

Abstract
A weakly coordinating monodentate heteroaryl thioether directing group has been developed for use in Pd(II) catalysis to orchestrate key elementary steps in the catalytic cycle that require conformational flexibility in a manner that is difficult to accomplish with traditional strongly coordinating directing groups. This benzothiazole thioether, (BT)S, directing group can be used to promote oxidative Heck reactivity of internal alkenes providing a wide range of products in moderate to high yields. To demonstrate the broad applicability of this directing group, an arene C–H olefination method was also successfully developed. Reaction progress kinetic analysis provides insights into the role of the directing group in each reaction, which is supplemented with computational data for the oxidative Heck reaction. Furthermore, this (BT)S directing group can be transformed into a number of synthetically useful functional groups, including a sulfone for Julia olefination, allowing it to serve as a “masked olefin” directing group in synthetic planning. In order to demonstrate this synthetic utility, natural products (+)-salvianolic acid A and salvianolic acid F are formally synthesized using the (BT)S directed C–H olefination as the key step.
Graphical Abstract
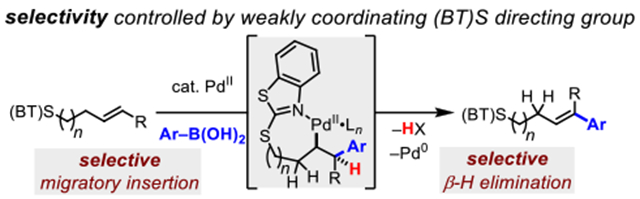
INTRODUCTION
Substrate directivity is a powerful approach for enhancing reactivity and controlling selectivity across numerous types of metal-catalyzed reactions.1 In substrate-directed reactions, pre-association of the catalyst to one or more Lewis basic sites on the substrate promotes a desired reaction outcome by perturbing the activation barriers for possible divergent pathways along the reaction coordinate. Kinetic reactivity is typically enhanced by virtue of induced intramolecularity as well as enforced proximity between the reactive group on the substrate and a free coordination site on the metal. Regioselectivity is dictated by preferential formation of a thermodynamically and/or kinetically stabilized metallacycle over an energetically disfavored constitutional isomer. Stereoselectivity can also be controlled in key bond-making and bond-breaking steps through stereochemical information on (or in the vicinity of) the coordinating atom(s).
Strategic application of substrate directivity in palladium-catalyzed alkene functionalization is a potent platform for achieving challenging modes of bond construction.1b Palladium-catalyzed Wacker oxidation and Mizoroki–Heck arylation of alkenes constitute the bedrock of modern homogeneous catalysis research, with the power of these reactions stemming from their ability to forge diverse C–C and C–heteroatom bonds from simple alkene starting materials.1d, 2 In the traditional reaction pathways, upon nucleometalation (or migratory insertion), the resulting alkylpalladium(II) intermediate undergoes facile β-H elimination to deliver the final oxidized alkene product. With substituted alkene starting materials, the regioselectivities of the initial addition step and subsequent (β-H elimination step are not straightforward to control in the absence of a strong steric or electronic bias between the two alkenyl carbon atoms. Our laboratory1e, 3 and others1d, 4 have studied substrate-directed palladium-catalyzed alkene functionalization reactions that initiate via Wacker- or Heck-type pathways. In these systems, strongly coordinating mono-, bi-, and tri-dentate directing auxiliaries can be used to control the regioselectivity of the alkene addition step and stabilize the resulting intermediate such that downstream elementary steps, such as protodemetalation, oxidative addition, or transmetalation, outcompete β-H elimination.
The design of new directing groups with distinct coordination properties constitutes a significant challenge and opportunity in this area of study, given the ability of the directing group to engender control of pathway selectivity during catalysis. In our past work we have used the strongly coordinating 8-aminoquinoline (AQ) and 2-pyridyl-8-aminoquinoline (PAQ) directing groups, which are efficient at suppressing β-H elimination to enable 1,2-difunctionalization and hydrofunctionalization. Based on these precedents, we hypothesized that a more weakly coordinating group may lead to a different scenario in which a β-H elimination step would nowbe kinetically accessible. Specifically, we sought to identify a directing group that would lie in the “goldilocks” region–possessing sufficient coordination strength to facilitate complete selectivity in the nucleopalladation step, even with especially challenging substrates (such as 1,2-dialkyl alkenes), yet stih weakly coordinating enough to allow selective β-H elimination. Such a reaction sequence would be synthetically valuable by allowing formal C(alkenyl)–H functionalization,2d enabling conversion of a simple alkene starting material into a regio- and stereodefined tri- or tetra-substituted alkene product (Scheme 1).
Scheme 1.

Background and Synopsis
To vahdate this general hypothesis, we elected to focus on the palladium(II)-catalyzed oxidative Heck reaction, anticipating that newly identified directing groups would also find utility in other palladium(II)-catalyzed reactions (vide infra). Despite extensive efforts over the past several decades, knowledge gaps persist in Heck-type chemistry. Directed variants of the pahadium(II)-catalyzed oxidative Heck reactions, for instance, are relatively rare, with White’s seminal study of heteroatom-directed oxidative Heck arylation of terminal alkenes a notable exception.5 A closely related body of literature has examined directed variants of classical-polarity palladium(0)-catalyzed Mizoroki–Heck reactions,1d with pioneering work here including contributions by Hallberg,5 Carreterro,6 Yoshida,7 and others.8 These studies have typically focused on heteroatom-substituted alkene substrates, including vinyl sulfides, ethers, or silanes, and in these designs, the coordinating group is built into the heteroatom linkage. For these systems, competitive binding of the phosphine ligand versus the directing group is an important consideration. Included in these studies are relatively few alkyl-substituted alkenes, with examples mostly limited to terminal allylic alcohols, amines, and their derivatives.
Across all Heck-type chemistry, internal asymmetrically substituted di- and tri-substituted alkenes remain difficult substrates. Sigman has developed elegant catalytic systems for both oxidative and non-oxidative redox relay asymmetric Heck arylation of internal alkenes.9 In these reactions, the highly electron-deficient palladium catalyst undergoes rapid β-H elimination/reinsertion (chain-walking) to an appropriate terminating group (e.g., an alcohol). Complementing such chain-walking Heck systems with those that preserve the alkene in a precise geometric relationship to the newly installed aryl group would be desirable in situations where the alkene is required in the resulting product. In parallel to this study, Shenvi reported an intermolecular Heck coupling that can direct arylation of electronically unbiased tri- and tetra-substituted alkenes and establish quaternary carbons while migrating the olefin only one position and not into conjugation with the carboxylic acid directing group as is common in redox-relay Heck reactions.10
RESULTS AND DISCUSSION
Directing Group and Reaction Optimization.
The investigation commenced by testing a series of potential directing groups for their ability to promote a classically challenging oxidative Heck reaction with a 1,2-substituted alkene. We focused on thioether-based directing groups (I) due to on their ease of installation via nucleophilic substitution chemistry, ability to be productively removed in a variety of preparatively useful reactions (e.g., Julia olefination), and established effectiveness as directing groups in palladium complex formation11 and catalysis12
A series of homoallylic thioether-based substrates containing internal (E)- or (Z)-alkenes were thus prepared. These substrates were subjected to Pd(II)-catalyzed oxidative Heck reaction conditions, which were optimized in parallel using a Design of Experiment workflow with automated reaction setup and sampling (see SI) (Table 1). As a control, a homoallylic alcohol substrate (I-A) was tested and did not deliver meaningful quantities of the desired product; similarly, simple aryl thioethers and sulfoxides (B-E) were ineffective, despite their previously established utility as directing groups in palladium(II) catalysis.13 We then turned our attention to azaheterocyclic thioethers, reasoning that the coordination strength of sulfur would be attenuated in these cases and that such substrates offered additional opportunities for coordination of the palladium catalysts to an N(sp2) atom during catalysis. Tetrazole directing group G afforded moderate yield of the desired product with both the E- and Z-alkene starting material; unfortunately, in this case the remainder of the starting material was completely converted into undesired byproducts. Based on the successful fiinctionali- zation of the internal alkene with directing group G, we tested F and H, but found minimal to no product. Benzoxazole thioether directing group I provided slightly diminished yields compared to directing group G. However, in this case the product selectivity was improved, as we did not observe significant byproduct formation. Fortunately, we discovered that two related directing groups, J and K, the latter of which we refer to as (BT)S throughout the remainder of the manuscript, offered high reactivity and selectivity. Yields in these cases were 86% and 80% for products (E)-II-J and (Z)-II-J, and 85% and 91% for products (Z)-II-K and (Z)-II-K. Substrates with similar thiazole-type directing groups, L–O, with different steric and electronic properties gave lower yields of II. We next tested oxidized versions of (BT)S, P and Q, as controls, but minimal to no product formation was observed in these cases.
Table 1.
Optimization of Directing Group for Oxidative Heck Coupling with 1,2-Dialkyl Alkenesa
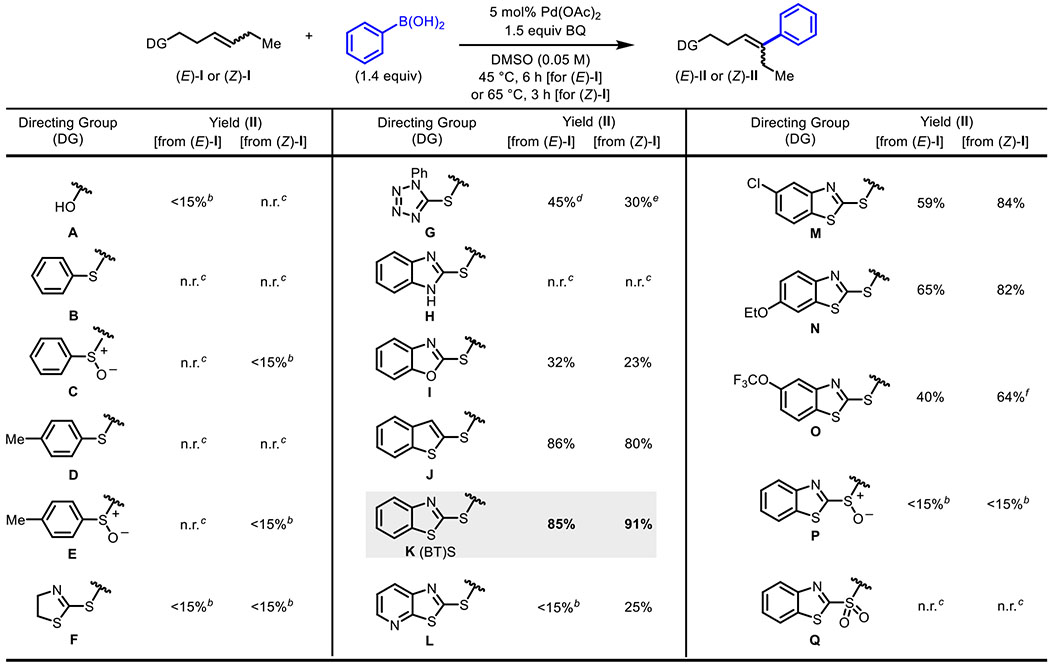 |
Isolated yields except where otherwise noted.
Determined by 1H NMR of the crude reaction using 1,3,5-trimethoxybenzene as internal standard.
n.r. = no reaction.
Inseparable from 22% of two unidentifiable isomers that contain a 1,2-disubstituted internal olefin motif.
Inseparable from 26% of two unidentifiable isomers that contain a 1,2-disubstituted internal olefin motif.
Isolated as a 7:1 mixture of isomers (Z:E).
Collectively, these results point to a somewhat complex relationship between structure and reactivity/selectivity, which was probed further through a detailed analysis of the possible coordination modes of K ((BT)S) (vide infra). Based on its effectiveness in this challenging oxidative Heck reaction and its potential utility in downstream chemistry (e.g., oxidation followed by modified Julia olefination), K ((BT)S) was selected for further investigation.
Scope of Oxidative Heck Reaction.
We proceeded to investigate the substrate and coupling partner scope of this directed oxidative Heck reaction (Scheme 2). Arylboronic acids with a variety of functional groups, including electron-withdrawing and electron-donating moieties, reacted under the standard conditions to give products 2a–k in high yields. Heteroarylboronic acids also gave the corresponding products 2l–p in reasonable yields. Notably, (E)-(3-phenylprop-l-en-l-yl)boronic acid provided product in reasonable yield; however both (E)- and (Z)-isomers, 2p, were formed in a 3:2 ratio. Two representative alkylboronic acids, methylboronic acid and neopentylboronic acid, were tested, but unfortunately these were not competent coupling partners in the reaction.
Scheme 2.
