

Abstract

Recently, chemoselective methods for the hydrogenation of fluorinated, silylated, and borylated arenes have been developed providing direct access to previously unattainable, valuable products. Herein, a comprehensive study on the employed rhodium-cyclic (alkyl)(amino)carbene (CAAC) catalyst precursor is disclosed. Mechanistic experiments, kinetic studies, and surface-spectroscopic methods revealed supported rhodium(0) nanoparticles (NP) as the active catalytic species. Further studies suggest that CAAC-derived modifiers play a key role in determining the chemoselectivity of the hydrogenation of fluorinated arenes, thus offering an avenue for further tuning of the catalytic properties.
Keywords: heterogeneous catalysis, rhodium, arene hydrogenation, fluoroarenes, CAAC, nanoparticles, on-surface
Arene hydrogenation is a powerful tool to transform readily available, planar starting materials into complex, three-dimensional building blocks.1 These are of interest, e.g., in pharmaceutical research, and can often not be synthesized efficiently with other methods.2 The control of the chemoselectivity, vital for the synthesis of functionalized cyclic saturated building blocks, is difficult, since very few catalysts are able to overcome the aromatic stabilization energy in absence of harsh conditions. Especially functional groups directly attached to the aromatic ring constitute a daunting challenge due to the generally prevalent hydrodefunctionalization side reaction. The bench-stable rhodium cyclic (alkyl)(amino)carbene (CAAC) precatalyst 1 in combination with a suitable additive (e.g., molecular sieves or silica gel) was essential in our recent studies on the hydrogenation of arenes bearing directly attached fluorine,3a,3b silyl3c and boryl substituents,3d and the consecutive dearomatization-hydrogenation of fluorinated pyridines.4 Building on research by the Bertrand group on the synthesis of CAACs5 and their rhodium complexes,6 this catalyst was first used by Zeng and co-workers in the preferential hydrogenation of arenes in the presence of carbonyl groups, showing a good tolerance of functional groups.7 Although some other catalysts have proven competent in our studies, the active catalyst derived from Rh-CAAC 1 has shown a superior functional group preservation, especially for difficult substrates with fluoro, silyl, and boryl moieties.3 Given the high synthetic utility of this precatalyst, we became interested in elucidating the catalytically active species. A general challenge in arene hydrogenation is the distinction between active homogeneous and heterogeneous species.8 In fact, many active catalysts that were initially described as homogeneous, as they are derived from homogeneous precursors, have since been shown to actually be heterogeneous in nature.9 The Zeng group originally assumed that the active catalyst derived from 1 is of homogeneous nature due to a mercury droplet test.7 However, during the course of our mechanistic investigation, two studies by the Bullock group appeared providing experimental evidence for rhodium nanoparticles (NP) as active catalysts.10 In their system, AgBF4 was used to abstract chloride from 1 and initiate NP formation in the absence of another additive. Based on rhodium K-edge X-ray absorption fine structure (XAFS) analysis and IR spectroscopy (among other experiments), the authors determined that rhodium(0) NPs, which are stabilized by pyrrolidinium cation 2 (derived from the CAAC 1a in the precursor), are the active catalyst in their case.10a Although Bullock and co-workers provided a thorough investigation of the active catalytic species derived from activation with AgBF4, the studied conditions differ significantly from the ones found optimal in our hydrogenation methods. Most importantly, no hydrogenation of fluorinated arenes was attempted and a silver salt was used to activate the precatalyst via a cationic pathway as opposed to molecular sieves or silica gel in our case. The resulting species derived from activation with AgBF4 was evaluated by us for the hydrogenation of fluorinated arenes and proved inefficient. In fact, multifluorinated substrates such as hexafluorobenzene failed to provide any product when using an AgBF4-activated precatalyst (see page S17).
Hence, a complementary study on the identity and understanding of the active catalyst system obtained in the presence of an additive like silica gel was needed. Ideally, such a study would provide a more detailed view on the chemoselective arene hydrogenation and allow for rational modifications of the catalyst in the future.
Differentiation between a Homogeneous and a Heterogeneous Active Catalyst
Inspired by the extensive work of the Finke group on the differentiation between heterogeneous and homogeneous active catalysts in arene hydrogenation,8a,8b,9 we began our studies with mechanistic experiments that aimed to distinguish an active heterogeneous from an active homogeneous catalyst (Figure 1). Our initial assumption of a heterogeneous reaction pathway was driven by various “telltale” signs observed during our previous studies, including the need of a stabilizing additive such as molecular sieves or silica gel, formation of a dark precipitate, and an observed induction period. In contrast to Zeng et al., we have observed a loss of catalytic activity in the presence of mercury, albeit under different reaction conditions. For the droplet test, see page S20. The conditions of the two droplet tests are adapted from the standard reaction conditions of either method, respectively, which are considerably different from one another, although both methods start with precatalyst 1. However, the challenging reproducibility and strong dependence on reaction conditions is a known, inherent problem for mercury droplet tests and thus neither of the two possible outcomes can be used to unequivocally differentiate hetero- from homogeneous catalysis pathways and should be complemented with further experiments.8a,8b,11 Substoichiometric poisoning with tetrahydrothiophene showed the loss of catalytic activity in the reaction (see Table S3).12 This points towards a heterogeneous active catalyst, since reactive sites would be buried inside the particles in such a system, thus requiring less than one equivalent for complete poisoning of reactivity. Furthermore, the dark gray to black solid residue obtained after hydrogenation (denoted as the residue 4) was isolated by filtration and washed extensively with the reaction solvent. The colorless supernatant was concentrated in vacuo, and no remaining residue could be detected. The residue 4 was used as catalyst in a new reaction (Maitlis’ test,13 see Table S4) and showed fully preserved yield and selectivity, suggesting that this solid residue contained the active catalyst. Furthermore, a 3-phase-test (Collman’s test)14 was performed, in which methyl 4-hydroxybenzoate was attached to a Wang-resin and submitted to hydrogenation conditions (see page S26). No hydrogenation of the phenyl groups in the solid resin phase occurred. This is the expected outcome for a heterogeneous catalysis pathway, since a reaction is highly unlikely to occur between three phases.
Figure 1.

Overview of the established Rh-CAAC catalyzed arene hydrogenation and the studies performed in this work in order to elucidate its active catalyst species. Dipp: diisopropylphenyl. FG: functional group.
We proceeded to investigate the kinetic behavior of the in situ prepared active catalyst derived from 1 and the preformed catalyst 4a by monitoring characteristic signals of the standard substrate 5 and of the corresponding product 6 as a function of the reaction time using kinetic 1H NMR measurements under hydrogen pressure (Figure 2). When using 1 as precatalyst in presence of silica gel, an induction period of 120–180 min was observed, which was absent when using preformed catalyst 4a(15) (Figure 2), thus indicating that the insoluble black residue obtained after hydrogenation contains the active catalyst. In agreement with all other performed experiments, this result strongly indicates that the active catalyst is heterogeneous in nature.16
Figure 2.
