K on LC3-II expression was examined. The expression of LC3-II protein was very low in untreated MDA-MB-453 breast cancer cells and untreated HMECs (Fig. 5A). JS-K, at 1- and 5-μM concentrations, increased the expression of LC3-II in MDA-MB-453 breast cancer cells by ~2-fold (Fig. 5A). On the other hand, JS-K did not increase the expression of LC3-II in HMECs (Fig. 5A). GFP-tagged LC3 was also used to study the autophagosome membrane association of LC3 in JS-K-treated cells. In untreated MDA-MB-231 cells, the fluorescent intensity of GFP-LC3 was very low, and GFP-LC3 was distributed in a diffuse pattern (Fig. 5B). In JS-K-treated MDA-MB-231 cells, the fluorescent intensity of GFP-LC3 was high, and GFP-LC3 was distributed in a punctate pattern (Fig. 5B). The number of cells with GFP-LC3 dots was counted. The percentage of untreated breast cancer cells with GFP-LC3 dots was <1% (Fig. 5C). JS-K increased the number of GFP-LC3 dots in breast cancer cells (Fig. 5C). The percentages of MDA-MB-231, SKBr3 and MDA-MB-453 cells with GFP-LC3 dots were 45, 19 and 58, respectively (Fig. 5C). However, JS-K did not increase the percentages of MCF-10A with GFP-LC3 dots (Fig. 5C). Together, these results indicate that JS-K selectively induces apoptosis and autophagy in breast cancer cells, but not in normal mammary epithelial cells.
Figure 5.
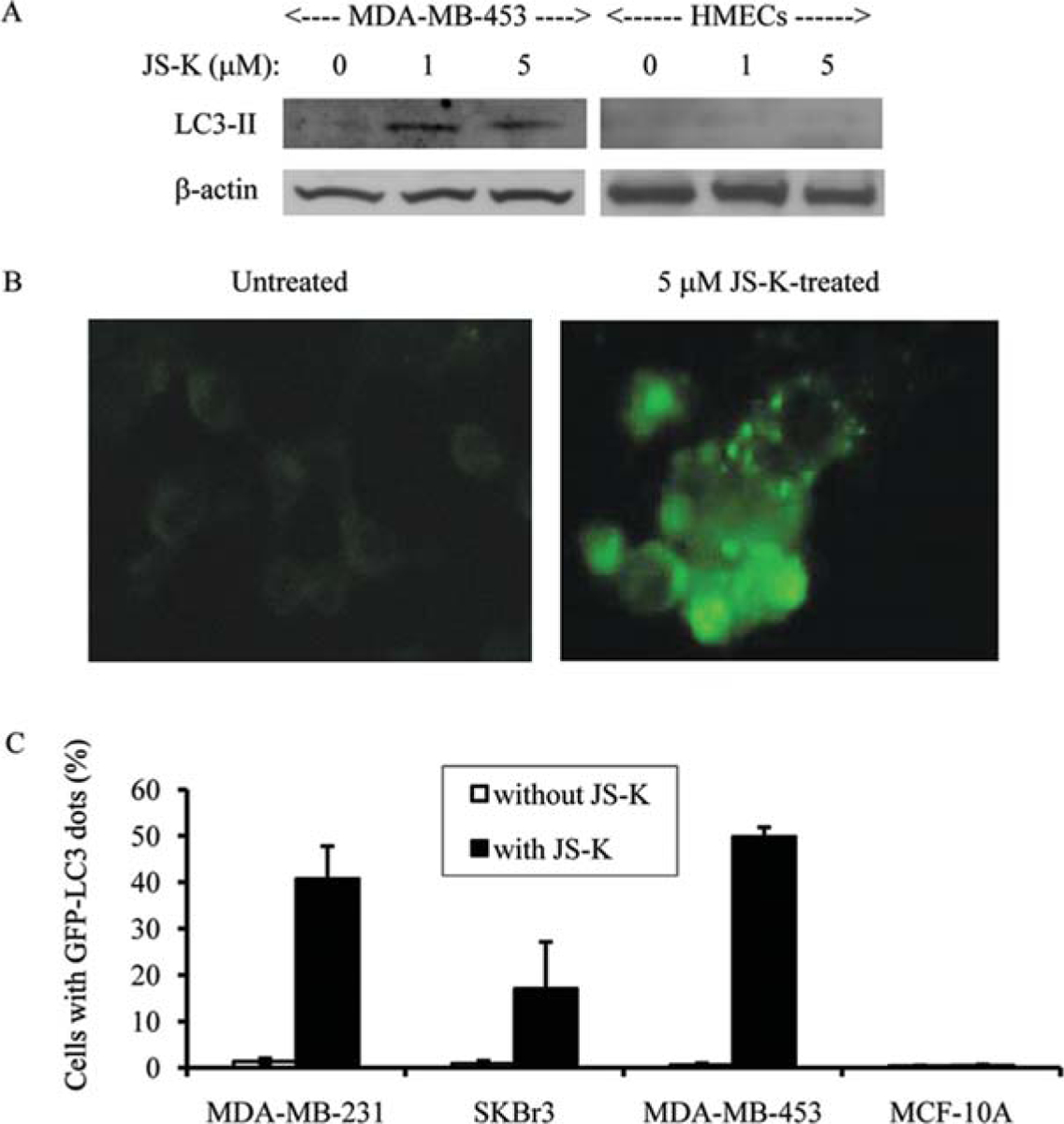
JS-K selectively increases the expression of LC3-II in breast cancer cells. MDA-MB-453 breast cancer cells and normal HMECs were incubated with JS-K (0, 1, 5 μM) for 3 days (A). Membranes were incubated with anti-LC3 antibody. ß-actin was used as a loading control. Protein bands were visualized by enhanced chemiluminescence. Distribution of GFP-LC3 in MDA-MB-231 cells treated with 0 or 5 μM JS-K for 3 days (B). The GFP-LC3 cDNA vector was transfected to MDA-MB-231 cells and treated with JS-K. The cells were observed under a fluorescent microscope. Quantification of breast cancer cells and MCF-10A mammary epithelial cells with characteristic GFP-LC3 dots that are indicative of autophagy (C). The proportion of the cells with GFP-LC3 dots was assessed in 200 GFP-positive cells in five different fields. Values are the means of experiments performed in triplicate; bars, SD.