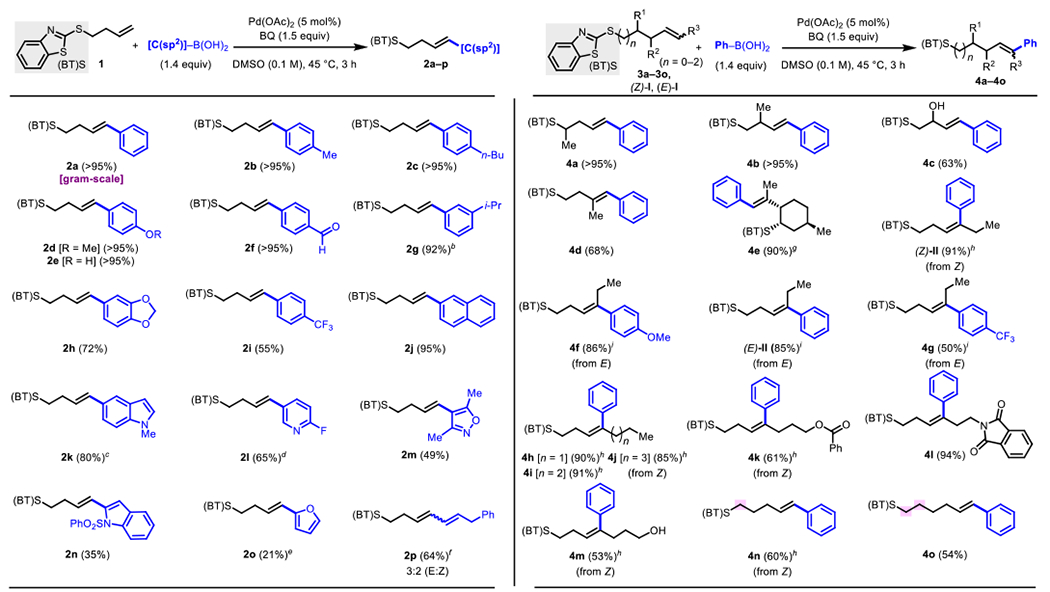
Scope for (BT)S-Directed Oxidative Heck Reactiona
aIsolated yields. bInseparable from 8% of an unknown isomer. c10:1 isomeric mixture of E and Z. dInseparable from 7% of an unknown isomer. eIsolated as a 6:1 mixture of inseparable isomers (E:Z). fIsolated as a 3:2 mixture; isomers separated by LC. gInseparable from 10% of an unknown isomer. hFrom the Z-olefin; 50 mM, 65 °C. gFrom the E-olefin; 50 m M, 6 h.
Next, we moved on to probe the alkene scope (Scheme 2). We found that alkyl branching was tolerated at both the α- and β-positions, as exemplified by products 4a and 4b, which were formed in nearly quantitative yields. We next tested a substrate containing an intervening alcohol, which is an established terminating group in Heck chain-walking processes. Nevertheless, in this case we observed that product 4c, with the alcohol intact, was formed in high yield. The fact that the corresponding ketone product is not formed in this case demonstrates that the alternative endo-β-H elimination pathway from the putative palladacycle intermediate is disfavored. Furthermore, 1,1-disubstituted alkenes were found to be tolerated, allowing for formation of 4d in moderately high yield and 4e, a derivative of the natural product isopulegol, in high yield. As observed in the directing group optimization table (Table 1), both E- and Z-configured 1,2-disubstituted alkenes gave high yield of the desired products (E)-II and (Z)-II, respectively, and importantly in both cases, the products were obtained as single alkene stereoisomers. With (E)-I as a representative internal alkene, other aryl boronic acids were tested, and it was found that an electron-rich aryl group led to higher reactivity compared to an electron-poor aryl group (vide infra). In the case of the p-CF3-substituted example, a 3:1 ratio of conjugated to non-conjugated products was obtained, suggesting that the identity of the aryl group can influence the energy barriers for competitive β-H elimination pathways following migratory insertion—at least to some extent. A series of other internal alkenes bearing different linear alkyl chains were tested (4h–4j) along with examples containing an additional functional group potentially capable of coordinating the catalyst or perturbing the electron properties of the alkene (4k–4m), and in all cases moderate to high yield of the desired, conjugated product was observed. Different tether lengths were also examined, and it was found that both γ,δ and δ,ε-unsaturated alkenes (4n and 4o) are competent in this system. No reaction was observed with the corresponding allylic substrate. Across this series of substrates, preference for aryl addition to the alkenyl carbon atom distal from the (BT)S group is consistent with the notion that the (BT)S group is coordinated during migratory insertion. Although some exceptions exist,6b,6c,8k directed Heck-type reactions typically favor formation of the regioisomer arising from a transition state where the migrating aryl or alkenyl group is exo-cyclic to incipient palladacycle,1d,5–8 even with internal non-conjugated alkenes,3h,4d,10 likely due to minimization of strain compared to the alternative possibility with the aryl group in an endo-cyclic orientation.
Tri- and tetra-substituted olefins (3p–3r) were also examined (Scheme 3). In these cases complex product mixtures were typically obtained, indicating that these substrate classes are generally incompatible with this method, likely due to increased steric hindrance and due to a greater number of possible side reactions.10 With tetrasubstitued substrate 3p, the formation of products 4r and 4s suggests that positional isomerization of the alkene takes place under the reaction conditions with this substrate. One interpretation of this result is that in cases where migratory insertion is sluggish due to steric hindrance, other off-cycle pathways can predominate. With substrates 3p and 3r, the requirement to undergo β-hydride elimination in an endo-fashion (towards the directing group and within the palladacycle) rather than the preferred exo-fashion is also likely a contributing factor to low yields. With alkene 3q, the product distribution suggests that the migratory insertion step was selective, but that subsequent β-H elimination was un- selective. Of the three tri/tetrasubstitued alkenes tested, 3r offered the highest selectivity. In this case, product 4w containing a quaternary carbon was formed as the main product along with regioisomer 4x in a 5:1 ratio and 49% combined yield.
Scheme 3.
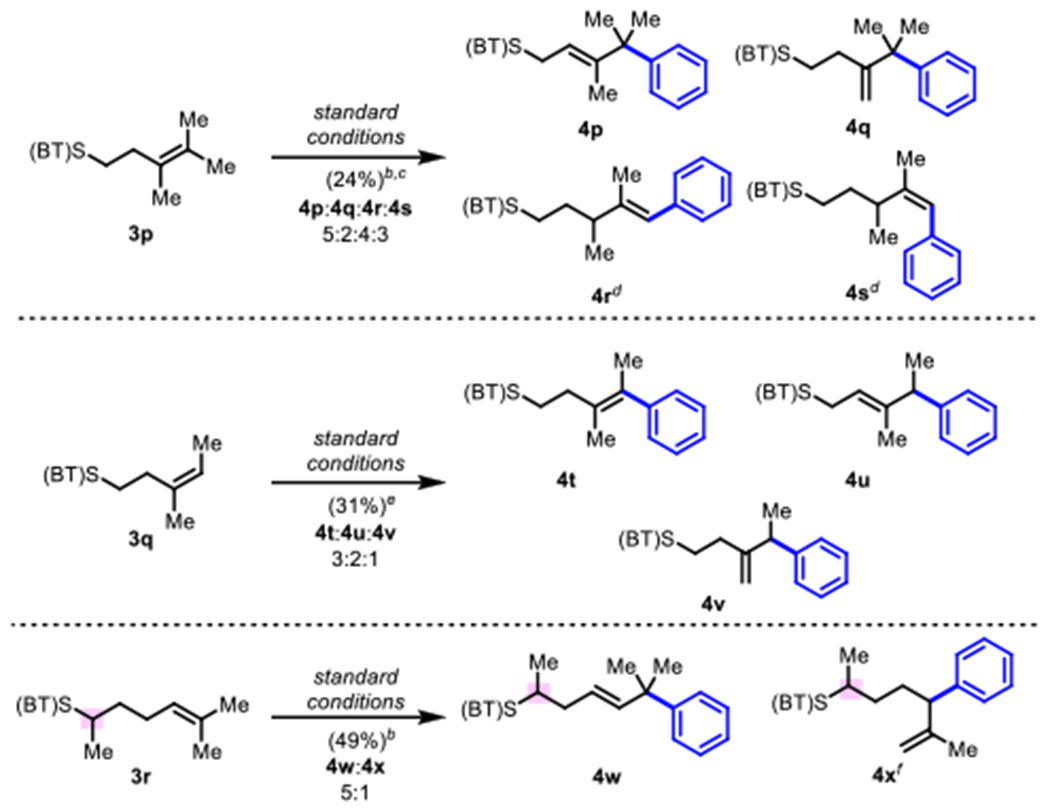
Results with Tri- and Tetra-substituted Olefinsa
aReaction conditions: alkene (0.1 mmol), phenylboronic acid (0.14 mmol), BQ(0.15 mmol), Pd(OAc)2 (5 mol%), DMSO (1 mL), 45 °C, 3 h, unless otherwise noted; isolated yields. bRun at 65 °C and in DMSO (2 mL). cIsolated as a 5:2:4:3 mixture of isomers; 50 mM at 65 °C. dIsomers separated by HPLC. eIsolated as a 3:2:1 mixture of isomers; isomers separated by HPLC. fObserved by NMR.
Reaction Progress Kinetic Analysis (RPKA).
The high regio- and stereo selectivity of this method with diverse 1,2-disubstituted alkenes prompted us to delve into the reaction mechanism by combining reaction kinetics and computational studies. First, to elucidate the kinetic features of catalytic process, we performed reaction progress kinetic analysis (RPKA)14 using representative (E)- and (Z)-alkene substrates. Insights for both substrate types were generally mutually consistent, so the ensuing analysis focuses on the (E) substrate, and information regarding the (Z)-alkene substrate can be found in the SI. Regarding general kinetic behavior, we noted that the reaction appears to contain two regimes: one that is operative until 35–50% conversion is reached (representing approximately the first 60 min for the reaction with the (E)-alkene and 30 min for the reaction with the (Z)-alkene under standard conditions), and a second regime from that point forward where the reaction proceeds more slowly. This could arise from several different root causes, including uncharacterized catalyst deactivation processes. Though both regimes are valuable to understand, we focused our attention on this initial stage of the reaction, as it is more likely to reflect the intrinsic kinetics before other off-cycle processes predominate.
First, we performed a “same-excess” experiment, which simulates entering the catalytic cycle after multiple catalyst turnovers in order to probe for catalyst activation or deactivation pathways (Figure 1A). The time-adjusted same-excess experiment without added product was observed to proceed at a faster rate than the standard reaction, pointing to possible catalyst deactivation or product inhibition. Results were similar with added product, suggesting that the catalyst is being deactivated through off-cycle processes. Catalyst deactivation in this reaction can take place if Pd(0) is not efficiently reoxidized, enabling precipitation of palladium black.
Figure 1.
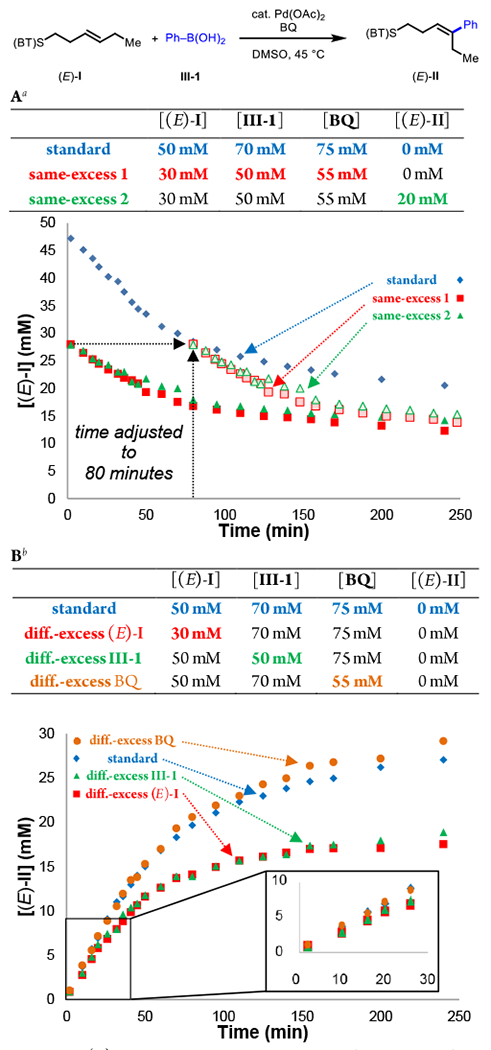
(A) Same-excess experiments with and without product. (B) Different-excess experiments of starting material, phenylboronic acid, and benzoquinone. aDue to the consumption of 1 equivalent of Benzoquinone per turnover, reactions were set up with Pd(OAc)2 (2.5 mM) and with (E)-I (variable), III, BQ, and (E)-II (for same-excess 3) corresponding to the expected amounts at time = 80 min for the standard reaction in DMSO (2.0 mL), at 45 °C, and in air. bReaction conditions: (E)-I (variable), III (variable), Benzoquinone (variable), Pd(OAc)2 (variable), DMSO (2.0 mL), 45 °C, air.
Next, driving forces were determined through a series of “different-excess” experiments (Figure 1B). We first varied catalyst concentration and found that the reaction rates increased at higher catalyst concentration. Visualizing the data using the Burés method (Figure 2)15 with an x-axis that is normalized by catalyst concentration reveals that the reaction is first-order in [Pd]. We then examined the effect of changing the concentration of others reaction components, namely decreased alkene concentration [(E)-I] (different-excess [(E)-I]), decreased phenylboronic acid concentration [III] (different-excess [III]), and decreased benzoquinone concentration [BQ] (different-excess [BQ]). Overlay between the standard reaction and the different-excess BQ experiment throughout the time course is consistent with apparent zero-order kinetics in [BQ]. The different-excess (E)-I and different-excess [III] experiments overlay reasonably well with the standard reaction at <30% conversion (first 30 min) and then deviate as the limiting reagents are consumed. This indicates apparent zero-order kinetics in [(E)-I] and [III] early in the reaction. First-order kinetics in [Pd] combined with zero-order kinetics in [(E)-I], [III], and [BQ], rule out substrate binding, transmetalation, and reoxidation, respectively as possible turnover-limiting steps, pointing to either migratory insertion of a substrate-bound [Pd(II)–Ar] species or β-H elimination as possible turnover-limiting steps. As mentioned above, electron-rich arylboronic acids were found to be more reactive (Figure 3). To quantify this trend, we measured initial rates of the three representative arylboronic acids with p- CF3, -H, and -OMe groups and found krel values of 1:3:8 across this series. This result does not necessarily allow one to disambiguate between the two mechanistic possibilities described above, as migratory insertion and β-H elimination could both potentially be facilitated by an electron-rich aryl group due to a more nucleophilic [Pd(II)–Ar] species and a more hydridic C–H bond, respectively. Since the turnover-limiting step cannot be distinguished based on the available rate dependencies, we next turned to complementary techniques to gain insight on these steps and other aspects of the catalytic cycle.
