

Abstract
Here, we develop an injection molded microfluidic approach for single cell analysis by making use of: 1) rapidly curing injectable hydrogels, 2) a high density microfluidic weir trap array, and 3) reversibly bonded PDMS lids that are strong enough to withstand the injection molding process, but which can be peeled off after the hydrogel sets. This approach allows for single cell patterns to be created with densities exceeding 40 cells per mm2, is amenable to high speed imaging, and creates microfluidic devices that enable efficient nutrient and gas exchange and the delivery of specific biological and chemical reagents to individual cells. We show that it is possible to organize up to 10,000 single cells in a few minutes on the device, and we developed an image analysis program to automatically analyze the single-cell capture efficiency. The results show single cell trapping rates were better than 80%. We also demonstrate that the genomic DNA of the single cells trapped in the hydrogel can be amplified via localized, multiple displacement amplification in a massively parallel format, which offers a promising strategy for analyzing single cell genomes. Finally, we show the ability to perform selective staining of individual cells with a commercial bioprinter, providing proof of concept of its ability to deliver tailored reagents to specific cells in an array for future downstream analysis. This injection molded microfluidic approach leverages the benefits of both closed and open microfluidics, allows multi-day single cell cultures, direct access to the trapped cells for genotypic endpoint studies.
Keywords: microfluidics, injection molding, single cell analysis, open microfluidics, hydrogel
Graphical Abstract
INTRODUCTION
The ability to analyze the degree of heterogeneity in cellular responses in the presence of chemical or physical perturbations requires a miniaturized platform that can simultaneously perform tens of thousands of identical experiments on single cells or small cellular communities, such as single cell derived clonal expansions or organoids.1–6 First and foremost, this platform should provide a hospital environment for cells in the absence of the perturbation7; however, it must also provide other functionalities, including: i) the ability to retain cells in the same locations during multi-day time-lapse imaging, ii) a footprint that is small enough such that all of the cell culture chambers can be imaged rapidly in multiple channels with a standard automated fluorescent microscope, and iii) compatibility with genomic assays at single cell resolution.
Despite this clear need, current technology falls short in meeting one or more of the above requirements. For example, 96- or 384- well plates are excellent at limiting cross-contamination of cells between different wells, however the imaging speed is too slow to achieve the necessary scale of observing tens of thousands of cellular responses in parallel,8,9 and additionally, single cells generally lose viability when cultured in isolation in the individual wells of a 96-well plate. Open microfluidic devices,10–13 such as the microraft array,14–16 are promising alternatives to achieve the required scale and recover the cells of interest for downstream genomic analysis; however they cannot adequately prevent cells from migrating away from their compartments due to the lack of physical confinement, which causes challenges for tracking individual cells (e.g., suspension cells) over multiple days. Closed microfluidic devices, such as microfluidic weir trap arrays,8,17–21 can achieve the required scale and also better confine cells to prevent their migration away from their initial positions; however these systems require continuous perfusion of cell culture medium, and it is challenging to implement these platforms to retrieve the molecular profiles of the arrayed cells in a high throughput manner.
In this work, we exploit the moldable property of hydrogels, which consist of crosslinked polymers that possess the unique physical, mechanical, and mass transport properties required to support cell growth, and thus have been widely used in microfluidics for cell culture and tissue engineering.22–24 Most prior hydrogel-based studies have been performed in microwell plates on cell populations as opposed to isolated single cells,25,26 and have primarily been used in studying differentiation of pluripotent stem cells,27 cultivating primary hepatocytes28 and analyzing cancer spheroid growth29. Droplet-based systems have also been used to encapsulate single cells in hydrogel beads.30 While most prior hydrogel bead studies have been limited to short-term experimental durations, a few studies have demonstrated longer-term cell culture studies31,32; however regardless of the experimental duration, mobile droplets have a different set of challenges in interfacing with external controllers, such as robotic liquid handlers, compared to open array-based platforms.
In order to combine the desirable biocompatibility and accessibility of an open format cell culture platform33 with the trapping and confinement capabilities of a closed format microfluidic trap array, we have developed a novel microfluidic injection molding approach that serves as a flexible method for entrapping single cells in a patterned hydrogel matrix for applications in high throughput cellular analysis. Injection molding is a low-cost, high-volume manufacturing process that is based on infusing liquid polymers into a cavity mold, allowing the polymer to set, and then opening of the mold to retrieve the part.34,35 Here, we emulate this approach in microfluidics to form single cell arrays by making use of biocompatible hydrogel crosslinkers that can be infused into devices containing an array of trapped single cells. Our approach leverages the ability of PDMS lids to reversibly bond to etched microfluidic channels, such that cells can be first organized in a high-density array format, after which a hydrogel is injected into the device to crosslink the array and stabilize the cellular pattern prior to peeling off the lid.
After peeling the lid, the open cell culture format is ideal for long-term nutrient delivery and gas exchange with the additional ability to use robotic liquid handlers to deliver unique biochemical reagents to specific cells necessary for downstream assays. To demonstrate one potential application of this injection-molding platform, we carried out multiple displacement amplification (MDA) to amplify the whole genome of single cells entrapped in the array. We also show how a bioprinter can be used to deposit fluorescent stains at precise locations on the cellular patterns, which provides “proof of principle” of myriad applications in local gene editing, immunostaining, and selective amplification of specific cells in the array. Finally, this approach is compatible with the 3D hydrogel-based cell culture techniques, which are more representative of in vivo conditions.25 In summary, our injection molded microfluidic platform demonstrates a series of novel capabilities, including >80% single cell capture efficiency, convenient long-term cell culture, easy access and reagent delivery to the cells and thus is an ideal platform for massively parallel and multiparameter single cell measurements combining phenotypic and genotypic characterizations.
EXPERIMENTAL METHODS
Device fabrication.
The microfluidic devices were fabricated by deep reactive ion etching of Silicon (Fig. S1), as previously described.20,36 Briefly, Shipley S1813 positive photoresist (MicroChem Corp., GmbH) was spin coated (4000 rpm, 1 min) onto 6-inch silicon wafers (University Wafer, Inc.), baked at 115 °C for 1 min, then exposed to UV irradiation (7.0 s at 12mW/cm2 intensity) from a mask aligner (MA6/BA6, Karl Süss) to define the pattern. The exposed wafer was developed in MF319 (MicroChem Corp., GmbH), then rinsed in DI water and blown dry in Nitrogen. Next, the wafer was etched with a deep reactive-ion etcher (Pegasus deep silicon etcher; SPTS Technologies, Ltd.) to a depth of 20 μm. To dice individual chips from the wafer, we used a second lithography step involving backside alignment and patterning of a photoresist, followed by a through silicon etch. The dice lines were aligned and patterned on the backside of the 6” wafer with AZ9260 photoresist (Micro- Chemicals, GmbH) that was spin coated at 1800 rpm for 60 s, baked at 110 °C for 3 min, then exposed to 3600 mJ of UV on the mask aligner. Next, the wafer was bonded to a carrier-wafer and a through-silicon etch was processed using deep reactive ion etching, which diced the wafer into individual devices. After that, the chips were cleaned in piranha solution. To seal the device, a 4 mm-thick PDMS lid with a 3 mm diameter inlet and 1 mm diameter outlet was attached to the chip (Fig. S1). The device was degassed by placing it in a vacuum container for ~20 min before using, which can avoid air bubbles during device priming.19
Cell culture.
