

Abstract
ΔFosB is a highly stable transcription factor that accumulates in specific brain regions upon chronic exposure to drugs of abuse, stress, or seizures, and mediates lasting behavioral responses. ΔFosB reportedly heterodimerizes with JunD forming a canonical bZIP leucine zipper coiled coil that clamps onto DNA. However, the striking accumulation of ΔFosB protein in brain upon chronic insult has brought its molecular status into question. Here, we demonstrate through a series of crystal structures that the ΔFosB bZIP domain self-assembles into stable oligomeric assemblies that defy the canonical arrangement. The ΔFosB bZIP domain also self-assembles in solution, and in neuron-like Neuro 2a cells it is trapped into molecular arrangements that are consistent with our structures. Our data suggest that, as ΔFosB accumulates in brain in response to chronic insult, it forms non-canonical assemblies. These species may be at the root of ΔFosB's striking protein stability, and its unique transcriptional and behavioral consequences.
Keywords: ΔFosB, Activator protein-1 (AP-1), Transcription factor, Basic leucine zipper, Protein accumulation, Neurological disorders
Graphical abstract

Highlights
-
•
ΔFosB, a transcription factor triggered by chronic brain insult, self-assembles.
-
•
ΔFosB leverages non-canonical arrangements of its leucine zipper.
-
•
ΔFosB self-assemblies are seen in solution and in neuron-like Neuro 2a cells.
-
•
Self-assembly may encode ΔFosB's striking stability and unique biological roles.
1. Introduction
ΔFosB is a member of the AP-1 (activator protein-1) family of transcription factors. In response to chronic exposure to drugs of abuse, stress, or seizures, ΔFosB protein accumulates to high levels in select brain regions including the nucleus accumbens and hippocampus (Nestler, 2015). The accumulated ΔFosB protein is extremely stable, remaining present in neurons even several weeks after the last insult (Carle et al., 2007, Hope et al., 1994, Ulery-Reynolds et al., 2009). Generated through alternative splicing of the parent FosB mRNA, the ΔFosB protein contains a disordered N-terminal region (Met1-Glu156), and a bZIP domain composed of a basic region (Lys157-Arg177) carrying a DNA-binding motif and a leucine zipper (Thr180-His218) that forms a coiled coil with a dimerization partner. However, ΔFosB lacks the 101 residue transactivation domain present in full-length FosB (Val238-Leu338) (Nestler, 2015) (Fig. 1a). AP-1 transcription factors bind to gene promoters containing AP-1 consensus sequences (TGA C/G TCA) and regulate their expression. Binding to DNA occurs when the leucine zippers dimerize, clamping the basic regions on either side of the DNA strand into the major groove like forceps (Glover and Harrison, 1995). The partner for ΔFosB, in vivo, is assumed to be JunD (Chen et al., 1997, Hiroi et al., 1998). However, in vitro, ΔFosB binds to AP-1 DNA sites alone in a specific manner (Jorissen et al., 2007, Wang et al., 2012). Thus, the exact nature of ΔFosB as it accumulates in vivo is unclear.
Fig. 1.

Canonical ΔFosB interactions. a) Domain structure of ΔFosB, FosB, and JunD. BR, basic region; L-Zip, leucine zipper; TAD, transactivation domain; b) ΔFosB and JunD bZIP domains broken down into heptad repeats. Residues at the d-, e-, and g-positions of heptads H1 through H5, and the cysteines are shown in color (aliphatic, brown; polar & neutral, cyan; basic, blue; acidic, red; cysteine, yellow). On the left, a cartoon representation indicates the basic region (dark blue), leucine zipper (red and cyan, respectively), and C-terminal residues (green); c) The ΔFosB/JunD bZIP heterodimer bound to DNA. The ruler indicates the heptad repeats. The leucine zipper region of ΔFosB is shown in red, and that of JunD in cyan; the DNA-binding regions are shown in blue. The C-terminal residues are shown in green. Leucine side chains at the d-position are shown in yellow; d) Helical wheel diagram showing the heptad composition and interactions. ‘N’ indicates the N-terminus of the helix; e) Example of a canonical d-position leucine interaction in the ΔFosB/JunD bZIP coiled coil shown in the structure and f) schematically.
We recently determined the three dimensional (3D) structure of ΔFosB in complex with JunD in the presence and absence of DNA (Yin et al., 2017). Each bZIP domain contains a long helix consisting of a series of heptad repeats [abcdefg]n (Fig. 1b). Characteristic leucine residues located at the d-positions of the repeats H1 through H5 form a leucine zipper with coiled coil geometry (Fig. 1c). These leucine side chains, together with the hydrophobic portions from side chains at the a-positions, form a hydrophobic core that aligns the two helices in a symmetrical and parallel manner (Fig. 1d). In this canonical mode of interaction, a leucine at the d-position of heptad i in one helix (a ‘d-leucine’) interacts with the facing helix by packing into a four-residue pocket like ‘knobs-into-holes’. Each d-leucine inserts between two sequential residues in the facing helix at the a’-position in heptads i and i+1 to define Setting 1 (residues a,b,c,d,e,f,g from one helix, and a’,b’,c’,d’,e’,f’,g’ from the facing helix) (Fig. 1e; Fig. 1f). The pocket is completed by two residues at the d’- and e’-position of heptad i, establishing the canonical packing, packing P1 (Fig. 1e; Fig. 1f). The d-leucines locate side-by-side equivalent d’-leucines on the facing helix. By contrast, the basic regions that contain the DNA-binding residues don't interact significantly.
AP-1 transcription factors like c-Fos and c-Jun have long been considered active only as dimers, and in the case of c-Fos and c-Jun, in particular, to show a clear preference for forming heterodimers over homodimers (Kohler and Schepartz, 2001, Newman and Keating, 2003). Despite extensive studies, the rules governing the assembly of bZIP domains are still not well understood (Lupas et al., 2017). Recent efforts to determine the dimerization profile of bZIPs experimentally, and the DNA sites they bind to, revealed an unanticipated breadth of interactions (Newman and Keating, 2003, Reinke et al., 2013, Rodríguez-Martínez et al., 2017). Their molecular architectures appear more diverse than previously assumed as well. For example, ΔFosB/JunD can adopt a tetrameric arrangement in the DNA-free form in four different crystal structures (Yin et al., 2017).
Because ΔFosB accumulates to high levels in brain, we investigated the hypothesis that it might form homomeric species. Self-assembly of ΔFosB would then be the product of its intrinsic affinity and its relative high abundance inside a particular cell compared to that for other partners. Even transient assembly of ΔFosB could be biologically important, for example to dynamically and sensitively regulate critical genes, such as those involved in regulating the excitability of neurons (Eagle et al., 2018). Upon decreasing ΔFosB protein levels, a transient assembly bound to DNA would fall apart, liberating the gene promoter. By contrast, unusually stable homomeric ΔFosB assemblies could mediate long lasting changes. Therefore, the exact molecular assemblies that ΔFosB forms are likely critically important for its function in brain, prompting this study to delineate them.
Here, we reveal that the ΔFosB bZIP domain assumes a portfolio of different molecular arrangements. In a series of crystal structures, ΔFosB is captured as tetramers as well as dimers. To form these non-canonical assemblies, ΔFosB uses a diverse set of novel arrangements for the d-leucines that differ from those in the ΔFosB/JunD heterodimer. Using analytical ultracentrifugation (AUC), we show that the ΔFosB bZIP domain can convert between different oligomeric species in solution. Finally, we demonstrate in Neuro 2a cells that ΔFosB likewise forms assemblies both in complex with itself as well as in complex with JunD. Hence, ΔFosB readily forms a range of stable macromolecular assemblies, in crystal structures, in solution, and in cells, with potential relevance in vivo.
2. Methods
2.1. Molecular cloning
The ΔFosB/FosB basic leucine zipper domain (FB, residues E153-K219) was sub-cloned into a pET21-NESG expression vector using the Mus musculus FosB cDNA and encodes the identical amino acid sequence as that in humans. The constructs have an N-terminal hexa-histidine (His) tag followed by a tobacco etch virus (TEV) protease cleavage site. Plasmids verified by DNA sequencing were transformed into E. coli Rosetta 2 (DE3) cells (Invitrogen).