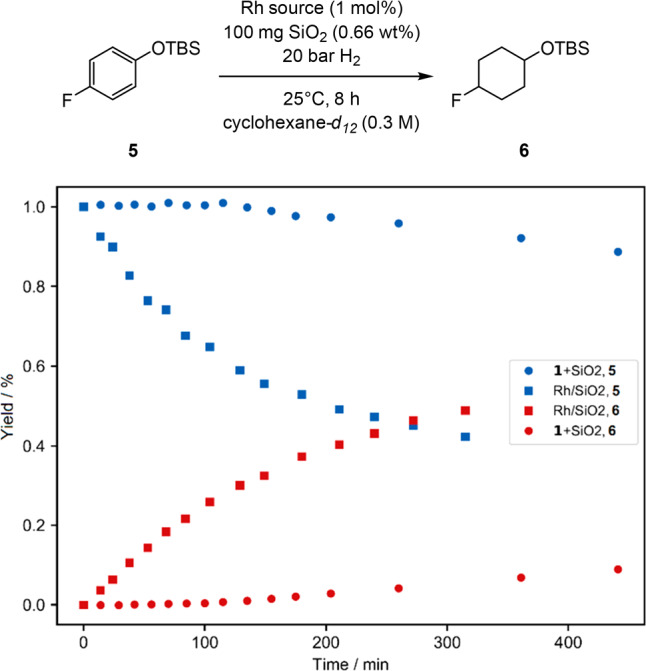
Yields of substrate 5 and product 6 using either precatalyst 1 + SiO2 or preformed catalyst 4a as a function of the reaction time. The reactions were performed in pressurized NMR tubes under hydrogen pressure and the yields were determined from the characteristic signals of 5 (proton signal at 6.77 ppm, 2H) and 6 (3.71 ppm, 1H, see pages S50−S52).
Observation and Characterization of Nanoparticles in Catalytic Residue 4a
The preformed catalyst 4a was further analyzed to deeply investigate the actual structure. High-resolution transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) with energy dispersive spectroscopy (EDS) analysis of 4a showed the presence of rhodium NPs (Figure 3a–d). The observed lattice distances of 0.19 and 0.22 nm in the particles were attributed to a Rh(200) and Rh(111) plane, respectively. This shows that the rhodium is in metallic form (Figure 3b and page S57). The average particle size of 4a was estimated to be 6.3 ± 2.1 nm from TEM particle size distribution histograms (Figure 3c). X-ray diffraction (XRD) of 4a (Figure S8) also exhibited two diffraction peaks at 41.0° and 47.4°, which were attributed to Rh(111) and Rh(200) planes of rhodium nanoparticles, respectively. The size of the rhodium NPs was also calculated by the Debye−Scherrer equation for the Rh(111) peak. The obtained value of 4.9 nm is in agreement with the TEM analysis. Rhodium K-edge X-ray absorption near edge structure (XANES, Figure 3e) indicated that the oxidation state of rhodium was close to zero. The Rh–Rh bond distance analyzed by rhodium K-edge extended X-ray fine structure (EXAFS, Figure 3f, Figure S7) was estimated to be 0.269 ± 0.003 nm (coordination number of rhodium being 8.7 ± 0.7). This is in accordance with that of rhodium metal (0.269 ± 0.001 nm), also supporting its metallic state. The Brunauer–Emmet–Teller (BET) surface area of 4a was measured to be 398 m2 g–1, which was comparable to that of the used silica gel (422 m2 g–1), implying that no severe aggregation of support occurred after the formation of rhodium NPs.
Figure 3.
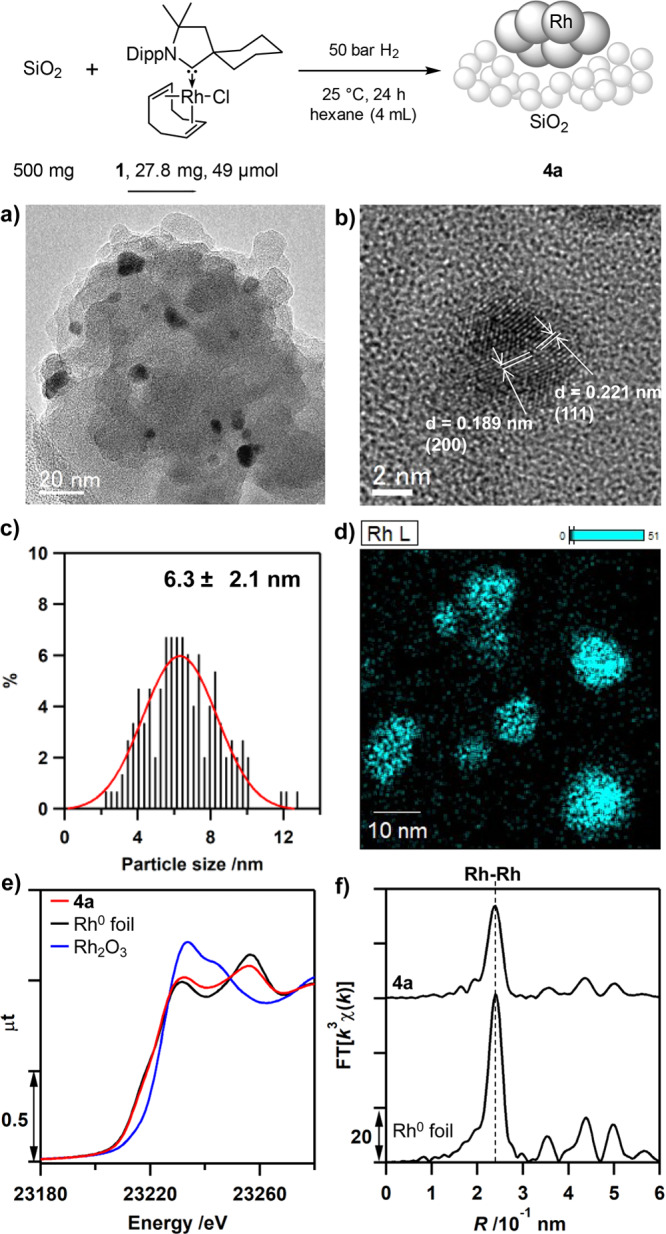
(a,b) TEM images of 4a with visible lattice structure (0.19 nm Rh(200), 0.22 nm Rh(111)). (c) Histogram showing the particle size distribution of 4a. (d) STEM-EDS image of 4a (blue, Rh Lα characteristic X-rays). (e) Rh K-edge XANES spectra of 4a (red), Rh(0) foil (black), and Rh2O3 (blue). (f) Rh K-edge EXAFS Fourier transforms of 4a and Rh(0) foil.