Figure 2.
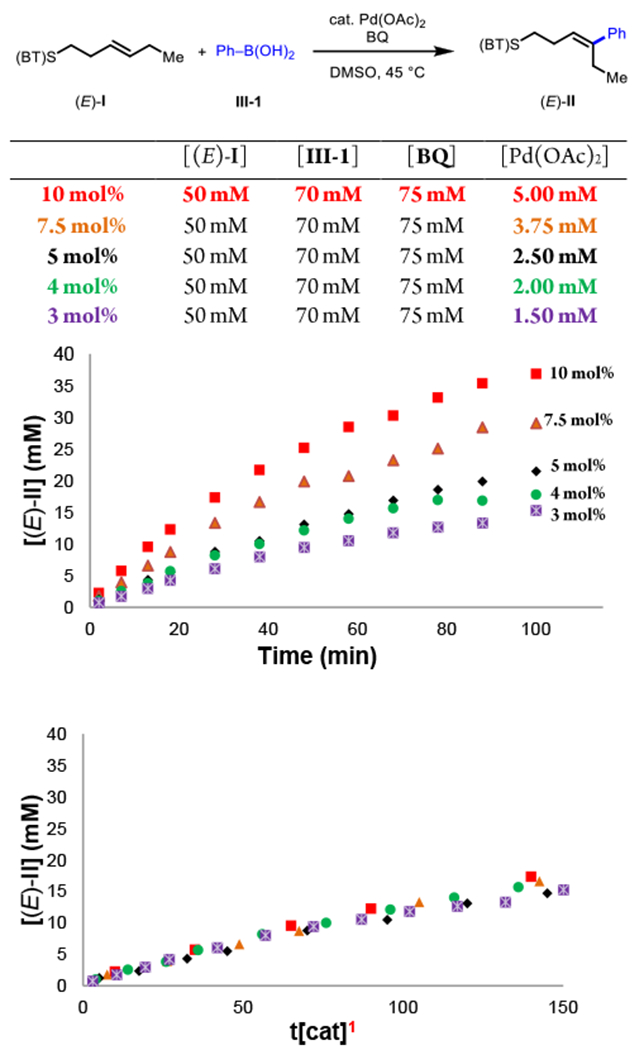
Reaction conditions (E)-I (50 mM), III-I (70 mM), Benzoquinone (75 mM), Pd(OAc)2 (variable), DMSO (2.0 mL), 45 °C, air
Figure 3.
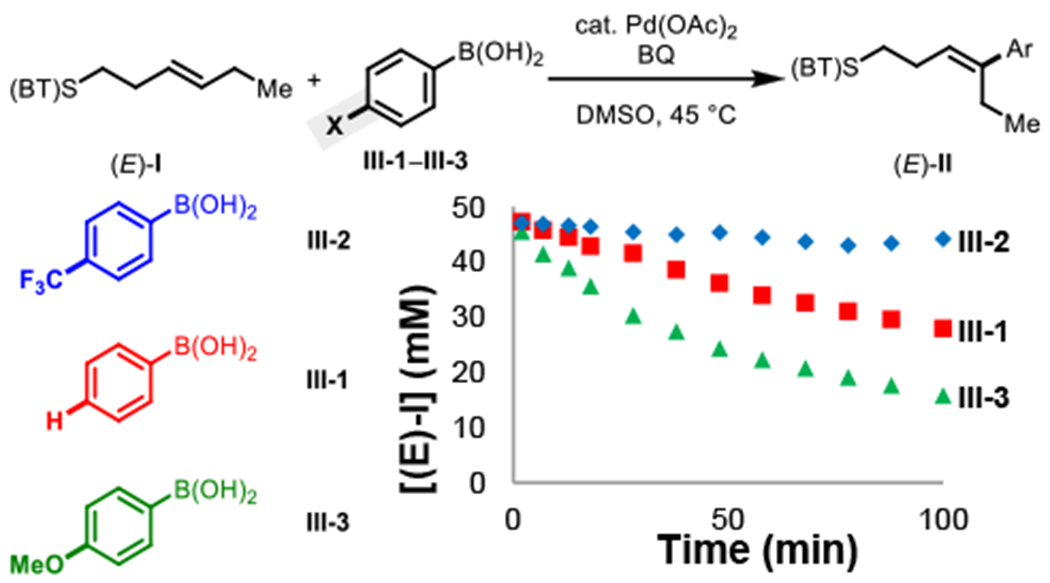
Reaction conditions (E)-I (50 mM), III-1–3 (70 mM), Benzoquinone (75 mM), Pd(OAc)2 (5 mol%), DMSO (2.0 mL), 45 °C, air.
Directing Group Coordination.
The results in Table 1 revealed the structural features that are required for a suitable directing group, demonstrating the importance of the embedded benzo-fused methylidene dithioacetal motif in J and K. Nevertheless, K((BT)S) also posses an N(sp2) capable of coordinating the palladium catalyst, complicating the potential coordination chemistry involved, which we sought to elucidate through crystallography and computation studies. Several different mono- and bidentate binding modes can be envisioned (Scheme 4), and to probe this, we first attempted to synthesize various palladium complexes of potential relevance to catalysis. Indeed, we were able to obtain a representative product-bound palladium species by combining 2a and Pd(tfa)2. The resulting 2:1 complex, Pd-1, was amenable to characterization by X-ray crystallography, and in the structure, the (BT)S directing group is coordinated in a monodentate fashion through the nitrogen atom, consistent with earlier literature precedents (Scheme 5).16 In this case the alkene was not bound to the palladium center. The same monodentate nitrogen binding mode was also later observed with a representative starting material–palladium complex for a related C–H activation reaction (vide infra).
Scheme4.
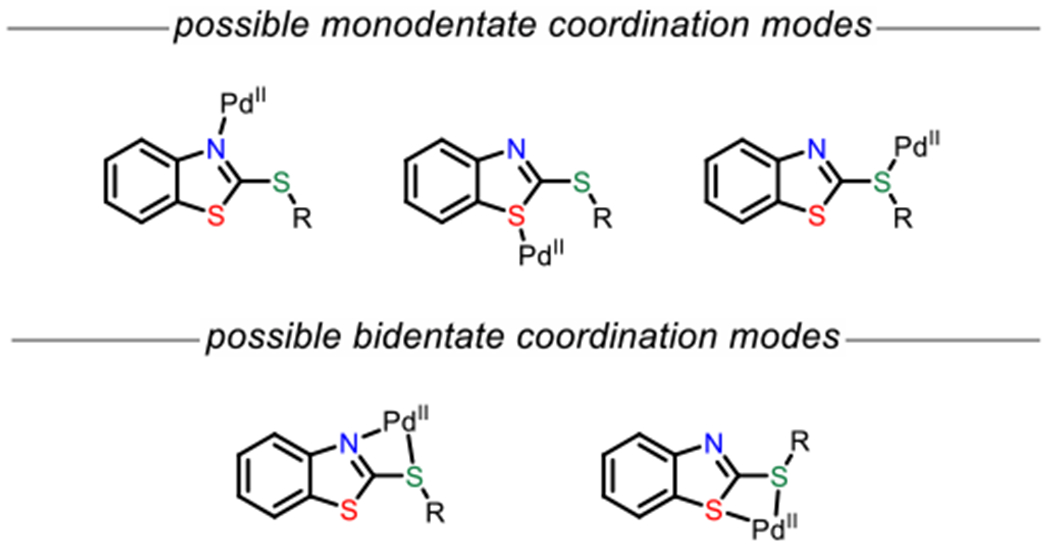
Potential Binding Modes of PdIIto (BT)S Group
Scheme 5.
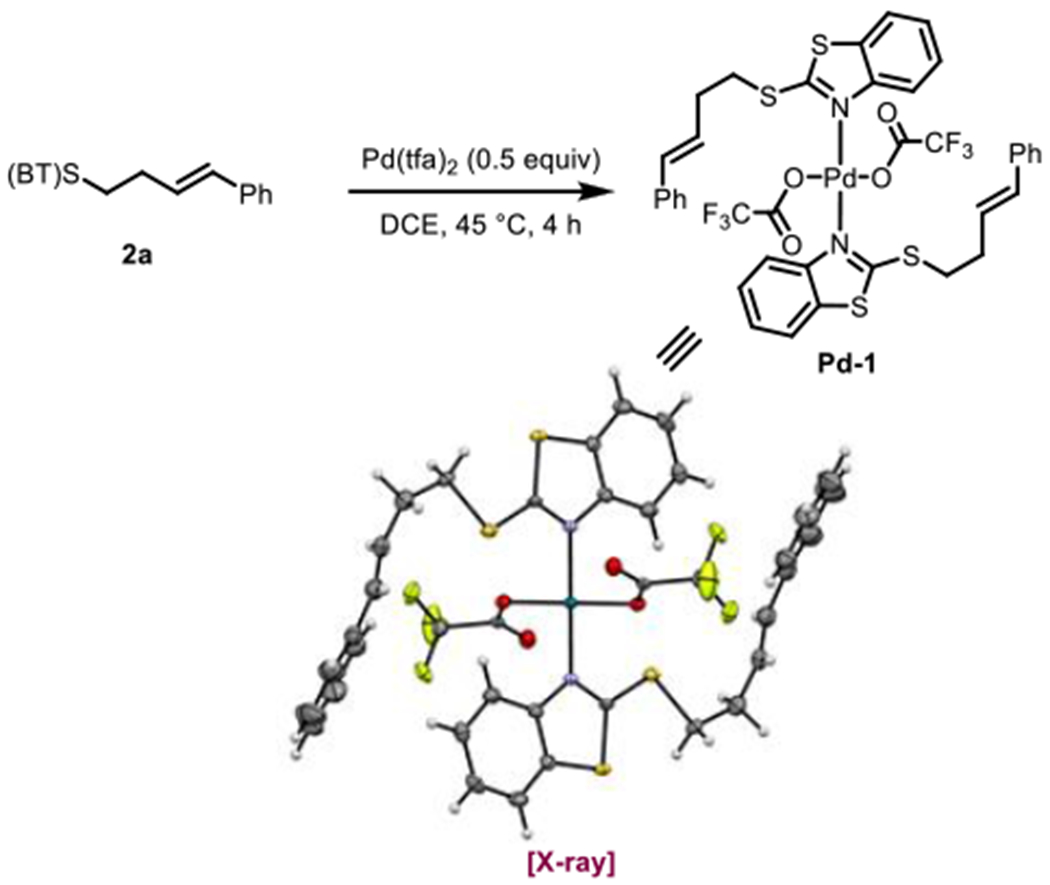
Crystal Structure of Pd(tfa)2o2 Complex
DFT Calculations.
Next, we performed density functional theory (DFT) calculations to further probe key aspects of the reaction mechanism. First, the possible monodentate coordination modes were evaluated using 1 as the model substrate. Geometry optimizations were performed on the intermediates of the proposed catalytic cycle at the migratory insertion step. Three coordination modes were investigated, i.e. coordination through N (INT1_N and INT2_N), coordination through S of the heterocycle (INT1_S, INT2_S) and coordination through S outside the heterocycle (INT1_S’, INT2_S’). The N-coordinated intermediates were lowest in energy, followed by the S (proximal)-coordinated intermediates (Scheme 6). The S(distal)-coordinated intermediates were highest in energy. These trends also held in the corresponding transition state energies for the migratory insertion step (see Figure S19 in the SI).
Scheme 6.
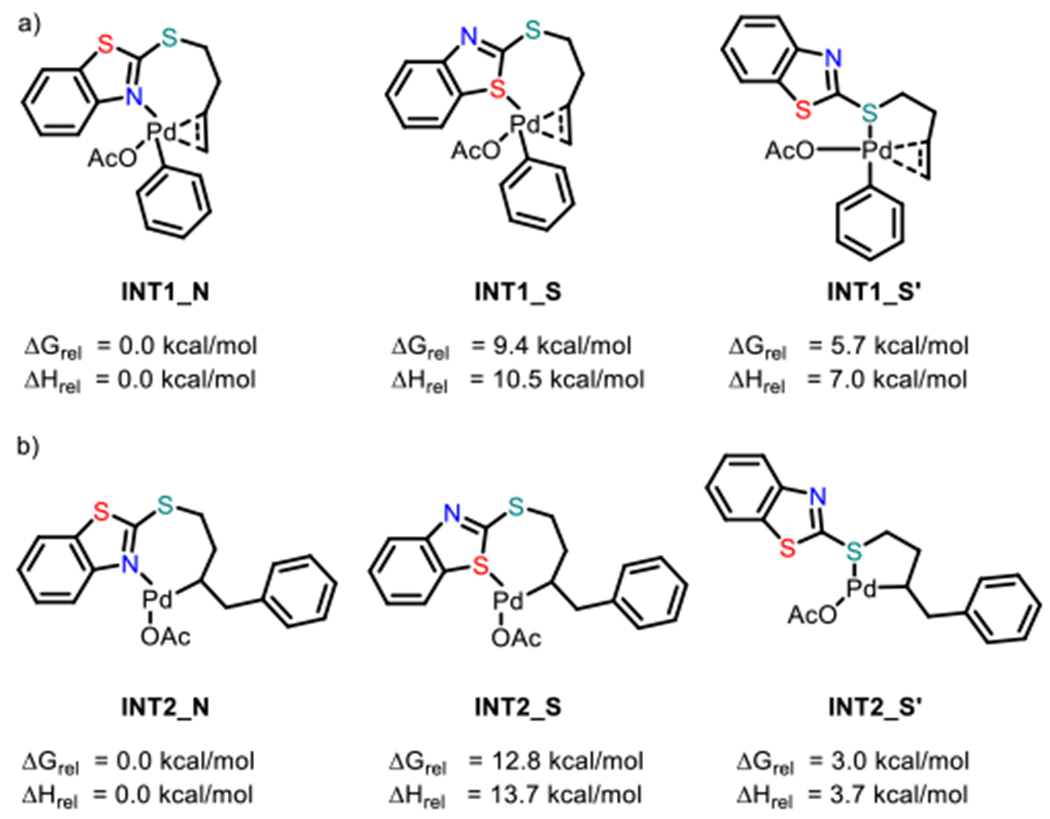
Relative energies of Possible Intermediates in the Oxidative Heck Reactionaa
aRelative free energies of the intermediates of the catalytic cycle a) before and b) after migratory insertion.