JS-K decreases heme oxygenase-1 expression.
The enzyme activity of heme oxygenase-1 (HO-1) may play a role in the basic resistance of mammalian cells to NO-mediated cytotoxicity (26). Primary cultures of spinal motor neurons and glia obtained from homozygous HO1-null mice are much more sensitive to NO cytotoxicity than are cells which express either one or two copies of a functional HO1 gene (26). In rats, HO-1 is expressed in the epithelial cells of the normal breast gland (27) but, during N-nitrosomethylurea-induced malignant transformation, HO-1 expression is decreased in the transformed breast cells (27). It is possible that HO-1 plays a role in defending against JS-K-mediated cytotoxicity in normal human mammary epithelial cells. We first investigated the expression of HO-1 in MCF-10A mammary epithelial cells and MDA-MB-231 breast cancer cells. The expression level of HO-1 appears to be slightly higher in MDA-MB-231 breast cancer cells than MCF-10A mammary epithelial cells (Fig. 6A). Next we investigated the effects of JS-K on HO-1 expression in MCF-10A and MDA-MB-231 cells. At 5-μM concentration, JS-K decreased HO-1 expression in MDA-MB-231 breast cancer cells by ~54% (Fig. 6A). However, JS-K, at 5-μM concentration, did not affect HO-1 expression in MCF-10A cells (Fig. 6A). We then determined whether inhibition of HO-1 by tin protoporphyrin (SnPP), a potent inhibitor of HO-1 (28), could increase the sensitivity of MCF-10A cells to JS-K. Although SnPP, at 12-μM concentration, inhibited HO-1 expression by 72% (Fig. 6B), it did not affect the sensitivity of MCF-10A cells to JS-K (Fig. 6C). These data indicate that JS-K could selectively decrease HO-1 expression in breast cancer cells. However, HO-1 appears to have a minor role in regulating JS-K resistance in normal mammary epithelial cells.
Figure 6.