Process of Nanoparticle Formation from Rhodium–CAAC Complex 1
In order to study the influence of the reaction time on the catalytic properties of the resulting rhodium NPs, preformed catalysts were prepared using reaction times of 3 h (4b) and 24 h (4c), respectively (Figure 4). In addition to chloride abstraction, the support is likely needed to stabilize and suppress the aggregation of growing NPs after the Rh(I) species are reduced to Rh(0).8b A comparison of the sizes of the formed particles with usual parameters such as the mean diameter was not possible, because the obtained size distributions were not uniform (Figure 4c). To extract information from the gathered data, we instead determined the fraction of small nanoparticles, which would be the most reactive catalytic species (blue-highlighted area in Figure 4c), by defining the following parameter: Ns/all = (number of counted particles with sizes below 3 nm)/(number of all counted particles). The fraction of the most reactive small particles was considerably larger after 3 h (4b, Ns/all = 32%, Figure 4c, left) than after 24 h reaction time (4c, Ns/all = 12%, Figure 4c, center). This fresh preparation of small reactive particles may explain the excellent reproducibility of our catalyst system in comparison with some commercial heterogeneous catalysts, which often show batch-dependent results for difficult reactions due to a varying particle size distribution. We also tested the influence of the amount of silica gel on the size of the rhodium NPs. Catalyst 4d, prepared with a 3-fold excess of silica gel compared to 4b and 4c, and 24 h reaction time, showed a considerably increased fraction of small NPs (Ns/all = 24%) compared to catalyst 4c (Ns/all = 12%). The larger number of small NPs when using more silica gel could offer one possible explanation for the observed and previously reported positive effect of increased amounts of silica gel for the hydrogenation of especially challenging highly fluorinated arenes such as hexafluorobenzene.3d In this previous study, increased amounts of silica led to lower amounts of defluorination and, thus, higher product yields (for a comparison see also Table S12). BET results of 4b (395 m2g–1), 4c (373 m2g–1), and 4d (379 m2g–1) are very similar to those of catalytic residue 4a. As a result, it is highly unlikely that the observed effect is caused by a surface area difference after rhodium NP formation.
Figure 4.
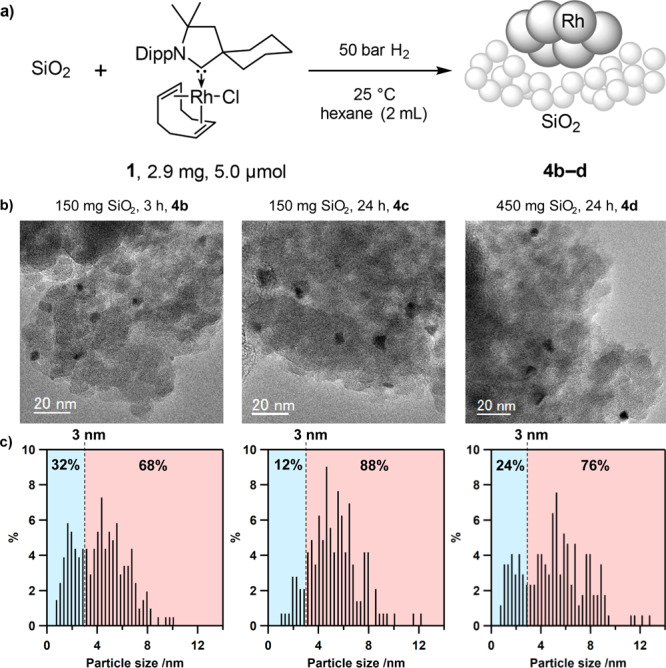
(a) Preparation of silica-supported NPs using different reaction times (catalysts 4b, 4c) or amounts of silica (catalyst 4d). (b) Representative TEM images of 4b–4d. Black dots represent areas of higher density and correspond to Rh, gray background is the silica support. (c) Histograms showing the particle size distribution of the prepared systems. Particle size distributions for all synthesized catalyst systems in this study are given in the Supporting Information.
Ligand Effect on Catalysis
We wondered if the observed high selectivity toward preservation of C−F bonds is introduced by the active ligand species derived from 1a or if a similar selectivity could be achieved with rhodium nanoparticles of similar size and loading within the identical support material. To test this, rhodium NPs 4e–4g on silica gel were prepared by impregnation of Rh(NO3)3·H2O on silica gel, followed by H2 reduction (synthesis page S6, characterization page S68; rhodium loadings were 0.87 wt % for 4e, 5.1 wt % for 4f, and 9.9 wt % for 4g, respectively). No organic ligand species were attached to these rhodium NPs. When using this method, the fractions of small NPs (<3 nm) (Ns/all(4e) = 58%, Ns/all(4f) = 41%, and Ns/all(4g) = 27%) were somewhat higher than those obtained when using catalysts derived from Rh–CAAC 1 as precursor (e.g., 4a–d). It is worth noting that we observed that an increased fraction of small NPs affects higher yields and that the fraction of small NPs decreases with an increased amount of rhodium down to 27%, which is less than that observed for catalyst 4b. The average particle size of 4e was estimated to be 2.8 ± 0.7 nm determined from particle size distribution histograms (Figure S12), which is slightly smaller than that of 4a with similar Rh loading. When the rhodium loading was increased, average particle sizes were slightly increased (3.2 ± 0.8 nm for 4f and 3.5 ± 1.0 nm for 4g, respectively).
Catalytic reaction results for the hydrogenation of the model substrate 5 are summarized in Table 1 (see also Table S11). 4e–4g all showed a significantly increased amount of defluorination when compared to the standard system (precursor 1 used, in situ generation of 4). In addition, the cis-selectivity of the NP systems is decreased. Therefore, the selectivity of the investigated system cannot be solely dependent on the NP size or rhodium loading.
Table 1. Comparison of Catalytic Performance of Different Rh/SiO2 Systems for the Hydrogenation of 5a.

| catalyst | Rh amount [wt %] | yield of 6 [%] (d. r.) | yield of 7 [%] |
|---|---|---|---|
| Rh–CAAC 1/SiO2 | 1.0 | 90 (94:6) | 3 |
| Rh–CAAC 1/SiO2 | 5.1 | 86 (94:6) | 4 |
| Rh/SiO24a | 1.0 | 89 (94:6) | 3 |
| Rh/SiO24e | 0.87 | 66 (87:13) | 18 |
| Rh/SiO24f | 5.1 | 59 (89:11) | 23 |
| Rh/SiO24g | 9.9 | 68 (88:12) | 23 |
Yields were determined via GC-FID against mesitylene as internal standard. Numbers do not add up to unity, because cyclohexane and cyclohexanol are formed as byproducts, which coelute with the solvent and thus cannot be detected.