2.2. Protein production and purification
To express recombinant FB, a culture of E. coli strain Rosetta 2 (DE3) grown in LB medium at 37 °C to a density of 0.6 (595 nm) was induced overnight with 0.5–1 mM isopropyl β-D-thiogalactoside (IPTG) at 16 °C. The harvested cells were re-suspended in lysis buffer (20 mM Tris pH 8, 1 M NaCl, 0.5 M NaBr, 20 mM imidazole, 1–2 mM Tris(2-carboxyethyl)phosphine (TCEP), adding optionally 0.5 mM PMSF), lysed by sonication, and centrifuged at 18,000 rpm (46,285×g) for 30 min. The supernatant was loaded onto a Ni-NTA agarose column (Invitrogen), washed with lysis buffer, and eluted in the same buffer containing 0.5 M imidazole. For structural studies, the His-tag was removed from the eluted protein with His-tagged TEV protease by incubating the protein overnight in 20 mM Tris pH 8, 1 M NaCl, and 2 mM TCEP; the protein was re-loaded on a Ni-NTA agarose column, and the flow-through containing the His-tag-free protein collected. The disulfide crosslinked FB (FBSS) was generated by dialyzing the protein overnight against 20 mM Tris pH 8, 0.2 M NaCl to remove reducing agent while exposing to ambient air, and further purified by size-exclusion chromatography using a HiLoad 16/600 Superdex 200 column in 20 mM Tris pH 8, 50 mM NaCl. Fractions containing FBSS were pooled, concentrated, and reapplied 3–5 times to improve homogeneity prior to crystallization. For structural studies, protein preparations were also made under reducing conditions by running the protein on a HiLoad 16/600 Superdex 200 column in 10 mM Tris pH 8, 0.5 M NaBr, 4 mM TCEP or 10 mM Tris pH 8, 50 mM NaCl, 2 mM TCEP and stored in that buffer at 10 mg/ml. For biophysical and biochemical studies, the reduced form of FB was generated by adding 2 mM dithiothreitol (DTT) to purified FBSS and then subjecting it to size-exclusion chromatography in 20 mM Tris pH 8, 0.2 M NaCl, and 2 mM DTT. Protein samples were concentrated to 10–20 mg/ml, aliquoted, and flash-frozen for storage. For AUC studies, the eluate from the Ni-NTA agarose column was diluted in 50 mM Tris pH 8, 0.5 M NaCl, 1 mM DTT, digested with TEV for 3 h at room temperature, and then incubated with Ni-NTA beads for 1 h at room temperature. The cleaved His-tag free protein was dialyzed overnight against 20 mM HEPES pH 7, 0.5 M NaCl, concentrated, and then further purified with size-exclusion chromatography using a HiLoad 16/600 Superdex 75 column (GE Healthcare) pre-equilibrated with the same buffer. Protein samples were concentrated to ca. 10 mg/ml, aliquoted, and flash-frozen for storage. The final protein samples were analyzed by SDS-PAGE to confirm that their purity was >95%.
2.3. Crystallization and X-ray data collection
FBSS crystals were grown at 293 K with the hanging-drop vapor-diffusion method by mixing 0.5–1 μl of FBSS (10 mg/ml protein in 20 mM Tris pH 8, 50 mM NaCl) with an equal volume of reservoir solution consisting of 2.5 M NaCl and 0.1 M sodium acetate pH 5.0. Serial streak seeding was performed to improve crystal morphology. Native FBSS crystals were cryo-protected in reservoir solution supplemented with 20% (v/v) ethylene glycol and then flash-cooled by plunging into liquid nitrogen. FBSS crystals were derivatized with platinum (FBSS–Pt) by soaking with 5 mM K2PtCl4 overnight. Diffraction data from a single native FBSS crystal and a single FBSS-Pt crystal was collected at the Advanced Photon Source (APS), Argonne National Laboratory on beamline 19-ID and 21-ID-F, respectively. FB Type I crystals were grown at 293 K with the hanging-drop vapor-diffusion method by mixing 1 μl of FB (10 mg/ml protein in 10 mM Tris pH 8, 0.4 M NaBr, 4 mM TCEP) with an equal volume of reservoir solution consisting of 17% (w/v) PEG 4000, 0.2 M (NH4)2SO4. Crystals were cryo-protected by transferring them to reservoir solution supplemented with 20% (v/v) ethylene glycol and then flash-cooled by plunging into liquid nitrogen. Diffraction data from a single FB Type I crystal was collected at APS, beamline 17-ID. FB Type II crystals were grown at 293 K with the sitting-drop vapor-diffusion method by mixing 0.5 μl of FB (10 mg/ml protein in 10 mM Tris pH 8, 50 mM NaCl, 2 mM TCEP) with 0.5 μl of reservoir solution consisting of 2 M (NH4)2SO4, 5% (v/v) isopropanol. Crystals were cryo-protected by transferring to reservoir solution supplemented with 15% (v/v) ethylene glycol and then flash-cooled by plunging into liquid nitrogen. Diffraction data from a single FB Type II crystal was collected at APS, beamline 19-ID.
2.4. Data processing, structure solution and refinement
Data was processed and reduced in HKL2000 (Otwinowski and Minor, 1997). The FBSS crystal structure was determined by experimental phasing with single-wavelength anomalous diffraction (SAD) using the anomalous diffraction data collected from an FBSS-Pt crystal. Eight heavy-atom sites were identified and an initial model was built using the program AutoSol in PHENIX (Adams et al., 2010). The initial model was used in molecular replacement with diffraction data from native FBSS crystals using PHASER (McCoy et al., 2007) in PHENIX. FB Type I and II crystal structures were determined by molecular replacement using FBSS as the search model. Iterative cycles of model building and refinement were carried out with COOT (Emsley et al., 2010) and PHENIX.refine. Hydrogens were added in the riding position to all models, and translation-libration-screw rotation (TLS) parameterization by TLSMD analysis was carried out in PHENIX (Adams et al., 2010), followed by validation with MolProbity (Williams et al., 2018). Ramachandran plot statistics (favored/allowed/disallowed) of the final models are: FBSS (99.5%/0.5%/0%), FB Type I (100%/0%/0%) and FB Type II (99.3%/0.7%/0%). The FB Type I crystal structure has relatively high Rwork/Rfree values of 0.271/0.287, presumably due to fiber diffraction patterns observed from the crystal that shadowed some of the reflections. For detailed statistics for data collection and refinement see Table 1. Coordinates and structure factors of FBSS, FB Type I and II can be accessed via Protein Data Bank IDs: codes 6UCI, 6UCL, and 6UCM, respectively.
Table 1.
Data collection and refinement statistics for the FB crystal structures.
| Crystallization conditions | FBss |
FBss-Pt |
FB, Type-I |
FB, Type-II |
|---|---|---|---|---|
| 2.5 M sodium chloride, 0.1 M sodium acetate, pH 5.0 | 2.5 M sodium chloride, 0.1 M sodium acetate, pH 5.0; soak in 5 mM K2PtCl4 | 17% (w/v) PEG 4000, 0.2 M ammonium sulfate | 2 M ammonium sulfate, 5% (v/v) isopropanol | |
| Data collectiona | ||||
| Wavelength (Å) | 1.008 Å | 0.9787 Å | 0.9795 Å | 1.006 Å |
| Space group | P21212 | P21212 | F222 | P3221 |
| Cell dimensions | ||||
| a, b, c (Å) | 72.28, 97.50, 45.20 | 69.79, 96.08, 45.86 | 39.51, 51.96, 150.45 | 101.93, 101.93, 51.35 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 35.67–2.09 (2.13–2.09) | 48.04–2.48 (2.54–2.48) | 30.78–2.21 (2.23–2.21) | 44.38–2.42 (2.47–2.42) |
| Unique reflections | 19,503 (958) | 10,902 (546) | 4,096 (194) | 11,940 (580) |
| Rmerge | 0.048 (0.746) | 0.117 (1.8) | 0.059 (0.845) | 0.086 (1.448) |
| Rpim | 0.027 (0.431) | 0.039 (0.59) | 0.030 (0.389) | 0.028 (0.495) |
| I/σ(I) | 26.7 (2.0) | 22.8 (2.2) | 25.0 (2.3) | 30.5 (2.1) |
| Completeness (%) | 99.5 (99.9) | 95.3 (95.0) | 99.6 (100.0) | 100.0 (100.0) |
| Multiplicity | 4.0 (3.9) | 10.5 (10.3) | 5.3 (5.2) | 10.5 (9.8) |
| Refinement | ||||
| Resolution (Å) | 35.67–2.09 (2.14–2.09) | 30.78–2.21 (2.78–2.21) | 44.38–2.42 (2.53–2.42) | |
| Rwork/Rfree | 0.207/0.241 (0.232/0.265) | 0.271/0.287 (0.300/0.340) | 0.208/0.245 (0.242/0.279) | |
| Total No. of Reflections | 18,902 (1072) | 2,929 (863) | 11,649 (1216) | |
| Reflections used Rfree | 1,809 (107) | 294 (86) | 1,172 (119) | |
| Non-hydrogen atoms | 2,008 | 472 | 1,312 | |
| Protein | 1,917 | 458 | 1,271 | |
| Ligands/ions | 15 | 3 | 25 | |
| Water | 70 | 11 | 16 | |
| B factors (Å2), overall | 47.1 | 40.1 | 52.5 | |
| Protein | 47.1 | 40.1 | 52.6 | |
| Ligands/ions | 69.0 | 59.2 | 54.2 | |
| Water | 41.9 | 35.0 | 42.4 | |
| r.m.s.d. bond lengths (Å) | 0.005 | 0.003 | 0.005 | |
| r.m.s.d. bond angles (°) | 0.58 | 0.48 | 0.58 | |
| Ramachandran plot (%) | ||||
| favored | 99.5 | 100 | 99.3 | |
| allowed | 0.5 | 0 | 0.7 | |
| disallowed | 0 | 0 | 0 | |
| Rotamer outliers, n (%) | 1 (0.51) | 1 (2.38) | 1 (0.76) | |
Numbers in parentheses refer to the highest-resolution shell.
data collected at 100 K.
2.5. Mass spectrometry
Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI TOF/TOF MS, Sciex 5800) was used to analyze protein molecular weights. Protein samples (1 μl) were spotted on a 384 Opti-TOF 123 mm × 81 mm SS plate, dried, sinapinic acid solution (1 μl) added and dried again. Cytochrome C solution was spotted next to the samples for external calibration. Intensity data were normalized with the highest peak set at 100%. Figures were made with GraphPad Prism v5.01 (GraphPad Software).
