

Abstract
The most common fermented beverage, lager beer, is produced by interspecies hybrids of the brewing yeast Saccharomyces cerevisiae and its wild relative Saccharomyces eubayanus. Lager-brewing yeasts are not the only example of hybrid vigor or heterosis in yeasts, but the full breadth of interspecies hybrids associated with human fermentations has received less attention. Here we present a comprehensive genomic analysis of 122 Saccharomyces hybrids and introgressed strains. These strains arose from hybridization events between two to four species. Hybrids with S. cerevisiae contributions originated from three lineages of domesticated S. cerevisiae, including the major wine-making lineage and two distinct brewing lineages. In contrast, the undomesticated parents of these interspecies hybrids were all from wild Holarctic or European lineages. Most hybrids have inherited a mitochondrial genome from a parent other than S. cerevisiae, which recent functional studies suggest could confer adaptation to colder temperatures. A subset of hybrids associated with crisp flavor profiles, including both lineages of lager-brewing yeasts, have inherited inactivated S. cerevisiae alleles of critical phenolic off-flavor genes and/or lost functional copies from the wild parent through multiple genetic mechanisms. These complex hybrids shed light on the convergent and divergent evolutionary trajectories of interspecies hybrids and their impact on innovation in lager-brewing and other diverse fermentation industries.
Introduction
Humans have been producing and consuming fermented beverages for thousands of years1. During this process, they have unwittingly shaped the evolutionary history of the microbes that are responsible for fermented products. The star of fermented beverage production is often Saccharomyces cerevisiae. Many studies have investigated the evolutionary impact of domestication in fermentation environments on the genomes of different lineages of this species2–13. These human-associated fermentation environments have also led to innovation through the hybridization of distantly related species.
Lager beers are made with hybrids between the distantly related species S. cerevisiae and Saccharomyces eubayanus14–16. These hybrids combine unique properties from each; S. cerevisiae’s carbon utilization and fermentation capabilities combined with S. eubayanus’s cryotolerance to produce yeasts that could ferment well in the cold17–22. Other interspecies hybrids of Saccharomyces have been associated, both favorably and unfavorably, with diverse fermentations. S. cerevisiae × Saccharomyces kudriavzevii hybrids are prized for their unique flavor profiles in beer and wine23. Conversely, hybrids and introgressed strains with large genomic contributions from S. eubayanus and Saccharomyces uvarum, are viewed as contaminants in breweries due to the production of off-flavors, while other strains have been associated with sparkling wine and cider fermentation16,24,25. Although these previous studies have hinted at the complexity of fermentation hybrids, their focus on a handful of strains or a handful of loci has only given us a fleeting glimpse of the diversity Saccharomyces hybrids, their total genomic compositions, and their evolution.
Here we identified, sequenced, and analyzed the genomes of 122 interspecies hybrids and introgressed strains in the genus Saccharomyces to understand their origins and evolutionary innovations. This collection contains pairwise hybrids, as well as more complex hybrids and introgressed strains with three or four parent species. We show that all genomic contributions from S. cerevisiae have arisen out of three domesticated lineages of S. cerevisiae, while all other parents belonged to Holarctic or European wild lineages of their respective species. We also analyzed inheritance of the mitochondrial genome and the genetic events generating functional diversity in genes relevant to fermented beverages. The genomic complexity of these hybrids provides insight into their origins and evolutionary successes in human-associated fermentation environments.
Results
Summary of Interspecies Hybrid Types
Here, we analyzed the genome sequences of 122 interspecies hybrids and introgressed strains of Saccharomyces, 63 strains of which are newly sequenced here, more than doubling the number of previously published hybrid genomes. Collectively, industrial settings dominated the isolation origins of all hybrids; 86% (n=105) were from beer, wine, cider, a distillery, or other beverages (Figure 1b, Table S1, Supplementary Text). We identified four types of hybrids: 1) lager-like (S. cerevisiae (Scer) × S. eubayanus (Seub)) (n=56); 2) S. cerevisiae × S. kudriavzevii (Skud) (n=15); 3) S. eubayanus × S. uvarum (Suva) (n=41); and 4) more complex hybrids, with three or four parent species (n=11 more than doubling those previously identified26) (Figure 1a, Table S1, Supplementary Text). These more complex hybrids fell into three groups: 4A) S. cerevisiae × S. kudriavzevii × S. eubayanus × S. uvarum (n=5), 4B) S. cerevisiae × S. eubayanus × S. uvarum (n=4), and 4C) one S. cerevisiae × S. kudriavzevii × S. eubayanus (Table S1). The lager-like hybrids were almost exclusively associated with beer (Figure 1b) and have genomic contributions that were consistent with previous observations in the two lineages (Saaz and Frohberg)27. The S. cerevisiae × S. kudriavzevii strains were associated with beer and wine (Figure1b). They had considerable differences in S. kudriavzevii genomic content, suggesting that these hybrids are of variable ages and evolutionary histories. The S. eubayanus × S. uvarum hybrids and introgressed strains were the most variable, both in isolation environment and genomic contributions (Figure 1, Table S1). The wide range in genomic contributions in these strains was likely influenced by their ability to backcross due to the low, but non-zero, spore viability of hybrids of these sister species16. These S. eubayanus × S. uvarum strains had the most total number of translocations (χ2 = 1250.1, p_adj = 2.64 E-15), as well as the most translocations shared with other hybrid types (χ2 = 15.964, p_adj = 0.0138) (Figure S2). The shared nature of some of these translocations in hybrids with more than two parents suggests that S. eubayanus × S. uvarum introgressed strains further hybridized to produce some of the complex three or four parent species hybrids. Thus, these four types of hybrids each show unique dynamics in genome evolution and are used for different products that range from several regional niche beverages to the globally dominant beer style, lagers.
Figure 1. Summary of genomic contributions and isolation environments for interspecies hybrids.
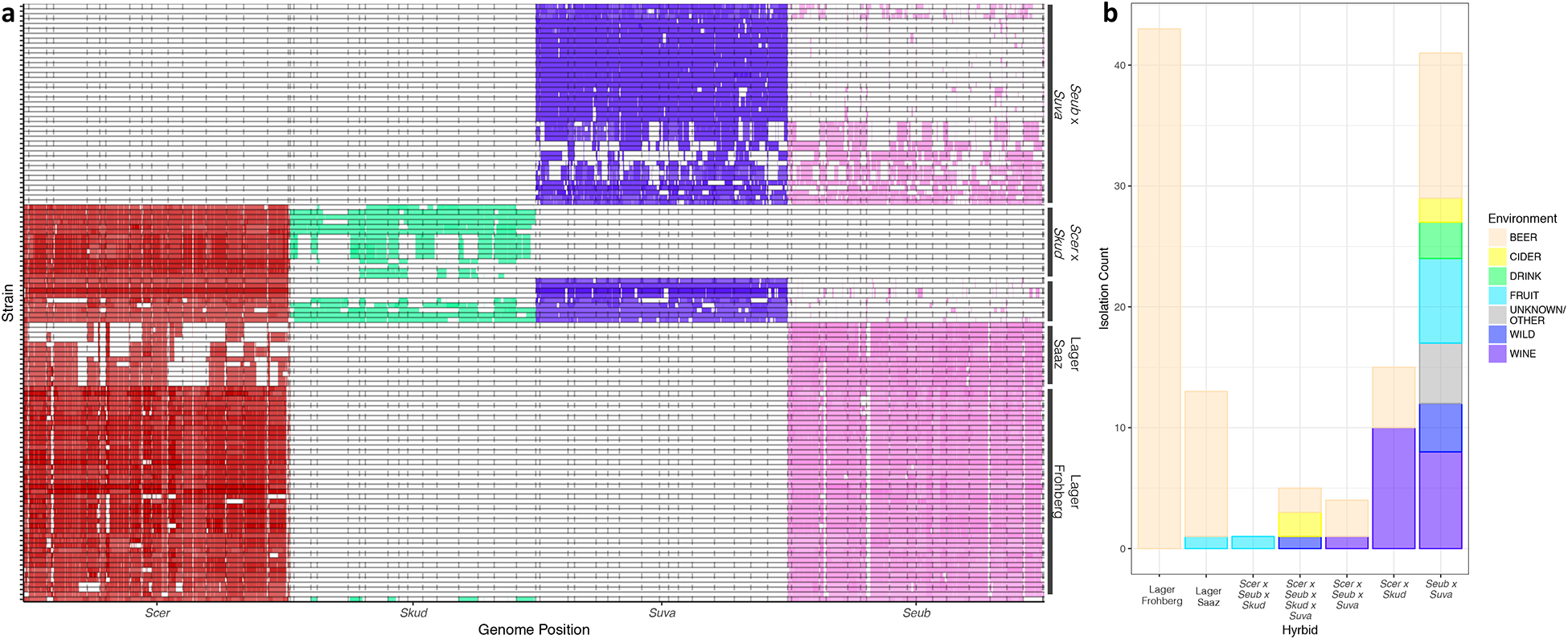
(a) Hybrids were clustered by genomic contributions. Lager strains are in the bottom half, S. uvarum × S. eubayanus strains are at the top, and most complex hybrids are in the middle, except for the single S. cerevisiae × S. eubayanus × S. kudriavzevii hybrid (very bottom). Individual hybrid strains are along the y-axis, and the genomes of the species contributing to hybrids are along the x-axis. S. cerevisiae (Scer) is in red, S. kudriavzevii (Skud) is in green, S. uvarum (Suva) is in purple, and S. eubayanus (Seub) is in pink. Dotted lines indicate chromosomes. Ploidy estimates are indicated by opacity, where darker regions are higher ploidy. (b) Counts of hybrids isolated from different environments. The lagers have been split into Saaz and Frohberg lineages. Other is grouped with Unknown and represents one isolate from a distillery. Tables S1 & S3 includes all isolation information and metadata.
Wild Parent Populations
Three out of four of the species contributing to these hybrids (S. kudriavzevii, S. uvarum, and S. eubayanus) have primarily been isolated from wild settings and have global distributions with populations that reflect their geography28,29. We used these established populations and phylogenomic and PCA approaches to evaluate the origins of these hybrids (Supplementary Text).
S. kudriavzevii has been isolated in Europe and Asia and consists of three described populations: Asia A, Asia B, and Europe23,30,31. The S. kudriavzevii sub-genomes of the hybrids all clustered with the European population as a monophyletic clade (Figure 2a, Figure S3, Table S2, File S1, Supplementary Text). These findings show that these hybrids were drawn from a closely related lineage of the European population of S. kudriavzevii.
Figure 2. Population and phylogenomic analyses of S. cerevisiae, S. kudriavzevii, S. uvarum, S. eubayanus, and their hybrid sub-genomes.
