

Abstract
The rise of sample multiplexing in quantitative proteomics for the dissection of complex phenotypic comparisons has been advanced by the development of ever more sensitive and robust instrumentation. Here, we evaluated the utility of the Orbitrap Eclipse Tribrid mass spectrometer (advanced quadrupole filter, optimized FTMS scan overhead) and new instrument control software features (Precursor Fit filtering, TurboTMT and Real-time Peptide Search filtering). Multidimensional comparisons of these novel features increased total peptide identifications by 20% for SPS-MS3 methods and 14% for HRMS2 methods. Importantly Real-time Peptide Search filtering enabled a ∼2× throughput improvement for quantification. Across the board, these sensitivity increases were attained without sacrificing quantitative accuracy. New hardware and software features enable more efficient characterization in pursuit of comparative whole proteome insights.
Graphical Abstract
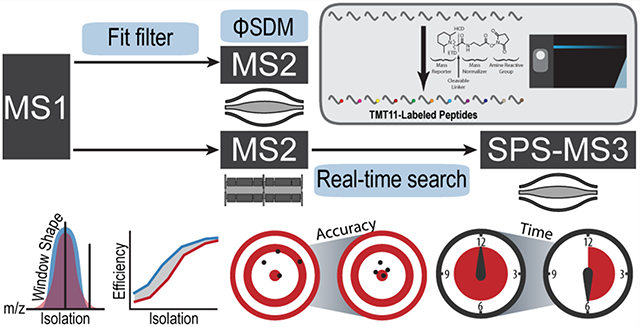
Mass spectrometry-based quantitative proteomics has undergone rapid development in recent years and has become a powerful tool in addressing questions in cell biology owing to the sensitivity and accuracy it provides.1 Yet improvements in sample preparation,2 chromatographic separations,3 bioinformatics,4,5 and particularly mass spectrometry instrumentation continue to advance the field. The Orbitrap Tribrid instrument series, since its first introduction, has enabled experimental flexibility in ion manipulations6 and scanning schemes7 coupled with parallel data acquisition for enhanced sensitivity.7,8
Among all quantitative proteomic strategies, isobaric tagging enables sample multiplexing capability to improve throughput and simultaneously provides improved precision while reducing missing values during sample comparison.9–11 Although the quantitative accuracy using isobaric tags can be distorted by cofragmenting peptide species within the isolation window,12,13 it can be restored by applying synchronous precursor selection technology coupled with MS3 (SPS-MS3) scans.14 The full SPS-MS3 scan scheme comprises isolation and fragmentation of a peptide precursor followed by a second round of ion selection and fragmentation to produce TMT reporters for quantitation. While the dedicated quantitative SPS-MS3 scan improves quantitative accuracy and precision, it results in a reduction in the spectral acquisition rate due to the additional isolation, fragmentation steps, along with the long transient times needed to resolve the TMT 11-plex isotopologues.15 Furthermore, only a small portion (e.g., as low as 25%) of all of the scans are retained in the final data set due to filtering for false-positive peptide identification and quantitative quality thresholds.16 As the need for analyzing large sample sizes continues to grow and the capability to do so in a short period of time is highly desired in clinical settings, approaches to retain quantitative accuracy while also improving throughput are in great demand.
One approach to addressing these analytical hurdles is to shorten the Orbitrap transient time. Grinfeld et al. described a computational approach termed the phase-constrained spectrum deconvolution method (ΦSDM) to improve high-resolution scan acquisition rates.17 ΦSDM with a transient time of 32 ms achieved comparable resolving power to the previously implemented enhanced FT (eFT) with a 96 ms transient, resulting in a boost in speed.18 However, to take full advantage of ΦSDM, shorter ion accumulation times are needed for ideal parallelization. To that end, the spectral processing algorithm is well suited to short gradients (e.g., 15 min) and MS2 level quantification, both of which have a high ion flux; however, the gain diminishes dramatically when applied to longer gradients or a scarce sample amount.18 By realizing a large portion of scans will not be retained in the final data set, another strategy to enhance speed is to avoid collecting SPS-MS3 scans with the knowledge that the preceding MS2 scans are not identifiable. Bailey et al. presented using real-time ion matching to inform downstream scan events.19 Erickson et al. introduced a real-time search (RTS) strategy using a binominal score. They demonstrated by continuously sequencing MS2 scans that arrive at the client and triggering SPS-MS3 acquisition only when a spectrum has been successfully matched to a peptide in real time a ∼50% boost in speed can be achieved.16 Schweppe et al. further developed a full-featured, real-time database searching platform, Orbiter,20 that takes advantage of the open source Comet search engine21 and incorporates FDR filtering on the fly to enable intelligent data acquisition. Orbiter achieved 2-fold faster acquisition speeds and improved quantitative accuracy compared to canonical SPS-MS3 methods (referred as SPS-MS3 hereafter). Now the RTS-MS3 strategy using Comet has been implemented on the new commercially available Orbitrap Eclipse platform, along with multiple hardware and software advancements.