K562, HL60 cells (ATCC, VA, USA) were cultured with 10% (vol/vol) fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS). MDA-MB-231/GFP (Cell Biolabs) and A549 (ATCC, VA, USA) cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% (vol/vol) FBS and 1% PS. All cells were grown in a cell-culture incubator with a humidified atmosphere of 5% (vol/vol) CO2 at 37 °C.
Hydrogel preparation.
For many studies, the basic hydrogel components consist of four-arm polyethylene glycol (PEG) acrylate (molecular weight=10,000) and HS-PEG-SH (MW=3,400) were obtained from Laysan Bio (Arab, AL). To prepare the solution, 6.4 mg four-arm PEG acrylate and 4.4 mg HS-PEG-SH were dissolved in 50 μL pH 7.4 phosphate buffered saline (PBS), respectively. Before using, 10 μL of the two components were mixed and vortexed to ensure complete mixing. The hydrogel components crosslink through the reaction between the thiol and acrylate groups in about 20 min at room temperature, however the hydrogel setting time was significantly faster at 37 °C. This PEG-based hydrogel was adopted in the MDA genome amplification due to its well characterized pore size.37–39
For long-term culture, on the other hand, we used HyStem® Cell Culture Scaffold Kit (HYS010, Sigma-Aldrich) due to its good performance for supporting cell growth.40 HyStem hydrogels are composed a thiol-modified hyaluronan and carboxymethyl hyaluronic acid-thiopropanoyl hydrazide that is mixed with a thiol-reactive crosslinker, polyethyleneglycol diacrylate, which together provides a suitable scaffold for 3D cell culture.41 The hydrogel was prepared by following the manufacturer protocol. Briefly, Hystem stock solutions (two bottles) were reconstituted by adding 1 mL degassed water into each bottle and vortexed until completely dissolved, and then the two solutions were mixed together. Next, the Extralink from the Hystem kit was reconstituted with 0.5 ml of degassed water and mixed with Hystem stock at 1:4 ratio (v/v) with gelation occurring within 20 minutes at room temperature.
Cell injection molding protocol.
Devices were primed with cell media by applying vacuum at the outlet with a syringe. To prevent cells from sticking to the chip during the loading process, we incubated the chip with 10% FBS prior to conducting the experiment, which allowed the proteins within the serum to coat the channel surface and reduce non-specific adhesion. Then 5 μL of the cell suspension (~1×106 cells/mL) was introduced at the inlet with flow driven by applying −25 mbar vacuum pressure at the outlet (OB1 Pressure Controller; Elveflow). After 3–5 min, the remaining cells at the inlet were rinsed with fresh medium and more medium was injected into the device to wash away excess cells in the microchannel. Subsequently, 20 μL of the hydrogel mixture was introduced to the device with the trapped cells. The device was maintained in a cell culture incubator for 20 min to allow the hydrogel to crosslink, and then the PDMS lid was carefully peeled off from the device. The hydrogel pattern has stronger adhesion to the Silicon chip than the PDMS lid, so the cells remain with the Silicon chip after the peeling process.
Automated imaging.
The chambers were imaged with a motorized Leica DMI-6000B microscope, MS-2000 Automated XY stage (Applied Scientific Instrumentation), and a Retiga 2000R camera (Photometrics). We used MetaMorph® software (Molecular Devices) to define a list of stage positions, then acquire, name, and save the images associated with each weir trap in the device in GFP, RFP, brightfield channels. We were able to visualize 20 – 30 weir traps in each image at 16× magnification, allowing full coverage of the array with several hundred images per channel at a speed of approximately 5 minutes per fluorescent channel. Imaging was performed both before and after the PDMS lid was removed.
On-chip MDA reaction.
First, the trapped cells were lysed by submerging the chip in a petri dish containing the denaturation buffer for 10 min at 65°C, which was then quenched with a stop solution for 5 min at room temperature. Next, the lysed MDA-MB-231/GFP cells were stained with 1 μM SYTO 64 Red Fluorescent Nucleic Acid Stain (Thermo Fisher Scientific, Waltham, MA) and imaged in the brightfield, GFP, and RFP channels (the GFP signal was quenched after the lysis process). The MDA reaction was then performed according to the QIAGEN® REPLI-G sc kit protocol, which involved submerging the chips in a petri dish containing the dNTPs, random hexmers, DNA polymerase, and other buffer components, and then incubating the dish on a heating block at 30 °C for 8 hours, followed by a terminal step at 65 °C for 3 min to inactivate the polymerase.
The images in RFP channel before and after the MDA reaction were analyzed to calculate the DNA amplification of single cells. The red fluorescence intensity around the cells before (A1) and after (A2) the reaction were extracted and subtracted by the relevant background (B1 and B2). DNA amplification was calculated as:
| (1) |
Localized reagent delivery.
A non-contact Piezorray Printer (S11 sciFlexarrayer, Scienion, Berlin, Germany) was used to selectively deliver precise amounts of reagents at specific locations of the device. The spots were deposited on a rectangular grid at the same spacing as the period of the weir trap array with different patterns used to demonstrate the printing resolution. Following protocols supplied by the vendor, we used the instrument’s camera and auto-alignment software to find fiducial marks on the chips, and define the grid of points to spot the printed molecules. Spot sizes of ~100 μm could be printed with this approach on a variety of substrates with good spatial registration relative to the target printing locations (+/− 10 μm was obtainable). Since the 300 pL spots rapidly dried within a few seconds after printing, the lateral diffusion of the molecules was negligible, leading to good reproducibility in the printing method. Our proof of concept work mainly involved printing fluorescent dyes and staining reagents. In these experiments, the cells were fixed for 30 min at 4 °C by immersing the device with the cells in a fixation solution (554655, BD Biosciences, San Diego, CA), and washed with a staining buffer containing fetal bovine serum (554656, BD Biosciences, San Diego, CA). The cells were maintained in the buffer at 4 °C (protected from light) until the printing. To process the printing, the device with the fixed cells was carefully blown dry with air. Following the standard imaging protocols, we were able to register and align the spots relative to known fiducial marks in the array. Fluorescein isothiocyanate (FITC, 10 μM, Sigma-Aldrich) was used for calibrating the printer resolution. Hoechst 33342 (10 μM; Thermo Fisher Scientific, Waltham, MA) was used to label the nuclei of specific cells, whereas SYTOX Green Nucleic Acid Stain (5 μM; Thermo Fisher Scientific, Waltham, MA) was used to stain all cells after printing.
Image analysis and cell counting.
The raw acquired images were analyzed with custom MATLAB® scripts (https://github.com/yellenlab/Cell-Array-Counter). This process involves aligning images using the template matching plugin of ImageJ, and then cropping the individual weirs in the bright field and fluorescent channels, respectively, which were saved and named according to the address location on the chip. The region of interest is defined by a user prompt to draw a bounding around the weir in one of the cropped images, which is applied to the rest of the images automatically with the previous image registration step. Within the regions of interest (ROIs), we used a circle-tracking algorithm adapted from the circular Hough transform to determine the number of cells based on the fluorescence pattern. To account for variation in cell shapes and sizes, a sensitivity coefficient is provided to cover a range of cellular pixel dimensions, which can be adjusted depending on the microscope magnification and camera settings. Using this approach, we construct heat maps to indicate the spatial distribution of cells in the array, as well as efficiency data quantifying the overall trapping performance across the chip. In some cases, we also use ImageJ to stich and overlay the image together to produce figures showing single-cell population across an entire chip or a desired sub-region.
Computational fluid dynamics (CFD) simulations.