One explanation that reconciles these results with the observation that both directing groups J and K ((BT)S) are similarly effective (Table 1) is that the catalytic reaction can take place with the directing group bound either through the proximal-S atom or through the N-atom. With BT(S), the latter pathway is computationally predicted to be lower-energy, though the former is also energetically accessible. The S(proximal)-coordinated pathway is likely to be operative in cases such as J, where the N-atom is absent.
Probing the Turnover-Limiting Step.
As experimental mechanistic investigations revealed the rate determining step to be either migratory insertion or β-hydride elimination, DFT calculations were performed to locate the transition states to determine the energetics of these two steps. A truncated (E) internal alkene was used as the model substrate to minimize computational time. The energy profile is shown in Figure 4. The syn-migratory insertion step (TS1) has an activation free energy barrier of 14.3 kcal/mol with respect to the π-alkene complex INT1. The subsequent β-hydride elimination step has a higher activation energy barrier of 15.7 kcal/mol (TS2) indicating that the β-hydride elimination is the turnover-limiting step. The optimized structures of TS1 and TS2 are shown in Figure 5. The weaker coordination of the (BT)S group is evidenced by the longer Pd…N distances of the transition states at both migratory insertion (2.18 Å) and β-hydride elimination (2.22 Å) steps. Shorter Pd…N distances have been reported for transition states with strongly coordinating directing groups such as 8-aminoquinoline and 2-pyridyl-8-aminoquinoline (2.02–2.08 Å).3e,17 The energy profile for the migratory insertion and β-hydride elimination was also calculated for the terminal alkene, 1 (see SI; Figure S17 and S18). The calculations revealed that the formation of the E-isomer is kinetically favored at the rate- and stereoselectivity-determining step, β-hydride elimination, consistent with experimental selectivity (see SI).
Figure 4.
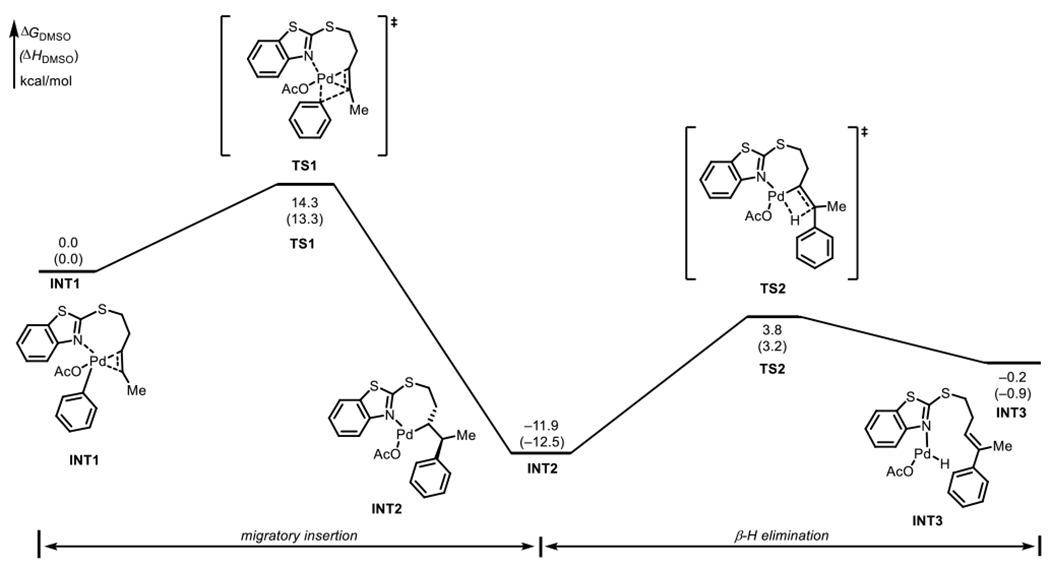
Computed energy profile for migratory insertion and β-hydride elimination steps.
Figure 5.
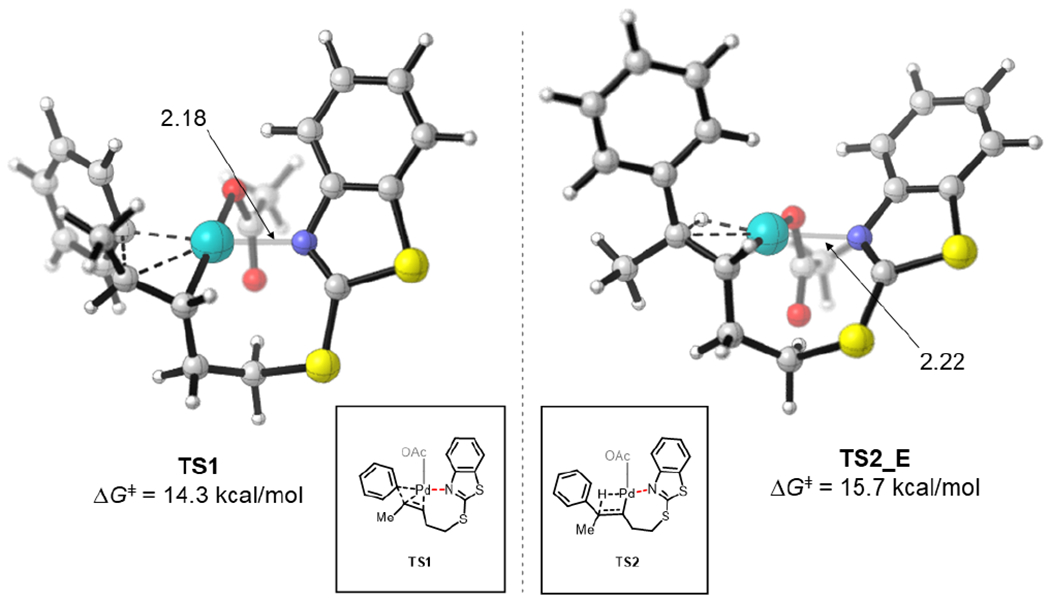
Computed transition states for migratory insertion (left) and β-hydride elimination (right) steps.
With the computational data suggesting that β-hydride elimination is containing deuterated ((E)-II-d2) andnon-deuterated ((E)-II) alkenyl positions (Figure 6). By monitoring desired product formation over the initial 45 min, we indeed found a primary KIE, kH/kD = 2.0. Additionally, we found that with the deuterated substrate the product distribution was not as clean, potentially due to other pathways (such as β-H elimination at other positions) being kinetically competitive when the typically favored β-H is replaced with a deuterium. Overall, the experimental and computational results are consistent with β-H elimination as the turnover-limiting step.
Figure 6.
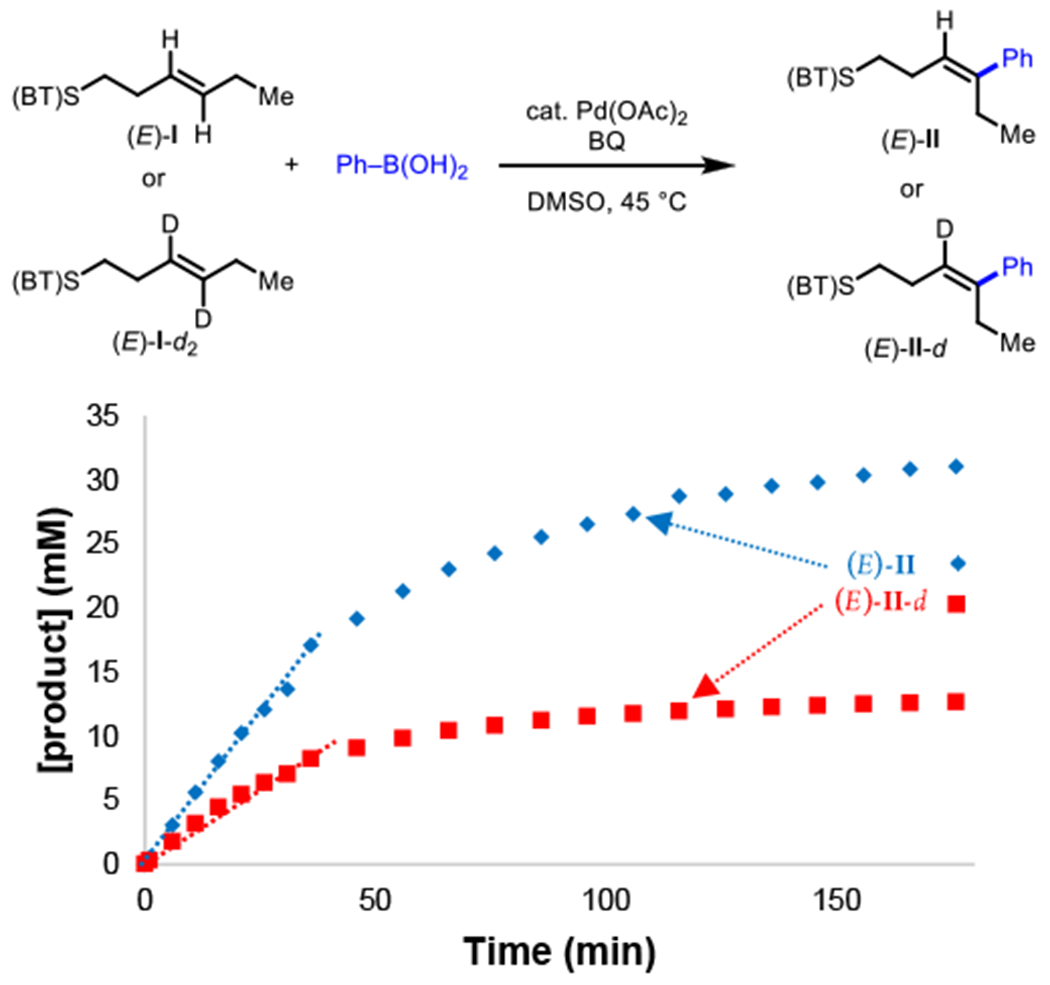
Deuterated starting material experiment for the directed oxidative Heck reaction. Reaction conditions (E)-I or (E)-I-d2 (50 mM), III-1(70 mM), Benzoquinone (75 mM), Pd(OAc)2 (5 mol%), DMSO (2.0 mL), 45 °C, air.
By combining the insights gathered from both DFT calculations and the kinetics experiments, we propose the general catalytic cycle as shown in Scheme 7. For simplicity, only the N-bound intermediates are shown, although as mentioned above S(proximal)-bound intermediates are also energetically accessible and could be active participants in the catalytic cycle. The reaction begins with substrate binding and transmetallation steps (in either potential order) to give intermediate Pd-B. Following this, a syn-migratory insertion gives the palladacycle Pd-C. Product is then generated via β-H elimination giving the Pd-hy- dride species Pd-D, which can undergo reductive elimination and reox- idation with BQ to provide the Pd(II) species required for turnover.
Scheme 7.
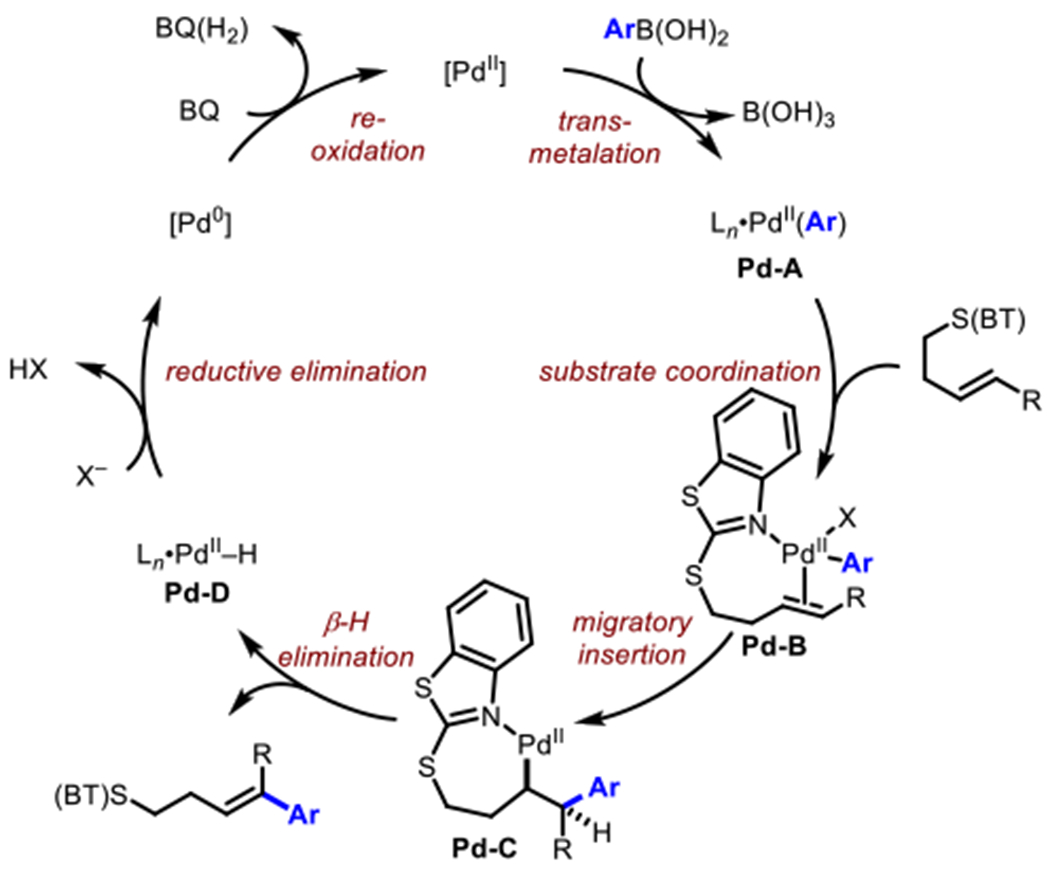
(BT)S-directed Oxidative Heck Catalytic Cycle
C(aryl)-H Olefination Background.