Herein we present the new Orbitrap Eclipse in the context of multiplexed, quantitative proteomics. A redesigned advanced quadrupole mass filter, optimized FTMS overhead, and higher-transmission ion optics resulted in ion transmission increases of 25–50%, which enabled shorter ion injection times and/or high signal-to-noise for quantitative measurements at a given isolation width. Novel instrument methods were built on the improved ion transmission, TurboTMT, and Real-time Peptide Search filtering (RTS). TurboTMT implements ΦSDM and resolves TMT reporter ions at lower nominal Orbitrap resolution (15 000 or 30 000). For HRMS2 methods at 15 000 or 30 000 resolution, quantitative accuracy was equivalent to spectra collected at the canonical HRMS2 resolution (50 000; necessary for TMT 11-plex isotopologue baseline resolution). The reduction in requisite resolution and injection times enabled a 14% increase in peptide identifications. Second, Tune 3.4 uses RTS to eliminate the acquisition of SPS-MS3 scans for precursors that do not result in a peptide identification (e.g., decoy and low scoring peptides). By eliminating the need to collect uninformative SPS-MS3 scans, the instrument has more time to collect additional MS2 scans, increasing the total peptide identifications. The RTS result also enables the improved quantitative accuracy relative to SPS-MS3 by selecting only matched fragment ions for MS3 quantitative scans. RTS increased the number of PSMs (with a quantitative SPS-MS3 scan) by 45% (5829 compared to 4022 for 60 min gradient) relative to canonical SPS-MS3. More strikingly, the number was virtually identical to that collected with HRMS2 (5829 compared to 5990 for 60 min gradient), yet providing improved quantitative accuracy. Finally, we assessed the new instrumentation by examining protein expression differences across a panel of cell lines (RKO, A549, U87 MG, HCT116, HEK293T, HeLa, MCF7, U2OS, SUM159, PANC1, and Jurkat) in an 11-plex experiment with HRMS2, SPS-MS3 and RTS-SPS-MS3 methods. Over 8000 proteins were quantified using any of the three, yet RTS-MS3 achieved the same 2× reduction in instrument time as HRMS2 and equal, if not better, accuracy as canonical SPS-MS3.