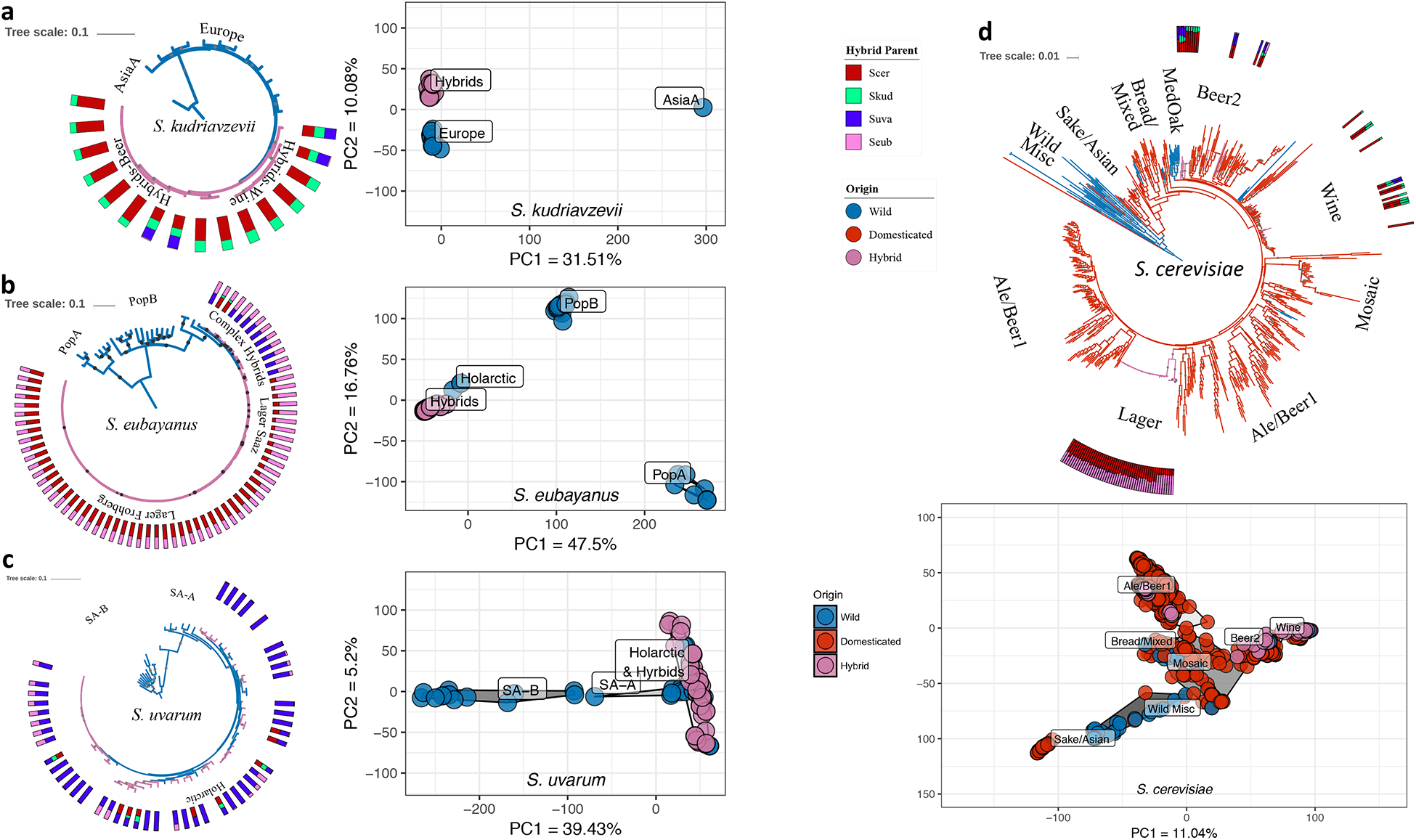
All phylogenies were built with RAxML with pure strains of a species and any hybrids with >50% complete sub-genome for given species. Bootstrap support values >70% are shown as gray dots. Branches are colored by the origin of isolation for each strain. Each hybrid has a stacked bar plot showing the genomic content for each species contributing to their genome; species colors are the same as in Figure 1a. For the Principal Component Analyses (PCA), dot colors represent strains’ origins, and color clouds represent populations or lineages. The axes of all PCAs are scaled to the same range. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figures S2, S4, S6, and S8; Table S2; and Files S1, S3, S5, and S7. (a) Left: Phylogeny of S. kudriavzevii with 30 strains and 38,992 SNPs from across the genome and rooted with an Asia B strain, IFO1803 (removed for clarity). Right: Principal component projection for PC1 and PC2, excluding Asia B. (b) Phylogeny of S. eubayanus with 92 strains and 18,878 SNPs from across the genome, rooted with Population A (PopA). Right: Principal component projection of PC1 and PC2. (c) Phylogeny for S. uvarum with 82 strains and 18,652 SNPs from across the genome, rooted with the Australasian lineage (removed for clarity). Right: Principal component projection for PC1 and PC2, excluding the Australasian lineage. (d) Top: Phylogeny for S. cerevisiae with 612 phased (for strains with >20K heterozygous sites) or unphased haplotypes and 21,222 SNPs from across the genome, rooted with the Taiwanese strain EN14S01 (removed for clarity). Previously identified wild lineages from West Africa, Malaysia, North America, Japan, and the Philippines are included in the Wild Misc group11,74. The other lineages are named in a similar manner to previous studies on ale-brewing and Mediterranean Oak (MedOak) strains8,9,71. Bottom: Principal component projection for PC1 and PC2 (including EN14S01, which groups with Sake/Asian).
In S. eubayanus, analysis of both large and small contributions, showed that these hybrids and introgressed strains clustered with the Holarctic lineage of S. eubayanus (Figure 2b, Figure S5, Table S2, File S3, Supplementary Text). Our vastly expanded dataset suggests that the Holarctic lineage is the closest known relative of all industrially relevant S. eubayanus hybrids and introgressed strains. The array of hybrids observed here requires that multiple hybridization events occurred between this lineage and other species. We also analyzed genetic diversity of the S. eubayanus contributions to industrial hybrids and introgressed strains (Supplementary Text). We found low nucleotide diversity in lager-like hybrids that shows that these widely used interspecies hybrids arose out of a narrow swath of S. eubayanus diversity, while the less frequently used hybrids and introgressed strains retained more nucleotide diversity.
S. uvarum has a parallel population structure to S. eubayanus26,32, with the exception of its increased isolation frequency in the Northern Hemisphere and the presence of pure strains isolated from Europe. Here we found that all contributions from S. uvarum arose out of the S. uvarum Holarctic lineage26. In contrast to our S. eubayanus findings, the S. uvarum sub-genomes of these hybrids and introgressed strains were interspersed with pure wild strains (Figure 2c, Figure S7 & S7, Table S2, File S5 & S6). These findings suggest that there have been multiple hybridization events and extensive backcrossing with wild lineages of S. uvarum, integrating wild diversity into these hybrids and leading to a diverse set of introgressed strains.
Domesticated S. cerevisiae Parent Lineages
Of the species contributing to domesticated interspecies hybrids, S. cerevisiae has the most extensive datasets, including industrial yeasts5,8–11. Through both phylogenomic and PCA approaches, we recapitulated the previously described domesticated S. cerevisiae clades8,9, and our 81 interspecies hybrids with S. cerevisiae contributions fell into three domesticated lineages: Wine, Ale/Beer1, and Beer2 (Figure 2d, Figure S9, Table S2, File S7).
The S. cerevisiae × S. kudriavzevii hybrids grouped with both Beer2 and Wine. Strains with contributions from three or four parent species fell into both clades (Beer2 and Wine), suggesting that these complex hybrids originated stepwise through iterative hybridization (Supplementary Text).
Interestingly, the only hybrids we detected in the Ale/Beer1 group were the lager-brewing yeasts (Figure 2d). The S. cerevisiae sub-genomes of the Saaz and Frohberg lager-brewing lineages formed distinct clades, and although we identified more Frohberg strains, Frohberg genetic diversity was lower (Supplementary Text). To determine if there was a particular clade of Ale/Beer1 that was the closest known relative to lager-brewing hybrids, we performed a targeted analysis of just the Ale/Beer1 S. cerevisiae strains and lager-brewing hybrids, (Figure S10 & S10, Table S2, File S8, Supplementary Text). Our concatenated phylogenomic analyses did not strongly support any recognized geographical clade of Ale/Beer1 S. cerevisiae strains as the closest outgroup to the lager-brewing yeasts. Our PCA analyses, which make no assumptions about consistent genome-wide signals, suggested several Stout beer, Wheat beer, and mosaic strains as sharing the most ancestry with lager-brewing yeasts, rather than any clade affiliated with a geographic style (Figure S9). Overall, our analyses clearly show that lager strains belong to the Ale/Beer1 lineage of S. cerevisiae and suggest affinity with a novel set of diverse beer yeasts, but they do not support any known extant strain as the sole closest relative.
Collectively, our data and analyses conclusively show that there have been multiple interspecies hybridization events between different domesticated lineages of S. cerevisiae and wild strains from three other Saccharomyces species (Figure 2d). The sheer number and diversity of hybrids analyzed here shows that evolutionary and industrial innovation through hybridization has happened on a scale and with a complexity beyond what previous smaller scale studies have suggested. In these diverse hybrids, the domesticated S. cerevisiae sub-genomes were likely preadapted with general industrial fermentation traits, while the wild parent likely contributed one or more traits advantageous in the specific new industrial fermentation niche being explored.
Mitochondrial Genome Inheritance
The classic example of yeast hybrid vigor comes from the cryotolerance of lager-brewing yeasts. S. eubayanus, S. kudriavzevii, and S. uvarum are all known to tolerate much colder temperatures33,34, and recent functional experiments have shown that the mitochondrial genome (mtDNA) plays a pivotal role in the cryotolerance of interspecies hybrids17,35. Strikingly, in our comprehensive dataset, a majority (94%) of the hybrids inherited a mtDNA from another species, rather than the S. cerevisiae mtDNA (Figure 3a).
Figure 3. Mitochondrial genome inheritance in interspecies hybrids.