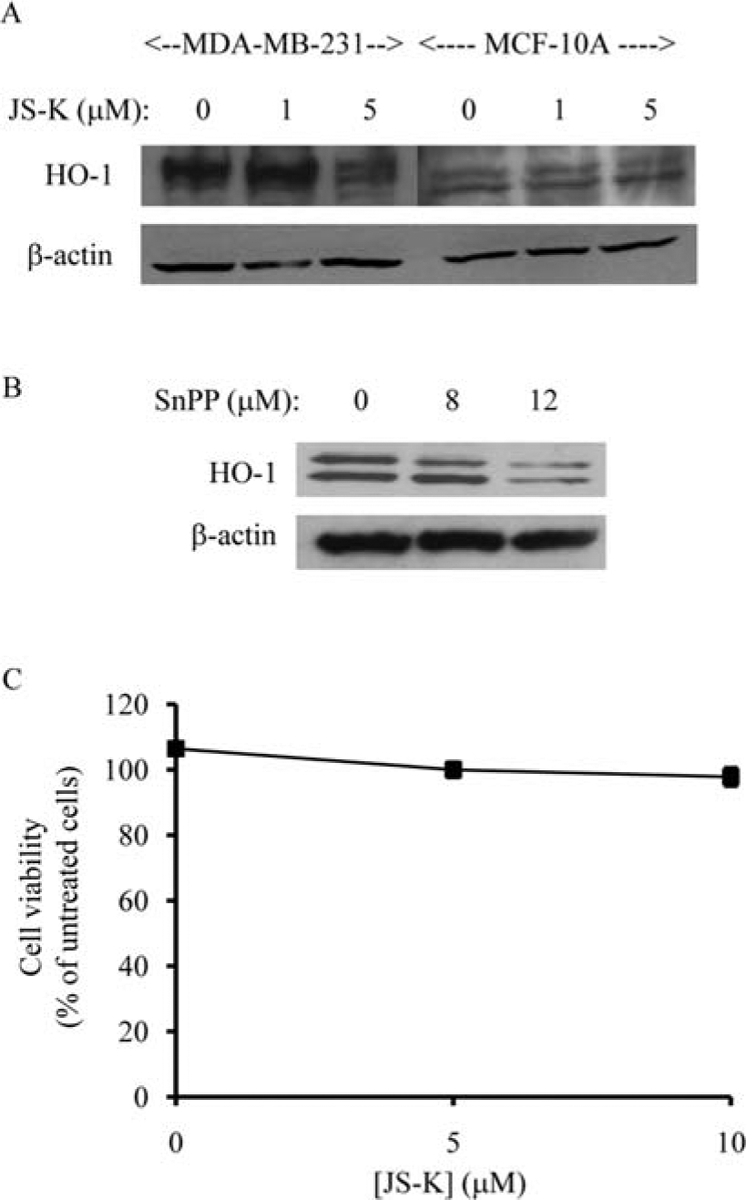
JS-K selectively decreases HO-1 expression in breast cancer cells. MDA-MB-231 breast cancer cells and MCF-10A mammary epithelial cells were treated with 0, 1, 5 μM of JS-K for 4 h (A). Membranes were incubated with anti-HO-1 antibody. ß-actin was used as a loading control. Protein bands were visualized by enhanced chemiluminescence. MCF-10A mammary epithelial cells were incubated with 0, 8, 12 μM of SnPP (HO-1 inhibitor) for 24 h (B). Membranes were incubated with anti-HO-1 antibody. ß-actin was used as a loading control. Protein bands were visualized by enhanced chemiluminescence. MCF-10A cells were preincubated with 12 μM of SnPP for 24 h before being treated with JS-K (0, 5, 10 μM) for 3 days (C). Cell viability was determined by the Promega Celltiter 96 AQueous non-radio-active proliferation assay. Data points represent the mean of five replicates. Data are expressed as the mean percentage of untreated cells ± standard deviation.
Discussion
Here we demonstrate that JS-K induces cytotoxicity in breast cancer cells. More importantly, we found that JS-K has minimal inhibitory effects on the normal HMECs or the non-malignant mammary epithelial cells. Thus, JS-K appears to have a selective cytotoxic effect against breast cancer cells. JS-K’s cytotoxic effect has mainly been attributed to its ability to release high levels of NO upon conjugation with glutathione by GST enzymes (14). Our observed selective cytotoxic effects of JS-K seem to support previous observation that oncogene-transformed fibroblasts, but not non-transformed fibroblasts, are sensitive to NO-mediated cell death (29). It is possible that normal cells express higher levels of enzymes, which are vital to the cellular defense against NO-mediated cytotoxicity, than malignant cells, thereby allowing NO to induce selective cytotoxicity against malignant cells. It has been suggested that HO-1 plays a vital role in the basic resistance of mammalian cells to NO-mediated cytotoxicity (27). Here we showed that JS-K decreased HO-1 expression in breast cancer cells while non-transformed mammary epithelial cells were resistant to such JS-K effect. However, reducing HO-1 expression did not sensitize the non-transformed cells to JS-K. HO-1 appears to play a minor role in conferring JS-K resistance in mammary epithelial cells. Other components of the JS-K molecule have been reported to contribute to its cytotoxicity. JS-K is capable of arylating glutathione and other cellular nucleophiles, irreversibly altering their normal function (30). Mammary epithelial cells may have high levels of enzymes that could defend the cells from cytoxicity induced by arylation, thereby allowing the cells to be resistant to JS-K.
