

Abstract
To report the clinical, hormonal, and genetic features of a female with multiple endocrine neoplasia type 1 (MEN1) with multiple uterine leiomyomas. The study was conducted at a tertiary care endocrinology unit. A 27-year-old female was diagnosed with prolactinoma, primary hyperparathyroidism (PHPT), and multiple uterine leiomyomas. In view of prolactinoma and PHPT, a clinical diagnosis of MEN1 syndrome was made. She also had multiple uterine leiomyomas for which myomectomy was done. Genetic analysis revealed a novel mutation c.1763C>T, p.S588L of MEN1 gene. The association of uterine leiomyomas with MEN1 is exceptionally rare. This is the first report of multiple uterine leiomyomas in a patient with MEN1 from our country and the first report of this mutation in the MEN1 gene in the world. We conclude that in the presence of multiple uterine leiomyomas and endocrine tumor, clinical examination and laboratory evaluation may uncover the diagnosis of MEN1 syndrome in these patients.
KEYWORDS: Multiple endocrine neoplasia type 1, multiple endocrine neoplasia type 1 gene, uterine leiomyomas
INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant disorder characterized by the presence of tumors in parathyroid, pancreatic islets, and anterior pituitary.[1] Other lesions associated with this syndrome include facial angiofibromas, collagenomas, adrenal tumors, carcinoid tumors, thyroid tumors, lipomas, and leiomyomas.[2,3] The association of uterine leiomyomas with MEN1 is exceptionally rare, with only four such cases reported in the literature.[3,4,5,6] Here, we describe a young female with genetically proven MEN1 syndrome from a novel mutation c.1763C>T, p.S588L of MEN1 gene, and multiple uterine leiomyomas.
SUBJECT AND METHODS
A 27-year-old female was referred to our center by a primary care physician for the evaluation of amenorrhea–galactorrhea syndrome. Investigations revealed hyperprolactinemia (serum prolactin 204 ng/ml); magnetic resonance imaging (MRI) pituitary revealed a 7 mm × 5 mm microadenoma. Oral cabergoline 0.5 mg/week was initiated which led to resumption of regular menstrual cycles, subsidence of galactorrhea, and normalization of serum prolactin. A year later, she developed menorrhagia; the menstrual bleeding got prolonged (7–9 days compared to her usual 3–4 days) and increased in amount. Ultrasonography and MRI of the pelvis [Figure 1] revealed multiple uterine leiomyomas – two subserosal leiomyomas measuring 8 cm × 6.5 cm and 2.7 cm × 2 cm located in the posterior uterine wall, an intramural leiomyoma measuring 1.5 cm × 1 cm located in the anterior uterine wall, and another intramural leiomyoma measuring 3 cm × 2.5 cm located in the posterior uterine wall. For leiomyomas, myomectomy was done, and histology was consistent with the diagnosis of uterine leiomyomas. Two years later, she developed nephrolithiasis of the left kidney. A workup for nephrolithiasis revealed hypercalcemia (serum calcium 11.77 mg/dl), hypophosphatemia (serum phosphate 2.3 mg/dl), hypercalciuria (24-h urinary calcium 296 mg), and elevated intact parathormone (iPTH, 107.5 pg/ml). Ultrasonography neck and technetium-99m sestamibi scan failed to localize any parathyroid lesion. Four-dimensional computed tomography of the neck was performed which revealed the presence of adenoma in the right superior parathyroid gland [Figure 2]. Parathyroid adenoma was removed; adenoma weighed 300 mg, and histology was consistent with diagnosis of parathyroid adenoma. After surgery, hypercalcemia subsided and iPTH normalized. She is on follow-up for more than a year with no recurrence of primary hyperparathyroidism (PHPT) or hyperprolactinemia.
Figure 1.

Contrast-enhanced magnetic resonance imaging of the pelvis showing multiple uterine leiomyomas (yellow arrows)
Figure 2.
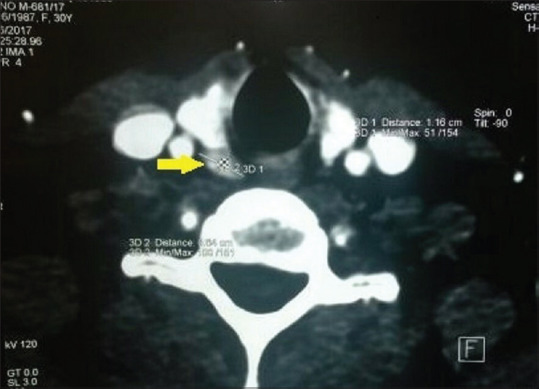
Four-dimensional computed tomography of the neck showing adenoma in the right superior parathyroid gland (yellow arrow)
Institutional ethical committee approval for the publication of this case report was requested and granted. Written informed consent was obtained for publication of this case report and any accompanying images.
RESULTS
In view of prolactinoma and PHPT, a clinical diagnosis of MEN1 syndrome was made. Serum gastrin levels were normal. From the peripheral blood of the patient, genomic DNA was extracted and sequencing of the MEN1 gene was performed. Sequencing analysis revealed a mutation in one of the alleles wherein cytosine at codon 1763 was replaced by thymidine, leading to replacement of serine by leucine at amino acid position 588 in the menin protein (c.1763C>T, p.S588L). Two siblings of the index case, an elder brother and a younger sister, are receiving treatment for microprolactinoma. No other family member has hypercalcemia or uterine leiomyoma at present. Due to lack of funding, we could not undertake genetic analysis of her siblings and parents.