Having established the efficacy ofthe (BT)S directing group in oxidative Heck chemistry, we were interested in exploring its utility in other palladium(II)-catalyzed reac-the turnover-limiting step, we next examined whether there was a kitions involving distinct elementary steps. Specifically, we were attracted netic isotope (KIE) in the rates between a representative substrations involving distinct elementary steps.Specifically ,we were attracted to directed ortho-C–H olefination (Fujiwara-Mortitani-type coupling) since the use of a versatile “masked olefin” directing group could allow rapid access to a number of conjugated natural products and materials.18 Weakly coordinating directing groups, including those distal to the C–H bond of interest, have provided a valuable solution in many arene C–H functionalization reactions catalyzed by palladium, including amination,19 formation of lactams,20 construction of dihydrobenzofurans,21 iodination,22 and olefination.23 We were encouraged to see that Zhang has shown the utility of using phenyl-, methyl-, and p-tolyl-thi- oethers24 and the corresponding sulfoxides25 for directed arene olefination, suggesting that the (BT)S directing group may provide a suitable directing group and then be able to serve as a modified Julia olefin precursor for easy transformation to the desired alkene. Furthermore, Maiti has developed an innovative sulfone directing group for meta-hydrox- ylation, which can subsequently serve as a Julia olefination precursor, and we hoped to provide a similar solution for ortho-C–H functionalization.26
C-H Functionalization Substrate Scope.
After reexamining the directing groups A-H and K-Q in Table 1 under optimized MPAA-accelerated C-H olefination reaction conditions,27 it was clear that (BT)S again was suitable in its abihty to successfully direct the desired reaction (Table S13 in supporting information).
Therefore, we next examined the scope of the (BT)S-directed C-H olefination reaction (Scheme 8). We were pleased to see that the electron-rich starting material 5a produced a nearly quantitative yield, and electron-rich, electron-poor, and alkyl functional groups were tolerated to yield 6b-h and 6k-n in moderate to high yields. The (BT)S directing group can also promote ortAo-olefination in polyaromatic ring systems, such as naphthalene producing 6i-j in moderate yields. As expected, a number of acrylates can be tolerated in this reaction to give 6o-q and 6s-t in high yields. We were excited to see that this (BT)S-directed ole- fination could also be expanded to other alkene classes such as acryla- mide giving 6r and styrene giving 6u. Interestingly, methyl methacrylate gave the non-conjugated product 6w, consistent with a mechanism in which β-H elimination proceeds exo to the putative metalacycle in this case. Other competent alkene coupling partners include phenyl vinyl sulfone (6x), dodecene (6y), ethenesulfonyl fluoride (6z)28, and ethyl crotonate (6aa).
Scheme 8.
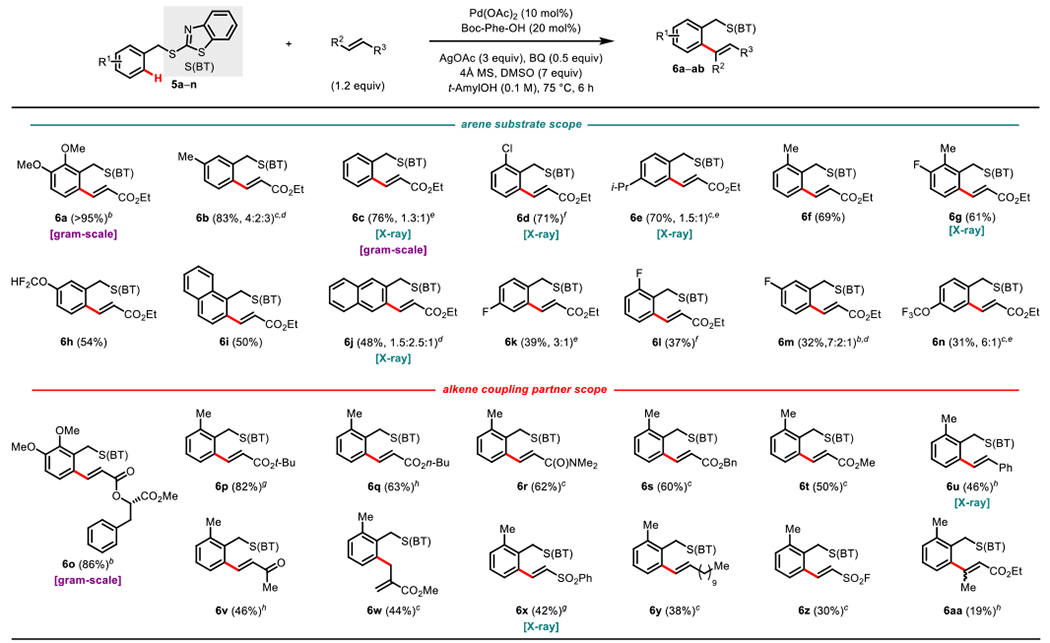
Scope for (BT)S-directed C-H Functionalizationa
aReaction conditions: 5a–aa (0.10 mmol), Alkene (1.2 equiv), AgOAc (3 equiv) Benzoquinone (0.5 equiv), Pd(OAc)2 (10 mol%), tAmylOH (1 mL), Boc-L-phenylalanine (0.2 equiv), DMSO (7 equiv), 4Å molecular sieves, 75 °C, air, 6 h. Percentages refer to isolated yields. b110 °C, nBu4NPF6 (2 equiv), 2h. cAlkene (2.2 equiv). dRatio of mono-, mono’-, and bis-olefinated products, respectively, with the major product depicted. Mono- and mono’-olefinated products isolated as a mixture. eRatio of mono- and bis-olefinated product, respectively, with the major product depicted. fAlkene (2.2 equiv), g24h. hAtkene (2.2 equiv), 24 h.
Notably, 6y represents a somewhat unique product in directed arene olefination, as the use of non-conjugated terminal alkenes remains rare.29 Notably, the thioether24 and sulfoxide25 directed reactions designed by Zhang were replicated with similar yield as previously reported for ethyl acrylate but yielded no product with dodecene showing the unique characteristics of (BT)S (Scheme 9).
Scheme 9.
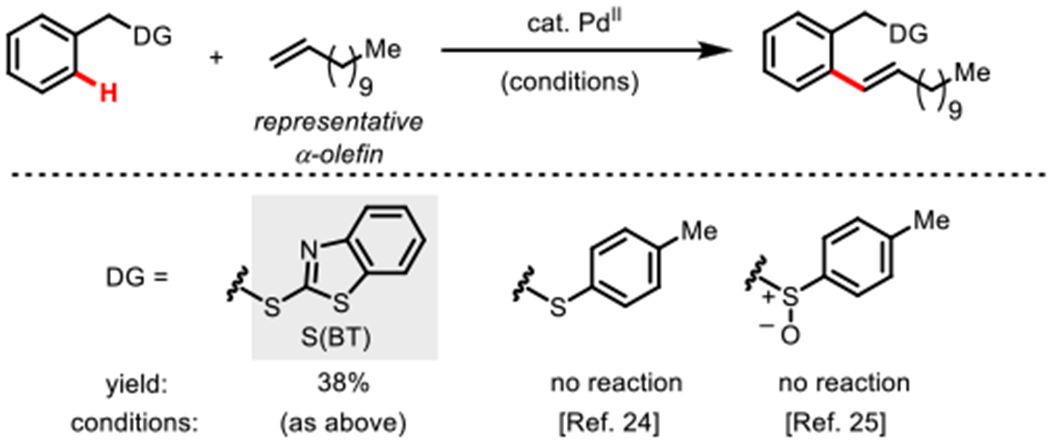
Unactivated Alkene Reactivitya
a(BT)S directed reaction run with the following conditions: starting material (0.10 mmol), Alkene (1.2 equiv), AgOAc (3 equiv) Benzoqui-none (0.5 equiv), Pd(OAc)2 (10 mol%), tAmylOH (1 ml), Boc-L-phenylalanine (0.2 equiv), DMSO (7 equiv), 4Å molecular sieves, 75 °C, air, 6 h; other reactions run both under above conditions and under originally reported conditions.
C-H Olefination RPKA.
We decided to probe the catalytic cycle of this reaction in an effort to better understand the role of (BT)S in directing C-H olefination. To this end, same excess and different excess experiments were performed using the simple, unsubstituted arene 5c as a standard substrate.
Due to unique facets of this reaction, several experimental modifications were made compared to the standard same-excess and different- excess protocols that are generally used. For both sets of data, starting material concentration, [5c], was plotted on the y-axis as opposed to product because both 6c and 6c’ are formed in the reaction, with 6c presumably being formed from which a second catalytic cycle to install an additional ethyl acrylate yields 6c’. Moreover, the presence of a secondary reaction converting 6c to 6c’ meant that the standard method of assuming a one-to-one coupling (and hence consumption) of starting material 5c and coupling reagent 7 for the design of the same- excess experiment would not accurately reflect reaction conditions at 45 min. Therefore, values for 5c, 7, 6c, and 6c’ as well as BQwere determined by qNMR at 45 min in the standard reaction, and those values were used when designing the same-excess experiments (see Figure S16 in the Supporting Information). The amount of AgOAc was also lowered to appropriately reflect oxidant consumption for each presumed cycle.
The same-excess data shows excellent overlay for the standard reaction and same-excess 2, which mimics the standard reaction at 45 min (Figure 7A). However, same-excess 1, which does not contain added product, shows an accelerated rate. This qualitatively implies that the decreased reaction rate is due to product inhibition rather than catalyst deactivation. This result, in combination with the result from the different-excess experiment discussed below, suggest a catalytic cycle in which palladium coordinates strongly to the products 6c and 6c’, and, due to this strong affinity, produces product 6c’ at a similar rate to the rate of formation of product 6c at around 0.040M concentration of 6c.
Figure 7.
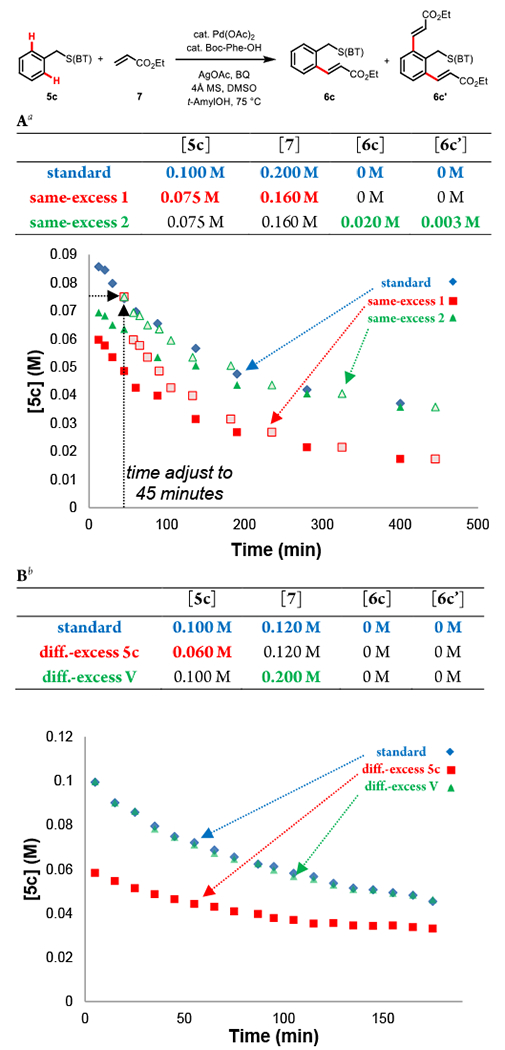
(A) Same-excess experiment. (B) Different-excess experiments of starting material and ethyl acrylate. aDue to secondary reaction of ethyl acrylate, same-excess 1 and 2 were set up with amounts of 5c, 7, 6c, 6c’, BQ(0.375 M), and AgOAc (2.250 M) to equal the amounts observed by qNMR at time = 45 min for the standard reaction (see Figure S16 in SI). bReaction conditions unless otherwise stated: 5c (variable), 7 (variable), AgOAc (3 equiv), Benzoquinone (0.5 equiv), Pd(OAc)2 (10 mol%), tAmylOH (1 ml), Boc-L-phenylalanine (0.2 equiv), DMSO (7 equiv), 4Å molecular sieves, 75 °C, air
Regarding the different excess experiments, overlay between the standard run and different-excess V shows a zero-order dependence in [V] suggesting that ethyl acrylate is not involved in the turnover-limiting step of this catalytic cycle (Figure 7B). Different-excess 5c has a reduced rate compared to the standard reaction, showing a positive-order dependence in [5c], This result is potentially consistent with either irreversible substrate binding between (BT)S and Pd or reversible substrate binding followed by turnover-limiting C–H functionalization. We favor the latter interpretation because substrate binding and dissociation is expected to be rapid. We were unable to detect the putative cy- clopalladated intermediate under a variety of conditions, consistent with the notion that C–H activation may be the slow step in catalysis.