Numerical simulations were conducted using a commercial CFD solver (Fluent 6.1) to solve the flow distribution inside the microfluidic channels42 subjected to a constant pressure drop. This software uses the 3D finite element method to solve the continuity equation, Navier-Stokes equation, and diffusion-convection relationships, as described by Mengeaud et al.43 The simulation assumes a Newtonian fluid with the properties of water at room temperature with channel boundaries assumed to be smooth and slip-free. The SIMPLE algorithm was implemented for pressure–velocity coupling, and the criterion for convergence was for the increment in each variable to fall below 1 ×10−5.44
RESULTS AND DISCUSSION
Overall working principle of the hydrogel injection molding approach.
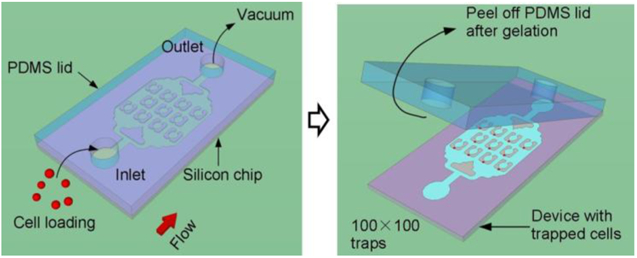
Closed microfluidic devices offer reliable control over fluid flow,45,46 but limits direct-access to the sample inside the microfluidic channels. Open microfluidic systems provide an alternative approach that allows direct access to the liquid sample or cells; however, the stochastic nature of prior cell trapping methods (i.e., random sedimentation into micro-wells47,48) leads to cell arrays with poor single-cell capture efficiency. We have combined the benefits of both open and closed microfluidic platforms in a novel injection molding approach that is described in the workflow shown in Fig.1. These devices are initially constructed as a closed system (Fig. 1a); however, instead of permanently bonding the PDMS lid to the channels, as is commonly done in conventional microfluidic devices, we used a reversible bonding approach by attaching a PDMS lid to the silicon chip without plasma bonding.49 Cells or reagents are loaded into the closed device through vacuum pressure applied to the outlet side, which prevents premature detachment of the lid (Fig. 1a, b). Successful priming of the device is achieved because we degassed the PDMS lid in a vacuum desiccator prior to conducting the experiment, such that during the blind filling process the trapped bubbles dissolve into the PDMS lid that acts as a gas sink. After cell loading and hydrogel crosslinking, the lid is peeled to open the device (Fig. 1c), and then the device is placed in a Petri dish for long-term cell culture or other biochemical analysis requiring direct access to the cells (Fig. 1d).
Figure 1. Working principle of the injection molded microfluidic approach.
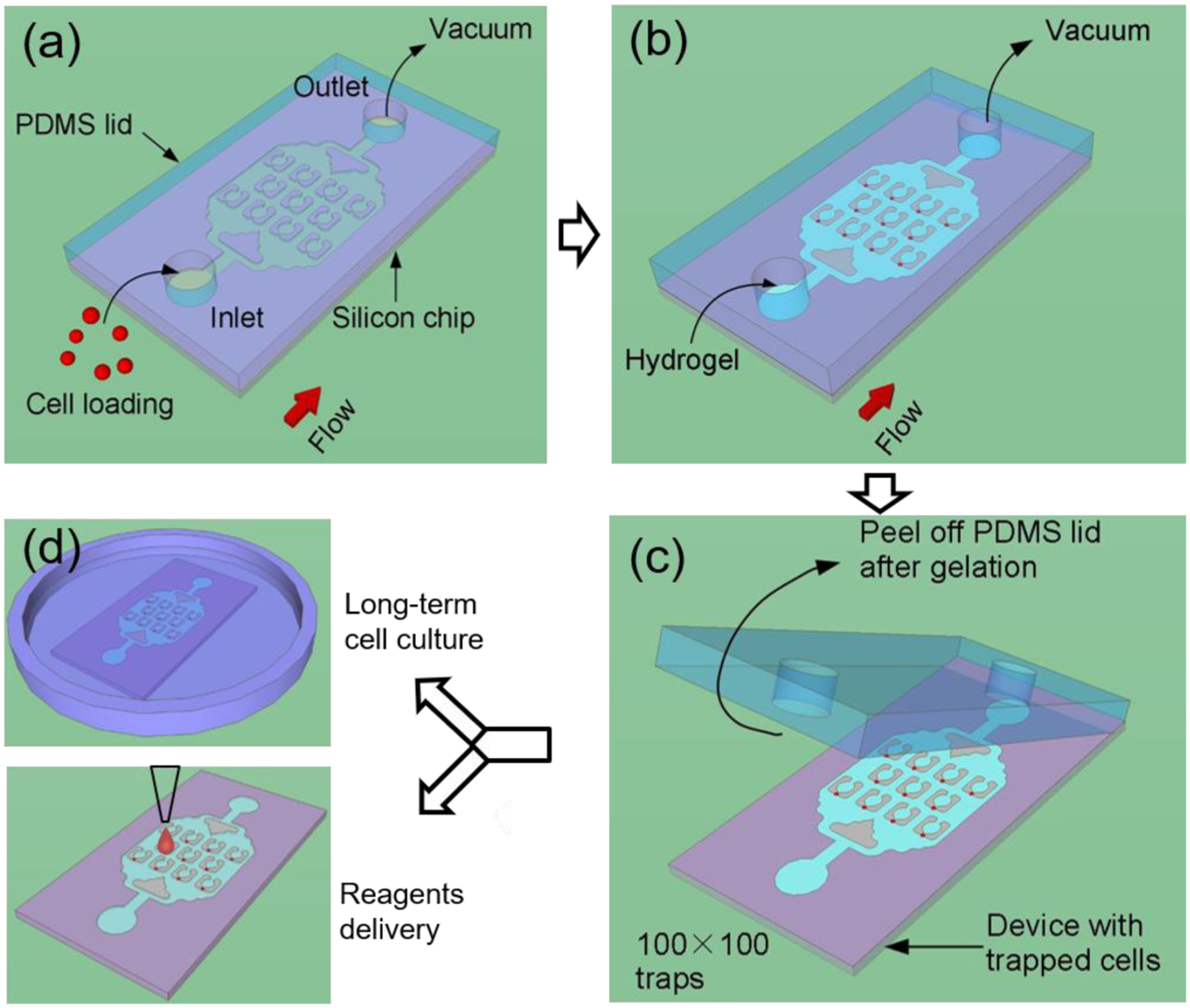
(a) Cells are loaded into the device by applying vacuum at the outlet to form a single cell array, after which (b) the hydrogel solution is injected into the device by vacuum and allowed to cure. (c) After curing, the PDMS lid is peeled off and finally (d) the device is placed in a cell culture flask for long term incubation, or alternatively, reagents are delivered to specific locations of the device.
Device design and capture efficiency.
The cell trapping devices were fabricated as previously described (Fig. S1).20,36 They consist of densely packed rectangular arrays of trapping elements arranged in a mesh microfluidic network (50 × 50 or 100 × 100 at a spatial density of ~40 traps/mm2). An image of the device, and SEM image of the individual traps are shown in Fig. 2. The x- and y- periods of the array are both 160 μm. Each apartment has a unique address, including a street number (left) and an apartment number (right), which allows the cells to be continuously tracked by image analysis (e.g., Street 22, Apartment 46 is indicated by the red circle in Fig. 2b).
Figure 2. Images of the devices.

An optical photograph of the assembled device is shown in (a). The device contains a 100×100 trap array organized in an area of 1.5×1.5 cm2. (b) An SEM image of the device is shown. Each weir has an individual address (indicated by the red circle) that allows for the identification of the trapped cells.