EXPERIMENTAL SECTION
Sample Preparation
TMT11-labeled triple knockout (TKO) yeast standards were obtained from Thermo Fisher Scientific (A40938). Pellets from 11 human cell lines (RKO, A549, U87 MG, HCT116, HEK293T, HeLa, MCF7, U2OS, SUM159, PANC1, and Jurkat) were lysed, reduced, and alkylated prior to digestion with LysC/Trypsin as described previously.2 Peptide digests were aliquoted to desired concentrations and labeled with TMT11 reagents. The labeled peptides were mixed and desalted (50 mg C18 SepPak, Waters) prior to basic pH reversed-phase fractionation. 96 fractions were collected and consolidated into 24 samples, out of which 12 nonadjacent ones were analyzed on the mass spectrometer.2
Mass Spectrometric Data Acquisition and Data Analysis
Experiments were performed on an EASY-nLC 1200 system coupled with an Orbitrap Fusion Lumos (referred as Lumos hereafter) or an Orbitrap Eclipse (referred as Eclipse hereafter) as further specified in respective experiments. Peptides were resuspended in 5% ACN/2% formic acid at 1 mg/mL, and 1 μg was loaded on an in-house packed C18 column (25 cm, 2.6 μm Accucore (Thermo Fisher Scientific), 100 μm i.d.). Peptides were separated with a linear gradient from 5% to 32% buffer B (95% ACN, 0.125% formic acid) and injected for MS analysis. LC gradients were run for 60 min unless otherwise noted. MS1 spectra were acquired in the Orbitrap (R = 120k; AGQ target = 400 000; MaxIT = 50 ms; RF Lens = 30%; mass range = 400–2000; centroid data). Dynamic exclusion was employed for 30 s excluding all charge states for a given precursor. MS2 spectra were collected in either the linear ion trap (rate = turbo; AGQ target = 20 000; MaxIT = 50 ms; NCECID= 35%) or the Orbitrap (R = 50k; first mass = 100 m/z; AGQ target = 250 000; MaxIT = 86 ms). Data were searched using a SEQUEST-based in-house pipeline. carbamidomethylation of cysteine residues (+57.021 Da), TMT of peptide N termini and lysine residues (+229.163 Da) were set as static modifications, while the oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) using a linear discriminant analysis and then assembled further to a final protein-level FDR of 1%.20,22–24
Statistical Analysis
All statistical analysis was performed using the R statistical scripting language.25 Unless otherwise noted, reported p-values were corrected using the Benjamini-Hochberg method.26
RESULTS AND DISCUSSION
The new Orbitrap Eclipse mass spectrometer has several new hardware and software improvements presumed to benefit isobaric tagging-based multiplexed quantitative proteomics, including but not limited to a redesigned quadrupole mass filter, a new on-the-fly Precursor Fit filter (referred as Fit filter hereafter), TurboTMT (ΦSDM) and RTS (Figure 1A).
Figure 1.

(A) New software features evaluated. Fit filter, TurboTMT, and real-time search are new software features and were evaluated, respectively. (B) Number of yeast (TKO) peptides identified and quantified based on different filtering criteria. Bars represent mean ± sd. (C) Illustration of isolation windows on the Lumos and Eclipse systems. The Eclipse has a larger quadrupole mass filter with improved isolation window shape and reduced peak “tailing”. When isolating a monoisotopic peak, the ratio between 13C peak and monoisotopic peak is higher with Lumos if the 13C peak is present in the “tail” region (left) whereas the ratio is higher if the 13C peak is covered by the isolation window (right). (D) Ratios between 13C and monoisotopic peaks (mean ± SEM), normalized to the maximal value observed for charge states 2 and 3. When the isolation width did not include 13C peak, the observed ratio was higher with the Lumos due to “tailing” whereas it was higher with the Eclipse when the 13C peak was included. (E) Illustration of the isolation window centered between two isotopologues. (F) Summed signal of monoisotopic and 13C peaks with various isolation width and offset settings for charge states 2 and 3, normalized to the maximal value observed. Isolation offsets were either 0 or half of the m/z distance between monoisotopic and 13C peaks.