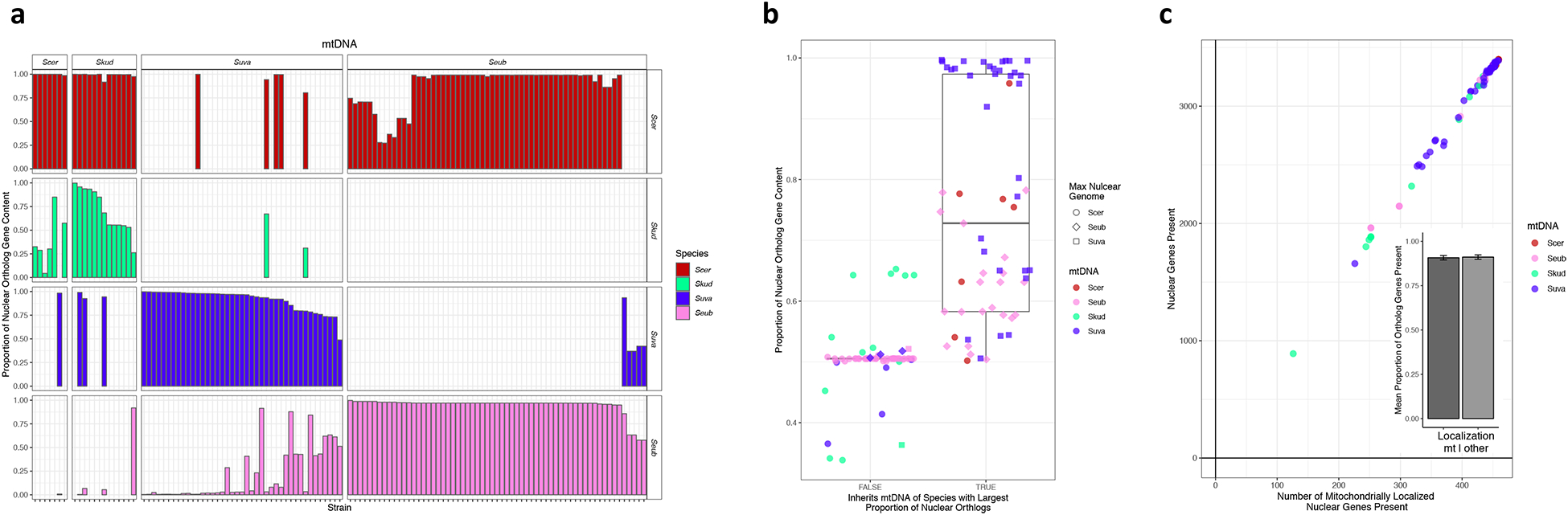
(a) The bar plots show proportion of 1:1:1:1 ortholog content for each sub-genome for each hybrid grouped by the mitochondrial genome (mtDNA) parent, which are labeled across the top. Colors represent different parent species and are that same as in of Figure 1a. (b) Analysis of concordance between which mtDNA was inherited and which parent contributed the most complete set of orthologous genes. “True” includes hybrids that inherited the most nuclear gene content from the same species as the mtDNA. “False” includes hybrids with mtDNA that did not match the species that contributed the most nuclear gene content. Colors represent the mtDNA parent, and shapes represent the largest nuclear genome contributor. The middle of the box plot corresponds to the median, the upper and lower limits are the 75th and 25th percentiles respectively, and the whiskers extend to the largest or smallest value no greater than 1.5 × the differences between the 75th and 25th percentiles. There was a significant correlation between the mtDNA parent and the largest nuclear genomic contributor (logistic regression p=3.58E-8, AIC= 118.21). Notably, the S. eubayanus × S. uvarum hybrids, which have often undergone many backcrossing events, follow this trend and are both cryotolerant species. (c) Linear relationship of the number of 1:1:1:1 orthologs versus the number of nuclear-encoded, mitochondrially localized genes present in the sub-genome that matches the mtDNA (linear regression p=2.0E-16, AIC= 1151.5). The inset shows the mean proportion of mitochondrially localized versus all other nuclear genes present in the sub-genome that matches the mitochondrial parent (p = 0.8612, odds ratio = 0.9653).
We tested if the parent that donated the mtDNA was also the parent that contributed the most nuclear gene content. We used a logistic regression to determine if the same parent species contributed both the mtDNA and the most complete set of orthologs. We found that this trend was generally true (p=8.0E-6, AIC= 83.75), but there were informative outliers (Figure 3b). In particular, more than half of the hybrids with S. kudriavzevii nuclear contributions inherited the S. kudriavzevii mtDNA, despite the fact that the S. kudriavzevii nuclear contribution was never in the majority. This discrepancy could be due to a fitness advantage conferred by the S. kudriavzevii mtDNA in colder fermentations, or it could be due to a fitness advantage conferred by the S. cerevisiae or other nuclear genomes36,37. Indeed, all outliers in our logistic regression analysis were in the direction of inheriting a cryotolerant parent’s mtDNA. These findings suggest that the inheritance of a cryotolerant mtDNA allowed these hybrids to thrive in colder environments where pure S. cerevisiae strains struggle, providing evolutionary and genetic innovation that enabled new fermentation techniques, such as lager brewing.
Hundreds of nuclear-encoded proteins localize to the mitochondria38. This interaction can be a source of genetic incompatibilities between the nuclear and mtDNAs, several of which have been characterized in Saccharomyces interspecies hybrids39–41. Therefore, we tested whether mitochondrially localized, nuclear-encoded genes were retained more often than other genes encoded in the nuclear genome matching the mtDNA parent. We found that more mitochondrially localized genes were retained in the same ratio as all other orthologs (p = 0.8612, odds ratio = 0.9653) (Table S3, Figure 3c). Although these results suggest that mitochondrial localization is not the main cause of the correlation between nuclear and mtDNA content, some nuance is warranted. First, only a small number of mitochondrially localized genes have been implicated in mito-nuclear incompatibilities39–41, and other factors that do not rely on protein localization could also play a role (e.g. metabolite exchange between the mitochondria and cytoplasm). Perhaps more importantly, these hybrids have often lost whole chromosomes or regions containing hundreds of genes at a time through chromosome mis-segregation or mitotic recombination events15; this restriction imposed by genetic linkage may prevent fine-scale retention or loss and obscure any signal driven by specific genes. Finally, some yet unmapped cryotolerant nuclear alleles might also be favored independently from the cryotolerant mtDNA. Overall, from this dataset, we conclude that there is a strong correlation between the amount of nuclear and mitochondrial DNA contributed by each parent species, but mitochondrially localized genes are not more affected than other genes.
Pan-Genome Analyses:
To characterize the core genome of these hybrids, we first analyzed the retention of 1:1:1:1 orthologs conserved in all four parent species and determined which parents contributed the least and most coding sequences to each hybrid. As few as 12 genes were retained in one strain, whereas some hybrids have retained almost complete sets of orthologs from all their parents (Figure S12, and Table S4). On average, these hybrids retained 56.2% of orthologs from the parent who contributed the least genomic material.
We preformed de novo genome assemblies to analyze the genomic content that was not present in the parent reference genomes (Figure S13). On average, these hybrids had 47.7 kbp of novel genomic content; the minimum was 2.2 kbp, and the maximum was 363.3 kbp. In addition to novel content that may come from the pan-genomes of other the Saccharomyces species, we detected previously characterized content from prior S. cerevisiae pan-genome analyses, including horizontally transferred genes (Supplemental Text)5,12,42. When we searched this material for Saccharomyces-like genes for which we could assign a function, we found an enrichment in genes associated with sugar transport, including the Gene Ontology43,44 terms: transporter activity (corrected p-val = 4.67E-08), sugar:proton symporter activity (corrected p-val = 6.04E-08), cation:sugar symporter activity (corrected p-val = 6.04E-08), and sugar transmembrane transporter activity (corrected p-val = 6.04E-08) (Table S5). The enrichment of sugar transport genes in the novel content of these hybrids and introgressed strains is consistent with strong selection for these activities in industrial fermentation environments.
Maltotriose Utilization Genes
We took a more detailed look at maltotrisoe utilizing genes because maltotriose is generally the second most abundant sugar in beer wort or malt extract, and Saccharomyces strains that utilize it are relatively rare outside of domesticated ale-brewing strains45–48. Our analyses of lager-brewing yeasts suggest that both S. cerevisiae and S. eubayanus contributed genes encoding functional maltotriose transporters to the hybrids, including alleles of S. cerevisiae MTT1 and S. eubayanus AGT1 previously shown to be functional18 (Figure 5b, Supplementary Text). We also recovered other predicted maltose/maltotriose transporter homologs in other interspecies hybrids and their parent species, which have yet to be explored functionally (Table S6). We conclude that the complexity and diversity of maltose transporter genes across Saccharomyces species is extensive and may have provided a source of functional diversity to fermentation hybrids.
Figure 5. Summary of hybrids and origin of lager traits.
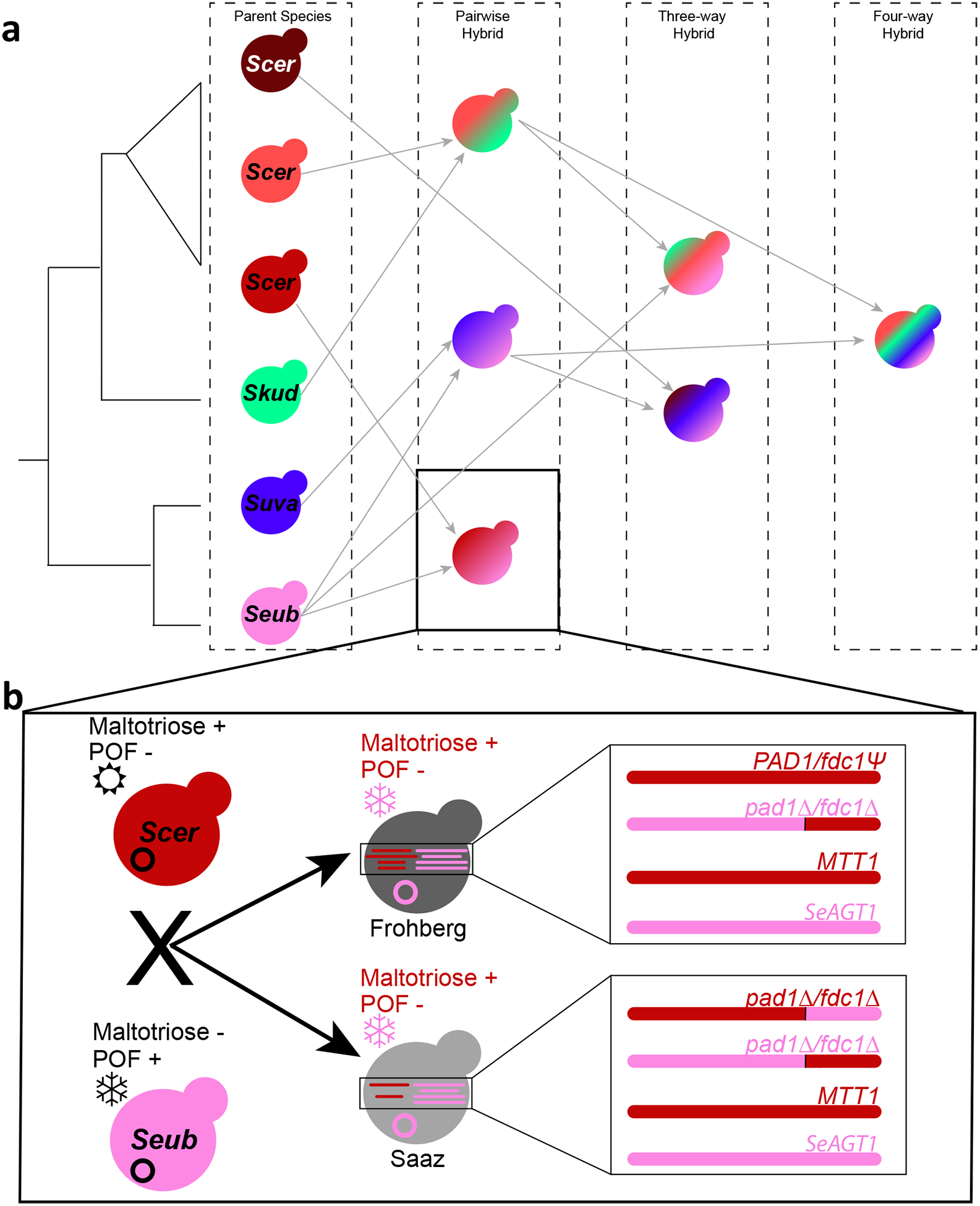
(a) Simplified summary of parents and resulting hybrids. On the left is a cladogram of just the Saccharomcyes species that have contributed to fermented beverage hybrids. Three distinct lineages of S. cerevisiae (Scer) have contributed to hybrids; for the wild parents (S. kudriavzevii (Skud), S. uvarum (Suva), and S. eubayanus (Seub)), Holarctic or European lineages gave rise to the hybrids. Gray lines point from each parent to the resulting hybrid. The order of secondary or tertiary hybridization events was inferred from genome composition. This simplified view does not show when multiple lineages of Scer have contributed to different hybrid types (e.g. Scer × Skud hybrids), backcrossing (e.g. Seub × Suva hybrids), or minor subtelomeric contributions (e.g. small Scer contributions to some Seub × Suva hybrids). (b) Summary of how lager-brewing yeasts acquired their unique trait profile. The two lager-brewing lineages, Saaz and Frohberg, arose out of hybridizations between domesticated Scer ale strains and wild Seub strains. The Scer strains could utilize maltotriose (+), did not produce phenolic-off-flavor (POF−), and preferred warmer temperatures (☼), while the Seub strains tolerated colder temperatures (❆), could not use maltotriose (−), and produced phenolic-off-flavors (POF+). The two lager-brewing lineages inherited the Seub mitochondrial genome (pink circle), which partly conferred cryotolerance. Both lineages also inherited maltotriose transporter genes from both parents (MTT1 from Scer and SeAGT1 from Seub). Finally, both lineages convergently became POF− through multiple distinct mechanisms, including pre-adaptation in the S. cerevisiae ale-brewing parent due to a mutated pseudogene (PAD1 | fdc1Ψ in red), aneuploidy removing functional S. eubayanus genes (pad1Δ | fdc1Δ in pink), and translocations in all Saaz strains and some Frohberg strains (pad1Δ | fdc1Δ in red).