Observation and Identity of Ligand Species on the Catalyst Surface
The comparison of the catalytic results obtained using 4a and 4e–4g suggested that catalyst performance is influenced by a ligand species. Therefore, we proceeded to characterize catalyst 4a and investigate if ligand species were present on the surface of the NPs. X-ray photoelectron spectroscopy (XPS) of catalyst 4a revealed a broad nitrogen signal at 400–402 eV (Figure 5). The best fitting to the given data is obtained with two signals in a 1:1 ratio, separated by more than 2 eV. While the intensity of the nitrogen signal (as well as that of the rhodium signal) is low due to the sparse dispersion within the silica, the significant distance between the fitted signals strongly suggests the existence of two different nitrogen species. Given that the CAAC ligand 1a is the sole nitrogen containing species in the precursor, two different nitrogen-containing species must have formed from that ligand under the reaction conditions. To elucidate the identity of the two unknown nitrogen-containing species in the sample, we turned to solid-state NMR spectroscopy. To overcome the problem of the sparse density of possible residues, complex 1 was prepared with a 13C-enriched CAAC precursor. The synthesis of the labeled CAAC precursor was conducted starting from commercially available isotope-labeled benzoic acid, using our own protocol for arene hydrogenation (see page S9 for the full synthetic route).3e,17
Figure 5.
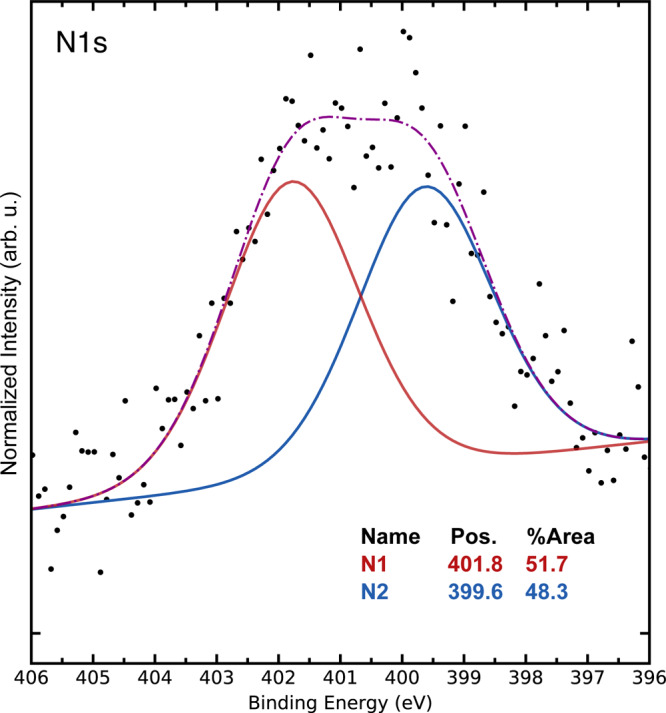
Plotted signal and fitting of the nitrogen-containing species, determined by XPS.
Samples for 13C{1H} CP/MAS NMR spectroscopy were prepared by stirring the labeled complex with silica gel under hydrogen atmosphere (cf. Figure 4a).18 The obtained 13C{1H} CP/MAS NMR spectrum shows two intense 13C signals at ∼186 and ∼65 ppm (Figure 6). Comparison with the spectrum of the labeled complex 1 confirmed that the signal at 186 ppm cannot be assigned to a ligated carbene species. Given our extensive experience in the field of carbenes on surfaces and given the high strength of carbene–metal bonds, this result was unexpected at first.19 However, the excellent overlap with the labeled signal of the 13C-enriched pyrrolidinium salt 2 suggested that this iminium cation is likely one of the two nitrogen-containing species.
Figure 6.
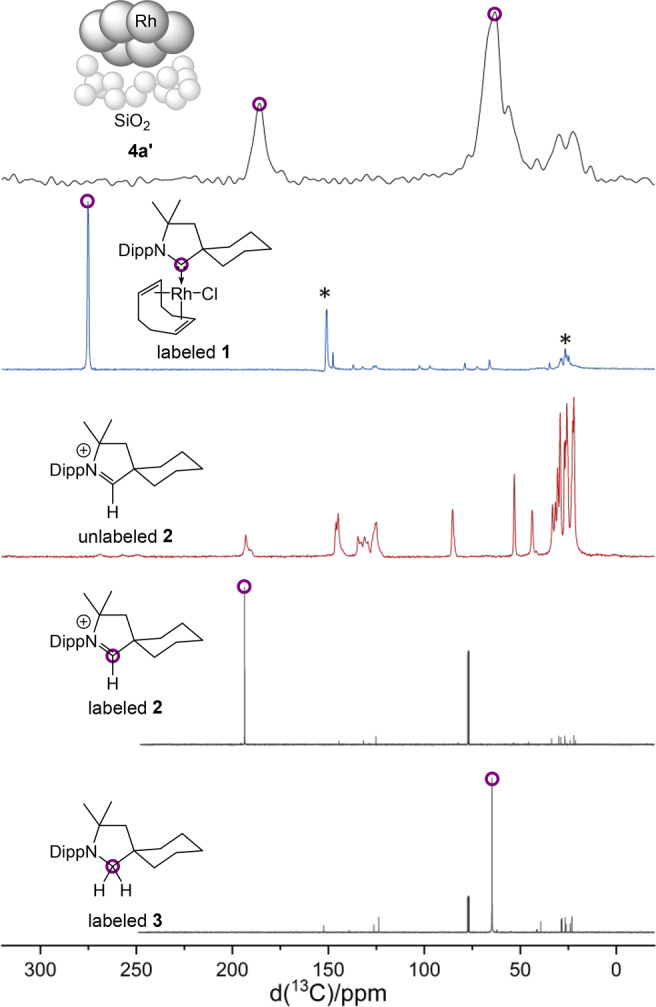
13C{1H} CP/MAS NMR spectra of 4a′ prepared from the 13C-labeled complex 1 (black), labeled complex 1 (blue), and unlabeled 2 (red). Peaks corresponding to the labeled carbon are highlighted. The two bottom spectra allow for a comparison with to the pure, 13C-labeled compounds 2 and 3 in solution. *Spinning side bands of the main signal.
That cation is likely generated by a reductive elimination of carbene and hydrogen from an intermediate carbene metal–hydride complex, as previously proposed by the Ananikov group.20 An alternative pathway could be the dissociation of the carbene ligand and subsequent protonation by acidic silanol groups present within the silica gel. However, a dissociation of a free carbene is unlikely regarding the strength of carbene–metal bonds. Chloride is most likely the counteranion, although deprotonated silanol groups are also imaginable. Given that the iminium moiety present in 2 is reducible under hydrogenation conditions, we assumed that the remaining species corresponding to the 13C signal at ∼65 ppm might be the amine 3. Hence, pyrrolidine 3 was synthesized from pyrrolidinium 2 (with chloride as a counteranion) by reduction with LiAlH4. In agreement with our assumption, pyrrolidine 3 showed a distinct signal at 64.9 ppm in deuterated chloroform. It is worth noting that amines are known ligands for nanoparticles.21 Comparison of spectra of labeled and unlabeled pyrrolidinium 2 further suggests that the smaller signals between 5 and 0 ppm are derived from the backbone of the pyrrolidinium cation and the pyrrolidine.