2.6. Circular dichroism spectroscopy
Circular Dichroism measurements were performed with a J-815 Spectropolarimeter (JASCO) equipped with a Peltier junction temperature controller at 20 °C using a quartz cuvette with 1 mm path length. Spectra were recorded at 0.5 nm intervals from 190 nm to 250 nm with a 1 nm bandwidth. Spectra were averaged over three scans, buffer spectra subtracted and then converted to mean residue ellipticity ([θ]). Protein concentration was determined by measuring UV absorbance at 215 nm (E 0.1% value of 11.7 for peptides) using a DeNovix DS-11+ spectrophotometer. Ellipticity of FBSS at 0.1 mg/ml (12 μM) in a buffer of 12.5 mM K2HPO4/KH2PO4 pH 7.0, 50 mM NaF were measured in the absence and presence of 1 mM TCEP. Reference values for [θ]222nm of 0 and -36,000 (deg cm2 dmol−1) are indicators of 0% and 100% helicity, respectively (Chen et al., 1974). Figures were made using Excel 2013 (Microsoft Corporation) and GraphPad Prism v5.01 (GraphPad Software).
2.7. Protein labelling
To label FBSS using amide coupling, a sample (~1 mg/ml, 120 μM) was incubated with 1 mM (5/6) TAMRA-SE (G-Biosciences) in 20 mM Tris pH 8, 50 mM NaCl at room temperature for 30 min. Labelled protein was separated from unreacted fluorophore by subjecting the sample twice to a SpinOUT GT-600 desalting column (G-Biosciences). The concentration of amide-coupled TAMRA was determined using UV absorption at 247 nm where TAMRA has an extinction coefficient of 80,000 cm−1 M−1. A labelling efficiency of 0.2 mol TAMRA per mol of FBSS was obtained.
2.8. Fluorescence-based equilibrium titration
Titration experiments were conducted in non-treated black 96-well plates (Costar) using a PHERAstar microplate reader (BMG Labtech) with the fluorescence polarization module for TAMRA (excitation/emission wavelengths 540 nm/590 nm, respectively). For each experiment, the gains of the parallel and perpendicular channels were calibrated with TAMRA in water to a polarization value of 50 mP and a fluorescence intensity close to that of the labelled protein. A blank control (buffer only) was subtracted from intensity readings for all experiments. Oligomerization of FB or FBSS was carried out by serially diluting 200 μM FBSS with buffer (20 mM Tris pH 8, 50 mM NaCl) supplemented with 0.1 μM TAMRA-labelled FBSS as a tracer. For experiments with FB, DTT was added to a final concentration of 2 mM. All titrations were carried out in duplicates. Fluorescence polarization data were analyzed with the MARS Data Analysis Software v3.01 (BMG Labtech). The data for FBSS oligomerization were fitted to the mathematical model for a dimer-tetramer equilibrium adapted from published methods (Veldkamp et al., 2005) using GraphPad Prism v5.01 (GraphPad Software).
2.9. Analytical ultracentrifugation
Sedimentation velocity experiments were performed at 4 °C in a Beckman Coulter Model XL-A analytical ultracentrifuge using 12-mm double sector (2-channel) charcoal filled epon centerpieces enclosed with quartz windows. FB at five concentrations (10 μM, 25 μM, 50 μM, 100 μM and 500 μM) in a sample volume of ~395 μl was loaded into the sample cells. For experiments with reduced protein, 1 mM TCEP was added. Proteins were prepared by dialyzing them against 1L of the desired buffer for 5h at 4 °C; for background subtraction, ~405 μl dialysis buffer after dialysis was used in the reference cells. The optimal wavelengths to measure the absorbance of the samples were determined with test scans carried out at 3000 rpm. Experiments were performed using a four-hole An-60 Ti rotor at 42,000 rpm (141,995×g) with a step size of 0.003 cm. Rotor and cuvette assembly were equilibrated to 4 °C prior to the experiments. We also performed one experiment at 20 °C which revealed the same species as at 4 °C. Solvent density, solvent viscosity, and estimates of the partial specific volume of FB (0.7184 ml/g at 4 °C and 0.7252 ml/g at 20 °C) were calculated using SEDNTERP (Laue et al., 1992). Data were fitted to a continuous size-distribution model using the SEDFIT software (Schuck, 2000). Standardization of S values (S20, w) and high resolution plots were generated using Gussi 1.0.8 (Brautigam and Cole, 2015).
Sedimentation equilibrium experiments were carried out to investigate which oligomeric species were present at 25 μM protein concentration in three different buffer conditions that showed major uniform peaks in sedimentation velocity runs. Experiments were performed in a four-hole An60-Ti rotor using absorbance optics to monitor protein concentration gradients at 236 nm. Experiments were performed at three different speeds of 7,000, 15,000, and 20,000 rpm for the high molecular weight species sedimenting at S20,w of 7.8, or at 18,000, 24,000, and 30,000 rpm for the oligomeric species sedimenting with lower S20,w values of 1.5 and 1.7.
Sedimentation equilibrium experiment data was processed using SEDFIT 9.3b (Schuck, 2000) and final analysis performed using SEDPHAT 12.1b (Vistica et al., 2004). For analysis, we used the model ‘Single Species of Interacting System’ by fitting the molecular weight followed by nonlinear least-squares refinement (NLSR) of the baseline noise level of 0.005 and a subsequent additional refinement of RI and TI noise, with TI noise rotor stretch. For the better fitting data observed under high salt conditions (500 mM NaCl, with or without 1 mM TCEP), a second analysis was performed using the model ‘Monomer-Dimer Self-Association’. Protein concentrations were fixed and mass conservation constraints enabled (Vistica et al., 2004) during the fit to both models. The fit was refined using NLS refinement and by adjusting the noise and bottom position. Data fitting was repeated with alternating optimization methods (Simplex and Marquardt-Levenberg). The quality of the fit was assessed using the local root mean square deviations (r.m.s.d.).
2.10. Cell-based assays
Cell culture and transfections were performed essentially as previously described (Cates et al., 2014). Neuro 2a cells (N2a; American Type Culture Collection) were cultured in EMEM (ATCC) supplemented with 10% heat-inactivated fetal bovine serum (ATCC) in a 5% CO2 humidified atmosphere at 37 °C. Cells were plated into 12-well plates. Twenty-four hours later (when cells were ~95% confluent) cells were transiently co-transfected with WT HA-ΔFosB, with or without FLAG-JunD, using Effectene (Qiagen). Constructs were expressed using the pCDNA3.1 plasmid backbone, and a total of 200 ng DNA was transfected per well. Approximately 48 h post transfection, cells were washed twice with 1 ml PBS, and whole-cell lysates were prepared in modified RIPA buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% (w/v) sodium dodecyl sulfate, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, pH 7.4, protease and phosphatase inhibitors [Sigma Aldrich]) with 10 μM diamide. Samples were prepared in Laemmli buffer with or without 1 mM DTT. Proteins were separated on 4–15% polyacrylamide gradient gels (Criterion System, BioRad), and Western blotting was performed using chemiluminescence (SuperSignal West Dura, Thermo Scientific). Total protein was assayed using Swift Membrane Stain (G-Biosciences) and quantified using ImageJ software (NIH). Primary antibodies were used to detect ΔFosB (Cell Signaling 5G4; 1:500), the FLAG-tag (Cell Signaling 14793S; 1:1000), or the HA-tag (Cell Signaling 3724S; 1:1000).
3. Results
3.1. Self-assembly of ΔFosB bZIP domains
The ΔFosB bZIP domain (FB), in the absence of reducing agent, migrates as a disulfide-bonded species by SDS-PAGE (Fig. 2a). Analysis by mass spectrometry and size exclusion chromatography confirms that, under reducing conditions, the disulfide-bonded species dissociates into monomers (Supplemental Fig. S1). This result is puzzling because it cannot easily be reconciled with the classical arrangement of a canonical AP-1 leucine zipper as seen in the ΔFosB/JunD heterodimer bound to DNA (Yin et al., 2017). There is only one cysteine residue in FB, Cys172, located at the g-position of heptad H(-1), in the DNA-binding region (Fig. 1c). In the canonical arrangement, these two cysteine residues fall on opposite sides of the interhelical core and are too far apart to form a disulfide bond (Fig. 1c; Fig. 1d). Also, in a canonical arrangement, the many glutamate residues at the e- and g-positions of the heptad repeats would approach each other causing electrostatic repulsion destabilizing a canonical coiled coil (Fig. 1b; Fig. 1d). We reasoned therefore, that ΔFosB self-assembles into different, non-canonical arrangements. Alternatively, of course, the DNA-binding region of FB where Cys172 is located, could also be disordered to such a large extent that its linkage via a disulfide bond would be possible.
Fig. 2.

FB (the ΔFosB bZIP) self-assembles in solution. a) Increasing amounts of purified FB (up to 20 μgr) assessed by SDS-PAGE under reducing (100 mM DTT) and non-reducing conditions. Markers shown in the first lane; b) FB (25 μM) adopts different assemblies as a function of ionic strength and reducing conditions in sedimentation velocity experiments; c) Different sedimentation coefficients are observed for FB as a function of protein concentration and buffer conditions by sedimentation velocity; d) Sedimentation equilibrium studies of FB in three different buffers: (1) 20 mM Tris pH 8, 500 mM NaCl, 1 mM TCEP (local r.m.s.d. for the fit 0.012811; Mw 17,116 Da); (2) 20 mM Tris pH 8, 500 mM NaCl (local r.m.s.d. 0.009223; Mw 17,388 Da); and (3) 20 mM Tris pH 8, 50 mM NaCl (local r.m.s.d. 0.043062; >120,000 Da depending on the radius of data used); e) Circular dichroism spectra of FBSS (12 μM, 0.1 mg/ml) in the absence and presence of 1 mM TCEP; f) Oligomerization equilibrium of FB as measured by fluorescence polarization. Unlabeled, oxidized FB, in the absence and presence of DTT, was titrated with 0.1 μM TAMRA-labelled oxidized FB. Dissociation constants (KD) are indicated. Error bars represent standard deviation of two replicates.