Phenolic Off-Flavor Genes
The introduction of genes from wild strains, especially the mitochondrial genome and S. eubayanus AGT1, may have been key to cold fermentations, but other genes likely negatively impacted products. 4-vinyl guaiacol (4VG) is perceived as a clove-like, phenolic, or smoky flavor and considered an undesirable off-flavor in most beers. Lager beers are known for their crisp flavor profiles that lack appreciable 4VG, while wild strains of S. eubayanus and other species produce 4VG49. Two genes, PAD1 and FDC1, are essential for the production of 4VG50. Studies in ale-brewing yeast show that this trait is under strong domestication selection (Supplementary Text), but the genotypes of PAD1 and FDC1 across diverse interspecies hybrids already in use by industry have not been investigated, nor have the evolutionary genetic events leading to these genotypes. In our large hybrid dataset, we analyzed both retention and predicted functionality of PAD1 and FDC1 alleles from their parent species (Figure 4).
Figure 4. Hybrid inheritance and functionality of genes responsible for 4-vinyl guaiacol (4-VG) production.
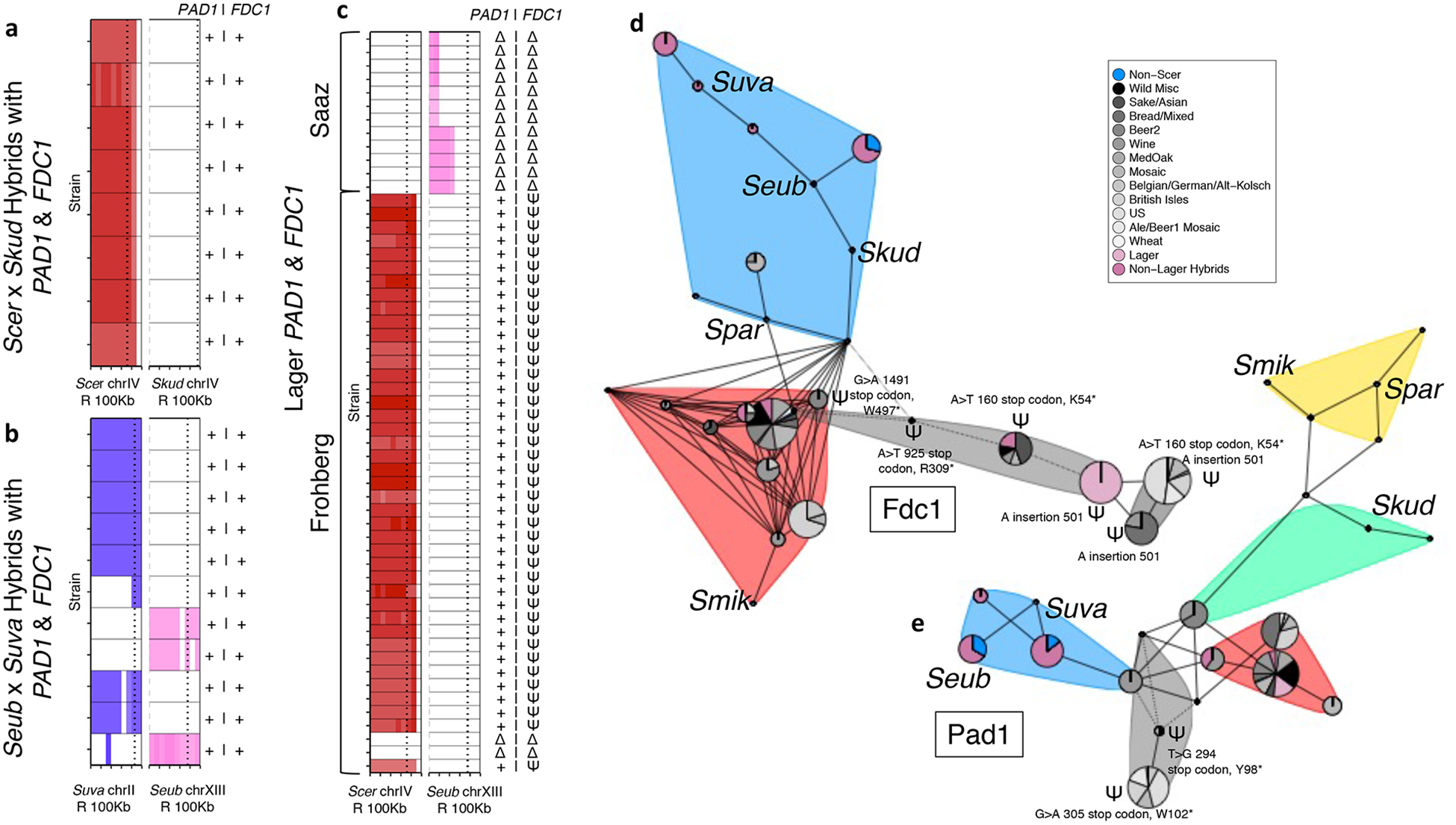
Retention of the regions where the adjacent PAD1 and FDC1 genes, which are both required for 4-VG production, are located in each parent species (a-c), shown as 10-kbp windows of ploidy estimates over last 100-kbp of the chromosome. Gene locations are represented by black dotted lines. Higher opacity represents higher ploidy. Species colors are that same as in Figure 1a. Scer = S. cerevisiae, Spar = Saccharomyces paradoxus, Smik = Saccharomyces mikatae, Skud = S. kudriavzevii, Suva = S. uvarum, and Seub = S. eubayanus. (a) Scer × Skud hybrids: all strains inherited versions of both PAD1 and FDC1 from Scer that are predicted to be functional, + | +, but they have lost the Skud alleles. (b) Suva × Seub hybrids: all strains inherited versions of PAD1 and FDC1, from either Suva or Seub, that are predicted to be functional, + | +. (c) All lager strains have completely lost the region in the Seub genome where these genes reside. Additionally, all Saaz strains have also completely lost the Scer versions of these genes, Δ | Δ. All but two Frohberg strains have retained versions of PAD1 from Scer that are predicted to be functional, but inherited Scer alleles of FDC1 that are predicted to be inactive due to a frameshift mutation, + | Ψ. Haplotype networks were built for the amino acid sequences for Fdc1 (d) and Pad1 (e). Colored pies correspond to Scer lineages, hybrids, or wild species with size representing the number of strains with that haplotype. Non-Scer nodes or groups of nodes are labeled by the species to which they correspond. Colored clouds correspond to communities: red is mostly Scer, blue is mostly non-Scer (including Seub and Suva), yellow is mostly Spar and Smik, green is mostly Skud, and gray is mostly loss-of-function alleles. Pseudogenes are marked as Ψ with additional information about the loss-of-function nucleotide and amino-acid changes. Dotted connections represent >100 amino acid differences.
In both S. cerevisiae × S. kudriavzevii and S. eubayanus × S. uvarum hybrids and introgressed strains, we found both FDC1 and PAD1 alleles that were predicted to be functional (Supplementary Text). These findings may reflect selection for diverse flavors, which are desirable in niche Trappist-style beers made with S. cerevisiae × S. kudriavzevii. In contrast S. eubayanus × S. uvarum are often viewed as contaminants in industrial brewing environments, and production of 4VG could contribute to this perception.
In the lager-brewing hybrids, we found that all strains have lost the ability to produce 4VG, but mechanism of this loss differed between Saaz and Frohberg (Supplementary Text). The Frohberg lager strains likely inherited a loss-of-function FDC1 allele from their domesticated S. cerevisiae parent and functional PAD1 and FDC1 alleles from their S. eubayanus parent. These functional wild alleles were then lost through translocations, likely due to break-induced replication. In contrast, the Saaz lineage has completely lost both the S. cerevisiae and S. eubayanus alleles of these genes through aneuploidy, an evolutionary trajectory facilitated by the fact that these subtelomeric genes reside on different chromosomes in these two species. The end result is that both Saaz and Frohberg lagers lack substantial phenolic off-flavors and have a crisp flavor profile. Even though Saaz and Frohberg strains evolved this trait through different final mutations that removed functional S. eubayanus alleles, the pre-adaptation of the domesticated S. cerevisiae parent, which already lacked functional genes, played a critical role by limiting the number of mutations needed. The contrast between Saaz and Frohberg strains highlights that there are many potential evolutionary trajectories open to interspecies hybrids to achieve a domestication trait.
Conclusions
Here, we characterized the genomes of 122 interspecies yeast hybrids and introgressed strains, the largest dataset of its kind to date. These hybrids have complex genomes with contributions from two to four species: S. cerevisiae, S. kudriavzevii, S. uvarum, and S. eubayanus (Figure 5a). The hybrids with S. cerevisiae contributions all arose out of three domesticated S. cerevisiae lineages: the wine lineage and two distinct beer clades. In contrast, all the S. kudriavzevii, S. uvarum, and S. eubayanus parents belonged to Holarctic or European wild lineages. Our results show how hybrid vigor also applies to microbes, with the domesticated S. cerevisiae parents providing genes and traits pre-adapted for industrial fermentations and the divergent species of Saccharomyces contributing new genes and traits that led to the successes of these hybrids in specific products. First, the frequent retention of mitochondrial genomes from cryotolerant parents likely conferred a fitness advantage during cold fermentation (Figure 5b). Second, although the S. cerevisiae genome is required for maltotriose utilization by hybrids, both S. eubayanus and S. cerevisiae contributed functional maltotriose transporter genes to lager-brewing yeasts. Third, phenolic off-flavor genes have been inactivated or eliminated from lager-brewing yeasts by multiple types of mutations (Figure 5b), while these genes have been retained in yeasts that ferment products where phenolic off-flavor is prized.