Evaluation of Precursor Fit Filter and TurboTMT
We first set out to evaluate the Fit filter and TurboTMT both separately or in combination for a typical 60 min HRMS2 experiment using the TKO standard (Figure 1B). The TKO standard consists of three knockout yeast strains (Δmet6, Δura2, and Δhis4) combined in triplicate with two wildtype yeast replicates. The knocked out strains should have no TMT reporter signal for the given proteins in corresponding channels and therefore provide a measure of interference.27 The Fit filter enables precursor ion selection for a defined precursor specificity. This is accomplished by comparing the observed isotopic envelope to a theoretical isotopic envelope. To queue a new MS2 scan the normalized similarity between these envelopes must be greater than a user-defined fit threshold. In this work we tested both a liberal (50%) and conservative (70%) precursor fit threshold. Another new feature to the Tribrid instruments is TurboTMT which enables faster FTMSn acquisition rates. As a reference we performed a classic HRMS2 experiment using eFT at an Orbitrap resolution setting of 50 000 and a maximum injection time of 86 ms. Two Fit filter thresholds, 50 and 70 which represent the agreement between the observed and expected targeted ion signals within the isolation window were tested. Experiments with TurboTMT used a resolving power of 30 000 and a maximum injection time of 54 ms to achieve optimal parallelization. It is worth noting although TurboTMT with 15 000 resolution is also possible, our preliminary results indicated that the injection times were too short to produce decent data quality. As expected, TurboTMT allows for an enhanced acquisition rate that translated into a ∼15% increase in total PSMs (Figure 1B). Conversely, adding the Fit filter slightly reduced PSMs–2% for Fit 50 and 4% for Fit 70, due to excluding precursors below the thresholds from triggering MS2 scans. However, the drop was not as large as expected, most likely because the inclusion of the Fit filter simply causes the same precursors to be selected at different points in their chromatograms. In addition, as the maximum injection time was capped at 54 ms instead of 86 ms, a ∼40% decrease in TMT summed signal-tonoise was observed (Figure S1A). We also calculated isolation purity and interference free index (IFI; IFI = 1 corresponds to no observed interference) as described previously.27
Our results agreed with previous reports that lower isolation specificity and/or summed signal-to-noise were more likely to produce low quality quantification with higher interference (Figure S1C, D).14,28 Therefore, we recommend applying such filters for improved accuracy. However, the sensitivity gain diminished rapidly when the filters were applied (Figure 1B).
Evaluation of Quadrupole Isolation Window Shape
The Eclipse uses a modified segmented quadrupole mass filter with an r0 of 5.25 mm instead of the 4 mm quadrupole currently used on the Lumos system. Increased r0 enlarges the area of acceptance, improves isolation efficiency, and refines the isolation window shape to achieve less “tailing”, as shown with a pure standard mixture.29 We hypothesized that this improvement could also apply to analyzing complex TMT-labeled samples. We designed two experiments with TKO to test this idea on both the Lumos and Eclipse platforms for comparison. First for a peptide precursor, a series of isolation events were performed at increasing isolation widths (i.e., 0.4, 0.5, 0.6, 0.8, 1, 1.2, and 2.5 m/z) centered at the monoisotopic peak. We assumed that if the Eclipse quadrupole had less tailing it would include less isotopic 13C peak when not in the window, and more when the window extended beyond the distance between two isotopologues. We calculated the intensity ratio of 13C to the monoisotopic peak and normalized it to the maximal observed for a given precursor, which, in most cases, was with the isolation window at 2.5 m/z (Figure 1C). We assumed when the window was large enough (>2 m/z), the ratios would be equal between the two systems, approximating the natural abundance of the isotopologues. When the isolation window did not include the 13C peak (ideally the 13C peak should be absent), the Lumos generated higher ratios due to window tailing (the 13C peak was isolated when it should have been excluded), whereas when the window encompassed both isotopes we observed higher ratios for the Eclipse, which was indicative of a more square isolation window (Figures 1C,D and S1E and Table S1). In the second experiment we offset the center of the isolation window to the middle of two isotopologues in a charge state-specific manner. We then measured the total ion intensity of the monoisotopic and 13C isotopes together. For example, we used an offset at 0.25 m/z for doubly charged precursors (isotopologue spacing Δm/z = 0.5) and used a series of isolation windows ranging in width from 0.5 to 1.2 (increment of 0.1). As a baseline for comparison, data were collected with no offset and an isolation width of 0.4 and 2.5. A perfectly square isolation window shape should enable complete isolation of both isotopologues. Therefore, we hypothesized that the Eclipse would generate higher summed signal compared to the Lumos (Figure 1E). Intensities from monoisotopic and 13C peaks were summed and normalized to the maximum observed for all isolation widths, which in most cases was acquired without an offset and with a width of 2.5 m/z. We compared the normalized total intensities under the assumption that when the window was large enough (>2 m/z), there would be no difference between the two systems. We observed more normalized total intensity with smaller windows on the Eclipse, suggesting that the squared isolation window improved ion transmission compared to the Lumos (Figure s1E,F and S1F and Table S2). As exemplified by doubly charged precursors, when the window width was below 0.9 m/z, the Eclipse reported higher normalized signal. Yet, when the window was over 0.9, the advantage diminished (Figure 1F, upper panel). Overall, the new quadrupole on the Eclipse mass spectrometer showed clearly improved quadrupole isolation transmission and refined isolation window shape.