Bottom-Up Synthesis of a Related Selective Hydrogenation Catalyst
Given the previous finding that the observed high chemoselectivity of the hydrogenation of fluorinated arenes is not solely determined by the particle size of the used rhodium catalyst, we wondered if the species 2 and 3 may have an influence on the catalytic activity and selectivity. In previous studies on the influence of ligands such as phosphines and phosphites on reactions catalyzed by Rh NPs, significant effects on the reactivity and selectivity of hydrogenation reactions were observed.22a−22c
Furthermore, imidazolium based ionic liquids, which were covalently anchored to the Al2O3-support, were shown to influence the catalytic hydrogenation of benzene with dispersed Ru NPs.22d Driven by these reports, we set out to prepare a catalyst system similar to 4a by adding 2 and 3 to synthesized rhodium NPs on silica (4e–4g) and to commercial rhodium on alumina (Table S9). These were tested for the hydrogenation of model substrate 5 using a catalyst loading of 1 mol %. We started by investigating the optimal ratio of both additives at a combined loading of 1.0 equivalents relative to the used rhodium but no clear trend emerged (see Table S11). The use of one equivalent of a single species only led to high amounts of defluorination (for pyrrolidine 3) or incomplete conversion (for pyrrolidinium 2). These results strongly suggest that both species 2 and 3 seem to play an important role in modifying the catalytic activity. For further experiments, we therefore kept a 1:1-ratio of these species, as suggested by the XPS studies. Next, varying equivalents of that 1:1 mixture with regard to rhodium were investigated (Figure 7). The studies showed an increase in conversion with decreased amounts of the modifiers. Full conversion was still achieved with as little as 0.3 equiv of the modifier mixture. Further decreasing the amount of the modifiers resulted in a significant increase in defluorination. The amount of defluorination and the obtained yield with 0.3 equivalents of a 1:1 modifier mixture was almost identical to the values observed when using the precursor 1 with silica gel and in situ generation of the active catalytic system (4).
Figure 7.

Yields of product 6 and byproduct 7 obtained by the hydrogenation of substrate 5 with Rh NPs on silica gel 4e as a function of the total equivalents of a 1:1 mixture of pyrroldinium 2 and pyrrolidine 3, which were added to the Rh NPs. Yields were determined by GC-FID analysis vs mesitylene.
As additional control experiments, preformed rhodium particles, obtained by submitting Rh(COD)Cl and silica gel to hydrogenation conditions, were tested untreated and treated with the modifiers 2 and 3. While reactivity was observed for the former case, a complete loss of activity was noted with one equivalent of the modifier mixture, irrespective of their ratio (see Table S10). We wondered whether the identity of the rhodium ligand, in this case CAAC 1a, may also influence the NP formation. To investigate this, different CAAC ligands 8 and 9 were used during the preparation of rhodium particles 4h and 4i, respectively (Figure 8). As expected, the size distribution of those rhodium particles differed from that of the active catalyst 4a derived from ligand 1a in the precursor complex. More importantly, the catalytic results differed significantly as well (see Figure S1, Table S6) with the residues 4h and 4i showing either strongly reduced or no activity at all (see page S29 for a more detailed discussion).
Figure 8.

Structures of alternative CAAC ligands 8 and 9 with representative TEM images of corresponding rhodium particles obtained from complex precursors and the size distribution.
The preformed catalyst 4e (with and without modifiers) was also investigated for the hydrogenation of hexafluorobenzene 10, since for this substrate the difference in chemoselectivity compared to other catalysts was especially pronounced. When applying the determined optimized amounts of modifiers to the preformed NPs the yield for hexafluorocyclohexane 11 could be increased from 4% (no modifiers) to 29% with modifiers. This constitutes 74% of the yield obtained with the established precursor Rh–CAAC 1 under otherwise identical conditions (Table 2).
Table 2. Comparing the Performance of Synthesized and Modified NPs with the Standard Catalyst System23,a.

| catalyst | yield 11 [%] | yield 12 [%] |
|---|---|---|
| Rh–CAAC 1/44 mg SiO2 | 39 | 22 |
| Rh/SiO2 (4e, 0.87 wt %) with 0.3 equiv of 2 and 3 (1:1) | 29 | 8 |
| Rh/SiO2 (4e, 0.87 wt %) | 4 | 57 |
Isolated yields are given.
In conclusion, we have elucidated the active catalyst derived from a molecular Rh–CAAC precursor, which has previously been shown to enable a highly chemoselective hydrogenation of a broad scope of (fluorinated) arenes and heteroarenes. By combining mechanistic and kinetic studies, we confirmed that the active catalyst is heterogeneous in nature. Various imaging, spectroscopic, and surface analysis techniques revealed silica gel-supported rhodium(0) NPs as active catalytic species. After excluding the presence of a metal-bound carbene species on the NPs, we further determined that CAAC-derived pyrrolidinium 2 and pyrrolidine 3 act as modifiers that are key in controlling the chemoselectivity of the hydrogenation of fluorinated arenes. Furthermore, the chemical identity of these modifiers and the amount of the silica gel support influence the size distribution of the NPs and thereby the catalytic properties. Finally, we were able to synthesize an active catalyst in a bottom-up approach that has a very similar reactivity compared to the established catalyst. We believe that this approach will enable much more facile means of tuning the catalytic activity of that catalyst system. Hence, the development of next-generation catalysts derived from the studied system is the subject of ongoing research in our laboratories.
Acknowledgments
We are grateful to the European Research Council (ERC Advanced Grant Agreement No. 788558), Fonds der Chemischen Industrie (Kekulé Ph.D. scholarship for D.M. and J.S.), the Studienstiftung des deutschen Volkes (Ph.D. scholarship for M.P.W.), the JST PRESTO Program (No. JPMJPR15S7), the JSPS KAKHENI (Grant No. 18K05144), and Core-to-Core program “Elements Function for Transformative Catalysis and Materials” for generous financial support. TEM and STEM-EDS measurements were conducted at the High Voltage Electron Microscope Laboratory, Institute of Materials and Systems for Sustainability, Nagoya University, supported by “Advanced Characterization Nanotechnology Platform” of MEXT, Japan. XAFS measurements were performed with the approval of PF-PAC (Grant 2017G534) and the Aichi Synchrotron Radiation Center. We are also grateful to C. Schlepphorst, V. Siozios, M. Wollenburg, T. Wagener, A. Heusler, H. Lund, and R. Eckelt for helpful scientific discussions and experimental support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01074.