To probe the ability of FB to self-assemble, we performed AUC. Sedimentation velocity experiments show that FB oligomerizes as a function of protein concentration, ionic strength, and the presence or absence of reducing agent (Fig. 2b; Fig. 2c). Under relatively low ionic strength (50 mM NaCl) and reducing conditions (1 mM TCEP), a small species (S20,w ~1.5) converts to a larger species (S20,w ~7.6) and then ultimately a population of large assemblies emerges as the protein concentration increases from 10 μM (0.082 mg/ml) to 100 μM (0.825 mg/ml) or more (Fig. 2c). Conversion to larger species occurs at high ionic strength (500 mM NaCl) as well, but at higher protein concentration (500 μM; 4.12 mg/ml). By contrast, in absence of reducing agent, the large S20,w ~7 species is seen already at 10 μM FB (0.082 mg/ml) at low ionic strength (50 mM NaCl). Thus, FB forms assemblies whereby the sole cysteine (Cys172) can form a disulfide bond. Sedimentation equilibrium experiments show that the small S20,w ~1.7 species corresponds to a dimer of FB, while the larger S20,w ~7 species corresponds to a large oligomeric assembly with a molecular weight of >120,000 Da (varying with the exact radius range of the data included) (Fig. 2d). We confirmed by circular dichroism spectroscopy that FB is folded in solution under both non-reducing and reducing conditions with a high helical content (Fig. 2e). To further confirm that FB forms homomeric oligomers in solution, we titrated fluorescently-tagged FB with untagged FB prepared under non-reducing conditions (‘oxidized FB’) in the absence and presence of reducing agent. Analysis of fluorescence polarization (FP) data suggests that FB forms oligomers with micromolar affinities in both cases that are stabilized by disulfide bonds, in agreement with the AUC data (Fig. 2f).
Given the unexpected assembly of FB oligomers, we pursued high-resolution 3D structural information and determined the crystal structures of FB under three different conditions (Table 1). In the oxidized form at high ionic strength, FB reveals a novel tetrameric assembly (FBSS) (Fig. 3a). At lower ionic strength, the reduced form of FB reveals the same tetrameric assembly, but it crystallizes in a different space group (FB Type I) (Fig. 3b). A third structure of FB, again in the reduced form but at high ionic strength, reveals a dimer that is structurally distinct from the other assemblies (FB Type II) (Fig. 3c).
Fig. 3.

3D-structures of FB self-assemblies. a) Crystal structure FBSS; b) Crystal structure FB Type I; c) Crystal structure FB Type II. In panel a) though c), each FB subunit is shown with the basic/DNA-binding region in blue (amino acids N-terminal to residue 179), the leucine-zipper region in red or grey (residues 180–214), and the C-terminal region in green (amino acids C-terminal to residue 215). Cys172 is shown as a yellow sphere. Helix chains are labeled A, B, C, C′, and D, respectively. Interactions between helices that are discussed in the text are labeled (Setting 2, Setting 3, and packing P2 through P5).
3.2. Structural features of ΔFosB bZIP self-assembly
Our crystal structures and the solution studies using AUC and FP demonstrate that FB can self-assemble. Strikingly, none of our structures feature a canonical leucine zipper coiled coil. The tetrameric assemblies FBSS and FB Type I can be interpreted as pairs of parallel coiled coils (helix A/B and helix C/D) or alternatively, as pairs of antiparallel coiled coils (helix A/D and helix B/C) (Fig. 3a; Fig. 3b). The complete tetrameric assembly is contained in the asymmetric unit in FBSS related by three molecular, non-crystallographic 2-fold symmetry axes. The two pairs of parallel helices align so that their Cys172 residues can form disulfide bonds (Fig. 3a; Supplemental Fig. S2). Heptads H1 and H2 of one helix (helix A) interact in a parallel fashion with heptads H1 and H2 of the facing helix (helix B) (see Fig. 1b and Fig. 1c for the nomenclature). In addition, heptad H2 interacts with heptad H5 of an anti-parallel helix (helix D). At the center of the assembly, all four helices come together and form a loose core of eight d-leucines from heptads H3 and H4 of each chain (i.e., LeuA197, LeuA204, LeuB197, LeuB204, LeuC197, LeuD197, LeuC204, and LeuD204). From here on, the helices swap partners, so that heptad H5 of helix A interacts with heptads H2 and H3 of the anti-parallel helix D, and no longer with the parallel helix B. The same tetrameric assembly is seen in FB Type I (Fig. 3b; Supplemental Fig. S3) with intact disulfide bonds even though the protein was initially reduced (Supplemental Fig. S2 and Methods). In this case, however, the three molecular axes of the tetramer coincide with crystallographic axes. FB can also form yet another distinct assembly under reducing conditions, an anti-parallel dimer (FB Type II) (Fig. 3c). Here, three FB subunits are found in the asymmetric unit. One FB subunit is well ordered and reveals a helical DNA-binding region; crystallographic symmetry generates a dimer with intimate interactions (helix C and helix C’). The two other FB subunits generate an identical dimer via non-crystallographic symmetry, but their DNA-binding regions are disordered. Because the helices are anti-parallel, Cys172 cannot form a disulfide bond in FB Type II (Supplemental Fig. S2).
The three crystal structures contain eight independent FB subunits. Even though no DNA is present and the FB subunits are incorporated into different assemblies, comparison reveals that the basic region (residues Lys157-Arg177) is ordered in five out of eight subunits, partially ordered in two others, and disordered in only one structure, while the DNA-binding motifs (residues Asn165-Arg173) are fully helical in six out of eight subunits, forming large continuous helices with the rest of the respective leucine-zipper regions. Superposition using heptads H3–H4 reveals that the long helices curve in different directions, with the curvatures becoming pronounced already within the leucine zipper affecting heptad H2 and the residues that are N-terminal (Fig. 4). Different curvatures are also observed for the individual FB monomers within a single crystal form. Our crystal structures thus reveal remarkable plasticity within the FB molecule and the ability to self-assemble into dramatically different arrangements.
Fig. 4.
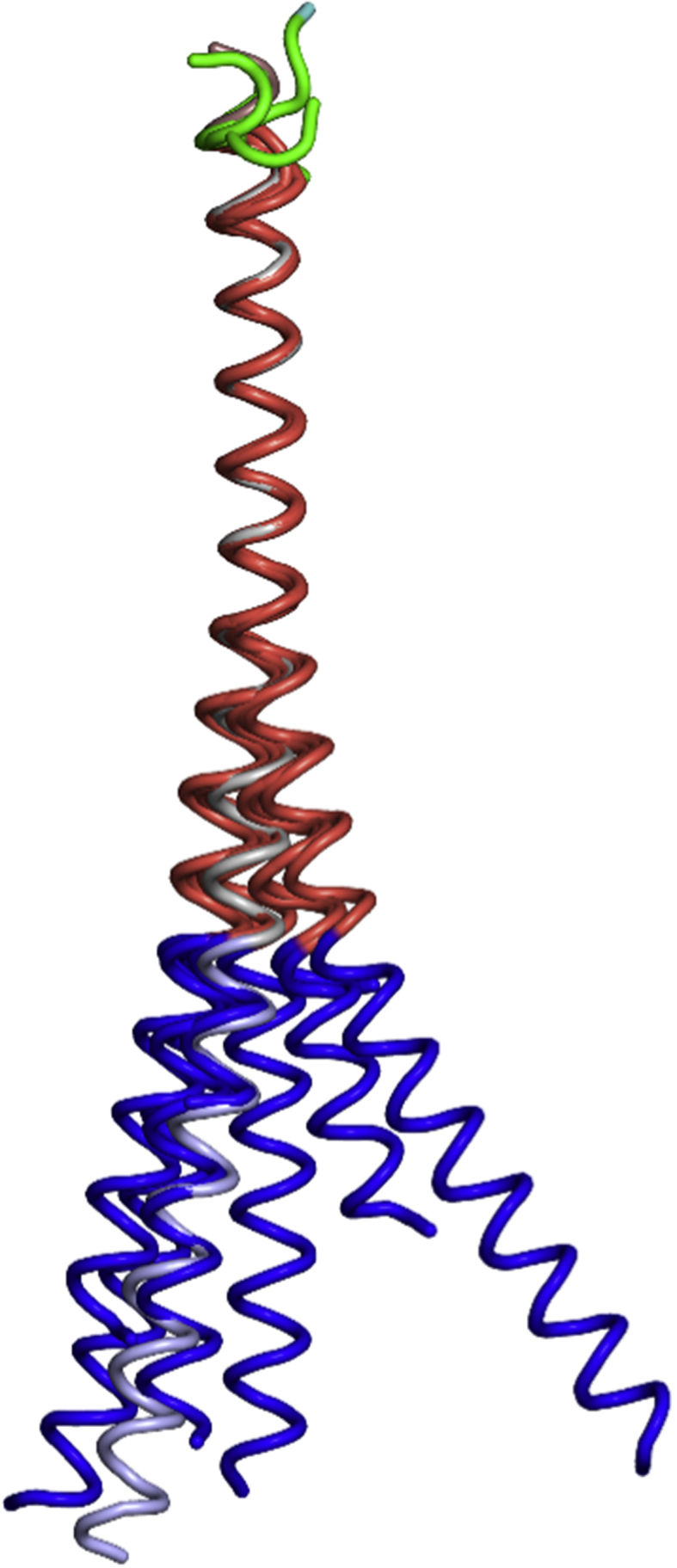
Superposition of FB subunits reveals conformational variability. Superposition of all eight independent FB monomers, FBSS (A, B, C, D), FB Type I (A) and FB Type II (A, B, C) using K194 – Arg210 (which contains H3 – H4). In grey, FB from the ΔFosB/JunD bZIP + DNA structure is also shown (PDB ID: 5VPE).