Evaluation of the Real-Time Search (RTS) Filter
Another critical component on the new system, the RTS filter, has already been proved remarkably advantageous in previous implementations.16,20 Herein we benchmarked its utility on the new Eclipse system. First, we analyzed the TKO standard with a 60 min gradient using HRMS2, SPS-MS3, and RTS-SPS-MS3 in triplicate. Not surprisingly, HRMS2 generated the most PSMs and unique peptides. However, after applying filters on the quantification, RTS-SPS-MS3 retained virtually the same number of quantified PSMs as HRMS2 (5990 with HRMS2, 4022 with SPS-MS3, 5829 with RTS-SPS-MS3 on average) and unique peptides (5392 with HRMS2, 3764 with SPS-MS3, and 5366 with RTS-SPS-MS3 on average; Figure 2A). Quantified PSMs are defined as those with a summed signal-to-noise ≥10014 and for HRMS2 and SPS-MS3, isolation purity must be ≥0.7.14,16,28,30 IFI calculations for the TKO proteins revealed higher interference with HRMS2, whereas SPS-MS3 and RTS-SPS-MS3 were comparable and better than HRMS2 (Figures 2B and S2).
Figure 2.

Performance evaluation with TKO standards using HRMS2, SPS-MS3, and RTS-MS3. (A) Numbers of total PSMs, unique peptide, quantified PSMs, and quantified unique peptide. Quantified PSMs are defined as PSMs with summed SN ≥ 100 and isolation specificity ≥0.7 for HRMS2 and SPS-MS3 and summed SN ≥ 100 for RTS. Bars represent mean ± sd. HRMS2 achieved the most total PSMs and unique peptides, whereas RTS retained the most quantified PSMs and unique peptides after filtering. (B) Interference-free index (IFI) of Met6 peptides (left) and the relative abundance of Met6 peptides in each TMT channel (right). Even after data filtering, HRMS2 still present excessive interference, whereas SPS-MS3 and RTS are much better in removing interference.
Real-Time Search (RTS) For Whole Proteome Quantitation
To allow a more comprehensive evaluation of the method, we collected a deep proteome data set on a fractionated sample consisting of 11 human cell lines labeled with TMT (Figure 3A). These cell line were selected from multiple tissues of origin (e.g., breast, lung, colon) to provide both large and small proteome differences necessary to compare the acquisition methods tested here (Figure S3). Twelve fractions were analyzed using HRMS2, SPS-MS3, and RTS-SPS-MS3. Fractions analyzed with HRMS2 and RTS-SPS-MS3 used a 90 min gradient method while SPS-MS3 used a 90 min (referred to as SPS90 hereafter) and a 180 min method (referred as SPS180 hereafter). In total, four data sets were collected (Figure 3A). The RTS-SPS-MS3 method collected the most quantified PSMs and unique peptides (Figure 3B). HRMS2, SPS180, and RTS-SPS-MS3 all quantified >8000 proteins (Figure 3B), although SPS180 took twice the instrument time (36 h). In contrast, SPS90 with equal time only quantified 7000 proteins, due to time-consuming SPS-MS3 scans on unidentifiable species.
Figure 3.

Deep proteome analysis with four different methods. (A) Eleven cell lines were digested with trypsin, labeled with TMT, separated into 12 fractions, and analyzed using HRMS2 or SPS-MS3 with two gradient length (i.e., 90 and 180 min) and RTS-SPS-MS3. (B) RTS quantified the most PSMs and unique peptides. (C) Number of quantified proteins. HRMS2, SPS180, and RTS quantified >8000 proteins.