Experimental procedures and spectroscopic and microscopic data (PDF)
Author Present Address
+ M.P.W.: Princeton University Frick Laboratory, Washington Road, Princeton, NJ 08544, United States.
Author Contributions
× M.P.W. and M.F. contributed equally.
The authors declare the following competing financial interest(s): The authors declare no conflict of interest. However, please, note that we patented the Process for Synthesizing Fluorinated Cyclic Aliphatic Compounds employing the investigated catalyst system: PCT/EP2018/054554.
Supplementary Material
References
- For an overview on homo- and heterogeneous arene hydrogenation see:; a Wiesenfeldt M. P.; Nairoukh Z.; Dalton T.; Glorius F. Selective Arene Hydrogenation for Direct Access to Saturated Carbo- and Heterocycles. Angew. Chem., Int. Ed. 2019, 58, 10460–10476. 10.1002/anie.201814471. [DOI] [PMC free article] [PubMed] [Google Scholar]; b The Handbook of Homogeneous Hydrogenation; de Vries J. G., Elsevier C. J., Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]; c Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds; Mortier J., Ed.; John Wiley & Sons, Inc: Hoboken, NJ, 2016. [Google Scholar]; d Gual A.; Godard C.; Castillón S.; Claver C. Soluble Transition-Metal Nanoparticles-Catalysed Hydrogenation of Arenes. Dalton Trans. 2010, 39, 11499–11512. 10.1039/c0dt00584c. [DOI] [PubMed] [Google Scholar]; e Qi S.-C.; Wei X.-Y.; Zong Z.-M.; Wang Y.-K. Application of Supported Metallic Catalysts in Catalytic Hydrogenation of Arenes. RSC Adv. 2013, 3, 14219–14232. 10.1039/c3ra40848e. [DOI] [Google Scholar]; f Giustra Z. X.; Ishibashi J. S. A.; Liu S.-Y. Homogeneous Metal Catalysis for Conversion between Aromatic and Saturated Compounds. Coord. Chem. Rev. 2016, 314, 134–181. 10.1016/j.ccr.2015.11.006. [DOI] [Google Scholar]; g Wang D.-S.; Chen Q.-A.; Lu S.-M.; Zhou Y.-G. Asymmetric Hydrogenation of Heteroarenes and Arenes. Chem. Rev. 2012, 112, 2557–2590. 10.1021/cr200328h. [DOI] [PubMed] [Google Scholar]; h Zhao D.; Candish L.; Paul D.; Glorius F. N-Heterocyclic Carbenes in Asymmetric Hydrogenation. ACS Catal. 2016, 6, 5978–5988. 10.1021/acscatal.6b01736. [DOI] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Previous arene hydrogenation methods from our group employing Rh-CAAC 1:; a Wiesenfeldt M. P.; Nairoukh Z.; Li W.; Glorius F. Hydrogenation of Fluoroarenes: Direct Access to All-Cis-(Multi)Fluorinated Cycloalkanes. Science 2017, 357, 908–912. 10.1126/science.aao0270. [DOI] [PubMed] [Google Scholar]; b See Tables S9 and S12 for a comparison to a selection of commercial heterogeneous catalysts for the hydrogenation of fluorinated arenes.; c Wiesenfeldt M. P.; Knecht T.; Schlepphorst C.; Glorius F. Silylarene Hydrogenation: A Strategic Approach That Enables Direct Access to Versatile Silylated Saturated Carbo- and Heterocycles. Angew. Chem., Int. Ed. 2018, 57, 8297–8300. 10.1002/anie.201804124. [DOI] [PubMed] [Google Scholar]; d Wollenburg M.; Moock D.; Glorius F. Hydrogenation of Borylated Arenes. Angew. Chem., Int. Ed. 2019, 58, 6549–6553. 10.1002/anie.201810714. [DOI] [PubMed] [Google Scholar]; e Arene hydrogenation methods employing Rh-CAACs from other groups:Ling L.; He Y.; Zhang X.; Luo M.; Zeng X. Hydrogenation of (Hetero)aryl Boronate Esters with a Cyclic (Alkyl)(amino)carbene-Rhodium Complex: Direct Access to cis-Substituted Borylated Cycloalkanes and Saturated Heterocycles. Angew. Chem., Int. Ed. 2019, 58, 6554–6558. 10.1002/anie.201811210. [DOI] [PubMed] [Google Scholar]; f Zhang X.; Ling L.; Luo M.; Zeng X. Accessing Difluoromethylated and Trifluoromethylated cis-Cycloalkanes and Saturated Heterocycles: Preferential Hydrogen Addition to the Substitution Sites for Dearomatization. Angew. Chem., Int. Ed. 2019, 58, 16785–16789. 10.1002/anie.201907457. [DOI] [PubMed] [Google Scholar]; g For a lead reference on the chemo- and enantioselective hydrogenation of fluorinated olefins, seePonra S.; Rabten W.; Yang J.; Wu H.; Kerdphon S.; Andersson P. G. Diastereo- and Enantioselective Synthesis of Fluorine Motifs with Two Contiguous Stereogenic Centers. J. Am. Chem. Soc. 2018, 140, 13878–13883. 10.1021/jacs.8b08778. [DOI] [PubMed] [Google Scholar]
- Nairoukh Z.; Wollenburg M.; Schlepphorst C.; Bergander K.; Glorius F. The Formation of all-cis-(Multi)Fluorinated Piperidines by a Dearomatization–Hydrogenation Process. Nat. Chem. 2019, 11, 264–270. 10.1038/s41557-018-0197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminal works on the synthesis of CAA(r)Cs by the Bertrand group:; a Lavallo V.; Canac Y.; Präsang C.; Donnadieu B.; Bertrand G. Stable Cyclic (Alkyl)(Amino)Carbenes as Rigid or Flexible, Bulky, Electron-Rich Ligands for Transition-Metal Catalysts: A Quaternary Carbon Atom Makes the Difference. Angew. Chem., Int. Ed. 2005, 44, 5705–5709. 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rao B.; Tang H.; Zeng X.; Liu L.; Melaimi M.; Bertrand G. Cyclic (Amino)(Aryl)Carbenes (CAArCs) as Strong σ-Donating and π-Accepting Ligands for Transition Metals. Angew. Chem., Int. Ed. 2015, 54, 14915–14919. 10.1002/anie.201507844. [DOI] [PubMed] [Google Scholar]; c Reviews:Soleilhavoup M.; Bertrand G. Cyclic (Alkyl)(Amino)Carbenes (CAACs): Stable Carbenes on the Rise. Acc. Chem. Res. 2015, 48, 256–266. 10.1021/ar5003494. [DOI] [PubMed] [Google Scholar]; d Melaimi M.; Jazzar R.; Soleilhavoup M.; Bertrand G. Cyclic (Alkyl)(Amino)Carbenes (CAACs): Recent Developments. Angew. Chem., Int. Ed. 2017, 56, 10046–10068. 10.1002/anie.201702148. [DOI] [PubMed] [Google Scholar]