DISCUSSION
MEN1 syndrome is defined by the presence of tumors in two or three endocrine glands, which include parathyroid, pancreatic islet cells, and anterior pituitary. The most common endocrine organ affected is parathyroid with a prevalence of 100% at 50 years of age.[1] The frequency of pituitary tumors in MEN1 syndrome ranges from 10% to 60%, and prolactinoma is the most frequent anterior pituitary tumor.[7] MEN1 tumors develop in several endocrine tissues and are multicentric and have high recurrence rate.[4]
Other so-called nonclassical neoplasms in MEN1 patients include facial angiofibromas, collagenomas, adrenal tumors, carcinoid tumors, thyroid tumors, lipomas, and leiomyomas. These tumors, like the typical MEN1 tumors, tend to be multiple. In MEN1 syndrome, leiomyomas have been reported in the esophagus, lung, uterus, skin, and ureter, and they differ from sporadic leiomyomas in being multicentric in many cases.[3,8]
Uterine leiomyomas are the most common solid tumors of the female genital tract, with a prevalence of 25%–70%.[9] Uterine leiomyomas in MEN1 and sporadic patients most likely develop through different pathogenetic mechanisms. McKeeby et al. in 2001 for the first time reported six uterine leiomyomas in two MEN1 patients, five of which exhibited 11q13 loss of heterozygosity (LOH); in contrast, only two out of 40 sporadic uterine leiomyoma exhibited 11q13 LOH.[3] This study documented that uterine leiomyoma in MEN1 patients develop through the inactivation of the MEN1 gene and therefore concluded that uterine leiomyomas should be considered an integral part of MEN1 syndrome. In the literature, there are two other reports of multiple uterine leiomyomas in MEN1.[5,6]
Uterine leiomyomas in our patient differ in important aspects from such tumors in previously reported patients with MEN1 syndrome. First, uterine leiomyomas in our patient were symptomatic, while in all the previous reports, uterine leiomyomas have been asymptomatic, incidentally found on abdominal imaging.[3,4,5,6] Second, in all the previous reports, uterine leiomyomas were diagnosed after the diagnosis of MEN1 was established, while in our patient, uterine leiomyomas were detected well before the diagnosis of MEN1 was established. It has been previously suggested that the late identification of uterine leiomyomas may indicate an altered pathogenesis and growth promotion in MEN1 associated smooth muscle tumors from that found in sporadic tumors, but our case reflects that this may not be the case.[3]
The MEN1 gene is located on the long arm of chromosome 11 (11q13) and encodes the 610 amino acid protein menin, which plays a critical role as a tumor suppressor.[10] It is involved in the regulation of gene transcription, apoptosis, and genome stability. Sequencing analysis of MEN1 gene in our patient revealed a novel mutation heterozygous for c.1763C>T, p.S588L. To the best of our knowledge, this mutation has not been reported previously.
CONCLUSION
We conclude that in women with multiple uterine leiomyomas and an endocrine tumor, careful history, clinical examination, and focused laboratory evaluation may uncover the diagnosis of MEN1 syndrome in these patients.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that her name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Almeida MQ, Stratakis CA. Solid tumors associated with multiple endocrine neoplasias. Cancer Genet Cytogenet. 2010;203:30–6. doi: 10.1016/j.cancergencyto.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doherty GM. Multiple endocrine neoplasia type 1. J Surg Oncol. 2005;89:143–50. doi: 10.1002/jso.20181. [DOI] [PubMed] [Google Scholar]
- 3.McKeeby JL, Li X, Zhuang Z, Vortmeyer AO, Huang S, Pirner M, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol. 2001;159:1121–7. doi: 10.1016/s0002-9440(10)61788-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi H, Kim S, Moon JH, Lee YH, Rhee Y, Kang ES, et al. Multiple endocrine neoplasia type 1 with multiple leiomyomas linked to a novel mutation in the MEN1 gene. Yonsei Med J. 2008;49:655–61. doi: 10.3349/ymj.2008.49.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall MJ, Innocent J, Rybak C, Veloski C, Scott WJ, Wu H, et al. Bilateral granulosa cell tumors: A novel malignant manifestation of multiple endocrine neoplasia 1 syndrome found in a patient with a rare menin in-frame deletion. Appl Clin Genet. 2015;8:69–73. doi: 10.2147/TACG.S72223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Maouche D, Welch J, Agarwal SK, Weinstein LS, Simonds WF, Marx SJ. A patient with MEN1 typical features and MEN2-like features. Int J Endocr Oncol. 2016;3:89–95. doi: 10.2217/ije-2015-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corbetta S, Pizzocaro A, Peracchi M, Beck-Peccoz P, Faglia G, Spada A. Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin Endocrinol (Oxf) 1997;47:507–12. doi: 10.1046/j.1365-2265.1997.3311122.x. [DOI] [PubMed] [Google Scholar]
- 8.Ikota H, Tanimoto A, Komatsu H, Ozawa Y, Matsushita H. Ureteral leiomyoma causing hydronephrosis in Type 1 multiple endocrine neoplasia. Pathol Int. 2004;54:457–9. doi: 10.1111/j.1440-1827.2004.01642.x. [DOI] [PubMed] [Google Scholar]
- 9.Medikare V, Kandukuri LR, Ananthapur V, Deenadayal M, Nallari P. The genetic bases of uterine fibroids; a review. J Reprod Infertil. 2011;12:181–91. [PMC free article] [PubMed] [Google Scholar]
- 10.Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A. 1998;95:1630–4. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]