Hundreds of years ago, a S. cerevisiae strain meeting a S. eubayanus strain sparked the cold-brewing revolution, and crisp refreshing lagers eventually overtook the global beer market. This extensive genomic dataset reveals the genetic mechanisms and distinct evolutionary trajectories followed by hybrid and introgressed strains associated with fermentation products. These diverse hybrids and introgressed strains highlight how dynamic and complex fermentation innovation has cascaded down divergent and convergent evolutionary trajectories.
Methods
Strain Selection and Sequencing
The strains newly published here are from wild or beverage isolations, the Agricultural Research Service (ARS) NRRL collection (https://nrrl.ncaur.usda.gov), and commercially available sources. Table S7 contains the full metadata for strains. Whole genome Illlumina paired-end sequencing was done as previously described using either 2X100 or 2X250 reads32,51. This short-read data is available through the NCBI SRA database under the accession number PRJNA522928. Short-read data for published genomes were downloaded from NCBI; Table S8 contains a full list of accession numbers and citations 8,9,11,16,26,30,32,42,52–72.
Hybrid Identification
We used sppIDer73, a hybrid detection and analysis pipeline, to identify new hybrids, pure species, and reconfirm the species and hybrid identities of published data. For sppIDer, we used a combination reference genome that included all published genomes for all the Saccharomyces species63,72,74,75 (https://www.yeastgenome.org/,www.saccharomycessensustricto.org). For S. kudriavzevii, we used the genome from the Portuguese strain ZP591. As previously noted72, the published S. uvarum genome has the labels for chromosomes X and XII swapped, so we manually corrected them. We ran sppIDer with parameters set to identify genomic contributions >1% of the total genome. As sppIDer is reference genome-based, inheritance of regions not in the reference genome was not analyzed. Therefore, interspecies hybrids with only minor or subtelomeric introgressions were missed with this method. We also detected some smaller introgressions through the pan-genome analyses (see below).
Hybrid isolation environment was classified based on marketed product type for commercial strains; for published strains or strains from the ARS NRRL collection, we used available metadata supplied by the authors or depositors. Full details on hybrid isolation environment classification can be found in Table S1. To determine if there was an association between hybrid type and isolation environment, we completed χ2 analyses of hybrid by environment and of environment by hybrid with a Bonferroni multiple test correction in R. We limited this test to our most common (n>15) hybrid types (S. cerevisiae × S. eubayanus, S. cerevisiae × S. kudriavzevii, and S. eubayanus × S. uvarum) and the most common (n>8) origins (beer, wine, and fruit).
Whole Genome Sequence Assembly Pipeline
Alignment and single nucleotide polymorphism (SNP) calling were done as described previously32. Briefly, short reads were mapped with bwa “mem” to a concatenated reference genome of just the contributing parents. Reference genomes used for concatenation were the same as used for sppIDer. Samtools “view” and “sort” were then used to prepare the mapped reads with a mapping quality greater than 20 for SNP calling. PCR duplicates were removed with picard “MarkDuplicates”, and read groups were set with picard “AddOrReplaceReadGroups”. SNPs were called with GATK’s haplotype caller. Genome coverage per base pair was assessed with bedtools “genomeCoverageBed”. Strain-specific FASTA files were created by replacing called SNPs in repeat-masked concatenated reference genomes. Variants called as indels were replaced with Ns. Regions of extremely high coverage, (i.e. the 99.9th percentile of genome-wide coverage) were masked as Ns. Regions that do not exist in hybrids were masked as Ns, and regions at low coverage (i.e. between 3X-10X, depending on where the 10th percentile of the distribution of depth of coverage across the concatenated genomes fell) were masked as Ns. The strain-specific FASTAs for hybrid genomes were split into their component sub-genomes to be analyzed with pure strains.
Genomic completeness was estimated as the percent of the reference genome with coverage above the low-coverage masking threshold. Ploidy was estimated across the combination genome in 10-kbp windows. We used the R package modes (version 0.7.0) to analyze the distribution of depth of coverage and determine the antimodes, which correspond to a change in ploidy state. Some manual curation was needed for strains with “smiley patterns”, a pattern of increased coverage at chromosome ends that has been noted in other depth-of-coverage analyses8,76 and may be due chromatin structure77. For these strains, we used only the coverages that fell below the 95th percentile to estimate the antimodes and then assigned the distal ends to the largest ploidy estimated. We also visually checked and corrected rare instances when a “smiley pattern” lowered the ploidy estimate for the middle of the chromosome. From this antimode analysis, we were able to assign each 10-kbp window a ploidy value. The total DNA base-pair content contributed by each parent could then be estimated as the sum of each ploidy value multiplied by 10k and the number of windows with that ploidy value. Correcting this total DNA content per species by the total sum of all contributing species gave us a measure of total genomic content per species. Genomic contribution to a hybrid genome can be viewed as genomic content and genomic completeness. To estimate genomic completeness, we determined what percent of a total parent sub-genome had at least one haploid copy. To estimate genomic content, we took into account both completeness and ploidy across the combination of subgenomes. Full details on hybrid genome contributions can be found in Table S1. For visualizations, we clustered the strains based on ploidy estimated across the combination genome using Ward’s method in the R package pvclust (v. 2.0–0)78.
For each strain, we calculated the number of sites called as heterozygous with GATK for each sub-genome. Strains with more than 20,000 heterozygous sites in any sub-genome were phased with GATK’s “ReadBackedPhasing” command79, which can phase short regions of the genome based on overlapping reads. We then split the output into two phases, one that retains more reference variants and one that contains more alternative variants in phased regions. This pseudo-phasing allowed us to investigate regions that are less similar to the published reference. We converted these phases into two strain-specific FASTA files and masked them for coverage as above. Both phases were included in all downstream analyses involving phased genomes, which are noted as “strainID 1” or “strainID 2”.
1:1:1:1 Orthologs
We identified genes that are orthologous across all parent genomes based on the annotations in the published gff files for each reference genome, which yielded a list of 3,856 genes. We used the coordinates to determine the coverage for each ortholog. Gene presence was noted if the mean coverage for that ortholog was >3X.
De Novo Genome Assembly and Pan-Genome Analyses
We assembled the hybrid genomes with the meta-assembler iWGS80 and choose the best assembly based on the largest N50 score. All hybrids, except DBVPG6257, were successfully assembled and are available under GenBank BioProject PRJNA522928.
We mapped the short-read data back to these assembled genomes and used the sppIDer output to classify to which parent reference genome each short read mapped. With this analysis, we determined which reads did not map to a parent reference genome but did assemble de novo into a contig of 1.5-kbp or greater. We classified these regions as “unmapped” and used a tBLASTx to search for S. cerevisiae-like genes using S288C ORFs and retaining hits with evalue < 10−10. To determine if this set of genes identified in these novel assembled regions were enriched for any functions, we used GO Term Finder (Version 0.86)43,44. To determine the potential origin of these novel regions, we used a BLASTn search of the NCBI nucleotide database (v5). The output of this was then parsed for number of hits with an e-value < 10−10. To determine the number of hits to different species, we completed χ2 analyses with a Bonferroni multiple test correction in R.
Translocation Identification
To detect shared breakpoints and translocations, we use LUMPY81 with the mapped short-read data. We masked for repetitive regions by excluding regions with coverage above twice the genome-wide mean. Each breakpoint call had to be supported by at least 4 reads to be included in downstream analyses. We parsed this output for species sub-genome, hybrid type, and the species pair between which the translocation was detected. We calculated the total number of called breakpoints, breakpoints that were shared in at least two hybrids of the same type, and breakpoints that were shared in multiple hybrid types. We compared these different categories with χ2 analyses and a Bonferroni multiple test correction in R.
We also identified translocations from the de novo assemblies. For this analysis, we used sppIDer results to assign regions of the de novo assemblies to a parent species. Some regions were unmapped with sppIDer, as noted above. Additionally, some regions had high coverage from multiple parents in the de novo assembly, where the donor species could not be unambiguously assigned; these regions are likely repetitive and difficult to assemble. Translocations were identified when regions that were >2-kbp came from different donor species and were assembled with <100-bp of unmapped or ambiguous data separating them. On average, we identified 17 translocations per strain. From this output, we counted the number of translocations identified in each hybrid type, the donor species, and the pair of species between which the translocations occurred. We compared hybrid type, species pair, and individual species with a χ2 analyses with a Bonferroni multiple test correction in R.
Mitochondrial Genome Analysis Pipeline
We use mitoSppIDer73 to determine the mitochondrial genome (mtDNA) parent for the hybrids. This analysis was done in a similar manner to the whole genome sppIDer analysis, except that mtDNAs for each Saccharomyces species were used72,82,83, except Saccharomyces jurei. GenBank accessions lacking full manuscripts included S. mikatae (KX707788) and S. kudriavzevii (KX707787).
To determine if the mtDNA parent was associated with retention of the nuclear genes, we performed a logistic regression in R. We used the set of 1:1:1:1 orthologs to determine which parent contributed the most complete set of orthologous genes. To determine if there was an enrichment for the retention of nuclear-encoded, mitochondrially interacting proteins, we used the set of genes products identified as localize to the mitochondria through the Yeast GFP Fusion Localization Database38. When we filtered for genes that were also 1:1:1:1 orthologs, our final list consisted of 459 genes. To determine if there was a linear relationship between retention of mitochondrially localized genes and all other orthologs, we performed a linear regression and to determine if there were more mitochondrially localized genes retained compared to all other genes, we used a Fisher’s Exact Test with a Bonferroni correction. Tests were performed in R.
Since past work has shown that reticulate evolution, introgression, and horizontal gene transfers are widespread in Saccharomyces mtDNAs84, we wanted to explore the inheritance of mitochondrially encoded genes in more depth. Due in part to their high AT content (~85%), mtDNAs are often poorly covered using Illumina sequencing. In particular, intergenic regions and coding sequencing with transposable elements (introns, homing endonucleases, and GC clusters) can be difficult to assemble. To explore the phylogenetic relationships of these mtDNAs, we used a bait-prey bioinformatic method to pull out the read sequences of coding sequences. We used HybPiper85 to pull out reads from the hybrid Illumina libraries that mapped to those mitochondrial genes using gene sequences from reference strains used in mitoSppIDer as baits. These extracted Illumina reads were aligned to the reference genes in Geneious (v. 6.1.6)86 and manually assembled. We successfully covered six mitochondrial genes (COX2, COX3, ATP6, ATP8, ATP9, and 15S rRNA), which were used to construct the mitochondrial phylogenetic haplotype network. This unique set of unambiguously completed genes was concatenated (4.7-kbp) by strain to produce the haplotype for each pure Saccharomyces or hybrid strain (Figure S14). Haplotypes and haplotype frequencies for each strain were encoded as a nexus-formatted file for PopART v1.7.287. The haplotype network was reconstructed using the TCS method88. Strains were assigned to each haplotype using DnaSP v589. For some strains, we could not assemble the 15S rRNA gene because of low-coverage data. For these strains, we inferred their haplotype designation based on an analysis where we removed the15S rRNA gene. This information is not included in Figure S14 but can be found in Table S9.