3.3. Novel interactions mediate self-assembly of ΔFosB
The d-leucines in the heptad repeats H1–H5 (Leu183, Leu190, Leu197, Leu204, and Leu211) play fundamental and defining roles in forming the non-canonical interfaces between helices in the FB structures. To delineate the binding pockets for these hallmark d-leucines, we assessed their setting, characterized by the two sequential residues on the facing helix in between which they insert, and their packing, characterized by the additional, nearby residues that complete each binding pocket (Fig. 3; Fig. 5). Compared to the canonical Setting 1 of the ΔFosB/JunD heterodimer, two novel settings are observed in the three crystal structures, Setting 2 and Setting 3. In Setting 2, found in FBSS and FB Type I between pairs of parallel helices, the d-leucines from one helix insert between sequential g’-position residues (Fig. 5b). This arrangement places the d-leucines from the two interacting helices side-by-side (forming a handshake, roughly perpendicular to the axes of the helices). Also, in Setting 2, the facing helix (helix B) rotates clockwise by about 55° with respect to helix A and docks its d-leucines on the other side of the d-position leucines of the parent helix (helix A). By contrast, in Setting 3, the d-leucines from one helix insert between two sequential d’-leucines on the facing helix, a setting found between antiparallel helices in FBSS, FB Type I and II (Fig. 5b). In this setting, the leucines form a vertical stack in a staggered fashion. Thus, the key difference between the canonical Setting 1 in ΔFosB/JunD, and the novel Setting 2 and Setting 3, is how the d-leucines insert into the facing helix; i.e., between sequential a’-positions in Setting 1, between g’-positions in Setting 2, and between d’-leucines in Setting 3, respectively, and the changes in relative disposition of the interacting helices (i.e. rotated, parallel, anti-parallel and/or translated).
Fig. 5.

Mechanisms of FB assembly. a) helical interactions observed in ΔFosB/JunD and in ΔFosB self-assemblies. From left to right, ΔFosB/JunD (packing P1; PDB ID:5VPE), FB Type I (packing P2, P3, P4 and P5) and FB Type II (packing P4). d-leucines are shown in yellow. Cys172 is shown as a yellow sphere. Helix chains are indicated as A, B, C, C′, and D; b) Close-up of the binding pockets for the d-leucines (each from a helical segment shown in red) as depicted above in a). Each d-leucine (L) is shown in gold together with its surrounding residues (≤5 Å away) from the facing helix (or helices) that complete the binding pocket. Coloring scheme for the heptad positions: a’ (green), c’ (magenta), d’ (red), e’ (cyan), and g’ (blue). Coloring of the helices as in a).
The pocket around the d-leucines is completed in dramatically different ways in the different settings. While the canonical packing, referred to as P1 (found in Setting 1) uses residues from the d’ and e’-positions from the facing parallel helix to complete the pocket around each d-leucine, packing P2 (found in Setting 2) recruits residues from the d’ and c’-positions on the facing parallel helix (Fig. 5b). Much more varied pockets are found between antiparallel helices, seen in the packings P3, P4 and P5 (Fig. 5b); key here is that a variable number of residues pack around each d-leucine (from two to eight), drawing from the d’, a’, c’, and/or g’-positions on one or two facing helices. In the most extreme case, LeuD190 at the core of the tetrameric assembly interacts with a total of eight different residues, four from each of the two facing helices. To accommodate these novel packing arrangements, many d-leucines change their rotamer conformation. Therefore, as the helices swap their interactions from one facing helix to another in the tetrameric assembly, the d-leucines bury into different pockets (packing P2, P3, P4, and P5). It is the ability of these d-leucines to form different hydrophobic, interhelical cores that permits the diverse and structurally drastically different assemblies to form.
3.4. ΔFosB assemblies in cells
To assess whether full-length ΔFosB self-assembles in a cellular environment, we leveraged the endogenous cysteine residues in a cell-based assay and exploited them as a ‘reporter of proximity’. Cys172 plays a key role in stabilizing the assemblies in the crystal structures FBSS and FB Type I, where it forms a disulfide bond between adjacent subunits (Fig. 3); non-reducing conditions also stabilize large species in solution (Fig. 2). In complex with JunD, ΔFosB Cys172 forms a disulfide bond as well, with JunD Cys285, pending a large conformational rearrangement that is only possible in the absence of DNA and underlies a putative redox switch governing DNA-binding (Yin et al., 2017). The cysteine residues in ΔFosB and JunD therefore provide an endogenous tool to probe their (self-)assembly in cells. To this end, we transfected hemagglutinin (HA)-tagged ΔFosB into Neuro 2a cells, a neuroblastoma line with neuron-like properties (LePage et al., 2005). To probe the formation of potential homomers versus heteromers, we also co-transfected FLAG-tagged JunD. During cell lysis, the samples were exposed to mildly oxidizing conditions (10 μM diamide) to test whether ΔFosB monomers were in such close proximity that their cysteine residues could form a disulfide bond covalently holding them together. Indeed, when the diamide-treated samples were analyzed by SDS-PAGE in the absence of reducing agents, ΔFosB was partly trapped in a ~75 kDa species, suggesting that ΔFosB adopts an arrangement in cells that permits a disulfide bond to form (Fig. 6). Under these same oxidizing conditions, HA-ΔFosB also covalently bound to JunD, forming an ~85–90 kDa complex. By contrast, under reducing conditions (1 mM DTT), ΔFosB reverted to a 37 kDa monomer and JunD to a ~47–50 kDa monomer (Fig. 6). Taken together, in neuron-like cells, ΔFosB can self-assemble generating species that are consistent with molecular arrangements that we see in our crystal structures, and it can assemble with JunD as well.
Fig. 6.
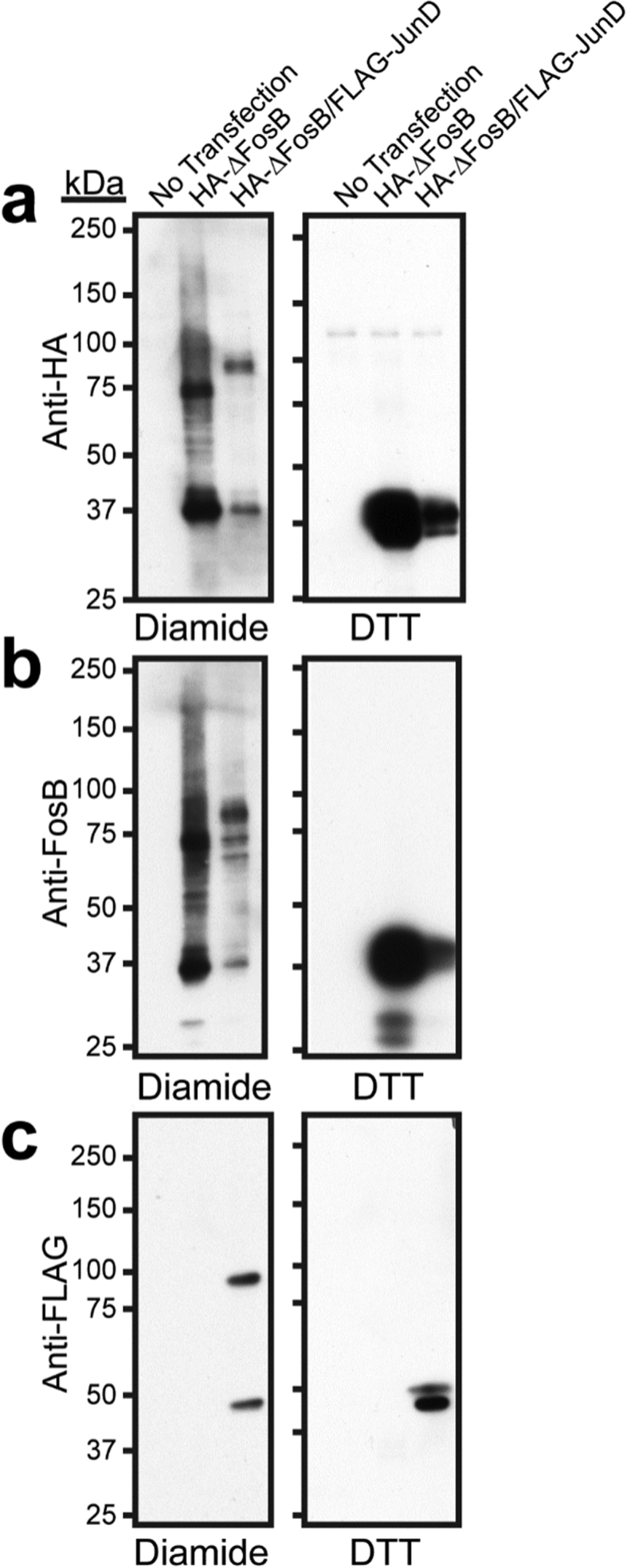
ΔFosB forms self-assemblies and assemblies with JunD in neuron-like cells. Neuro 2a cells were either not transfected or transiently transfected with HA-tagged ΔFosB, with or without FLAG-tagged JunD. Cells were homogenized and samples prepared in mildly oxidizing (diamide) or reducing (DTT) conditions. a) Western blot analysis using anti-HA antibodies (Cell Signaling 3724S; 1:1000) reveals HA-tagged ΔFosB species; b) Western blot analysis using anti-ΔFosB/FosB antibodies (Cell Signaling 5G4; 1:500) reveals ΔFosB; c) Western blot analysis using anti-FLAG antibodies (Cell Signaling 14793S; 1:1000) reveals FLAG-tagged JunD.