Directing Group Coordination.
Similar to our mechanistic studies in the oxidative Heck reaction, we attempted to synthesize various complexes relevant to catalysis, and in this case were able to successfully isolate a starting-material-bound palladium trifluoroacetate complex Pd-2, which we could characterize by X-ray crystallography. As with the above crystal for the oxidative Heck reaction, the complex shows coordination of palladium through the nitrogen of the benzothiazole (Scheme 10).
Scheme 10.
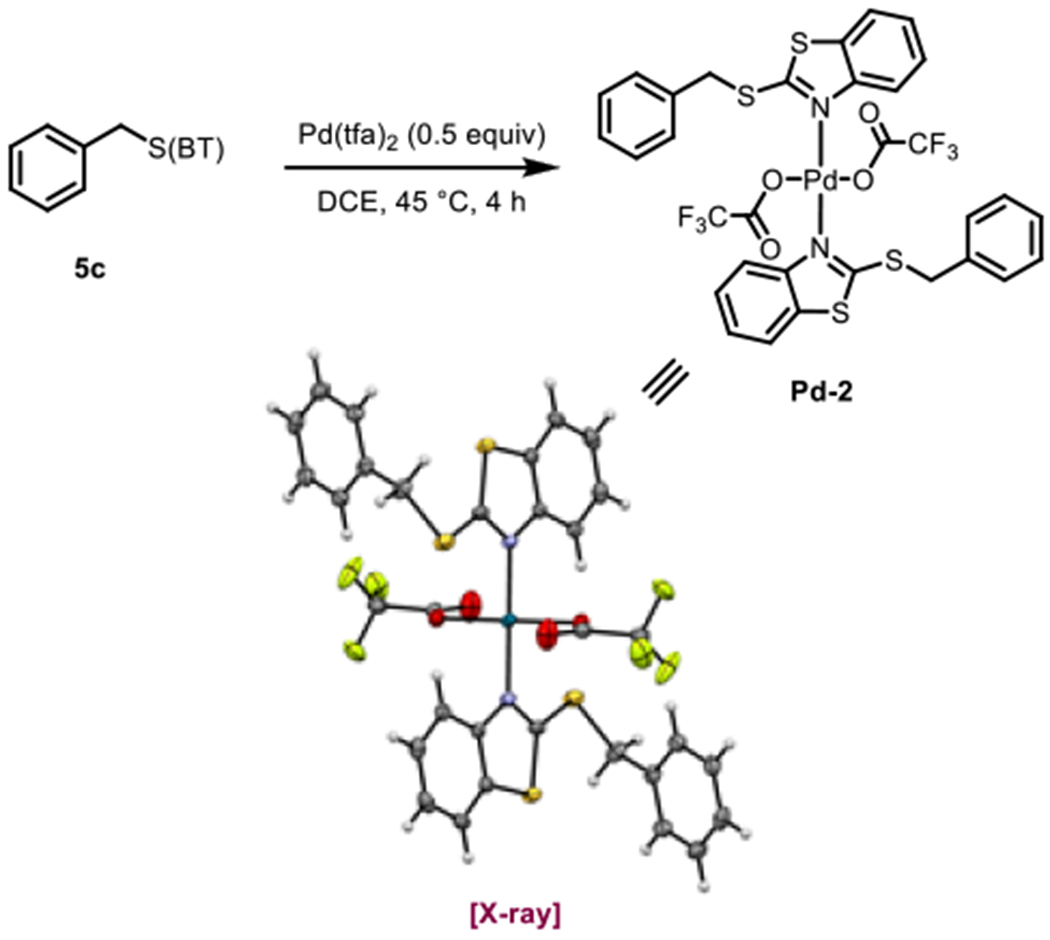
Crystal structure of Pd(tfa)2o 5c complex
Productive Removal and Transformation.
Product 2a from the oxidative Heck reaction was used as a model substrate for directing group conversion and removal (Scheme 11). Moderate yield was observed in a palladium-mediated isocyanide insertion, the product of which was isolated as 8 after acidic workup.30 Fortunately, by treating 2a with ammonium molybdate tetrahydrate, the sulfone product 9 could be formed in quantitative yield with no apparent oxidation of the olefin.31 Sulfone 9 can be subsequently modified through several methods. Treatment with sodium borohydride affords a nearly quantitative yield of 10 containing the versatile sulfinic acid functional group.32 A modified Julia olefination with vertraldehyde gave product 11 in 54% isolated yield as the (E)-alkene.33 Sulfone 9 was also transformed to the carboxylic ester 12 in high yield via treatment with ethyl cyanoformate in the presence of LiHMDS and subsequent removal of the of the ben- zothiazole-sulfone using Zn-dust from the isolated intermediate.34 Oxidation of 2a with mCPBA also gave high yield of sulfoxide 13. Successive treatment of 13 with ammonium acetate gave the sulfanone product 14 in 61 % yield.35 A representative C-H olefination product, 6c, was also oxidized and subsequently transformed to the (E)-alkene 18 with veratraldehyde in 78% isolated yield.
Scheme 11.
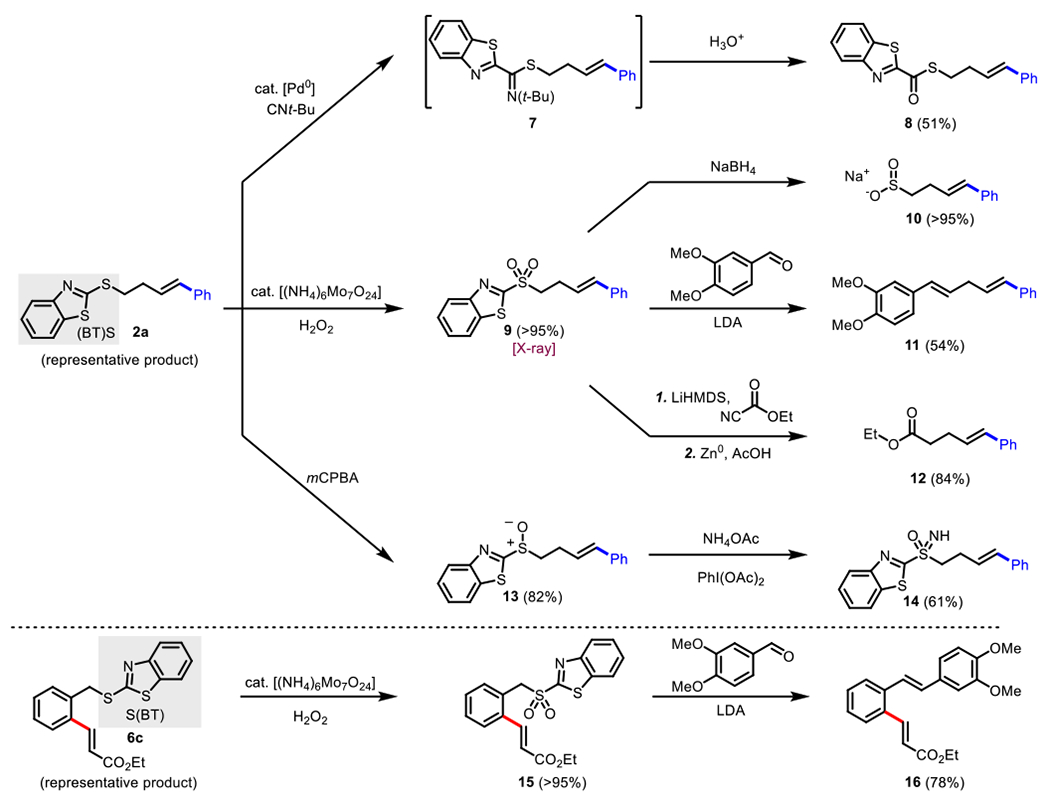
Productive Removal and Transformation of the (BT)S Directing Group
Synthetic Applications.
To demonstrate the utility of the (BT) S directing group in complex molecule synthesis, two natural products, salvianolic acid F and (+)-salviano!ic acid A were formally synthesized using the (BT)S-directed C-H olefination reaction as the key step following a strategy inspired by the carbonyl directed synthesis by Xuan (Scheme 12).36 Through this approach, salvianolic acid F was synthesized in 5 steps with a projected 19.2% overall yield from mercap- tobenzothiazole, which represents the highest overall yield and lowest step count synthesis yet published.37 (+)-Salvianolic acid A was also formally synthesized in 9 steps overall in 14.1% projected yield with the longest linear route being from the mercaptobenzothiazole representing 7 steps with a projected 16.6% yield. This synthesis of (+)-salvi- anolic acid A has a similar step-count and slightly elevated yield compared to the most recent, highest yielding synthesis yet published.36
Scheme 12.
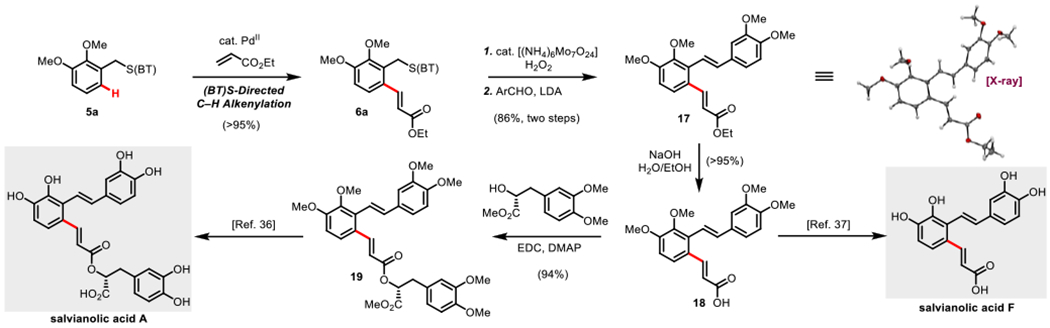
Formal Synthesis of (+)-Salvianolic acid A and Salvianolic acid F
CONCLUSION
In conclusion, we have demonstrated the utility of a new weakly-coordinating heteroaryl thioether directing group, (BT)S, that can facilitate catalytic transformations that require conformational flexibility for integral steps, such as β-hydride elimination (Scheme 13). This new directing group, which can be readily introduced by nucleophilic substitution of a hydroxyl or bromo functional group, serves as a versatile functional group for downstream manipulation. We probed each of the two reactions by performing RPKA and DFT, which helped to elucidate aspects of the mechanism for both catalytic cycles. We also demonstrated the utility of the (BT)S directing group by achieving high overall yields in the synthesis of two natural products.
Scheme 13.
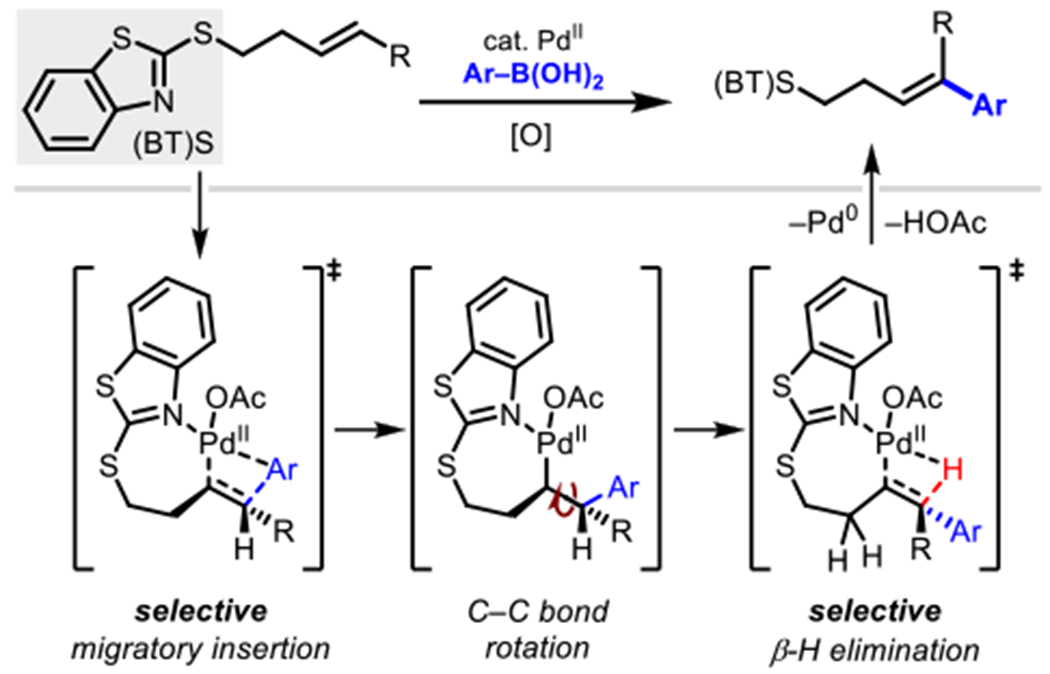
Illustration of the Selectivity of (BT)S
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by The Scripps Research Institute (TSRI) and the National Institutes of Health (5R35GM125052-02). We further acknowledge the NSF for a Graduate Research Fellowship (NSF/DGE-1842471, A.M.R). We thank Prof. Ryan Shenvi (TSRI) and Tucker Huffman (Shenvi Lab, TSRI) for helpful discussions and Prof. Donna G. Blackmond (TSRI) for guidance with RPKA experiments. We also thank Prof. Arnold L. Rheingold, Dr. Milan Gembicky, and Dr. Curtis E. Moore for X-ray crystallographic analysis (UCSD). Brittany Sanchez (ASF, TSRI) is acknowledged for HPLC and HRMS analysis. We thank John A. Gurak, Jr. (Engle Lab, TSRI) and Zhen Liu (Engle Lab, TSRI) for their assistance of proofreading the manuscript. We also thank Ziehen Wang (Engle Lab, TSRI) for his preliminary exploration of directed oxidative Heck reations. We further thank Umi- core for their generous donation of the Z-selective olefin metathesis catalyst Grubbs C675.