Genes of Functional Interest Analysis Pipeline
To assemble the sequences of genes relevant to brewing, we again used HybPiper85. To be included for further analyses, the assembled length had to be at least as long as the bait gene and had to have a minimum 10X depth of coverage. For the baits, we used either gene sequences from the S. cerevisiae strain S288C found on the Saccharomyces Genome Database (https://www.yeastgenome.org); from the S. eubayanus type strain, CBS12357T 72; or the lager strain W34/7090. For the PAD1 analysis in S. eubayanus × S. uvarum hybrids, we used the PAD1 gene sequence from the S. uvarum reference genome, CBS700163. To get precise gene locations for PAD1 and FDC1, we used a tBLASTn search of the S. eubayanus, S. kudriavzevii, and S. uvarum reference genomes with the S. cerevisiae sequences for these genes as the query.
The assembled genes were aligned with MAFFT v.791, allowing for reverse complementation. The alignments were manually trimmed to the protein-coding sequences. For PAD1 and FDC1, the alignments were conceptually translated to amino acid sequences, and haplotype networks were built with a modified minimum-spanning network and visualized with iGraph92 in R. The haplotype networks were split into communities as previously described93.
Pairwise distances between sequences were calculated using the trimmed MAFFT nucleotide sequence alignments and the p-distance method as implemented in MEGA-X94 with the following parameters: substitutions to include Transitions + Transversions, assuming uniform rates among sites, and using pairwise deletion of gaps. The percent identity of hits to the bait sequence was organized by species, and hybrid status was recorded in Table S6, along with the origin of the bait gene and tallies of sequences whose translations were visually identified as being incomplete or containing premature stop codons.
Phylogenomic and Population Structure Analyses
We masked regions with no coverage as Ns, which is interpreted as missing data by most tools; therefore, for downstream whole genome analyses, we only included sub-genomes that were >50% complete (i.e. major contributions). To include the minor contribution hybrids in the non-S. cerevisiae analyses, we used reduced genomes that were concatenations of the regions of the genome that existed in at least one minor hybrid (Table S10). This procedure allowed us include strains with minor introgressions and only use regions of the genome that had been contributed by the minor parent. To balance some of our analyses for Saaz and Frohberg lager strains, we used a random subset of Frohberg strains to match the number of Saaz strains. Phylogenomic trees were built with RAxML v8.195 using SNPs from the whole genome for the major analyses or the reduced genome for the minor analyses. Trees were visualized with iTOL96. The PCA analyses were done with the adegenet package in R97 and visualized with ggPlot298. Estimates of adjusted π (π *100) were calculated with the PopGenome package in R99.
Data and Code Availability
References and accession numbers for the published data used can be found in Table S8. Short-read data newly published here is available through the NCBI SRA database under the accession number PRJNA522928. Custom R and Python scripts used for this publication can be found on GitHub (https://github.com/qlangdon/hybrid-ferment-invent).
Supplementary Material
Figure S1. Genomic contribution comparison of Muri and WLP351.
Modified sppIDer plot, where the y-axis is estimated ploidy, rather than coverage, for the S. cerevisiae (50%) × S. eubayanus (5%) × S. uvarum (45%) strains Muri61 and WLP351.
Figure S2. Summary of total genomic coverage and shared translocations.
The minimum and maximum normalized coverage of all strains that contain each chromosome are shown as colored bars. Darker chromosomes mean that chromosome is present in more strains. Vertical dotted lines represent translocations that are shared in at least four strains, including between hybrid types. The color of the line represents the reciprocal species. (a) Only lager strains and translocations found only in lagers. (b) All 122 hybrids and interspecies translocations.
Figure S3. Phylogenomic trees for S. kudriavzevii with strains labeled.
(a) Phylogeny identical to Figure 2a with strains labeled. (b) Phylogeny identical to Figure S4 with strains labeled. Newick files are available as Files S1 & S2.
Figure S4. Phylogenomic and population placement of hybrids with minor S. kudriavzevii contributions.
(a) Phylogenomic tree built with 36 strains and 12,424 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. (b) PCA using whole genome data for European S. kudriavzevii strains and all major contributor hybrids. (c) PCA using a reduced genome (67%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S3, Table S2, and File S2.
Figure S5. Phylogenomic trees for S. eubayanus with strains labeled
(a) Phylogeny identical to Figure 2b with strains labeled. (b) Phylogeny identical to Figure S6 with strains labeled. Newick files available as Files S3 & S4.
Figure S6. Phylogenomic and population placement of hybrids with minor S. eubayanus contributions.
(a) Phylogenomic tree built with 112 strains and 69,631 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. Long branches are biased by the extensive missing data in hybrids with very small contributions from S. eubayanus. (b) PCA using whole genome data for Holarctic S. eubayanus strains and all major contributor hybrids. (c) PCA using a reduced genome (25%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S5, Table S2, and File S4.
Figure S7. Phylogenomic trees for S. uvarum with strains labeled.
(a) Phylogeny identical to Figure c with strains labeled. (b) Phylogeny identical to Figure S8 with strains labeled. Newick files are available as Files S5 & S6.
Figure S8. Phylogenomic and population placement of hybrids with minor S. uvarum contributions.
(a) Phylogenomic tree built with 69 strains and 36,541 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1A. (b) PCA using whole genome data for Holarctic S. uvarum strains and all major contributor hybrids. (c) PCA using a reduced genome (84%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S7, Table S2, and File S6.
Figure S9. Phylogenomic tree for full S. cerevisiae analysis with strains labeled. Phylogeny identical to Figure 2d with strains labeled. A Newick file is available as File S7.
Figure S10. Phylogenomic and population placement of lagers within the Ale/Beer1 clade.
(a) Phylogenomic tree built with 267 strains and 21,953 SNPs from the whole genome. The total number of Frohberg strains was down-sampled to match the same number of Saaz strains. The tree was rooted with the Wine strain DBVPG1106. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. (b) PCA using whole genome data for Ale/Beer1 strains, all Saaz strains, and the down-sampled set of Frohberg strains. The two lineages of lager strains form separate groups, but they do not cluster with any described geographical lineage of the Ale/Beer1 clade. Pure S. cerevisiae Ale/Beer1 strains outside of the labeled lineages are unplaced, including a cluster of Stout strains, Wheat strains, and mosaic strains that our analyses suggest share the most ancestry with lager-brewing yeasts. (c) PCA using all lager strains. The low diversity in the Frohberg lager strains drives PC1, which led us to balance the dataset by down-sampling this lineage. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S11, Table S2, and File S8.
Figure S11. Phylogenomic tree for Ale/Beer1 S. cerevisiae analysis with strains labeled. Phylogeny identical to Figure S10 with strains labeled. A Newick file is available as File S8.
Figure S12. 1:1:1:1 orthologs present in hybrid genomes.
(a) Stacked bar chart of all 1:1:1:1 orthologs present in hybrids. Strains are sorted from most to least ortholog content. Completeness of the ortholog set from the species that contributed the most (b) or least (c) orthologs to the strains. Strains are ordered independently in all panels.
Figures S13. Complete de novo genome assembly for all strains.
Total assembled genome for each strain. Regions are colored by which parent could be assigned in the de novo assembly based on the sppIDer results. “Multi” are regions where reads from many species mapped at high coverage. “Unmapped” are novel regions assembled from reads that do not map to parent reference genomes. For each assembly, contigs are ordered from largest to smallest from left to right.
Figure S14. Mitochondrial genome haplotype network.
Six mitochondrial genes were concatenated in 364 wild Saccharomyces strains and interspecies hybrids and used to build a TCS88 phylogenetic network. Haplotype classification is provided in Table S9. Haplotypes are represented by circles, and circle size is scaled according to the haplotype frequency. Pie charts show the frequency of haplotypes based on species or hybrid designation. The number of mutations separating each haplotype are indicated by lines on the edges connecting the haplotype circles.
Figure S15. Labeled (in turquoise) haplotype networks for PAD1 and FDC1. Edge numbers are the number of amino acid changes. Networks correspond to those used in Figure 4 for the amino acid sequences of (a) Fdc1 and (b) Pad1. (b) A different haplotype network orientation of Figure 4E that increases the visibility of each community and haplotype. Table S9 contains the key to which strains belong to which haplotype.
File S1. Newick formatted file of the S. kudriavzevii phylogeny with major hybrids.
Table S2. PCA analyses.
Table S1. All hybrids and their parent contributions.
File S3. Newick formatted file of the S. eubayanus phylogeny with major hybrids.
File S2. Newick formatted file of the S. kudriavzevii phylogeny with minor hybrids.
File S4. Newick formatted file of the S. eubayanus phylogeny with minor hybrids.
File S5. Newick formatted file of the S. uvarum phylogeny with major hybrids.
File S6. Newick formatted file of the S. uvarum phylogeny with minor hybrids.
File S7. Newick formatted file of the S. cerevisiae phylogeny with all strains analyzed.
File S8. Newick formatted file of the S. cerevisiae phylogeny of just the Ale/Beer1 clade.
Table S3. Results of Fisher's Exact Test and Bonferroni correction of mitochondrially localized genes.
mtInteracting = nuclear-encoded but mitochondrially localized gene.
Table S4. Summary of number of 1:1:1:1 orthologs present in each sub-genome.
Table S6. Brewing relevant gene summaries.
"-" Indicates when HybPiper failed to recover and assemble genes for this group or that these assemblies failed our length and coverage cutoffs.
Table S5. GO term results of genes found in novel regions of the de novo assembled genomes.
Table S7. Metadata for all strains newly sequenced in this study.
The “New hybrid” column denotes hybrid genome sequences that are newly published in this study.
Scer = S. cerevisiae, Spar = Saccharomyces paradoxus, Smik = Saccharomyces mikatae, Skud = S. kudriavzevii, Suva = S. uvarum, and Seub = S. eubayanus.
Table S8. Published data accession information.
Table S9. Haplotype key for mitochondrial genomes, PAD1, and FDC1.
Dataset A only includes strains where 15S rRNA could be assembled, while Dataset B has 15S rRNA removed.
Table S10. Regions used for minor contribution analyses.