Based on our previous work,19,20 the microfluidic architecture was designed to simultaneously optimize several key parameters, namely, (1) high flow velocity through the weir traps to increase the cell capture efficiency, (2) high density of the trap array to enable more cells to be analyzed, and (3) wide channels to accommodate a range of cell sizes. The design that worked best over a range of different cell types is shown in Figure 3. The widths of the weir trap, Wt, the constriction, Wc, and the bypass channel, WB, are 16 μm, 5 μm, and 35 μm, respectively, and the depth of the microfluidic channels is 20 μm (Fig. 3a, b). The width of the bypass is designed to be significantly larger than the typical diameter of human cancer cells, which helps to prevent blockages due to the presence of cell clumps. However, the bypass width must not be so large that it reduces flow through the weir trap. We additionally decreased the flow resistance through the trap by including large circular regions behind the weir traps. Although these features may appear like cell culture chambers, their actual purpose was to increase the cell trapping efficiency by balancing the hydrodynamic flow through the trap relative to the bypass (Fig. S2). The fluid flow throughout the chip appears to be evenly distributed, since no pronounced spatial bias was observed in the trapping statistics (Fig. 3c).
Figure 3. Evaluation of the trapping efficiency.
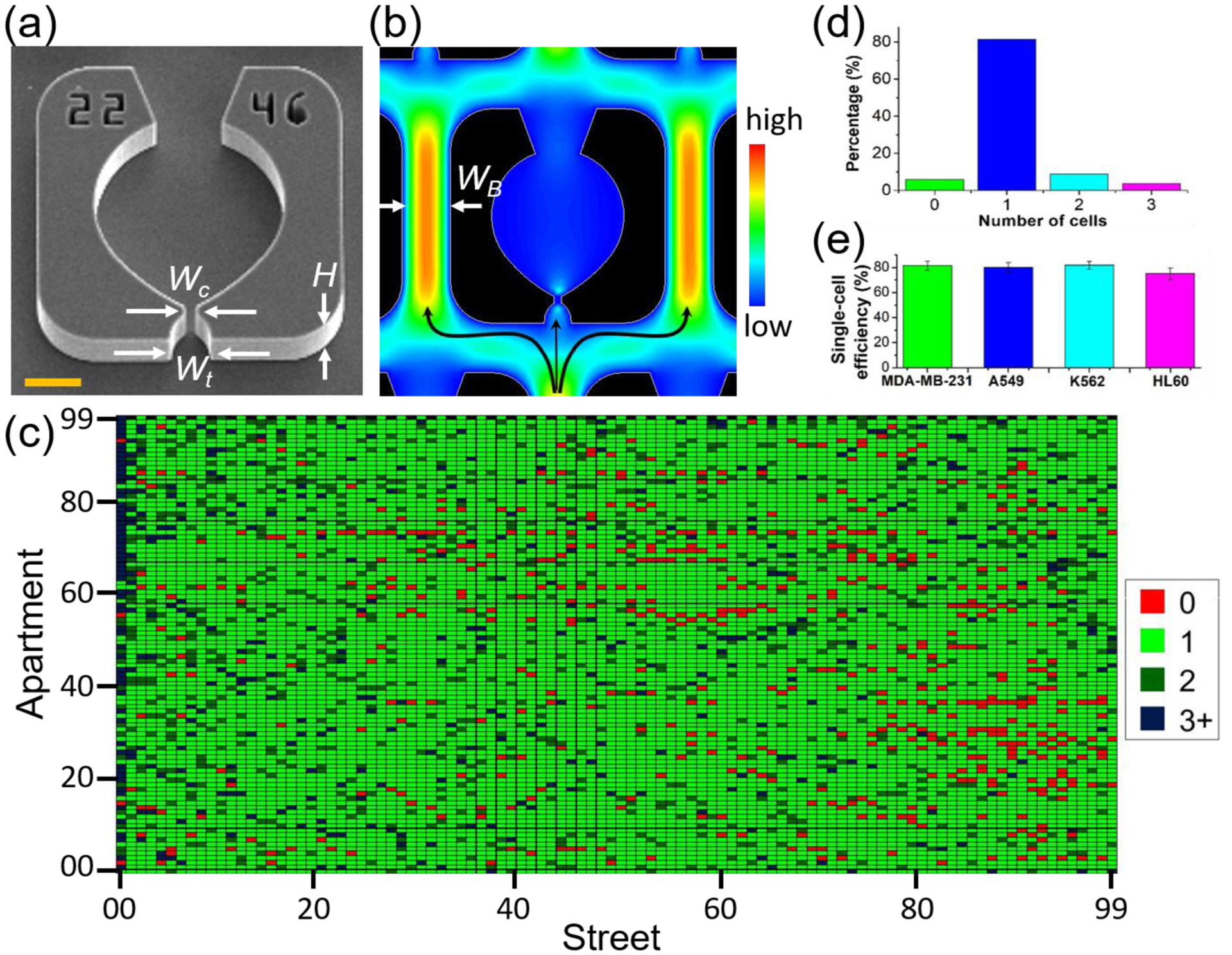
(a) SEM image of a single apartment is shown, where the critical dimensions are labeled for the trap width (Wt = 16 μm) and the constriction width (Wc = 5 μm) and the channel height (H= 20 μm). Scale bar is 20 μm. (b) Simulated flow velocity profile in a single trap. In simulations and experiments, the width of the bypass WB is fixed at 35 μm. (c) The single-cell trapping efficiency is determined by a custom MATLAB code which uses fluorescent intensity analysis combined with circle tracking to determine the number of cells in each apartment. (d) Bar-plotting of the result in (c) to show the percentage of different numbers of cells trapped in each trapping area. (e) Average single-cell trapping efficiency for different cell lines on the 100×100 array (N=5 devices for each cell line).
To evaluate the capture efficiency, we first tested the devices with 10 μm fluorescent beads (Sigma-Aldrich), and found that very high trapping efficiencies >90% could be obtained, as shown in Figures S3 and S4. Next, we tested a combination of adhesive cell lines (MDA-MB-231, breast cancer; A549, lung cancer) and suspension cell lines (K562, chronic myeloid leukemia; HL60, acute myeloid leukemia) in order to evaluate the trapping efficiency across a span of different cell diameters, and on cells with different biochemical and mechanical properties of different cells. Fig. S5 shows an example of MDA-MB-231/GFP cells trapped in the 100×100 device. Like other closed microfluidic devices with similar weir trapping architectures,8,19,20,50 this approach leads to highly efficient single cell capture rates. For example, MDA-MB-231, A549 and K562 cells have a similar average size of ~16–20 μm, however the distribution of cell sizes can be quite large depending on the cell cycle state and can range in size from 12 – 30 μm in diameter. Despite the size differences, we obtained similar capture efficiencies for all three cell lines of 81.4 ± 3.5)%, (80 ± 4)% and (81.8± 3)% respectively (N=5 devices for each), as shown in Fig. 3e. For significantly smaller cells, such as the HL60 cell line which has an average diameter of 13 μm, the single cell trapping efficiency is a little lower at ~75% with a greater tendency for doublets. These results suggest that some optimization may yield better trapping statistics for a given cell type, however the improvements are not likely to be substantial.
Hydrogel injection molding and biocompatibility.