4. Discussion
4.1. Summary
ΔFosB has unique, well-described physiological roles in drug addiction, stress resilience, and cognition (Nestler, 2015, Eagle et al., 2015, Corbett et al., 2017). With more than 50 bZIP-domain containing DNA-binding proteins (bZIPs) found in humans, bZIPs assemble in a myriad of different combinations, and their choice of their partner dictates which genes they regulate and how they impact gene expression (Newman and Keating, 2003, Reinke et al., 2013, Reinke et al., 2013, Lupas et al., 2017, Rodríguez-Martínez et al., 2017, Wilson and Filipp, 2018). However, the molecular properties and precise partners of ΔFosB are not well understood.
Here, we reveal that ΔFosB can self-assemble, in addition to forming heterodimers with JunD. Our crystal structures demonstrate that the ΔFosB bZIP domain can pack in dramatically different molecular architectures. The characteristic leucines of the leucine zipper still mediate key interactions between the helices but deviate from the canonical bZIP heterodimers seen in ΔFosB/JunD and also c-Fos/c-Jun. Strikingly, the basic regions that include the DNA-binding motifs are fully helical and not disordered. In solution, ΔFosB self-assembles and can transition between different species in vitro following changes in the protein concentration, oxidizing-reducing environment, and ionic strength that we used to mimic cellular triggers. By exploiting its putative redox switch to trap oligomers, we show that ΔFosB self-assembles in Neuro 2a cells as well. Taken together, our data suggest that ΔFosB can form stable molecular assemblies on its own and that, as the protein accumulates in response to chronic insult, these assemblies may form in vivo as well.
4.2. ΔFosB bZIP domains adopt multiple molecular architectures
In total, nine different crystal structures of the ΔFosB bZIP are now available, alone and in complex with JunD (Fig. 3; Yin et al., 2017). In complex with JunD and bound to DNA, the ΔFosB bZIP aligns in parallel so that the helices can clamp onto double-stranded DNA like forceps (Yin et al., 2017). In the absence of DNA, the ΔFosB/JunD bZIP heterodimer can further assemble into tetramers (Yin et al., 2017). Here, we reveal that the ΔFosB bZIP domain on its own, in absence of DNA, can also generate a tetrameric assembly with both parallel and anti-parallel subunits, as well as a dimer with anti-parallel subunits (Fig. 3). Capturing such structural diversity for a single bZIP transcription factor is unprecedented. In GCN4, architectural heterogeneity was achieved, but required strategic mutagenesis (Grigoryan and Keating, 2008). For example, the mutations Glu20Cys and Glu20Ser (e-position) alter the balance between parallel and antiparallel coiled coils (Yadav et al., 2006), while Asn16Gln (buried at the dimerization interface) generates dimers as well as trimers of GCN4 (Gonzalez et al., 1996). The leucine zipper of cell cycle-regulated Nek2 kinase also appears to adopt two different assemblies although their structural basis is not known (Croasdale et al., 2011). Yet ΔFosB produces diverse assemblies using solely its native amino acid sequence. bZIP assemblies are thought to be energetically very close and separated by low energy barriers (Grigoryan and Keating, 2008, Lupas et al., 2017), generating an efficient way to produce a portfolio of species with dramatically different functional consequences.
4.3. ΔFosB utilizes multiple interaction modes
The wealth of structural information for ΔFosB represented in nine different crystal structures (Fig. 3; Yin et al., 2017) permits the elucidation of different assembly mechanisms. In canonical bZIP dimers, the a- and d-position residues form the interface between the helices and promote stability by forming a hydrophobic core, exploiting van der Waals interactions that are particularly favorable when the packing is highly complementary (Lupas et al., 2017, Grigoryan and Keating, 2008, Miller, 2009, Vinson et al., 2002). The residues at the g- and e-positions are suggested to determine which bZIPs will bind each other (e.g., through electrostatic steering) and guide assembly (Lupas et al., 2017, Grigoryan and Keating, 2008, Miller, 2009, Vinson et al., 2002, Vinson et al., 2006). For instance, repulsive interhelical interactions between g-position residues of one helix and those at the e’-position of the facing helix in c-Jun and c-Fos homodimers are thought to promote preferential c-Fos/c-Jun heterodimers formation over c-Fos homodimers (Miller, 2009, Vinson et al., 2002, Vinson et al., 2006, Mason et al., 2006, O'Shea et al., 1992). ΔFosB overcomes this barrier, however, by utilizing novel interactions between the helices. The characteristic d-leucines still play a dominant role in mediating the interaction between helices, and they do so by harnessing helical rotations and translations, different leucine rotamers, and/or recruiting residues from more than one helix to complete their binding pockets. The ΔFosB bZIP does indeed contain a relatively high number of negatively charged glutamate residues at the e- and g-positions (7 out of the 10 e-/g-positions in heptads H1–H5) (Fig. 1b). In the ΔFosB/JunD bZIP heterodimer, six of these residues are neutralized in salt bridges with lysine or arginine residues from either JunD or ΔFosB. In the homomeric ΔFosB tetramer, because the helices are rotated, the e- and g-positions do not approach each other. The glutamate residues at the e-position line the length of the helical bundle on the outside, while the glutamate residues at the g-position line the inside of the tetrameric core where they are largely disordered perhaps to avoid electrostatic repulsion (Supplemental Fig. S4). The intrinsic flexibility of the ΔFosB bZIP helix (Fig. 4) likely also aids assembly because it allows ΔFosB to splay apart to engage in energetically favorable and stable packing interactions in the different assemblies. General ‘rules of engagement’ for canonical bZIP dimers have been identified over the past two decades, based on Setting 1, packing P1, and they typically involve at least four or five heptads to form a coiled coil (Vinson et al., 2006). Yet, it has proven challenging to use these general rules to accurately predict interactions and to engineer them (Vinson et al., 2002, Newman and Keating, 2003, Mason et al., 2006, Grigoryan and Keating, 2006, Grigoryan et al., 2009). Our results demonstrate that the ΔFosB bZIP domain can readily assemble into completely novel architectures that deviate from these rules, suggesting that computational approaches to design bZIP-based coiled coils should be expanded.
4.4. Role of Cys172 and Cys222 in ΔFosB
ΔFosB Cys172 has been implicated as part of a redox switch regulating binding to DNA (Yin et al., 2017). Releasing the disulfide bond between Cys172 (ΔFosB) and Cys279 (JunD; mouse numbering) triggers a large conformational change in ΔFosB that enables the two DNA-binding motifs to separate and insert into the major groove on either side of DNA (Yin et al., 2017). The ΔFosB bZIP also forms a disulfide bonded species, in solution in the absence of JunD (Fig. 2a). In the ΔFosB tetramer, pairs of helices rotate and align with their basic regions in parallel so that their Cys172 can form disulfide bonds with each other, and the DNA-binding regions are positioned so that they could also insert into the major groove of DNA if they flex apart sufficiently, i.e., the ΔFosB tetramers may be DNA-binding compatible. However, we cannot exclude that ΔFosB will form a completely different assembly when it binds DNA. In addition, our structures suggest a functional role for ΔFosB Cys222 as well, which is directly C-terminal to the leucine zipper. In vivo, Cys222 may regulate the assemblies of ΔFosB by locking the bZIP domains into parallel dimers that are compatible with DNA-binding, thereby preventing the formation of other assemblies, such as anti-parallel dimers and/or tetramers that trigger other functional consequences. There is precedence for disulfide bonds to stabilize the bZIP dimers of AP-1 transcription factors, because ATF4 and ATF5 form homodimers and disulfide bonds that span the leucine zipper region (via a Cys residue at the a-position of heptad H2 (Fig. 1b; Ciaccio et al., 2008). In ΔFosB, such a ‘zip-lock’ cysteine could prevent the exchange of subunits or partners at cellular locations where an oxidative environment is maintained.