With four separate data sets, quantitative accuracy and precision were investigated for any systematic errors caused by each method (Figure S4). We calculated log2 ratios between a representative pair of cell lines and examined how these ratios correlated between different methods (Figures 4 and S5). MCF7 and HCT116 cell line data produced a Pearson R2 greater than 0.9 when comparing protein quantification using SPS90 or SPS180 versus RTS-SPS-MS3. These data corroborated evidence (e.g., Figure 2) of greater accuracy and reproducibility using MS3-based methods (Figure 4B,C).20 However, a skewed slope leaning toward RTS-SPS-MS3 when compared to HRMS2 and reduced correlation coefficient (R2 = 0.88) suggested ratio compression with HRMS2 (Figure 4A).14,31 Beyond examining two representative cell lines, we kept proteins with ≥3 quantified PSMs and calculated the coefficient of variation (CV) among peptides assigned to the same protein per cell line. Ideally the CV should be minimal because these sets of peptides reflect the same protein abundance in a respective cell line. We observed that HRMS2 produced higher CVs per protein per cell line, most likely due to the presence of higher levels of quantitative interference relative to the other methods tested (Figure 5A). From another perspective, we calculated the variance of relative abundance per protein among 11 channels. The cell lines included in the experiment were biochemically distinct (Figure S3) and large variance were expected in a substantial fraction of proteins. The median variances for SPS90, SPS180, and RTS were 17.4, 17.9, and 18.3, respectively, whereas it was 10.6 for HRMS2 (p-value <2 × 10−16, FDR-corrected ANOVA; Figure 5B). These differences were likely caused by interference which compressed all protein quantitation values toward unity.12–14 The result of which was fewer total proteins with high abundance fold changes used to determine altered protein abundance across cell lines (Figure S6).
Figure 4.

Quantitative comparison. Log2 ratio (MCF7/HCT116) comparison between (A) HRMS2 vs RTS-SPS-MS3, (B) SPS90 vs RTS-SPS-MS3, and (C) SPS180 vs RTS-SPS -MS3. SPS90 and SPS180 achieved R2 > 0.9 with RTS-SPS-MS3. Skewed slope in (A) suggests ratios from HRMS2 are noticeably compressed due to interference. Gene symbols are labeled if compared ratios present a difference (log2 values) greater than +3 or less than −3.
Figure 5.

Quantitative variance per protein within and across cell lines. (A) Proteins with three peptides are included in the comparison and distributions of coefficient of variation (CV) among peptides per protein per cell line are plotted. Ideally, peptides for a particular protein and cell line should have identical quantitative values. Larger CV indicates more variation for the measurement of a protein within a given cell line, most likely due to interference from coisolated ion species. (B) Variance of relative abundance per protein across 11 cell lines. Quantitative proteomics reveals distinct composition of the 11 cell lines (Figure S3), so consequently a substantial fraction of proteins is expected to have large variation across cell lines. Reduced variations from HRMS2 relative to SPS90, SPS180, and RTS indicate ratio compression toward uniform values. Variance for the HRMS2 analysis was on average 1.7-fold lower than the other three methods.
Proteins commonly quantified by all methods were included in the comparison where correlations between the same proteins and pairs of methods were calculated. In general, quantification by HRMS2 had lower correlation with any other method (Figure 6A). The quantitative deviation of the HRMS2 methods from the three other independent, whole-proteome analyses suggested that the HRMS2 quantification deviated more from the true values. Exemplified by HSPA2, although HRMS2 revealed similar general trend as the other SPS-MS3-based methods, it presented noticeable ratio compression and higher variation among contributing peptides (Figure 6B). Another example with excessive interference was BRAF. We observed a distinct quantitative profile for HRMS2 compared to the other methods, with all channels having nearly equal abundance and a variance of just 3.1. In contrast, SPS90, SPS180 and RTS-SPS-MS3 all suggested Jurkat expressed 3× more BRAF relative to A549, with a variance of 12.4, 15.0, and 25.1 across 11 channels, respectively (Figure 6C). The two BRAF peptides quantified by HRMS2 had a Pearson correlation of −0.41, whereas the three peptides quantified by RTS-SPS-MS3 had an average Pearson correlation of 0.95 (Figure 6C). SPS180 quantified four BRAF peptides with an average Pearson correlation of 0.63 (SPS90 on the other hand only quantified a single peptide). We partly attributed the enhancement in precision to RTS’s ability to only select matched b- and y-ions instead of randomly selecting high intensity peaks for the subsequent quantitative SPS-MS3 scans.20
Figure 6.