ABBREVIATIONS
- (BT)S
Benzothiazole thioether
- RPKA
Reaction Progress Kinetic Analysis
- BQ
benzoquinone
- PTFE
Polytetrafluoroethylene
- LCMS
Liquid Chromatography—Mass Spectrometry
- HPLC
High Pressure Liquid Chromatography
Footnotes
The authors declare no competing financial interest.
Supporting Information
Experiment details, spectra data, copies of NMR spectra, X-ray crystallographic data, and computational details. These materials are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Brown JM; Directed Homogeneous Hydrogenation [New Synthetic Methods (65)]. Angew. Chem. Int. Ed. Engl 1987, 26, 190–203. [Google Scholar]; (b) Hoveyda AH; Evans DA; Fu GC; Substrate- Directable Chemical Reactions. Chem. Rev 1993, 93, 1307–1370. [Google Scholar]; (c) Breit B; Controlling Stereoselectivity with the Aid of a Reagent- Directing Group: Hydroformylation, Cuprate Addition, and Domino Reaction Sequences. Chem. Eur. J 2000, 6, 1519–1524. [DOI] [PubMed] [Google Scholar]; (d) Oestreich M; Neighbouring-Group Effects in Heck Reactions. Eur. J. Org. Chem 2005, 2005, 783–792. [Google Scholar]; (e) Gurak JA; Engle KM; Regioselective Hydroamination Using a Directed Nucleopalladation/Protodepalladation Strategy. Synlett 2017, 28, 2057–2065. [Google Scholar]
- 2.(a) McDonald RI; Liu G; Stahl SS; Palladium(II)-Catalyzed Alkene Functionahzation via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jensen KH; Sigman MS; Mechanistic Approaches to Palladium-Catalyzed Alkene Difunctionahzation Reactions. Org. Biomol. Chem 2008,6,4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee AL; Enantioselective Oxidative Boron Heck Reactions. Org. Biomol Chem 2016,14, 5357–5366. [DOI] [PubMed] [Google Scholar]; (d) Sigman MS; Werner EW;Imparting Catalyst Control upon Classical Palladium-Catalyzed Alkenyl C-H Bond Functionahzation Reactions. Acc. Chem. Res 2012, 45, 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Heck RF; Palladium-Catalyzed Reactions of Organic Hahdes with Olefins. Acc. Chem. Res 1979, 12, 146–151. [Google Scholar]; (f) Dounay AB; Overman LE; The Asymmetric Intramolecular Heck Reaction in Natural Product Total Synthesis. Chem. Rev 2003, 103, 2945–2964. [DOI] [PubMed] [Google Scholar]
- 3.(a) Gurak JA; Yang KS; Liu Z; Engle KM; Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc 2016,138, 5805–5808. [DOI] [PubMed] [Google Scholar]; (b) Yang KS; Gurak JA; Liu Z; Engle ICM; Catalytic, Regioselective Hydrocarbofunctionalization of Unactivated Alkenes with Diverse C- H Nucleophiles. J. Am. Chem. Soc 2016,138,14705–14712. [DOI] [PubMed] [Google Scholar]; (c) Liu Z; Zeng T; Yang KS; Engle KM; β,γ-Vicinal Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Directed Nucleopalladation. J. Am. Chem. Soc 2016, 138, 15122–15125. [DOI] [PubMed] [Google Scholar]; (d) Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM; Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. . Am. Chem. Soc 2017, 139, 11261–11270. [DOI] [PubMed] [Google Scholar]; (e) O’Duill ML; Matsuura R; Wang Y; Turnbull JL; Gurak JA; Gao D-W; Lu G; Liu P; Engle KM; Tridentate Directing Groups Stabilize 6-Membered Palladacycles in Catalytic Alkene Hydrofunctionalization. J. Am. Chem. Soc 2017, 139, 15576–15579. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu Z; Ni H-Q; Zeng T; Engle KM; Catalytic Carbo- and Aminoboration of Alkenyl Carbonyl Compounds via Five- and Six-Membered Palladacycles. J. Am. Chem. Soc 2018, 140, 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zeng T; Liu Z; Schmidt MA; Eastgate MD; Engle ICM; Directed, Palladium(II)-Catalyzed Intermolecular Aminohydroxylation of Alkenes Using a Mild Oxidation System. Org. Lett 2018,20, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Matsuura R; Jankins TC; Hill DE; Yang KS; Gallego GM; Yang S; He M; Wang F; Marsters RP; McAlpine I; Engle KM; Palladium(II)-Catalyzed γ-Selective Hydroarylation of Alkenyl Carbonyl Compounds with Arylboronic Acids. Chem. Sci 2018, 9,8363–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Neufeldt SR; Sanford MS; Asymmetric Chiral Ligand-Directed Alkene Dioxygenation. Org. Lett 2013, 15, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Talbot EPA; Fernandes, T de A.; McKenna JM; Toste FD; Asymmetric Palladium-Catalyzed Directed Intermolecular Fluoroarylation of Styrenes. J. Am. Chem. Soc 2014, 136, 4101–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang H; Bai Z; Jiao T; Deng Z; Tong H; He G; Peng Q; Chen G; Palladium-Catalyzed Amide-Directed Enantioselective Hydrocarbofunctionahzation of Unactivated Alkenes Using a Chiral Monodentate Oxazoline Ligand. J. Am. Chem. Soc 2018, 140, 3542–3546. [DOI] [PubMed] [Google Scholar]; (d) Wang C; Xiao G; Guo T; Ding Y; Wu X; Loh TP; Palladium-Catalyzed Regiocontrollable Reductive Heck Reaction of Unactivated Aliphatic Alkenes. J. Am. Chem. Soc 2018, 140, 9332–9336. [DOI] [PubMed] [Google Scholar]
- 5.Delcamp JH; Brucks AP; White MC; A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc 2008,130,11270–11271. [DOI] [PubMed] [Google Scholar]
- 6.(a) Larhed M; Carl-Magnus A; Hallberg A; Chelation-Controlled, Palladium-Catalyzed Arylation of Vinyl Ethers. Acta Chem. Scand 1993,47,212–217. [Google Scholar]; (b) Larhed M; Andersson C-M; Hallberg A; Chelation-Controlled, Palladium-Catalyzed Arylation of Enol Ethers with Aryl Triflates. Ligand Control of Selection for α-or β-Arylation of [2-(Dimethylamino)ethoxy]ethene. Tetrahedron 1994, 50, 285–304. [Google Scholar]; (c) Olofsson IC; Sahlin H; Larhed M; Hallberg A; Regioselective Palladium-Catalyzed Synthesis of β-Arylated Primary Allylamine Equivalents by an Efficient Pd-N Coordination. J. Org. Chem 2001, 66, 544–549. [DOI] [PubMed] [Google Scholar]; (d) Nilsson P; Larhed M; Hallberg A; A New Highly Asymmetric Chelation-Controlled Heck Arylation. /. J. Am. Chem. Soc 2003,125,3430–3431. [DOI] [PubMed] [Google Scholar]
- 7.(a) Buezo ND; Alonso I; Carretero JC; Sulfinyl Group as a Novel Chiral Auxiliary in Asymmetric Heck Reactions. J. Am. Chem. Soc 1998, 120, 7129–7130. [Google Scholar]; (b) Buezo ND; Mancheno OG; Carretero JC; The 2-(N,N-Dimethylamino)phenylsulfinyl Group as an Efficient Chiral Auxiliary in Intramolecular Heck Reactions. Org. Lett 2000, 2, 1451–1454. [DOI] [PubMed] [Google Scholar]; (c) de la Rosa JC; Diaz N Carretero JC; Asymmetric Synthesis of 1-Aryl and 1,3-Diarylcyclopentenes by the Heck Reaction of 1-Sulfinylcyclopentenes with Iodoarenes. Tetrahedron Lett. 2000,41,4107–4111. [Google Scholar]; (d) Diaz Buezo N de la Rosa JC; Priego J; Alonso I; Carretero JC; Sulfoxides as Stereochemical Controllers in Intermolecular Heck Reactions. Chem. Eur. J 2001, 7, 3890–3900. [DOI] [PubMed] [Google Scholar]; (e) Alonso I; Carretero JC; Highly Stereoselective Synthesis of Trisubstituted α,β-Unsaturated Sulfoxides by Heck Reaction. J. Org. Chem 2001, 66,4453–4456. [DOI] [PubMed] [Google Scholar]
- 8.(a) Itami K; Mitsudo IC; Kamei T; Koike T; Nokami T; Yoshida J; Highly Efficient Carbopalladation Across Vinylsilane: Dual Role of the 2-PyMe2Si Group as a Directing Group and as a Phase Tag. J.Am. Chem. Soc 2000,122, 12013–12014. [Google Scholar]; (b) Itami K; Nokami T; Ishimura Y; Mitsudo K; Kamei T; Yoshida J; Diversity-Oriented Synthesis of Multisubstituted Olefins through the Sequential Integration of Palladium-Catalyzed Cross-Coupling Reactions. 2- Pyridyldimethyl(vinyl)silane as a Versatile Platform for Olefin Synthesis. J. Am. Chem. Soc 2001, 123, 11577–11585. [DOI] [PubMed] [Google Scholar]; (c) Itami IC; Ushiogi Y; Nokami T; Ohashi Y; Yoshida J; Stereoselective Synthesis of Multisubstituted Butadienes through Directed Mizoroki—Heck Reaction and Homocouphng Reaction of Vinyl (2-pyridyl)silane. Org. Lett 2004, 6, 3695–3698. [DOI] [PubMed] [Google Scholar]; (d) Itami K; Mineno M; Muraoka N; Yoshida J; Sequential Assembly Strategy for Tetrasubstituted Olefin Synthesis Using Vinyl 2-Pyrimidyl Sulfide as a Platform. J.Am. Chem. Soc 2004,126,11778–11779. [DOI] [PubMed] [Google Scholar]; (e) Bernocchi E Cacchi S; Ciattini PG; Morera E; Ortar G; Palladium-Catalysed Vinylation of Allylic Alcohols with Enol Triflates. A Convenient Synthesis of Conjugated Dienols. Tetrahedron Lett. 1992, 33, 3073–3076. [Google Scholar]; (f) Filippini L; Gusmeroli M j Riva R; Palladium-Catalyzed Coupling of Organic Halides and Tertiary Allylic Amines. Tetrahedron Lett. 1993, 34, 1643–1646. [Google Scholar]; (g) Badone D Guzzi U; Palladium-Catalyzed β-Arylation of Modified Vinyl Ethers with Aryl Triflates. Tetrahedron Lett. 1993,34,3603–3606. [Google Scholar]; (h) Ono K; Fugami K; Tanaka S; Tamaru Y; Palladium Catalyzed Arylation of N-alkyl O-allyl Carbamates: Synthesis of Cinnamyl Alcohols via Heck Arylation. Tetrahedron Lett. 1994, 35, 4133–4136. [Google Scholar]; (i) Kang S-K; Jung K-Y; Park C-H; Namkoong E-Y; Kim T-H; Palladium-Catalyzed Arylation of Allylic Diols: Highly Selective Synthesis of Phenyl- Substituted Allylic Diols. Tetrahedron Lett. 1995, 36, 6287–6290. [Google Scholar]; (j) Kang S-K; Lee H-W Jang S-B; Kim T-H; Pyun S-J; Complete Regioselection in Palladium-Catalyzed Arylation and Alkenylation of Allylic Alcohols with Hypervalent Iodonium Salts. . Org. Chem 1996, 61,2604–2605. [DOI] [PubMed] [Google Scholar]; (k) Oestreich M; Sempere-Culler F; Machotta AB; Catalytic Desymmetrizing Intramolecular Heck Reaction: Evidence for an Unusual Hydroxy-Directed Migratory Insertion. Angew. Chem. Int. Ed 2004,44,149–152. [DOI] [PubMed] [Google Scholar]
- 9.(a) Werner EW; Mei T-S; Burckle AJ; Sigman MS; Enantioselective Heck Arylations of Acychc Alkenyl Alcohols Using a Redox-Relay Strategy. Science 2012, 338, 1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mei T-S; Werner EW; Burckle AJ; Sigman MS; Enantioselective Redox- Relay Oxidative Heck Arylations of Acychc Alkenyl Alcohols using BoronicAcids. J.Am. Chem. Soc 2013,135, 6830–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mei T-S; Patel HH; Sigman MS; Enantioselective Construction of Remote Quaternary Stereocentres. Nature 2014, 508, 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu L;Hilton MJ; Zhang X; Norrby P-O; Wu Y-Dj Sigman MS; Wiest O; Mechanism, Reactivity, and Selectivity in Palladium-Catalyzed Redox-Relay Heck Arylations of Alkenyl Alcohols. J. Am. Chem. Soc 2014, 136,1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang C; Santiago CB; Kou L; Sigman MS; Alkenyl Carbonyl Derivatives in Enantioselective Redox Relay Heck Reactions: Accessing α,β-Unsaturated Systems. J. Am. Chem. Soc 2015,137,7290–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Larock RC; Leung W-Y; Stolz-Dunn S; Synthesis of Aryl-Substituted Aldehydes and Ketones via Palladium- Catalyzed Couphng of Aryl Hahdes and Non-Allylic Unsaturated Alcohols. Tetrahedron Lett. 1989, 30, 6629–6632. [Google Scholar]; (g) Berthiol F; Doucet H; Santelh M; Heck Reaction of Aryl Bromides with Pent-4- en-2-ol, 2-Phenylpent-4-en-2-ol, or Hept-6-en-3-ol Catalysed by a Palladium-Tetraphosphine Complex. Synthesis 2005, 37, 3589–3602. [Google Scholar]