4.5. The DNA-binding regions of ΔFosB are helical in absence of DNA
The basic regions (Lys157-Arg177) and their DNA-binding motifs (Asn165-Arg173) are helical in the majority of the eight independent ΔFosB subunits. Previously, the DNA-binding motifs in bZIPs have been assumed to be disordered without the negatively charged phosphate groups of DNA neutralizing the high number of positively charged residues (Miller, 2009, Das et al., 2012). In ΔFosB, while they do make interactions with nearby chains in the crystal packing and thus may be stabilized somewhat, they also exist in solution as indicated by our circular dichroism spectroscopy experiments (Fig. 2e). The DNA-binding motifs in the ΔFosB/JunD bZIP heterodimer in the DNA free-form are also helical in all seven independent copies from four different crystal structures (Yin et al., 2017). Therefore, the basic region of ΔFosB and its DNA-binding motifs appear to have a high intrinsic propensity for helix formation (Fig. 2e). The ability of the basic regions in ΔFosB to form stable helices in the absence of DNA is important, because helical propensity is an integral driver of coiled coil stability (Mason et al., 2006). It may also be a crucial biological variable to regulate the assembly and function of different bZIPs. For example, in GCN4 and C/EBPβ, the DNA motifs are unfolded in solution in absence of DNA (Miller, 2009), while they are helical in ATF4 (PDB ID: 1CI6) (Podust et al., 2001). Therefore, the DNA-binding motifs do not need to be buried in the major groove of DNA to adopt an α-helical conformation. The propensity to form α-helices is enhanced by residues directly N-terminal to the DNA-binding motifs as well as perhaps by co-factor proteins that bind in proximity to these residues (Das et al., 2012). Furthermore, phosphorylation of Ser/Thr residues has been shown to impact the stability of α-helices (Szilák et al., 1997a, Szilák et al., 1997b, Hendus-Altenburger et al., 2017). Indeed, ΔFosB Thr149 located N-terminally to the DNA-binding motif is phosphorylated by CAMKII, and the phosphomimetic mutant Thr149Asp increases the transcriptional activity in reporter assays (Cates et al., 2014). ΔFosB Thr180 is buried at the fulcrum of the forceps in the ΔFosB/JunD bZIP structure where it would not be readily available to kinases as a substrate without prior protein conformational rearrangements. However, in the context of the ΔFosB tetramer, Thr180 is solvent accessible and could easily be phosphorylated, supporting the existence of such tetrameric assemblies in vivo. Taken together, the high helical propensity of the basic region of ΔFosB may promote self-assembly by enabling the subunits to pack in a variety of energetically favorable assemblies.
4.6. Physiological relevance of structural plasticity in ΔFosB
ΔFosB is an unusual member of the AP-1 class of bZIPs because it is extremely stable; its protein accumulates to high levels in brain and remains high in neurons even after the stimuli (i.e., chronic exposure to drugs of abuse, stress or seizures) have stopped (Nestler, 2015). Some of ΔFosB's stability can be attributed to the absence of two C-terminal degron domains found in the much shorter lived FosB and to phosphorylation of a serine in its disordered N-terminal domain (Nestler, 2015). However, these features likely do not fully account for all of the increased stability, and the exact nature of the accumulated ΔFosB species in vivo has remained unknown. Certainly in vitro, ΔFosB can both homodimerize and heterodimerize with JunD and both forms bind to AP-1 DNA consensus sequences (Jorissen et al., 2007, Wang et al., 2012). Our present studies also show that the ΔFosB bZIP domain is sufficient to self-assemble already at low protein concentration (i.e., 10 μM; 0.082 mg/ml, the lowest concentration we could monitor with our AUC system) generating dimers at high ionic strength and larger assemblies at low ionic strength (Fig. 2). These results are consistent with our previous studies revealing that full-length ΔFosB protein can form dimers in the range of 1–55.4 μM (0.026–1.465 mg/ml) (Jorissen et al., 2007). In neuron-like Neuro 2a cells, we show that assemblies of ΔFosB alone as well as in complex with JunD are indeed formed as ~75 kDa and ~85–90 kDa complexes, respectively (Fig. 6). Co-transfection of ΔFosB with JunD resulted in predominantly ~85–90 kDa ΔFosB/JunD complexes being formed with little ~75 kDa ΔFosB self-assemblies, suggesting that ΔFosB preferentially forms heteromeric assemblies with JunD when sufficient JunD is available, in agreement with previous studies of c-Fos/c-Jun (Miller, 2009, Vinson et al., 2002, Vinson et al., 2006, Mason et al., 2006, O'Shea et al., 1992). We cannot, however, assess what percentage of ΔFosB is oligomeric inside cells, because very mild oxidizing conditions (10 μM diamide) were used to trap ΔFosB assemblies in order to prevent artificial crosslinking. This precaution was necessary, because mouse ΔFosB used here has four cysteine residues (Cys172 and Cys222 in the bZIP domain but also two cysteine residues in the intrinsically disordered N-terminal region). Mouse JunD has only two cysteine residues that are equivalent to Cys172 and Cys222 in ΔFosB. Also interactions within assemblies that are not covalently attached fall apart during SDS-PAGE analysis and hence escape detection, e.g., a tetramer such as FBSS/FB Type I would resolve into a dimer. The broad range of molecular species formed by ΔFosB, alone or in combination with JunD, is likely unique to this alternative splice form of FosB. The absence of the extensive C-terminal region seen in full-length FosB (but also other Fos proteins such as c-Fos, FRA-1, and FRA-2) (Fig. 1a) not only removes destabilizing degron domains (Carle et al., 2007), but also may boost the formation of higher order self-assemblies by removing steric hindrance thereby promoting the domain-swapped helices seen in the ΔFosB tetrameric assemblies. Indeed, removing the C-terminal 71 a.a. from FosB (to 1–317 a.a.) doubles the half-life of the protein in a proteosomally-dependent way, while removing an additional 40 a.a. (to 1–277 a.a.) again doubles the half-life but now in a proteosomally-independent way, though the remaining protein is still half as stable as ΔFosB (1–237 a.a.) (Carle et al., 2007).
5. Conclusions
Our findings are intriguing in light of the high levels of ΔFosB protein that accumulate in the brain upon chronic administration of drugs of abuse, stress, or seizures. It is increasingly recognized that (aberrant) oligomerization and/or aggregation of particular proteins can be associated with a disease. For example, a wide portfolio of different proteins can generate amyloid-like structures that are associated with different diseases (Iadanza et al., 2018). Oligomerization can be regulated not just by the exact amino acid sequence of the monomeric building block (further altered via alternative splicing, mutations, and/or post-translational modifications), but also via its protein levels, leading to the disease state (Iadanza et al., 2018). Specific structural assemblies associate with particular disease states, for example, distinct complexes of tau protein are isolated from post-mortem brains of people with Alzheimer's versus Pick's Disease (Fitzpatrick et al., 2017, Iadanza et al., 2018, Falcon et al., 2018). Hence, it is possible that, as protein levels of ΔFosB rise, specific molecular assemblies form in select brain regions that associate with the pathogenesis of distinct pathological states (e.g., drug addiction, cognitive decline linked to Alzheimer's disease, etc.). The concentration of ΔFosB versus that of other partners, e.g., JunD, would then be a key determinant of which complex is favored and its stability. For this reason, it will be very important in future work to manipulate ΔFosB protein levels in cell culture and in vivo to assess whether accumulation of ΔFosB leads to stable self-assemblies that amass as a function of protein concentration (Fig. 7). Determining these parameters in vivo and correlating them with the transcriptional and functional states of neurons in the brain will be major challenges in future studies, but could also provide an enormous leap in our understanding of activity dependent changes in neuronal gene expression and brain function.
Fig. 7.

Model of ΔFosB accumulation and self-assembly. The ΔFosB protein accumulates in specific regions of the brain upon exposure to chronic insult such as drugs of abuse, stress, or seizures. Once a cellular threshold level of ΔFosB protein is reached, swamping ambient concentrations of JunD, self-assembly occurs generating several non-canonical molecular arrangements. The functional impact of these species must now be elucidated. For clarity, the disordered regions are not portrayed.
Data Deposition
Atomic coordinates and structure factors for the FBSS, FB Type I and FB Type II crystal structures are deposited in the Protein Data Bank under accession codes 6UCI, 6UCL, and 6UCM, respectively. The authors declare no competing financial interests. Correspondence and requests for materials should be addressed to G.R. (garudenk@utmb.edu).
Funding
This study was funded by NIH R01 DA040621, the Brain and Behavior Research Foundation, and the Sealy Center for Structural Biology at the University of Texas Medical Branch (UTMB).
CRediT authorship contribution statement
Zhou Yin: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Resources, Writing - original draft, Visualization, Writing - review & editing. Harikanth Venkannagari: Methodology, Validation, Formal analysis, Investigation, Visualization, Writing - review & editing. Haley Lynch: Methodology, Investigation, Resources, Visualization. Galina Aglyamova: Methodology, Investigation, Resources. Mukund Bhandari: Methodology, Investigation, Resources. Mischa Machius: Validation, Formal analysis, Investigation, Writing - review & editing, Visualization, Supervision. Eric J. Nestler: Conceptualization, Resources, Writing - review & editing, Funding acquisition. Alfred J. Robison: Conceptualization, Resources, Writing - review & editing, Supervision. Gabby Rudenko: Conceptualization, Resources, Data curation, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare no competing interests.