- Lavallo V.; Canac Y.; DeHope A.; Donnadieu B.; Bertrand G. A rigid Cyclic (Alkyl)(amino)carbene Ligand Leads to Isolation of Low-Coordinate Transition Metal Complexes. Angew. Chem., Int. Ed. 2005, 44, 7236–7239. 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Rao B.; Cong X.; Zeng X. Highly Selective Hydrogenation of Aromatic Ketones and Phenols Enabled by Cyclic (Amino)(Alkyl)Carbene Rhodium Complexes. J. Am. Chem. Soc. 2015, 137, 9250–9253. 10.1021/jacs.5b05868. [DOI] [PubMed] [Google Scholar]
- For reviews on the distinction between homo- and heterogeneous catalyst systems see; a Widegren J. A.; Finke R. G. A Review of Soluble Transition-Metal Nanoclusters as Arene Hydrogenation Catalysts. J. Mol. Catal. A: Chem. 2003, 191, 187–207. 10.1016/S1381-1169(02)00125-5. [DOI] [Google Scholar]; b Widegren J. A.; Finke R. G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A: Chem. 2003, 198, 317–341. 10.1016/S1381-1169(02)00728-8. [DOI] [Google Scholar]; c Dyson P. J. Arene Hydrogenation by Homogeneous Catalysts: Fact or Fiction?. Dalton Trans. 2003, 15, 2964–2974. It can be concluded that no singular experiment can unambiguously distinguish between a homo- or heterogeneous nature of the active catalyst and a definitive elucidation requires a series of experiments 10.1039/b303250g. [DOI] [Google Scholar]
- For selected examples of studies on the distinction between a homo- or heterogeneous nature of the active catalyst see; a Lin Y.; Finke R. G. A More General Approach to Distinguishing “Homogeneous” from “Heterogeneous” Catalysis: Discovery of Polyoxoanion- and Bu4N+-Stabilized, Isolable and Redissolvable, High-Reactivity Ir∼190–450 Nanocluster Catalysts. Inorg. Chem. 1994, 33, 4891–4910. 10.1021/ic00100a012. [DOI] [Google Scholar]; b Weddle K. S.; Aiken J. D.; Finke R. G. Rh(0) Nanoclusters in Benzene Hydrogenation Catalysis: Kinetic and Mechanistic Evidence That a Putative [(C8H17)3NCH3]+[RhCl4]- Ion-Pair Catalyst Is Actually a Distribution of Cl- and [(C8H17)3NCH3]+ Stabilized Rh(0) Nanoclusters. J. Am. Chem. Soc. 1998, 120, 5653–5666. 10.1021/ja973045h. [DOI] [Google Scholar]; c Bayram E.; Linehan J. C.; Fulton J. L.; Szymczak N. K.; Finke R. G. Determination of the Dominant Catalyst Derived from the Classic [RhCp*Cl2]2 Precatalyst System: Is It Single-Metal Rh(1)Cp*-Based, Subnanometer Rh4 Cluster-Based, or Rh(0)n Nanoparticle-Based Cyclohexene Hydrogenation Catalysis at Room Temperature and Mild Pressures?. ACS Catal. 2015, 5, 3876–3886. 10.1021/acscatal.5b00315. [DOI] [Google Scholar]
- a Tran B. L.; Fulton J. L.; Linehan J. C.; Lercher J. A.; Bullock R. M. Rh(CAAC)-Catalyzed Arene Hydrogenation: Evidence for Nanocatalysis and Sterically Controlled Site-Selective Hydrogenation. ACS Catal. 2018, 8, 8441–8449. 10.1021/acscatal.8b02589. [DOI] [Google Scholar]; b Tran B. L.; Fulton J. L.; Linehan J. C.; Balasubramanian M.; Lercher J. A.; Bullock R. M. Operando XAFS Studies on Rh(CAAC)-Catalyzed Arene Hydrogenation. ACS Catal. 2019, 9, 4106–4114. 10.1021/acscatal.8b04929. [DOI] [Google Scholar]
- Chernyshev V. M.; Astakhov A. V.; Chikunov I. E.; Tyurin R. V.; Eremin D. B.; Ranny G. S.; Khrustalev V. N.; Ananikov V. P. Pd and Pt Catalyst Poisoning in the Study of Reaction Mechanisms: What Does the Mercury Test Mean for Catalysis?. ACS Catal. 2019, 9, 2984–2995. 10.1021/acscatal.8b03683. [DOI] [Google Scholar]
- For poisoning studies of metals with sulfur-containing compounds see the following book chapter:; Bartholomew C. H.; Agrawal P. K.; Katzer J. R. Sulfur Poisoning of Metals. In Advances in Catalysis; Eley D. D., Pines H., Weisz P. B., Eds.; Academic Press, 1982; Vol. 31, pp 135–242;. [Google Scholar]; For a case study seeGarbarino G.; Romero Perez A.; Finocchio E.; Busca G. A Study of the Deactivation of Low Loading Ni/Al2O3 Steam Reforming Catalyst by Tetrahydrothiophene. Catal. Commun. 2013, 38, 67–73. 10.1016/j.catcom.2013.04.007. [DOI] [Google Scholar]
- Hamlin J. E.; Hirai K.; Millan A.; Maitlis P. M. A Simple Practical Test for Distinguishing a Heterogeneous Component in a Homogeneously Catalyzed Reaction. J. Mol. Catal. 1980, 7, 543–544. [Google Scholar]
- Collman J. P.; Kosydar K. M.; Bressan M.; Lamanna W.; Garrett T. Polymer-bound Substrates: A Method to Distinguish between Homogeneous and Heterogeneous Catalysis. J. Am. Chem. Soc. 1984, 106, 2569–2579. 10.1021/ja00321a014. [DOI] [Google Scholar]
- The catalytic system of Rh on SiO2 can be prepared by stirring 1 with silica gel under hydrogen pressure with or without any substrate. In our studies we have found no difference in reactivity between both cases (see page S23). Different amounts of silica gel can be used, which we have found to have an impact on reactivity. Where appropriate, letters have been used in addition to number 4 in this manuscript to differentiate between catalyst systems prepared under slightly different conditions.