Cells were first loaded into the weir traps, and next a hydrogel is infused into the channels and crosslinked prior to removing the PDMS lid. In most experiments, we used PEG-based hydrogels; however, we also demonstrate this approach with a few designer hydrogels that incorporate gelatin, hyaluronic acid, and other methacrylate-based precursors, which can be crosslinked by various means.51 After the hydrogel fully sets, the lid is peeled off, and then the cell array can be cultured or further processed according to the flow chart of Fig. 1. One of the main benefits of using hydrogels is that it allows this platform to accommodate both suspension cells and adherent cells because the cells are spatially constrained in the hydrogel after crosslinking.
To support long-term cell culture, the hydrogel must 1) be biocompatible, 2) have low enough viscosity to enable introduction into the microfluidic channels, and 3) must crosslink at an appropriate time scale that is slow enough to allow the gels to fully replace the media inside the chip (minutes) but quick enough to crosslink shortly after the media was replaced (hours). Many types of hydrogels can work with this approach, including those containing an acrylate functionality, such as poly (ethylene glycol) diacrylate (PEGDA), gelatin methacryloyl (GelMA), methacrylated hyaluronic acid (MeHA) and poly-N-isopropylacrylamide (PNIPAAm), which can be crosslinked with UV or chemical initiators and also by including thiolated crosslinking reagents.51–54 These types of hydrogels are frequently used for establishing 3D cellular micro-environments,24, 40–41 making it possible to culture both suspension and adherent cells with this hydrogel-based approach.
To verify that the hydrogel confinement approach is biocompatible, we tested this approach with two suspension cell lines of K562 and HL60. As shown in Fig. 4a, K562 cells are trapped in the device and remain in place after the PDMS lid is removed (see right panel of Fig. 4a). A larger area view of a 50×50 cell array with and without the PDMS lid is shown in supplementary materials (Fig. S6). Next, we cultured the arrayed K562 cells embedded in the hydrogel for 48 hours. The high porosity of the hydrogel allowed efficient diffusion of nutrients and waste, enabling the cellular array to be maintained over several days, consistent with the expected cell growth rates in a normal Petri dish. Fig. 4b shows two examples of dividing K562 cells which remain confined near their initial trapping positions. This microfluidic architecture has a bistable trapping configuration, which depends on the direction of flow. When the flow direction is reversed, the trapped cells are pushed back into the larger circular regions, (Fig. S7) and were cultured there for 48 hours (Fig. S8). Fig. S9 shows four examples of on-chip culture of HL60 cells. In summary, we foresee that the ability to create well defined cellular patterns with this hydrogel injection molding technique will have immediate applications in drug discovery, tissue engineering, wound healing, and drug delivery.55
Figure 4. Biocompatibility of the injection-molded single cell arrays.
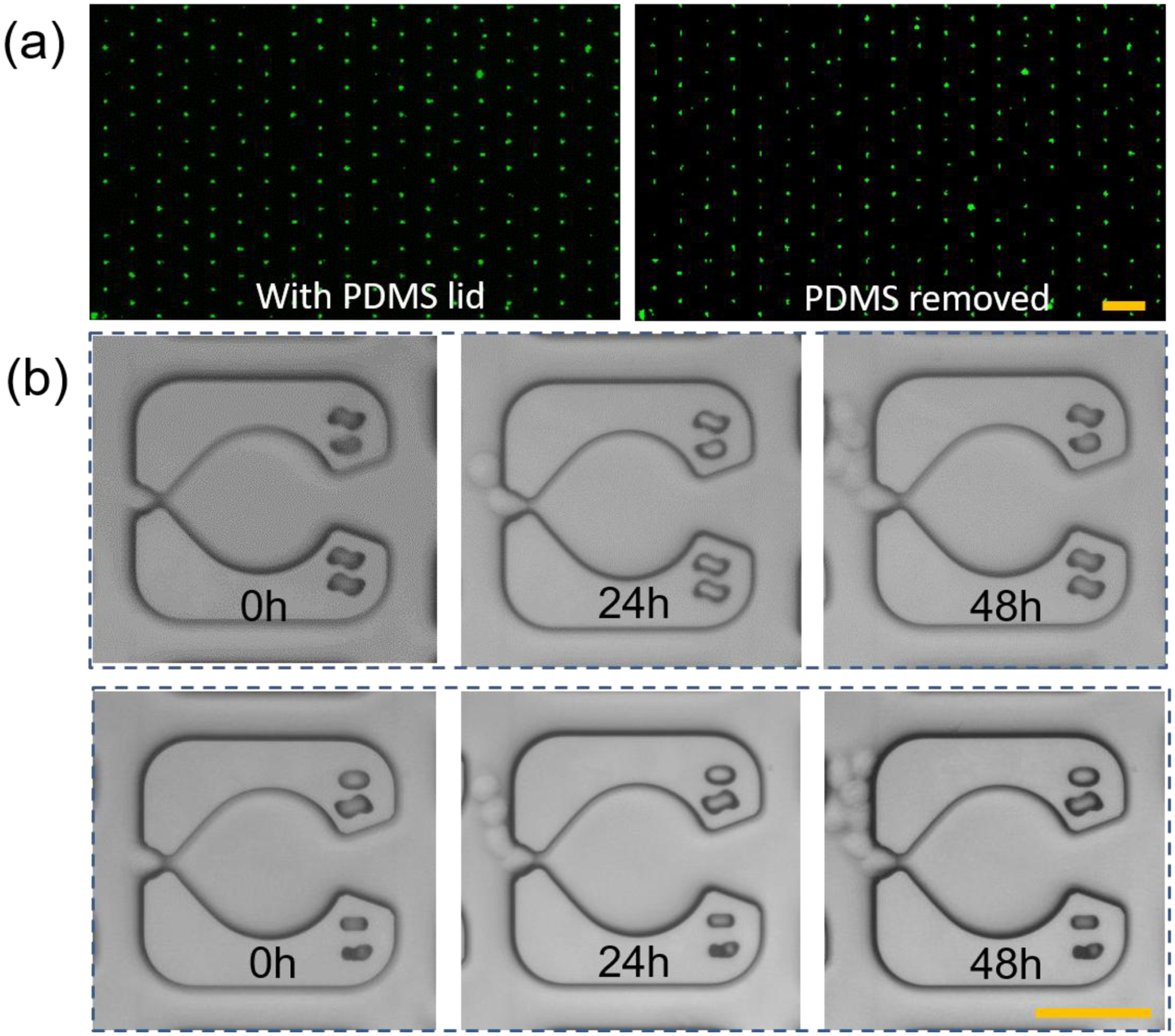
(a) The trapped K562 cells on the device with (left) and without (right) the PDMS lid. Scale bar, 200 μm. This result showed that the crosslinked hydrogel was stable in the device and it could retain the cells after removing the PDMS lid. (b) Long-term culture of the single cells encapsulated in the hydrogel. Scale bar, 50 μm.
Massively parallel local MDA reaction for single-cell whole-genome amplification.
Next, we aimed to demonstrate the ability of this injection molding approach to amplify the genomes of single cells.39 Whole genome amplification (WGA) is one of the required sample preparation steps that is often used to analyze clonal diversity in tumor populations.2,56 Many promising microfluidic approaches have been developed for single-cell WGA57,58 with MDA being one of the most popular methods due to its ability to be conducted at room temperature.39 Here, we performed MDA on up to 10,000 single cells in parallel using this injection molding approach (Fig. 5a). MDA-MB-231 cells were organized inside the microchip in single cell per trap format and compartmentalized in PEG hydrogel interfaces that remain stable after peeling off the PDMS lid (Fig. 5b). At the hydrogel concentration of 10% (w/v), the pore size of PEG gels is ~25 nm, which is large enough to allow free passage of small molecules (e.g., lysis buffer, enzymes, and primers) but small enough to retain the larger nucleic acids.37–39 Thus, amplification of the resulting genomic DNA remains spatially localized. As shown in Fig. 5, the fluorescent signal from the hydrogel remains spatially localized and increases by ~10 times after the MDA reaction in comparison to that before MDA (calculated based on Eq. 1). If desired, these gels can be digested 1 M potassium hydroxide39 in order to retrieve the MDA reaction products for downstream sequencing analysis.