Acknowledgements
We thank the staff at the Advanced Photon Source Beamlines 17-ID, 19-ID, and 21-ID for assistance during data collection. DNA-sequencing and protein mass-spectrometry services were provided by the Next Generation Sequencing Core Facility and the Mass Spectrometry Core at UTMB. Drs. M. White and L. Holthauzen at the Sealy Center for Structural Biology and Molecular Biophysics at UTMB are thanked for helpful discussions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crstbi.2019.12.001.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam C.A. Chapter Five - Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. In: Cole J.L., editor. Methods in Enzymology. Academic Press; 2015. pp. 109–133. [DOI] [PubMed] [Google Scholar]
- Carle T.L., Ohnishi Y.N., Ohnishi Y.H., Alibhai I.N., Wilkinson M.B., Kumar A., Nestler E.J. Proteasome-dependent and -independent mechanisms for FosB destabilization: identification of FosB degron domains and implications for DeltaFosB stability. Eur. J. Neurosci. 2007;25:3009–3019. doi: 10.1111/j.1460-9568.2007.05575.x. [DOI] [PubMed] [Google Scholar]
- Cates H.M., Thibault M., Pfau M., Heller E., Eagle A., Gajewski P., Bagot R., Colangelo C., Abbott T., Rudenko G. Threonine 149 phosphorylation enhances ΔFosB transcriptional activity to control psychomotor responses to cocaine. J. Neurosci. 2014;34:11461–11469. doi: 10.1523/JNEUROSCI.1611-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.H., Yang J.T., Chau K.H. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- Chen J., Kelz M.B., Hope B.T., Nakabeppu Y., Nestler E.J. Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J. Neurosci. 1997;17:4933–4941. doi: 10.1523/JNEUROSCI.17-13-04933.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaccio N.A., Moreno M.L., Bauer R.L., Laurence J.S. High-yield expression in E. coli and refolding of the bZIP domain of activating transcription factor 5. Protein Expr. Purif. 2008;62:235–243. doi: 10.1016/j.pep.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett B.F., You J.C., Zhang X., Pyfer M.S., Tosi U., Iascone D.M., Petrof I., Hazra A., Fu C.-H., Stephens G.S. ΔFosB Regulates Gene Expression and Cognitive Dysfunction in a Mouse Model of Alzheimer's Disease. Cell Rep. 2017;20:344–355. doi: 10.1016/j.celrep.2017.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croasdale R., Ivins F.J., Muskett F., Daviter T., Scott D.J., Hardy T., Smerdon S.J., Fry A.M., Pfuhl M. An undecided coiled coil: the leucine zipper of Nek2 kinase exhibits atypical conformational exchange dynamics. J. Biol. Chem. 2011;286:27537–27547. doi: 10.1074/jbc.M110.196972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.K., Crick S.L., Pappu R.V. N-terminal segments modulate the α-helical propensities of the intrinsically disordered basic regions of bZIP proteins. J. Mol. Biol. 2012;416:287–299. doi: 10.1016/j.jmb.2011.12.043. [DOI] [PubMed] [Google Scholar]
- Eagle A.L., Gajewski P.A., Yang M., Kechner M.E., Al Masraf B.S., Kennedy P.J., Wang H., Mazei-Robison M.S., Robison A.J. Experience-Dependent Induction of Hippocampal ΔFosB Controls Learning. J. Neurosci. 2015;35:13773–13783. doi: 10.1523/JNEUROSCI.2083-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle A.L., Williams E.S., Beatty J.A., Cox C.L., Robison A.J. ΔFosB Decreases Excitability of Dorsal Hippocampal CA1 Neurons. eNeuro. 2018;5 doi: 10.1523/ENEURO.0104-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B., Zhang W., Murzin A.G., Murshudov G., Garringer H.J., Vidal R., Crowther R.A., Ghetti B., Scheres S.H.W., Goedert M. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. 2018;561:137–140. doi: 10.1038/s41586-018-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A.W.P., Falcon B., He S., Murzin A.G., Murshudov G., Garringer H.J., Crowther R.A., Ghetti B., Goedert M., Scheres S.H.W. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547:185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.N., Harrison S.C. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez L., Woolfson D.N., Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat. Struct. Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- Grigoryan G., Keating A.E. Structure-based prediction of bZIP partnering specificity. J. Mol. Biol. 2006;355:1125–1142. doi: 10.1016/j.jmb.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Grigoryan G., Keating A.E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan G., Reinke A.W., Keating A.E. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendus-Altenburger R., Lambrughi M., Terkelsen T., Pedersen S.F., Papaleo E., Lindorff-Larsen K., Kragelund B.B. A phosphorylation-motif for tuneable helix stabilisation in intrinsically disordered proteins - Lessons from the sodium proton exchanger 1 (NHE1) Cell Signal. 2017;37:40–51. doi: 10.1016/j.cellsig.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Hiroi N., Marek G.J., Brown J.R., Ye H., Saudou F., Vaidya V.A., Duman R.S., Greenberg M.E., Nestler E.J. Essential role of the fosB gene in molecular, cellular, and behavioral actions of chronic electroconvulsive seizures. J. Neurosci. 1998;18:6952–6962. doi: 10.1523/JNEUROSCI.18-17-06952.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope B.T., Nye H.E., Kelz M.B., Self D.W., Iadarola M.J., Nakabeppu Y., Duman R.S., Nestler E.J. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Iadanza M.G., Jackson M.P., Hewitt E.W., Ranson N.A., Radford S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell. Biol. 2018 doi: 10.1038/s41580-018-0060-8. [DOI] [PubMed] [Google Scholar]
- Jorissen H.J.M.M., Ulery P.G., Henry L., Gourneni S., Nestler E.J., Rudenko G. Dimerization and DNA-binding properties of the transcription factor DeltaFosB. Biochemistry. 2007;46:8360–8372. doi: 10.1021/bi700494v. [DOI] [PubMed] [Google Scholar]
- Kohler J.J., Schepartz A. Kinetic studies of Fos.Jun.DNA complex formation: DNA binding prior to dimerization. Biochemistry. 2001;40:130–142. doi: 10.1021/bi001881p. [DOI] [PubMed] [Google Scholar]
- Laue T.M., Shah B.D., Ridgeway T.M., Pelletier S.L. Analytical Ultracentrifugation in Biochemistry and Polymer Science. In: Harding S., Rowe A., editors. Royal Society of Chemistry. 1992. pp. 90–125. [Google Scholar]
- LePage K.T., Dickey R.W., Gerwick W.H., Jester E.L., Murray T.F. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit. Rev. Neurobiol. 2005;17:27–50. doi: 10.1615/critrevneurobiol.v17.i1.20. [DOI] [PubMed] [Google Scholar]
- Lupas A.N., Bassler J., Dunin-Horkawicz S. The Structure and Topology of α-Helical Coiled Coils. In: Parry D.A.D., Squire J.M., editors. Fibrous Proteins: Structures and Mechanisms. Springer International Publishing; 2017. pp. 95–129. [Google Scholar]
- Mason J.M., Schmitz M.A., Müller K.M., Arndt K.M. Semirational design of Jun-Fos coiled coils with increased affinity: Universal implications for leucine zipper prediction and design. Proc. Natl. Acad. Sci. USA. 2006;103:8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. The importance of being flexible: the case of basic region leucine zipper transcriptional regulators. Curr Protein Pept Sci. 2009;10:244–269. doi: 10.2174/138920309788452164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler E.J. ΔFosB: a transcriptional regulator of stress and antidepressant responses. Eur. J. Pharmacol. 2015;753:66–72. doi: 10.1016/j.ejphar.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.R.S., Keating A.E. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science. 2003;300:2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- O'Shea E.K., Rutkowski R., Kim P.S. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell. 1992;68:699–708. doi: 10.1016/0092-8674(92)90145-3. [DOI] [PubMed] [Google Scholar]
- Podust L.M., Krezel A.M., Kim Y. Crystal structure of the CCAAT box/enhancer-binding protein beta activating transcription factor-4 basic leucine zipper heterodimer in the absence of DNA. J. Biol. Chem. 2001;276:505–513. doi: 10.1074/jbc.M005594200. [DOI] [PubMed] [Google Scholar]
- Reinke A.W., Baek J., Ashenberg O., Keating A.E. Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science. 2013;340:730–734. doi: 10.1126/science.1233465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Martínez J.A., Reinke A.W., Bhimsaria D., Keating A.E., Ansari A.Z. Combinatorial bZIP dimers display complex DNA-binding specificity landscapes. eLife. 2017;6 doi: 10.7554/eLife.19272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilák L., Moitra J., Vinson C. Design of a leucine zipper coiled coil stabilized 1.4 kcal mol-1 by phosphorylation of a serine in the e position. Protein Sci. 1997;6:1273–1283. doi: 10.1002/pro.5560060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilák L., Moitra J., Krylov D., Vinson C. Phosphorylation destabilizes alpha-helices. Nat. Struct. Biol. 1997;4:112–114. doi: 10.1038/nsb0297-112. [DOI] [PubMed] [Google Scholar]
- Ulery-Reynolds P.G., Castillo M.A., Vialou V., Russo S.J., Nestler E.J. Phosphorylation of DeltaFosB mediates its stability in vivo. Neuroscience. 2009;158:369–372. doi: 10.1016/j.neuroscience.2008.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp C.T., Peterson F.C., Pelzek A.J., Volkman B.F. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005;14:1071–1081. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C., Myakishev M., Acharya A., Mir A.A., Moll J.R., Bonovich M. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell. Biol. 2002;22:6321–6335. doi: 10.1128/MCB.22.18.6321-6335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C., Acharya A., Taparowsky E.J. Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim. Biophys. Acta. 2006;1759:4–12. doi: 10.1016/j.bbaexp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R.A., Rouault T.A., Schuck P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Wang Y., Cesena T.I., Ohnishi Y., Burger-Caplan R., Lam V., Kirchhoff P.D., Larsen S.D., Larsen M.J., Nestler E.J., Rudenko G. Small molecule screening identifies regulators of the transcription factor ΔFosB. ACS Chem. Neurosci. 2012;3:546–556. doi: 10.1021/cn3000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S., Filipp F.V. A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst. Biol. Appl. 2018;4:24. doi: 10.1038/s41540-018-0061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M.K., Leman L.J., Price D.J., Brooks C.L., Stout C.D., Ghadiri M.R. Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochemistry. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z., Machius M., Nestler E.J., Rudenko G. Activator Protein-1: redox switch controlling structure and DNA-binding. Nucleic Acids Res. 2017;45:11425–11436. doi: 10.1093/nar/gkx795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.