Quantitative correlation and ratio compression. (A) Correlation of protein relative abundance between pairs of data acquisition methods. Distribution of Pearson r values are plotted. Lower correlation coefficients between HRMS2 and other methods are most likely due to interference. Outlier points are not shown. (B and C) Relative protein (bars) and contributing peptide (lines) abundance across the 11 cell lines for each method. In both cases protein values are compressed toward the median (1:1) for HRMS2. (B) HSPA2 protein quantitation across the four methods had high correlation (r = 0.999 vs RTS). (C) Braf protein quantitation correlated poorly between the HRMS2 and MS3-based methods (r = −0.575 vs RTS). On the peptide level, in addition to reduced ratio compression, high correlation (Braf: rHRMS2 = −0.15; rRTS = 0.95) among individual peptides from RTS reinforces the importance of correct SPS ion selection.
CONCLUSIONS
We evaluated the utility of the new Orbitrap Eclipse mass spectrometer for multiplexed proteomic analysis. We observed improved acquisition efficiency and quantitative accuracy. Furthermore, the potential to couple this new instrument with recent hardware advances (e.g., FAIMS) and the extended reporter series available with the TMTPro 16-plex reagents will provide new avenues to greatly improve the speed and performance of multiplexed quantitative workflows.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank members of the Gygi lab for helpful discussion. Funding sources include GM132129 (J.A.P.) and GM67945 (S.P.G.), and J.N.P. is partially fund by the Mexican Council for Science and Technology (289937).
Footnotes
The authors declare the following competing financial interest(s): G.C.M., J.D.C., D.J.B, A.M.R., R.H., and V.Z. are employees of ThermoFisher Scientific, the manufacturer of the Orbitrap Eclipse Tribrid mass spectrometer.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05685.
Contributor Information
Qing Yu, Harvard Medical School, Boston, Massachusetts 02115, United States.
Joao A. Paulo, Harvard Medical School, Boston, Massachusetts 02115, United States
Jose Naverrete-Perea, Harvard Medical School, Boston, Massachusetts 02115, United States.
Graeme C. McAlister, Thermo Fisher Scientific, San Jose, California 95134, United States
Jesse D. Canterbury, Thermo Fisher Scientific, San Jose, California 95134, United States
Derek J. Bailey, Thermo Fisher Scientific, San Jose, California 95134, United States
Aaron M. Robitaille, Thermo Fisher Scientific, San Jose, California 95134, United States
Romain Huguet, Thermo Fisher Scientific, San Jose, California 95134, United States.
Vlad Zabrouskov, Thermo Fisher Scientific, San Jose, California 95134, United States.
Steven P. Gygi, Harvard Medical School, Boston, Massachusetts 02115, United States.
Devin K. Schweppe, Harvard Medical School, Boston, Massachusetts 02115, United States.