Figure 5. Massively parallel single cell MDA.
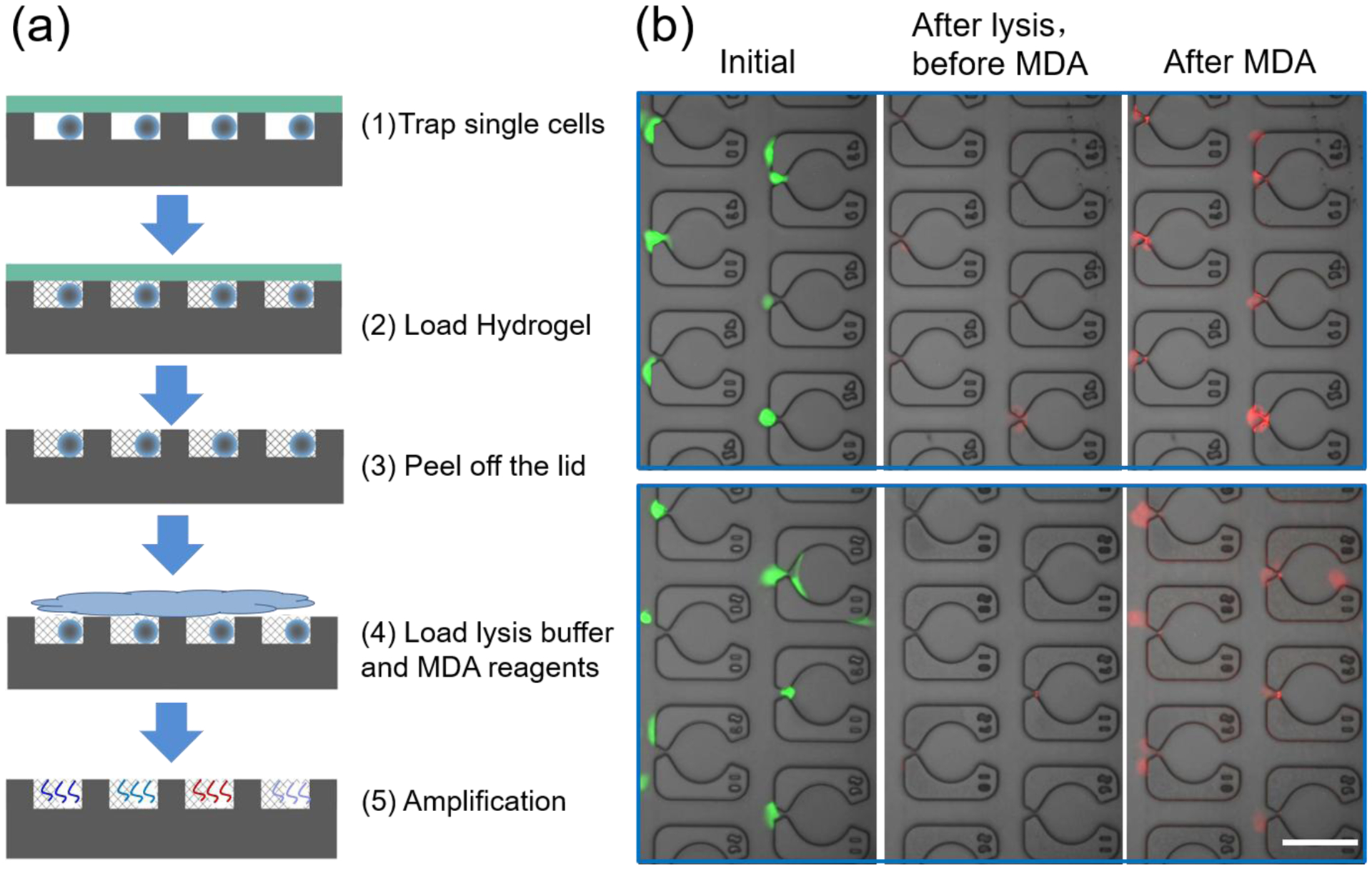
(a) Single cells are trapped and allowed to attach to the device, followed by hydrogel loading/crosslinking and lid removal. Then cells are lysed, and MDA is carried out to amplify the whole genome of single cells at 30 °C for 8 hours. (b) Merged images for the single cells at the initial state (left panel), after lysis but before MDA reaction (central panel) and after the MDA reaction (right panel). The red fluorescent signal increased significantly after MDA in comparison to that before MDA, indicating that amplification is occurring in a site selective manner. Scale bar, 100 μm.
Localized printing of biochemical reagents onto single cells.
Lastly, we aimed to demonstrate proof of concept of the ability to deliver reagents to specific spatial addresses in the open device using a commercially available bioprinter. To evaluate the printing resolution, we first deposited drops of FITC solution (~356 pL in average, Movie S1) at specific locations in the device at a printing speed of ~5 spots/second (Movie S2). Some example micropatterns are shown in Fig. 6a and Fig. S10. Based on this result, we next printed reagents that enable selective staining of cell-specific features. For example, we printed Hoechst 33342 solution on specific cells in the array to selectively stain certain cell nuclei (Fig. 6b,c and Fig. S11 and S12). These results demonstrated that the distance between two apartments (160 μm) is sufficient to avoid contamination during the printing process.
Figure 6. Localized printing of biochemical reagents on the device and cell-array.