Apoptosis was the main mechanism by which JS-K induces cytotoxicity in HL-60 leukemia cells, Hep 3B hepatoma cells and MM.1S multiple myeloma cells (14,16,31). Here we demonstrate that JS-K induced apoptosis and autophagy in breast cancer cells. Various cancer therapies, such as radiation, rapamycin, arsenic trioxide, ceramide, etoposide, tamoxifen, and resveratrol, have been shown to induce apoptotic as well as autophagic cell death (32). Furthermore, apoptosis and autophagy appear to be interconnected; the phosphoinositol-3 kinase family (33–36) and the Bcl-2 family (37,38) have been shown to regulate both of these processes.
In conclusion, JS-K uses two programmed cell death mechanisms to selectively kill breast cancer cells. JS-K holds great promise as a novel therapeutic agent against breast tumors.
Acknowledgements
This work was conducted in The University of Texas M.D. Anderson Cancer Center, and was supported by the Cancer Research and Prevention Foundation (A.M.T.), the Cancer Center Core Grant NCI CA16672, NCI Contract NO1-CO12400 with SAIC-Frederick Inc. (J.E.S.) and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (L.K.K.). We thank Wendy Schober (The University of Texas M.D. Anderson Cancer Center Department of Blood and Marrow Transplantation Core Lab), and Kenneth Dunner Jr (The University of Texas M.D. Anderson Cancer Center Department of Cancer Biology Core Lab) for their technical assistance.
References
- 1.Dhar A, Brindley JM, Stark C, Citro ML, Keefer LK and Colburn NH: Nitric oxide does not mediate but inhibits transformation and tumor phenotype. Mol Cancer Ther 2: 1285–1293, 2003. [PubMed] [Google Scholar]
- 2.Xie K, Huang S, Dong Z, et al. : Transfection with the inducible nitric oxide synthase gene suppresses tumorigenicity and abrogates metastasis by K-1735 murine melanoma cells. J Exp Med 181: 1333–1343, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hussain SP, Trivers GE, Hofseth LJ, et al. : Nitric oxide, a mediator of inflammation, suppresses tumorigenesis. Cancer Res 64: 6849–6853, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Tatemichi M, Tazawa H, Masuda M, et al. : Suppression of thymic lymphomas and increased nonthymic lymphomagenesis in Trp53-deficient mice lacking inducible nitric oxide synthase gene. Int J Cancer 111: 819–828, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Gauthier N, Lohm S, Touzery C, et al. : Tumour-derived and host-derived nitric oxide differentially regulate breast carcinoma metastasis to the lungs. Carcinogenesis 25: 1559–1565, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Le X, Wei D, Huang S, Lancaster JR Jr and Xie K: Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors. Proc Natl Acad Sci USA 102: 8758–8763, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simeone A-M, Broemeling L, Rosenblum J and Tari AM: Her2/neu reduces the sensitivity of breast cancer cells to N-(4-hydroxyphenyl)retinamide. Oncogene 22: 6739–6747, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Simeone A-M, Li YJ, Broemeling LD, Johnson MM, Tuna M and Tari AM: Cyclooxygenase-2 is essential for HER2/neu to suppress N-(4-hydroxyphenyl)retinamide apoptotic effects in breast cancer cells. Cancer Res 64: 1224–1228, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Howie AF, Miller WR, Hawkins RA, Hutchinson AR and Beckett GJ: Expression of glutathione S-transferase B1, B2, Mu and Pi in breast cancers and their relationship to oestrogen receptor status. Br J Cancer 60: 834–837, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shea TC, Claflin G, Comstock KE, et al. : Glutathione transferase activity and isoenzyme composition in primary human breast cancers. Cancer Res 50: 6848–6853, 1990. [PubMed] [Google Scholar]
- 11.Gilbert L, Elwood LJ, Merino M, et al. : A pilot study of pi-class glutathione S-transferase expression in breast cancer: correlation with estrogen receptor expression and prognosis in node-negative breast cancer. J Clin Oncol 11: 49–58, 1993. [DOI] [PubMed] [Google Scholar]
- 12.Silvestrini R, Veneroni S, Benini E, et al. : Expression of p53, glutathione S-transferase-pi, and Bcl-2 proteins and benefit from adjuvant radiotherapy in breast cancer. J Natl Cancer Inst 89: 639–645, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, Tan PH, Thiyagarajan J and Bay BH: Prognostic significance of glutathione S-transferase-pi in invasive breast cancer. Mod Pathol 16: 558–565, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Shami PJ, Saavedra JE, Wang LY, et al. : JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther 2: 409–417, 2003. [PubMed] [Google Scholar]