- Schwartz J. Alkane Activation by Oxide-Bound Organorhodium Complexes. Acc. Chem. Res. 1985, 18, 302–308. Following Schwartz’s definition, a heterogeneous catalyst is described by having multiple active sites, as opposed to one active site in a homogeneous catalyst. This definition includes insoluble, supported catalysts, as well as soluble nanoclusters 10.1021/ar00118a004. [DOI] [Google Scholar]
- An alternative pathway for the amine synthesis is a direct hydrogen splitting starting from the free carbene:; Frey G. D.; Lavallo V.; Donnadieu B.; Schoeller W. W.; Bertrand G. Science 2007, 316, 439–441. 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]
- For studies analyzing carbenes on surfaces with solid-state NMR techniques, see; a Baquero E. A.; Tricard S.; Flores J. C.; de Jesús E.; Chaudret B. Highly Stable Water-Soluble Platinum Nanoparticles Stabilized by Hydrophilic N-Heterocyclic Carbenes. Angew. Chem., Int. Ed. 2014, 53, 13220–13224. 10.1002/anie.201407758. [DOI] [PubMed] [Google Scholar]; b Ernst J. B.; Muratsugu S.; Wang F.; Tada M.; Glorius F. Tunable Heterogeneous Catalysis: N-Heterocyclic Carbenes as Ligands for Supported Heterogeneous Ru/K-Al2O3 Catalysts to Tune Reactivity and Selectivity. J. Am. Chem. Soc. 2016, 138, 10718–10721. 10.1021/jacs.6b03821. [DOI] [PubMed] [Google Scholar]; c Ernst J. B.; Schwermann C.; Yokota G.; Tada M.; Muratsugu S.; Doltsinis N. L.; Glorius F. Molecular Adsorbates Switch on Heterogeneous Catalysis: Induction of Reactivity by N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2017, 139, 9144–9147. 10.1021/jacs.7b05112. [DOI] [PubMed] [Google Scholar]; d Baquero E. A.; Tricard S.; Coppel Y.; Flores J. C.; Chaudret B.; de Jesús E. Water-Soluble Platinum Nanoparticles Stabilized by Sulfonated N-Heterocyclic Carbenes: Influence of the Synthetic Approach. Dalton Trans. 2018, 47, 4093–4104. 10.1039/C8DT00240A. [DOI] [PubMed] [Google Scholar]
- For recent reviews on the field of carbenes on surfaces, see; a Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–596. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]; b Zhukhovitskiy A. V.; MacLeod M. J.; Johnson J. A. Carbene Ligands in Surface Chemistry: From Stabilization of Discrete Elemental Allotropes to Modification of Nanoscale and Bulk Substrates. Chem. Rev. 2015, 115, 11503–11532. 10.1021/acs.chemrev.5b00220. [DOI] [PubMed] [Google Scholar]; c Smith C. A.; Narouz M. R.; Lummis P. A.; Singh I.; Nazemi A.; Li C.-H.; Crudden C. M. N-Heterocyclic Carbenes in Materials Chemistry. Chem. Rev. 2019, 119, 4986–5056. 10.1021/acs.chemrev.8b00514. [DOI] [PubMed] [Google Scholar]; d Selected studies:MacLeod M. J.; Goodman A. J.; Ye H.-Z.; Nguyen H. V.-T.; Van Voorhis T.; Johnson J. A. Robust gold nanorods stabilized by bidentate N-heterocyclic-carbene-thiolate ligands. Nat. Chem. 2019, 11, 57–63. 10.1038/s41557-018-0159-8. [DOI] [PubMed] [Google Scholar]; e Narouz M. R.; Osten K. M.; Unsworth P. J.; Man R. W. Y.; Salorinne K.; Takano S.; Tomihara R.; Kaappa S.; Malola S.; Dinh C.-T.; Padmos J. D.; Ayoo K.; Garrett P. J.; Nambo M.; Horton J. H.; Sargent E. H.; Hakkinen H.; Tsukuda T.; Crudden C. M. N-heterocyclic carbene functionalized magic-number gold nanoclusters. Nat. Chem. 2019, 11, 419–425. 10.1038/s41557-019-0246-5. [DOI] [PubMed] [Google Scholar]
- a Astakhov A. V.; Khazipov O. V.; Chernenko A. Yu.; Pasyukov D. V.; Kashin A. S.; Gordeev E. G.; Khrustalev V. N.; Chernyshev V. M.; Ananikov V. P. A New Mode of Operation of Pd-NHC Systems Studied in a Catalytic Mizoroki–Heck Reaction. Organometallics 2017, 36, 1981–1992. 10.1021/acs.organomet.7b00184. [DOI] [Google Scholar]; b Khazipov O. V.; Shevchenko M. A.; Chernenko A. Yu.; Astakhov A. V.; Pasyukov D. V.; Eremin D. B.; Zubavichus Y. V.; Khrustalev V. N.; Chernyshev V. M.; Ananikov V. P. Fast and Slow Release of Catalytically Active Species in Metal/NHC Systems Induced by Aliphatic Amines. Organometallics 2018, 37, 1483–1492. 10.1021/acs.organomet.8b00124. [DOI] [Google Scholar]
- Kunz S. Supported, Ligand-Functionalized Nanoparticles: An Attempt to Rationalize the Application and Potential of Ligands in Heterogeneous Catalysis. Top. Catal. 2016, 59, 1671–1685. 10.1007/s11244-016-0687-7. [DOI] [Google Scholar]
- a Castelbou J. L.; Gual A.; Mercade E.; Claver C.; Godard C. Ligand effect in the Rh-NP catalyzed partial hydrogenation of substituted arenes. Catal. Sci. Technol. 2013, 3, 2828–2833. 10.1039/c3cy00388d. [DOI] [Google Scholar]; b Llop Castelbou J.; Bresó-Femenia E.; Blondeau P.; Chaudret B.; Castillón S.; Claver C.; Godard C. Tuning the Selectivity in the Hydrogenation of Aromatic Ketones Catalyzed by Similar Ruthenium and Rhodium Nanoparticles. ChemCatChem 2014, 6, 3160–3168. 10.1002/cctc.201402524. [DOI] [Google Scholar]; c Castelbou J. L.; Blondeau P.; Claver C.; Godard C. Surface Characterisation of Phosphine and Phosphite Stabilised Rh Nanoparticles: A Model Study. RSC Adv. 2015, 5, 97036–97043. 10.1039/C5RA21835G. [DOI] [Google Scholar]; d Foppa L.; Luza L.; Gual A.; Weibel D. E.; Eberhardt D.; Teixeira S. R.; Dupont J. Sputtering-deposition of Ru nanoparticles onto Al2O3 modified with imidazolium ionic liquids: synthesis, characterisation and catalysis. Dalton Trans. 2015, 44, 2827–2834. 10.1039/C4DT03039G. [DOI] [PubMed] [Google Scholar]
- The conditions for the hydrogenation of hexafluorobenzene were chosen to match the optimal conditions for the bottom-up designed system as closely as possible and differ slightly from the best conditions for this system found in our earlier reports (see Table S12 and ref (3c)).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.