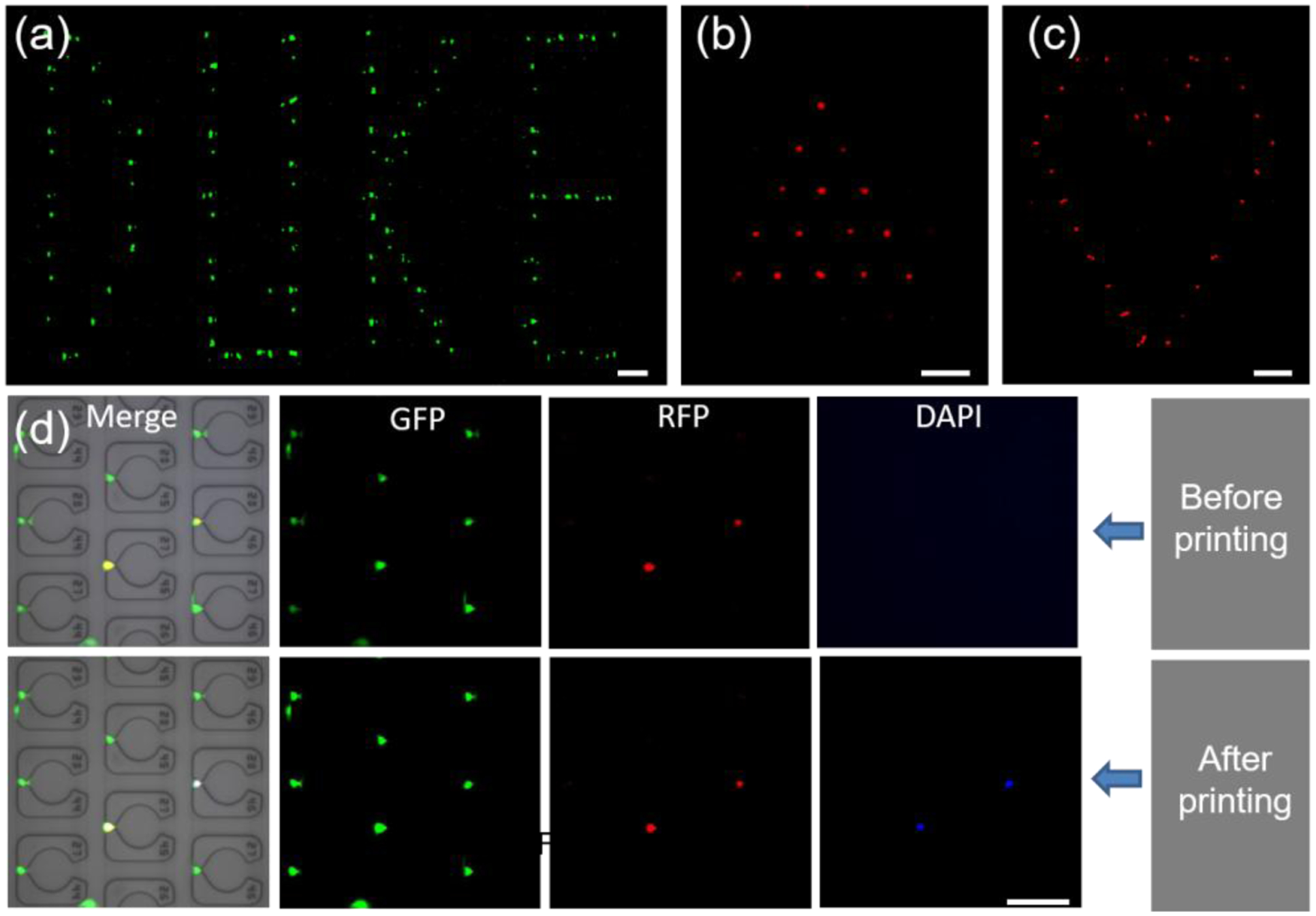
(a) A pattern of ‘DUKE’ is generated by printing FITC on a blank chip. (b-c) ‘Filled’ triangle and ‘unfilled’ heart cell-patterns obtained by printing Hoechst 33342 on an injection molded single-cell array. Red is used as a pseudo-color for Hoechst 33342 to show the pattern more clearly. See also supporting data in Figure S10–S12. (d) Precise printing of staining dye on the selected MDA-MB-231 cells in a single-cell array. Cells showed both green and red fluorescence were selected as target cells to print Hoechst 33342. After the printing, the target cells showed green, red and blue fluorescence. The scale bar is 200 μm for (a-c) and 100 μm for (d).
One of the unique advantages of this open hydrogel molded microfluidic device is the ability to selectively analyze cells that possess desirable features through a combination of imaging and selective deposition of unique reagents. For example, it may be possible to analyze only the drug-resistant cells from patient biopsy samples through time-lapse imaging,59 and then selectively analyze the small fraction of drug resistant cellular genomes.60 For proof of concept, we stained cells with a dye that fluoresces in the RFP channel and spiked them into the parental MDA-MB-231/GFP cell line at a 10:1 ratio. This system was used as a model of phenotypic heterogeneity that can be distinguished in multi-channel fluorescent imaging, where in this example, the RFP+ cells are models of cells with the phenotypic traits of interest (Fig. 6d, top row), and we recorded their spatial locations in the array. Next, we printed Hoechst 33342 onto the desired subset of RFP+/GFP+ cells, as shown in the bottom row of Fig. 6d. After printing, these cells could be identified in the DAPI channel, which proves the fundamental concept that individual cells in the hydrogel can be selectively identified and chemically manipulated (See Fig. S13 for another printing example). These results further motivate the use of this injection molding approach in modifying or characterizing the cells of interest with high precision and resolution, which has many potential applications in single cell analysis.
CONCLUSIONS
There is an urgent need for more high throughput methods to organize and characterize single cells in order to analyze the functional and genomic heterogeneity within biological systems. In this work, we utilized the concept of injection molding to establish a novel microfluidic single cell analysis approach that leverages the controllability of closed systems in terms of cellular pattern formation with the accessibility of open systems in terms of biocompatibility and local reagent delivery. The device can assemble an array of up to 10,000 single cells within minutes, and allows long-term cell culture like in conventional Petri dishes. In addition, we demonstrated massively parallel whole genome amplification of single cells trapped in the hydrogel, which enables efficient sample preparation for downstream genomic analysis. Finally, we showed the ability to precisely and efficiently print chemicals onto target cells by combining a bioprinter with our cellular analysis platform. We thus expect that this injection molded microfluidic system forms an important first step towards a more comprehensive single-cell analysis approach, which has the ability to combine high throughput phenotypic characterization with preparation of single cell libraries for next generation sequencing.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Holly Leddy for her help on the operation of the Scienion printer. The authors are thankful to support by NIH awards 1R01GM123542 and 1R21GM131279. This work was performed in part at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (Grant ECCS-1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). J.D.M. was supported by an NIH biotechnology training grant (T32GM008555).
Footnotes
CONFLICTS OF INTEREST
Y.L., J.D.M., and B.B.Y. are named inventors on a pending patent application.