- 15.Udupi V, Yu M, Malaviya S, Saavedra JE and Shami PJ: JS-K, a nitric oxide prodrug, induces cytochrome c release and caspase activation in HL-60 myeloid leukemia cells. Leukemia Res 30: 1279–1283, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Kiziltepe T, Hideshima T, Ishitsuka K, et al. : JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood 110: 709–718, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basolo F, Elliott J, Tait L, et al. : Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog 4: 25–35, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Santner SJ, Dawson PJ, Tait L, et al. : Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat 65: 101–110, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Simeone A-M, Colella S, Krahe R, Johnson MM, Mora E and Tari AM: N-(4-Hydroxyphenyl)retinamide and nitric oxide prodrugs exhibit apoptotic and anti-invasive effects against bone metastatic breast cancer cells. Carcinogenesis 27: 568–577, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Akar U, Chaves-Reyez A, Barria M, et al. : Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 4: 669–679, 2008. [DOI] [PubMed] [Google Scholar]
- 21.He H, Dang Y, Dai F, et al. : Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem 278: 29278–29287, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Simeone AM, McMurtry V, Nieves-Alicea R, et al. : TIMP-2 mediates the anti-invasive effects of the nitric oxide-releasing prodrug JS-K in breast cancer cells. Breast Cancer Res 10: R44, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren Z, Kar S, Wang Z, Wang M, Saavedra JE and Carr BI: JS-K, a novel non-ionic diazeniumdiolate derivative, inhibits Hep 3B hepatoma cell growth and induces c-Jun phosphorylation via multiple MAP kinase pathways. J Cell Physiol 197: 426–434, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Kitagaki J, Yang Y, Saavedra JE, Colburn NH, Keefer LK and Perantoni AO: Nitric oxide prodrug JS-K inhibits ubiquitin E1 and kills tumor cells retaining wild-type p53. Oncogene 28: 619–624, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabeya Y, Mizushima N, Ueno T, et al. : LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop A, Yet SF, Lee ME, Perrella MA and Demple B: A key role for heme oxygenase-1 in nitric oxide resistance in murine motor neurons and glia. Biochem Biophys Res Commun 325: 3–9, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Hill M, Pereira V, Chauveau C, et al. : Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J 19: 1957–1968, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Breslow E, Chandra R and Kappas A: Biochemical properties of the heme oxygenase inhibitor, Sn-protoporphyrin. Interactions with apomyoglobin and human serum albumin. J Biol Chem 261: 3135–3141, 1986. [PubMed] [Google Scholar]
- 29.Heigold S, Sers C, Bechtel W, Ivanovas B, Schafer R and Bauer G: Nitric oxide mediates apoptosis induction selectively in transformed fibroblasts compared to nontransformed fibroblasts. Carcinogenesis 23: 929–941, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Shami PJ, Maciag AE, Eddington JK, et al. : JS-K, an arylating nitric oxide (NO) donor, has synergistic anti-leukemic activity with cytarabine (ARA-C). Leuk Res 33: 1525–1529, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bagga D, Wang L, Farias-Eisner R, Glaspy J and Reddy S: Differential effects of prostaglandin derived from w-6 and w-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci USA 100: 1751–1756, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondo Y, Kanzawa T, Sawaya R and Kondo S: The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5: 726–734, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Kihara A, Kabeya Y, Ohsumi Y and Yoshimori T: Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2: 330–335, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima N, Yamamoto A, Hatano M, et al. : Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152: 657–668, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275: 992–998, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Wishart MJ, Taylor GS and Dixon JE: Phoxy lipids: revealing PX domains as phosphoinositide binding modules. Cell 105: 817–820, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu S, Kanaseki T, Mizushima N, et al. : Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6: 1221–1228, 2004. [DOI] [PubMed] [Google Scholar]
- 38.Pattingre S, Tassa A, Qu X, et al. : Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939, 2005. [DOI] [PubMed] [Google Scholar]