- 10.Huffman TR; Wu Y; Emmerich A; Shenvi RA; Intermolecular Heck Couphng with Hindered Alkenes Directed by Potassium Carboxylates. Angew. Chem. Int. Ed 2019, 58, 2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For representative examples, see:; (a) Perales JB; Van Vranken DL; Thioether-Directed Platinum-Catalyzed Hydrosilylation of Olefins. J. Org. Chem 2001, 66, 7270–7274. [DOI] [PubMed] [Google Scholar]; (b) Dupont J; Basso NR; Meneghetti MR; Chloropalladation of Propargyl Thioethers: A Facile Synthesis of Cyclopalladated Compounds. Polyhedron 1996,15, 2299–2302. [Google Scholar]; (c) Rao GK; Kumar A; Kumar S; Dupare UB; Singh AK; Palladacycles of Thioethers Catalyzing Suzuki-Miyaura C-C Coupling: Generation and Catalytic Activity of Nanoparticles. Organometallics 2013, 32, 2452–2458. [Google Scholar]; (d) Mann SE; Aliev AE; Tizzard GJ; Sheppard TD; Sulfur-Directed Olefin Oxidations: Observation of Divergent Reaction Mechanisms in the Palladium-Mediated Acetoxylation of Unsaturated Thioacetals. Organometallics 2011,30, 1772–1775. [Google Scholar]
- 12.For representative examples, see:; (a) Yao J; Yu M; Zhang Y; Thioethers as Directing Group for the Palladium-Catalyzed Direct Arylation of Arenes. Adv. Synth. Catal 2012, 354, 3205–3210. [Google Scholar]; (b) Wang B; Lin C; Liu Y; Fan Z; Liu Z; Zhang Y; Thioether-Directed Acetoxylation of C(sp2)-H Bonds of Arenes by Palladium Catalysis. Org. Chem. Front 2015, 2, 973–977. [Google Scholar]; (c) Likun J; Wang J; Dong G; Palladium-Catalyzed γ-C(sp3)-H Arylation of Thiols via a Detachable Protecting/Directing Group. Angew. Chem. Int. Ed 2018, 57, 12352–12355; for related example with platinum, see: [DOI] [PubMed] [Google Scholar]; (d) Perales JB; Van Vranken DL; Thioether-Directed Platinum-Catalyzed Hydrosilylation of Olefins. J. Org. Chem 2001, 66, 7270–7274. [DOI] [PubMed] [Google Scholar]
- 13.(a) Zheng Y Song W; Zhu Y; Wei B; Xuan L; Pd-Catalyzed Acetoxylation of γ-C(sp3)-H Bonds of Amines Directed by a Removable Bts-Protecting Group. J. Org. Chem 2018,83,2448–2454. [DOI] [PubMed] [Google Scholar]; (b) Petrova E; Rasina D; Jirgensons A; N-Sulfonylcarboxamide as an Oxidizing Directing Group for Ruthenium-Catalyzed C–H Activation/Annulation. Eur. J. Org. Chem 2017,2017, 1773–1779. [Google Scholar]
- 14.(a) Blackmond DG; Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed 2005,44,4302–4320. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG; Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015,137,10852–10866. [DOI] [PubMed] [Google Scholar]
- 15.Burés J; A Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem. Int. Ed. Engl 2016,55, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Al-Hayaly L. a. J.; Buttrus NH; Al-Allaf TAK; Homobimetallic Complexes of Platinum(II) Group Metals with Sulphur Containing Ligands R2C = CR2 (R = 5-Phenyl-1,3,4-oxadiazole-2-thiol; 4,5-Diphenyl-1,2,4-triazole-3-thiol or Mercaptobenzothiazole). Asian J. Chem 2002, 14, 1421–1426. [Google Scholar]; (b) Song R-F; Liu J-H; Qian H; Zhao K-Y; Dichloro[8-(1,3-benzothiazol-2-ylsulfanylmethyl)-quinoline-jN,N00]palladium(II). Acta Crystallogr. Sect. Sect. E: Struct. Rep. Online 2005, 61, 2142–2144. [Google Scholar]; (c) Pornsuriyasak P; Gangadharmath UB; Rath NP; Demchenko AV; A Novel Strategy for Oligosaccharide Synthesis via Temporarily Deactivated S-Thiazolyl Glycosides as Glycosyl Acceptors. Org. Lett 2004, 6, 4515–4518. [DOI] [PubMed] [Google Scholar]; (d) Li Y-W; Gu G-B; Liu H-Y; Sung HHY; Williams ID; Chang C-K; A New iso-Amyl Benzothiazolyl Sulfoxide as an Extractant for Palladium and the Crystal Structure of Its Palladium (II) Complex. Molecules 2005,10,912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hughes MN Rutt KJ; The Far Infrared Spectra of Some Complexes of Palladium (II) with Thiazoles. Spectrochim. Acta, Pt. A: Mol. Spectrosc 1971, 27, 924–926. [Google Scholar]; (f) Fuchita Y Yoshinaga K; Kusaba H; Mori M; Hiraki K; Takehara K; Synthesis and Characterisation of the Six-Membered Cyclopalladated Complexes of 2-Benzylbenzothiazole. Inorg. Chim. Acta 1995,239,125–132. [Google Scholar]
- 17.For coordinates of the transition states, see SI in: (a) Liu M; Yang P; Karunananda MK; Wang Y; Liu P; Engle KM C(alkenyl)-H Activation via Six-Membered Palladacycles: Catalytic 1,3-Diene Synthesis, J. Am. Chem. Soc 2018,140, 5805–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carbon-carbon unsaturated bonds have also been utilized as directing groups for arene C-H olefination, but the scope and utility of these transformations tend to be limited due to the difficulty of installing the directing group, reduction of the directing group during the course of the reaction, and the inability to remove or controllably transform after the C-H functionalization reaction. For a review, see: Minami Y; Hiyama T; Collaborative Interaction of Carbon-Carbon Unsaturated Bond Groups with Transition-Metal Catalysts for C-H Bond Functionalization. Chem. Lett 2018,47,1–8. [Google Scholar]
- 19.Mei T-S; Wang X; Yu J-Q; Pd(II)-Catalyzed Amination of C-H Bonds Using Single-Electron or Two-Electron Oxidants. J. Am. Chem. Soc 2009,131,10806–10807. [DOI] [PubMed] [Google Scholar]
- 20.Wasa M; Yu J-Q; Synthesis of β-, γ-, and δ-Lactams via Pd(II)-Catalyzed C-H Activation Reactions. J. Am. Chem. Soc 2008, 130,14058–14059. [DOI] [PubMed] [Google Scholar]
- 21.Wang X; Lu Y Dai H-X; Yu J-Q; Pd(II)-Catalyzed Hydroxyl-Directed C-H Activation/C-O Cyclization: Expedient Construction of Dihydrobenzofurans. J. Am. Chem. Soc 2010, 132, 12203–12205. [DOI] [PubMed] [Google Scholar]
- 22.Mei T-S; Wang D-H Yu J-Q; Expedient Drug Synthesis and Diversification via ortho-C-H Iodination using Recyclable PdI2 as the Precatalyst. Org. Lett 2010,12, 3140–3143. [DOI] [PubMed] [Google Scholar]
- 23.(a) Li G; Leow D; Wan L; Yu J-Q; Ether-Directed ortho-C-H Olefination with a Palladium(II)/Monoprotected Amino Acid Catalyst. Angew. Chem. Int. Ed 2013, 52, 1245–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li G; Wan L; Zhang G; Leow D; Spangler J; Yu J-Q; Pd(II)-Catalyzed C-H Functionalizations Directed by Distal Weakly Coordinating Functional Groups. J. Am. Chem. Soc 2015,137,4391–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu M; Xie Y; Xie C; Zhang Y; Palladium-Catalyzed C–H Alkenylation of Arenes Using Thioethers as Directing Groups. Org. Lett 2012,14,2164–2167. [DOI] [PubMed] [Google Scholar]
- 25.Wang B; Shen C; Yao J; Yin H; Zhang Y; Palladium(II)-Catalyzed ortho-Olefination of Arenes Applying Sulfoxides as Remote Directing Groups. Org. Lett 2014,16,46–49. [DOI] [PubMed] [Google Scholar]
- 26.Maji A; Bhaskararao B; Singha S; Sunoj RB; Maiti D; Directing Group Assisted Meta-Hydroxylation by C-H Activation. Chem. Sci 2016, 7,3147–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engle KM; The Mechanism of Palladium(II)-Mediated C-H Cleavage with Mono-N-Protected Amino Acid (MPAA) Ligands: Origins of Rate Acceleration. Pure Appl. Chem; 2016, 88,119–138. [Google Scholar]
- 28.For previous examples of enthenesulfonyl fluoride (ESF) in C-H olefination, see:; (a) Wang S-M; Li C; Leng J; Bukhari SNA; Qin H-L; Rhodium(III)-Catalyzed Oxidative Coupling of N-Methoxybenzamides and Ethenesulfonyl fluoride: a C-H Bond Activation Strategy for the Preparation of 2-Aryl Ethenesulfonyl Fluorides and Sulfonyl Fluoride Substituted γ-Lactams. Org. Chem. Front 2018, 5,1411–1415. [Google Scholar]; (b) Liu M; Yang P; Karunananda MK; Wang Y; Liu P; Engle KM; C(alkenyl)-H Activation via Six-Membered Palladacycles: Catalytic 1,3-Diene Synthesis. J. Am. Chem. Soc; 2018,140, 5805–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li C; Wang S-M; Qin H-L; A Rh-Catalyzed Air and Moisture Tolerable Aldehyde (Ketone)-Directed Fluorosulfonylvinylation of Aryl C(sp2)-H Bonds. Org. Lett 2018, 20, 4699–4703. [DOI] [PubMed] [Google Scholar]; (d) Wang S-M; Moku B; Leng J; Qin H-L; Rh-Catalyzed Carboxylates Directed C-H Activation for the Synthesis of ortho-Carboxylic 2-Arylethenesulfonyl Fluorides: Access to Unique Electrophiles for SuFEx Click Chemistry. Eur. J. Org. Chem 2018,2018, 4407–4410. [Google Scholar]; (e) Ncube G; Huestis MP; Directed Cp*RhIII-Catalyzed Fluorosulfonylvinylation of Arenes. Organometallics 2019, 38, 76–80. [Google Scholar]; (f) Chen X-Y; Wu Y; Zhou J; Wang P; Yu J-Q; Synthesis of β-Arylethenesulfonyl Fluoride via Pd-Catalyzed Nondirected C-H Alkenylation. Org. Lett 2019,21,1426–1429. [DOI] [PubMed] [Google Scholar]
- 29.(a) Engle KM; Wang D-H; Yu J-Q; Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc 2010, 136, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deb A; Bag S; Kancherla R; Maiti D; Palladium-Catalyzed Aryl C-H Olefination with Unactivated, Aliphatic Alkenes. J. Am. Chem. Soc 2014, 136, 13602–13605. [DOI] [PubMed] [Google Scholar]
- 30.Otsuka S; Nogi K; Yorimitsu H; Palladium-Catalyzed Insertion of Isocyanides into the C–S Bonds of Heteroaryl Sulfides. Angew. Chem. Int. Ed 2018,57, 6653–6657. [DOI] [PubMed] [Google Scholar]
- 31.Baudin JB; Stereochemistry of Direct Olefin Formation from Carbonyl Compounds and Lithiated Heterocyclic Sulfones. Bull. Soc. Chim. Fr 1993,130, 856–878. [Google Scholar]
- 32.Day JJ; Neill DL; Xu S; Xian M; Benzothiazole Sulfinate: A Sulfinic Acid Transfer Reagent under Oxidation-Free Conditions. Org. Lett 2017,19, 3819–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Modified Julia olefinations are known to exhibit variable E/Z selectivity as a function of reaction solvent, base, and coupling partner, and the high E-selectivity in this case is a result of the reaction conditions and the identity of the arylaldehyde coupling partner, as has been described previously. Blakemore PR; The Modified Julia Olefination: Alkene Synthesis via the Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. J. Chem. Soc.; Perkin Trans. 1 2002, 2563–2585. [Google Scholar]
- 34.Bon DJYD; Kováč O; Ferugová V; Zálešák F; Pospíšil J; One and Two-Carbon Homologation of Primary and Secondary Alcohols to Corresponding Carboxylic Esters Using β-Carbonyl BT Sulfones as a Common Intermediate. J. Org. Chem 2018, 83,4990–5001. [DOI] [PubMed] [Google Scholar]
- 35.Zenzola M; Doran R; Degennaro L; Luisi R; Bull JA; Transfer of Electrophilic NH Using Convenient Sources of Ammonia: Direct Synthesis of NH Sulfoximines from Sulfoxides. Angew. Chem. Int. Ed 2016,55,7203–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y; Song W-B; Xuan L-J; The Asymmetric Total Synthesis of (+)-Salvianolic Acid A. Tetrahedron 2016, 72, 5047–5050. [Google Scholar]
- 37.Dalla V; Cotelle P; The Total Synthesis of Salvianolic Acid F. Tetrahedron 1999,55, 6923–6930. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.