Acknowledgments
We thank Kevin J. Verstrepen for coordinating publication with their study; Amanda B. Hulfachor and Martin Bontrager for preparing a subset of Illumina libraries; the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing Illumina sequencing facilities and services; Marc-André Lachance, Ashley Kinart, Drew T. Doering, Randy Thiel, and Dan Carey for strains; and Margaret Langdon, Amanda B. Hulfachor, and Kayla Sylvester for collecting fermentation samples and/or isolating strains. This material is based upon work supported by the National Science Foundation under Grant Nos. DEB-1253634 (to CTH) and DGE-1256259 (Graduate Research Fellowship to QKL), the USDA National Institute of Food and Agriculture Hatch Project No. 1003258 to CTH, and in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science Nos. DE-SC0018409 and DE-FC02-07ER64494 to Timothy J. Donohue). QKL was also supported by the Predoctoral Training Program in Genetics, funded by the National Institutes of Health (5T32GM007133). DP is a Marie Sklodowska-Curie fellow of the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 747775). EPB was supported by a Louis and Elsa Thomsen Wisconsin Distinguished Graduate Fellowship. DL was supported by CONICET (PIP 392), FONCyT (PICT 3677), and Universidad Nacional del Comahue (B199). CTH is a Pew Scholar in the Biomedical Sciences, Vilas Faculty Early Career Investigator, and H. I. Romnes Faculty Fellow, supported by the Pew Charitable Trusts, Vilas Trust Estate, and Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF), respectively.
References
- 1.Hornsey IS Alcohol and Its Role in the Evolution of Human Society. (RSC Publishing, 2012). [Google Scholar]
- 2.Fay JC & Benavides JA Evidence for Domesticated and Wild Populations of Saccharomyces cerevisiae. PLoS Genet. 1, e5 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liti G, Peruffo A, James SA, Roberts IN & Louis EJ Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex. Yeast 22, 177–192 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Gallone B et al. Origins, evolution, domestication and diversity of Saccharomyces beer yeasts. Curr. Opin. Biotechnol 49, 148–155 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Legras JL et al. Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Mol. Biol. Evol 35, 1712–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodríguez ME et al. Saccharomyces uvarum is responsible for the traditional fermentation of apple chicha in Patagonia. FEMS Yeast Res. 17, fow109 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Barbosa R et al. Multiple Rounds of Artificial Selection Promote Microbe Secondary Domestication—The Case of Cachaça Yeasts. Genome Biol. Evol 10, 1939–1955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallone B et al. Domestication and Divergence of Saccharomyces cerevisiae Beer Yeasts. Cell 166, 1397–1410.e16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonçalves M et al. Distinct Domestication Trajectories in Top- Fermenting Beer Yeasts and Wine Yeasts. Curr. Biol 26, 1–12 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Duan SF et al. The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peter J et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556, 339–344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsit S & Dequin S Diversity and adaptive evolution of Saccharomyces wine yeast: a review. FEMS Yeast Res. 15, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almeida P, Barbosa R, Bensasson D, Gonçalves P & Sampaio JP Adaptive divergence in wine yeasts and their wild relatives suggests a prominent role for introgressions and rapid evolution at noncoding sites. Mol. Ecol 26, 2167–2182 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Hittinger CT, Steele JL & Ryder DS Diverse yeasts for diverse fermented beverages and foods. Curr. Opin. Biotechnol 49, 199–206 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Gibson B & Liti G Saccharomyces pastorianus: genomic insights inspiring innovation for industry. Yeast 32, 17–27 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Libkind D et al. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc. Natl. Acad. Sci. U. S. A 108, 14539–44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker EP et al. Mitochondrial DNA and temperature tolerance in lager yeasts. Sci. Adv 5, eaav1869 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker EP & Hittinger CT Evolution of a novel chimeric maltotriose transporter in Saccharomyces eubayanus from parent proteins unable to perform this function. PLOS Genet. 15, e1007786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hebly M et al. S. cerevisiae × S. eubayanus interspecific hybrid, the best of both worlds and beyond. FEMS Yeast Res. 15, 1–14 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Gibson BR, Storgårds E, Krogerus K & Vidgren V Comparative physiology and fermentation performance of Saaz and Frohberg lager yeast strains and the parental species Saccharomyces eubayanus. Yeast 30, 255–266 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Gorter de Vries A et al. Laboratory evolution of a Saccharomyces cerevisiae x S. eubayanus hybrid under simulated lager-brewing conditions: genetic diversity and phenotypic convergence. bioRxiv 31, 1–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monerawela C & Bond U Brewing up a storm: The genomes of lager yeasts and how they evolved. Biotechnol. Adv 35, 512–519 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Peris D, Pérez-Torrado R, Hittinger CT, Barrio E & Querol A On the origins and industrial applications of Saccharomyces cerevisiae × Saccharomyces kudriavzevii hybrids. Yeast 35, 51–69 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Nguyen HV & Boekhout T Characterization of Saccharomyces uvarum (Beijerinck, 1898) and related hybrids: Assessment of molecular markers that predict the parent and hybrid genomes and a proposal to name yeast hybrids. FEMS Yeast Res. 17, 1–19 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Nguyen HV, Legras JL, Neuvéglise C & Gaillardin C Deciphering the hybridisation history leading to the lager lineage based on the mosaic genomes of Saccharomyces bayanus strains NBRC1948 and CBS380 T. PLoS One 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almeida P et al. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat. Commun 5, 4044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunn B & Sherlock G Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 18, 1610–1623 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hittinger CT Saccharomyces diversity and evolution: a budding model genus. Trends Genet. 29, 309–17 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Boynton PJ & Greig D The ecology and evolution of non-domesticated Saccharomyces species. Yeast 31, 449–462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hittinger CT et al. Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 464, 54–58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampaio JP & Gonçalves P Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl. Environ. Microbiol 74, 2144–52 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peris D et al. Complex Ancestries of Lager-Brewing Hybrids Were Shaped by Standing Variation in the Wild Yeast Saccharomyces eubayanus. PLoS Genet. 12, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salvadó Z, Arroyo-López FN, Barrio E, Querol A & Guillamón JM Quantifying the individual effects of ethanol and temperature on the fitness advantage of Saccharomyces cerevisiae. Food Microbiol. 28, 1155–61 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Gonçalves P, Valério E, Correia C, de Almeida JMGCF & Sampaio JP Evidence for divergent evolution of growth temperature preference in sympatric Saccharomyces species. PLoS One 6, e20739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li XC, Peris D, Hittinger CT, Sia EA & Fay JC Mitochondria-encoded genes contribute to evolution of heat and cold tolerance in yeast. Sci. Adv 5, eaav1848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortiz-Tovar G, Pérez-Torrado R, Adam AC, Barrio E & Querol A A comparison of the performance of natural hybrids Saccharomyces cerevisiae × Saccharomyces kudriavzevii at low temperatures reveals the crucial role of their S. kudriavzevii genomic contribution. Int. J. Food Microbiol 274, 12–19 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Tronchoni J, Medina V, Guillamón JM, Querol A & Pérez-Torrado R Transcriptomics of cryophilic Saccharomyces kudriavzevii reveals the key role of gene translation efficiency in cold stress adaptations. BMC Genomics 15, 1–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huh K et al. Global analysis of protein localization in budding yeast. (2003). [DOI] [PubMed] [Google Scholar]
- 39.Chou JY, Hung YS, Lin KH, Lee HY & Leu JY Multiple molecular mechanisms cause reproductive isolation between three yeast species. PLoS Biol. 8, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HY et al. Incompatibility of Nuclear and Mitochondrial Genomes Causes Hybrid Sterility between Two Yeast Species. Cell 135, 1065–1073 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Hou J & Schacherer J Negative epistasis: a route to intraspecific reproductive isolation in yeast? Curr. Genet 62, 25–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novo M et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc. Natl. Acad. Sci 106, 16333–16338 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ashburner M et al. Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Consortium TGO The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han E-K, Cotty F, Sottas C, Jiang H & Michels CA Characterization of AGT1 encoding a general alpha-glucoside transporter from Saccharomyces. Mol. Microbiol 17, 1093–1107 (1995). [DOI] [PubMed] [Google Scholar]
- 46.Salema-Oom M, Pinto VV, Gonçalves P & Spencer-Martins I Maltotriose Utilization by Industrial. Society 71, 5044–5049 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horák J Regulations of sugar transporters: insights from yeast. Curr. Genet 59, 1–31 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Dietvorst J, Londesborough J & Steensma HY Maltotriose utilization in lager yeast strains: MTTI encodes a maltotriose transporter. Yeast 22, 775–788 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Diderich JA, Weening SM, van den Broek M, Pronk JT & Daran J-MG Selection of Pof-Saccharomyces eubayanus Variants for the Construction of S. cerevisiae × S. eubayanus Hybrids With Reduced 4-Vinyl Guaiacol Formation. Front. Microbiol 9, 1640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukai N, Masaki K, Fujii T, Kawamukai M & Iefuji H PAD1 and FDC1 are essential for the decarboxylation of phenylacrylic acids in Saccharomyces cerevisiae. J. Biosci. Bioeng 109, 564–569 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Shen X-X et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175, 1533–1545.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bing J, Han P-J, Liu W-Q, Wang Q-M & Bai F-Y Evidence for a Far East Asian origin of lager beer yeast. Curr. Biol 24, R380–1 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Borneman AR, Forgan AH, Pretorius IS & Chambers PJ Comparative genome analysis of a Saccharomyces cerevisiae wine strain. FEMS Yeast Res. 8, 1185–1195 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Borneman AR et al. Whole-Genome Comparison Reveals Novel Genetic Elements That Characterize the Genome of Industrial Strains of Saccharomyces cerevisiae. PLoS Genet. 7, e1001287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borneman AR, Forgan AH, Kolouchova R, Fraser JA & Schmidt SA Whole Genome Comparison Reveals High Levels of Inbreeding and Strain Redundancy Across the Spectrum of Commercial Wine Strains of Saccharomyces cerevisiae. G3 6, 957–971 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunn B, Richter C, Kvitek DJ, Pugh T & Sherlock G Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res. 22, 908–924 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gayevskiy V & Goddard MR Saccharomyces eubayanus and Saccharomyces arboricola reside in North Island native New Zealand forests. Environ. Microbiol 18, 1137–1147 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Gayevskiy V, Lee S & Goddard MR European derived Saccharomyces cerevisiae colonisation of New Zealand vineyards aided by humans. FEMS Yeast Res. 16, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hewitt SK, Donaldson IJ, Lovell SC & Delneri D Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS One 9, e92203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hose J et al. Dosage compensation can buffer copynumber variation in wild yeast. Elife 4, 1–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krogerus K, Preiss R & Gibson B A unique Saccharomyces cerevisiae × Saccharomyces uvarum hybrid isolated from norwegian farmhouse beer: Characterization and reconstruction. Front. Microbiol 9, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okuno M et al. Next-generation sequencing analysis of lager brewing yeast strains reveals the evolutionary history of interspecies hybridization. DNA Res. 1, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scannell DR et al. The Awesome Power of Yeast Evolutionary Genetics: New Genome Sequences and Strain Resources for the Saccharomyces sensu stricto Genus. G3 1, 11–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skelly DA et al. Integrative phenomics reveals insight into the structure of phenotypic diversity in budding yeast. Genome Res. 23, 1496–1504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strope PK et al. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 125, 762–774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van den Broek M et al. Chromosomal copy number variation in Saccharomyces pastorianus is evidence for extensive genome dynamics in industrial lager brewing strains. Appl. Environ. Microbiol 81, 6253–6267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yue JX et al. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat. Genet 49, 913–924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng DQ et al. Genome sequencing and genetic breeding of a bioethanol Saccharomyces cerevisiae strain YJS329. BMC Genomics 13, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bergström A et al. A high-definition view of functional genetic variation from natural yeast genomes. Mol. Biol. Evol 31, 872–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akao T et al. Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7. DNA Res. 18, 423–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Almeida P et al. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol. Ecol 24, 5412–5427 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Baker E et al. The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeasts. Mol. Biol. Evol 32, 2818–2831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langdon QK, Peris D, Kyle B & Hittinger CT sppIDer: A Species Identification Tool to Investigate Hybrid Genomes with High-Throughput Sequencing. Mol. Biol. Evol 35, 2835–2849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liti G et al. Population genomics of domestic and wild yeasts. Nature 458, 337–341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liti G et al. High quality de novo sequencing and assembly of the Saccharomyces arboricolus genome. BMC Genomics 14, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peris D et al. Biotechnology for Biofuels Hybridization and adaptive evolution of diverse Saccharomyces species for cellulosic biofuel production. Biotechnol. Biofuels 10, 1–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Teytelman L et al. Impact of Chromatin Structures on DNA Processing for Genomic Analyses. PLoS One 4, e6700 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki R & Shimodaira H Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006). [DOI] [PubMed] [Google Scholar]
- 79.McKenna A et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou X et al. In Silico Whole Genome Sequencer and Analyzer ( iWGS ): a Computational Pipeline to Guide the Design and Analysis of de novo Genome Sequencing Studies. G3 6, 3655–3662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Foury F, Roganti T, Lecrenier N & Purnelle B The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 440, 325–331 (1998). [DOI] [PubMed] [Google Scholar]
- 83.Sulo P et al. The evolutionary history of Saccharomyces species inferred from completed mitochondrial genomes and revision in the ‘yeast mitochondrial genetic code’. DNA Res. 24, 571–583 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peris D et al. Molecular Phylogenetics and Evolution Mitochondrial introgression suggests extensive ancestral hybridization events among Saccharomyces species. Mol. Phylogenet. Evol 108, 49–60 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Johnson MG et al. HybPiper: Extracting Coding Sequence and Introns for Phylogenetics from High- Enrichment. Appl. Plant Sci 4, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kearse M et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leigh JW & Bryant D POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol 6, 1110–1116 (2015). [Google Scholar]
- 88.Clement M, Snell Q, Walke P, Posada D & Crandall K TCS: estimating gene genealogies. in Proceedings 16th International Parallel and Distributed Processing Symposium 7 pp (IEEE, 2002). doi: 10.1109/IPDPS.2002.1016585 [DOI] [Google Scholar]
- 89.Librado P & Rozas J DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009). [DOI] [PubMed] [Google Scholar]
- 90.Walther A, Hesselbart A & Wendland J Genome Sequence of Saccharomyces carlsbergensis, the World’s First Pure Culture Lager Yeast. G3 4, 783–793 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Csardi G & Nepusz T The igraph software package for complex network research. InterJournal 1695, 1–9 (2006). [Google Scholar]
- 93.Opulente DA et al. Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol. 16, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kumar S, Stecher G, Li M, Knyaz C & Tamura K MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol 35, 1547–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stamatakis A RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Letunic I & Bork P Interactive tree of life ( iTOL ) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jombart T adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008). [DOI] [PubMed] [Google Scholar]
- 98.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag New York, 2009). [Google Scholar]
- 99.Pfeifer B & Wittelsbuerger U Package ‘PopGenome ‘. (2015). doi: 10.1111/rssb.12200 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genomic contribution comparison of Muri and WLP351.