SUPPORTING INFORMATION
Supporting Information includes the description of device operation and evaluation and relevant supporting data. This material is available free of charge via the Internet at http://pubs.acs.org
REFENCES
- (1).Bhatia SN; Ingber DE Nat Biotechnol 2014, 32, 760–772. [DOI] [PubMed] [Google Scholar]
- (2).Navin NE Genome Biol 2014, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang K; Chou CK; Xia X; Hung MC; Qin L Proc Natl Acad Sci U S A 2014, 111, 2948–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Papalexi E; Satija R Nat Rev Immunol 2018, 18, 35–45. [DOI] [PubMed] [Google Scholar]
- (5).Dhar M; Lam JN; Walser T; Dubinett SM; Rettig MB; Di Carlo D Proc Natl Acad Sci U S A 2018, 115, 9986–9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jiang H; Zhang XW; Liao QL; Wu WT; Liu YL; Huang WH Small 2019, e1901787. [DOI] [PubMed] [Google Scholar]
- (7).Chen P; Chen D; Li S; Ou X; Liu B-F TrAC Trends in Analytical Chemistry 2019. [Google Scholar]
- (8).Skelley AM; Kirak O; Suh H; Jaenisch R; Voldman J Nat Methods 2009, 6, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Berry SB; Zhang TZ; Day JH; Su XJ; Wilson IZ; Berthier E; Theberge AB Lab on a Chip 2017, 17, 4253–4264. [DOI] [PubMed] [Google Scholar]
- (10).Hsu CH; Chen C; Folch A Lab Chip 2004, 4, 420–424. [DOI] [PubMed] [Google Scholar]
- (11).Juncker D; Schmid H; Delamarche E Nat Mater 2005, 4, 622–628. [DOI] [PubMed] [Google Scholar]
- (12).Casavant BP; Berthier E; Theberge AB; Berthier J; Montanez-Sauri SI; Bischel LL; Brakke K; Hedman CJ; Bushman W; Keller NP; Beebe DJ P Natl Acad Sci USA 2013, 110, 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cheng JW; Chang TC; Bhattacharjee N; Folch A Biomicrofluidics 2016, 10, 024122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gracz AD; Williamson IA; Roche KC; Johnston MJ; Wang F; Wang Y; Attayek PJ; Balowski J; Liu XF; Laurenza RJ; Gaynor LT; Sims CE; Galanko JA; Li L; Allbritton NL; Magness ST Nat Cell Biol 2015, 17, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Welch JD; Williams LA; DiSalvo M; Brandt AT; Marayati R; Sims CE; Allbritton NL; Prins JF; Yeh JJ; Jones CD Nucleic acids research 2016, 44, 8292–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).DiSalvo M; Harris DM; Kantesaria S; Peña AN; Allbritton-King JD; Cole JH; Allbritton NL Anal Chem 2018, 90, 4792–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Di Carlo D; Wu LY; Lee LP Lab Chip 2006, 6, 1445–1449. [DOI] [PubMed] [Google Scholar]
- (18).Dura B; Servos MM; Barry RM; Ploegh HL; Dougan SK; Voldman J Proc Natl Acad Sci U S A 2016, 113, E3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ying L; Hee JJ; Crystal W; Bangshun H; Kai Z; Pengchao Z; Timothy V; Lidong Q Advanced Biosystems 2017, 1, 1700085. [Google Scholar]
- (20).Ohiri KA; Kelly ST; Motschman JD; Lin KH; Wood KC; Yellen BB Lab Chip 2018, 18, 2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dura B; Dougan SK; Barisa M; Hoehl MM; Lo CT; Ploegh HL; Voldman J Nat Commun 2015, 6, 5940. [DOI] [PubMed] [Google Scholar]
- (22).Yanagawa F; Sugiura S; Kanamori T Regen Ther 2016, 3, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goy CB; Chaile RE; Madrid RE Reactive and Functional Polymers 2019, 104314. [Google Scholar]
- (24).Choe G; Park J; Park H; Lee JY Polymers (Basel) 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Duval K; Grover H; Han LH; Mou Y; Pegoraro AF; Fredberg J; Chen Z Physiology (Bethesda) 2017, 32, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (26).Fukuda J; Khademhosseini A; Yeo Y; Yang XY; Yeh J; Eng G; Blumling J; Wang CF; Kohane DS; Langer R Biomaterials 2006, 27, 5259–5267. [DOI] [PubMed] [Google Scholar]
- (27).Cosson S; Lutolf MP Sci Rep 2014, 4, 4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).You J; Shin DS; Patel D; Gao Y; Revzin A Adv Healthc Mater 2014, 3, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Li Y; Kumacheva E Sci Adv 2018, 4, eaas8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kamperman T; Karperien M; Le Gac S; Leijten J Trends Biotechnol 2018, 36, 850–865. [DOI] [PubMed] [Google Scholar]
- (31).Zhu Z; Yang CJ Acc Chem Res 2017, 50, 22–31. [DOI] [PubMed] [Google Scholar]
- (32).Kamperman T; Henke S; Visser CW; Karperien M; Leijten J Small 2017, 13. [DOI] [PubMed] [Google Scholar]
- (33).Mao S; Zhang W; Huang Q; Khan M; Li H; Uchiyama K; Lin J-M Angewandte Chemie International Edition 2018, 57, 236–240. [DOI] [PubMed] [Google Scholar]
- (34).Fernandes C; Pontes AJ; Viana JC; Gaspar-Cunha A Advances in Polymer Technology 2018, 37, 429–449. [Google Scholar]
- (35).Maghsoudi K; Jafari R; Momen G; Farzaneh M Materials Today Communications 2017, 13, 126–143. [Google Scholar]
- (36).Shields C. W. t.; Cruz DF; Ohiri KA; Yellen BB; Lopez GP J Vis Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Raeber GP; Lutolf MP; Hubbell JA Biophys J 2005, 89, 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wu YB; Joseph S; Aluru NR J Phys Chem B 2009, 113, 3512–3520. [DOI] [PubMed] [Google Scholar]
- (39).Xu L; Brito IL; Alm EJ; Blainey PC Nat Methods 2016, 13, 759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Engel BJ; Constantinou PE; Sablatura LK; Doty NJ; Carson DD; Farach-Carson MC; Harrington DA; Zarembinski TI Adv Healthc Mater 2015, 4, 1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mannino RG; Santiago-Miranda AN; Pradhan P; Qiu Y; Mejias JC; Neelapu SS; Roy K; Lam WA Lab Chip 2017, 17, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Liu C; Li Y; Liu B-F Talanta 2019, 205, 120136. [DOI] [PubMed] [Google Scholar]
- (43).Mengeaud V; Josserand J; Girault HH Anal Chem 2002, 74, 4279–4286. [DOI] [PubMed] [Google Scholar]
- (44).Hu R; Liu C; Xuan J; Xu Y; Li T; Liu B-F; Li Y; Yang Y Sensors and Actuators B: Chemical 2019. [Google Scholar]
- (45).Chen P; Yan S; Wang J; Guo Y; Dong Y; Feng X; Zeng X; Li Y; Du W; Liu BF Anal Chem 2019, 91, 1619–1626. [DOI] [PubMed] [Google Scholar]
- (46).Zhang P; Zhou X; He M; Shang Y; Tetlow AL; Godwin AK; Zeng Y Nature Biomedical Engineering 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yang L; Wang Z; Deng Y; Li Y; Wei W; Shi Q Anal Chem 2016, 88, 11077–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Han X; Wang R; Zhou Y; Fei L; Sun H; Lai S; Saadatpour A; Zhou Z; Chen H; Ye F; Huang D; Xu Y; Huang W; Jiang M; Jiang X; Mao J; Chen Y; Lu C; Xie J; Fang Q, et al. Cell 2018, 172, 1091–1107 e1017. [DOI] [PubMed] [Google Scholar]
- (49).Park JW; Vahidi B; Taylor AM; Rhee SW; Jeon NL Nat Protoc 2006, 1, 2128–2136. [DOI] [PubMed] [Google Scholar]
- (50).Di Carlo D; Lee LP Anal Chem 2006, 78, 7918–7925. [DOI] [PubMed] [Google Scholar]
- (51).Ahmed EM J Adv Res 2015, 6, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Idota N; Tsukahara T; Sato K; Okano T; Kitamori T Biomaterials 2009, 30, 2095–2101. [DOI] [PubMed] [Google Scholar]
- (53).Jang K; Sato K; Mawatari K; Konno T; Ishihara K; Kitamori T Biomaterials 2009, 30, 1413–1420. [DOI] [PubMed] [Google Scholar]
- (54).Tanaka N; Moriguchi H; Sato A; Kawai T; Shimba K; Jimbo Y; Tanaka Y RSC Advances 2016, 6, 54754–54762. [Google Scholar]
- (55).Chung BG; Lee KH; Khademhosseini A; Lee SH Lab Chip 2012, 12, 45–59. [DOI] [PubMed] [Google Scholar]
- (56).Prakadan SM; Shalek AK; Weitz DA Nature Reviews Genetics 2017, 18, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lan F; Demaree B; Ahmed N; Abate AR Nat Biotechnol 2017, 35, 640.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Yu ZL; Lu SJ; Huang YY Anal Chem 2014, 86, 9386–9390. [DOI] [PubMed] [Google Scholar]
- (59).Housman G; Byler S; Heerboth S; Lapinska K; Longacre M; Snyder N; Sarkar S Cancers (Basel) 2014, 6, 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Masuda T; Song W; Nakanishi H; Lei W; Noor AM; Arai F PLoS One 2017, 12, e0174937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.