REFERENCES
- (1).Gillet LC; Leitner A; Aebersold R Annu. Rev. Anal. Chem. 2016, 9 (1), 449–72. [DOI] [PubMed] [Google Scholar]
- (2).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA J. Proteome Res. 2018, 17 (6), 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shishkova E; Hebert AS; Westphall MS; Coon JJ Anal. Chem. 2018, 90 (19), 11503–11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Myers SA; Klaeger S; Satpathy S; Viner R; Choi J; Rogers J; Clauser K; Udeshi ND; Carr SA J. Proteome Res. 2018, 18 (1), 542–547. [DOI] [PubMed] [Google Scholar]
- (5).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacLean BX; Villen J; MacCoss MJ Nat. Commun. 2018, 9 (1), 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Brunner AM; Lossl P; Liu F; Huguet R; Mullen C; Yamashita M; Zabrouskov V; Makarov A; Altelaar AF; Heck AJ Anal. Chem. 2015, 87 (8), 4152–8. [DOI] [PubMed] [Google Scholar]
- (7).Senko MW; Remes PM; Canterbury JD; Mathur R; Song Q; Eliuk SM; Mullen C; Earley L; Hardman M; Blethrow JD; Bui H; Specht A; Lange O; Denisov E; Makarov A; Horning S; Zabrouskov V Anal. Chem. 2013, 85 (24), 11710–4. [DOI] [PubMed] [Google Scholar]
- (8).Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ Mol. Cell. Proteomics 2014, 13 (1), 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rauniyar N; Yates JR 3rd J. Proteome Res. 2014, 13 (12), 5293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Savitski MM; Reinhard FBM; Franken H; Werner T; Savitski MF; Eberhard D; Molina DM; Jafari R; Dovega RB; Klaeger S; Kuster B; Nordlund P; Bantscheff M; Drewes G Science 2014, 346 (6205), 1255784. [DOI] [PubMed] [Google Scholar]
- (11).Savitski MM; Mathieson T; Zinn N; Sweetman G; Doce C; Becher I; Pachl F; Kuster B; Bantscheff MJ Proteome Res. 2013, 12 (8), 3586–98. [DOI] [PubMed] [Google Scholar]
- (12).Ting L; Rad R; Gygi SP; Haas W Nat. Methods 2011, 8 (11), 937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wenger CD; Lee MV; Hebert AS; McAlister GC; Phanstiel DH; Westphall MS; Coon JJ Nat. Methods 2011, 8 (11), 933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McAlister GC; Nusinow DP; Jedrychowski MP; Wuhr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP Anal. Chem. 2014, 86 (14), 7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hogrebe A; von Stechow L; Bekker-Jensen DB; Weinert BT; Kelstrup CD; Olsen JV Nat. Commun. 2018, 9 (1), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Erickson BK; Mintseris J; Schweppe DK; Navarrete-Perea J; Erickson AR; Nusinow DP; Paulo JA; Gygi SP J. Proteome Res. 2019, 18 (3), 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Grinfeld D; Aizikov K; Kreutzmann A; Damoc E; Makarov A Anal. Chem. 2017, 89 (2), 1202–1211. [DOI] [PubMed] [Google Scholar]
- (18).Kelstrup CD; Aizikov K; Batth TS; Kreutzman A; Grinfeld D; Lange O; Mourad D; Makarov AA; Olsen JV J. Proteome Res. 2018, 17 (11), 4008–4016. [DOI] [PubMed] [Google Scholar]
- (19).Bailey DJ; Rose CM; McAlister GC; Brumbaugh J; Yu P; Wenger CD; Westphall MS; Thomson JA; Coon JJ Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (22), 8411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP, Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. Proteome Res. 2020, in press DOI: 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eng JK; Jahan TA; Hoopmann MR Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (22).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villen J; Haas W; Sowa ME; Gygi SP Cell 2010, 143 (7), 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Savitski MM; Wilhelm M; Hahne H; Kuster B; Bantscheff M Mol. Cell. Proteomics 2015, 14 (9), 2394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Elias JE; Gygi SP Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (25).R Core Team R A Language and Environment for Statistical Computing; https://www.R-project.org.
- (26).Benjamini Y; Hochberg Y Journal of the royal statistical society. Series B (Methodological) 1995, 57, 289–300. [Google Scholar]
- (27).Paulo JA; O’Connell JD; Gygi SP J. Am. Soc. Mass Spectrom. 2016, 27 (10), 1620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).O’Brien JJ; O’Connell JD; Paulo JA; Thakurta S; Rose CM; Weekes MP; Huttlin EL; Gygi SP J. Proteome Res. 2018, 17 (1), 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McAlister GC; Goodwin M; Mathur R; Earley L; Lange O; Huguet R; Zabrouskov V; Senko MW In Modifying the Ion Optics and Scan Sequences on a Tribrid MS to Improve Sensitivity, Duty Cycle, and Overall Instrument Ease-of-Use. 67th ASMS Conference on Mass Spectrometry and Allied Topics; American Society for Mass Spectrometry: Atlanta, GA, 2019. [Google Scholar]
- (30).Wuhr M; Haas W; McAlister GC; Peshkin L; Rad R; Kirschner MW; Gygi SP Anal. Chem. 2012, 84 (21), 9214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach J-J; Paulo JA; Gygi SP Anal. Chem. 2019, 91 (6), 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.