Modified sppIDer plot, where the y-axis is estimated ploidy, rather than coverage, for the S. cerevisiae (50%) × S. eubayanus (5%) × S. uvarum (45%) strains Muri61 and WLP351.
Figure S2. Summary of total genomic coverage and shared translocations.
The minimum and maximum normalized coverage of all strains that contain each chromosome are shown as colored bars. Darker chromosomes mean that chromosome is present in more strains. Vertical dotted lines represent translocations that are shared in at least four strains, including between hybrid types. The color of the line represents the reciprocal species. (a) Only lager strains and translocations found only in lagers. (b) All 122 hybrids and interspecies translocations.
Figure S3. Phylogenomic trees for S. kudriavzevii with strains labeled.
(a) Phylogeny identical to Figure 2a with strains labeled. (b) Phylogeny identical to Figure S4 with strains labeled. Newick files are available as Files S1 & S2.
Figure S4. Phylogenomic and population placement of hybrids with minor S. kudriavzevii contributions.
(a) Phylogenomic tree built with 36 strains and 12,424 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. (b) PCA using whole genome data for European S. kudriavzevii strains and all major contributor hybrids. (c) PCA using a reduced genome (67%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S3, Table S2, and File S2.
Figure S5. Phylogenomic trees for S. eubayanus with strains labeled
(a) Phylogeny identical to Figure 2b with strains labeled. (b) Phylogeny identical to Figure S6 with strains labeled. Newick files available as Files S3 & S4.
Figure S6. Phylogenomic and population placement of hybrids with minor S. eubayanus contributions.
(a) Phylogenomic tree built with 112 strains and 69,631 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. Long branches are biased by the extensive missing data in hybrids with very small contributions from S. eubayanus. (b) PCA using whole genome data for Holarctic S. eubayanus strains and all major contributor hybrids. (c) PCA using a reduced genome (25%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S5, Table S2, and File S4.
Figure S7. Phylogenomic trees for S. uvarum with strains labeled.
(a) Phylogeny identical to Figure c with strains labeled. (b) Phylogeny identical to Figure S8 with strains labeled. Newick files are available as Files S5 & S6.
Figure S8. Phylogenomic and population placement of hybrids with minor S. uvarum contributions.
(a) Phylogenomic tree built with 69 strains and 36,541 SNPs from regions of the genome that exist in at least one minor contributing hybrid. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1A. (b) PCA using whole genome data for Holarctic S. uvarum strains and all major contributor hybrids. (c) PCA using a reduced genome (84%) but including additional minor hybrids. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S7, Table S2, and File S6.
Figure S9. Phylogenomic tree for full S. cerevisiae analysis with strains labeled. Phylogeny identical to Figure 2d with strains labeled. A Newick file is available as File S7.
Figure S10. Phylogenomic and population placement of lagers within the Ale/Beer1 clade.
(a) Phylogenomic tree built with 267 strains and 21,953 SNPs from the whole genome. The total number of Frohberg strains was down-sampled to match the same number of Saaz strains. The tree was rooted with the Wine strain DBVPG1106. Bootstrap support values >70% are shown as gray dots. Branch colors represent origin of isolation. The inner colors correspond to origin or population. Outer stacked bar plots show the genomic content for each of the hybrids; species colors match Figure 1a. (b) PCA using whole genome data for Ale/Beer1 strains, all Saaz strains, and the down-sampled set of Frohberg strains. The two lineages of lager strains form separate groups, but they do not cluster with any described geographical lineage of the Ale/Beer1 clade. Pure S. cerevisiae Ale/Beer1 strains outside of the labeled lineages are unplaced, including a cluster of Stout strains, Wheat strains, and mosaic strains that our analyses suggest share the most ancestry with lager-brewing yeasts. (c) PCA using all lager strains. The low diversity in the Frohberg lager strains drives PC1, which led us to balance the dataset by down-sampling this lineage. Phylogenies with strain names, Newick formatted files, and data frames used to build PCAs are available as Figure S11, Table S2, and File S8.
Figure S11. Phylogenomic tree for Ale/Beer1 S. cerevisiae analysis with strains labeled. Phylogeny identical to Figure S10 with strains labeled. A Newick file is available as File S8.
Figure S12. 1:1:1:1 orthologs present in hybrid genomes.
(a) Stacked bar chart of all 1:1:1:1 orthologs present in hybrids. Strains are sorted from most to least ortholog content. Completeness of the ortholog set from the species that contributed the most (b) or least (c) orthologs to the strains. Strains are ordered independently in all panels.
Figures S13. Complete de novo genome assembly for all strains.
Total assembled genome for each strain. Regions are colored by which parent could be assigned in the de novo assembly based on the sppIDer results. “Multi” are regions where reads from many species mapped at high coverage. “Unmapped” are novel regions assembled from reads that do not map to parent reference genomes. For each assembly, contigs are ordered from largest to smallest from left to right.
Figure S14. Mitochondrial genome haplotype network.
Six mitochondrial genes were concatenated in 364 wild Saccharomyces strains and interspecies hybrids and used to build a TCS88 phylogenetic network. Haplotype classification is provided in Table S9. Haplotypes are represented by circles, and circle size is scaled according to the haplotype frequency. Pie charts show the frequency of haplotypes based on species or hybrid designation. The number of mutations separating each haplotype are indicated by lines on the edges connecting the haplotype circles.
Figure S15. Labeled (in turquoise) haplotype networks for PAD1 and FDC1. Edge numbers are the number of amino acid changes. Networks correspond to those used in Figure 4 for the amino acid sequences of (a) Fdc1 and (b) Pad1. (b) A different haplotype network orientation of Figure 4E that increases the visibility of each community and haplotype. Table S9 contains the key to which strains belong to which haplotype.
File S1. Newick formatted file of the S. kudriavzevii phylogeny with major hybrids.
Table S2. PCA analyses.
Table S1. All hybrids and their parent contributions.
File S3. Newick formatted file of the S. eubayanus phylogeny with major hybrids.
File S2. Newick formatted file of the S. kudriavzevii phylogeny with minor hybrids.
File S4. Newick formatted file of the S. eubayanus phylogeny with minor hybrids.
File S5. Newick formatted file of the S. uvarum phylogeny with major hybrids.
File S6. Newick formatted file of the S. uvarum phylogeny with minor hybrids.
File S7. Newick formatted file of the S. cerevisiae phylogeny with all strains analyzed.
File S8. Newick formatted file of the S. cerevisiae phylogeny of just the Ale/Beer1 clade.
Table S3. Results of Fisher's Exact Test and Bonferroni correction of mitochondrially localized genes.
mtInteracting = nuclear-encoded but mitochondrially localized gene.
Table S4. Summary of number of 1:1:1:1 orthologs present in each sub-genome.
Table S6. Brewing relevant gene summaries.
"-" Indicates when HybPiper failed to recover and assemble genes for this group or that these assemblies failed our length and coverage cutoffs.
Table S5. GO term results of genes found in novel regions of the de novo assembled genomes.
Table S7. Metadata for all strains newly sequenced in this study.
The “New hybrid” column denotes hybrid genome sequences that are newly published in this study.
Scer = S. cerevisiae, Spar = Saccharomyces paradoxus, Smik = Saccharomyces mikatae, Skud = S. kudriavzevii, Suva = S. uvarum, and Seub = S. eubayanus.
Table S8. Published data accession information.
Table S9. Haplotype key for mitochondrial genomes, PAD1, and FDC1.
Dataset A only includes strains where 15S rRNA could be assembled, while Dataset B has 15S rRNA removed.
Table S10. Regions used for minor contribution analyses.
Data Availability Statement
References and accession numbers for the published data used can be found in Table S8. Short-read data newly published here is available through the NCBI SRA database under the accession number PRJNA522928. Custom R and Python scripts used for this publication can be found on GitHub (https://github.com/qlangdon/hybrid-ferment-invent).